
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
S-Allylmercaptocysteine induces G2/M phase arrest and apoptosis via ROS-mediated p38 and JNK signaling pathway in human colon cancer cells in vitro and in vivo†
Xiaosong Zhua,
Xiaoyan Jiangc,
Chonggang Duand,
Ang Lia,
Yueyue Suna,
Qiuchen Qia,
Yan Liua,
Siying Li*a and
Zhongxi Zhao *ab
*ab
aSchool of Pharmaceutical Sciences, Shandong University, 44 West Wenhua Road, Jinan, Shandong 250012, P. R. China. E-mail: zxzhao@sdu.edu.cn; uszxzhao@gmail.com; li-siying@hotmail.com; Fax: +86-531-88382548; Tel: +86-531-88382187
bShandong Provincial Key Laboratory of Mucosal and Transdermal Drug Delivery Technologies, Shandong Academy of Pharmaceutical Sciences, 989 Xinluo Street, Jinan, Shandong 250101, P. R. China
cDepartment of Pharmacy, Qilu Hospital of Shandong University, 107 West Wenhua Road, Jinan 250012, China
dShandong Key Laboratory of Chemical Drugs, Shandong Academy of Pharmaceutical Sciences, Jinan 250101, China
First published on 20th October 2017
Abstract
S-Allylmercaptocysteine (SAMC), a water-soluble organosulfur garlic derivative, has exhibited potential antitumor activity in various malignancies. Here we explored the effects of SAMC on colon cancer in vitro and in vivo and further elucidated the underlying molecule mechanisms. In this study, we found that SAMC potently suppressed cell viability and induced G2/M phase arrest and apoptosis in colon cancer HCT116 cells. Further studies showed that reactive oxygen species (ROS) attributed to SAMC-induced cell cycle arrest and apoptosis, which was attenuated by N-acetyl cysteine (NAC), an ROS scavenger. Moreover, we found that SAMC activated p38 and c-Jun N-terminal kinase (JNK) signal pathway, which was also blocked by NAC. Finally, SAMC administration in mice effectively delayed the growth of HCT116 xenografts without signs of toxicity. In conclusion, SAMC induced cell cycle G2/M phase arrest and apoptosis via ROS-mediated p38 and JNK signaling pathway in colon cancer HCT116 cells. In light of these results, SAMC may be a promising agent for anticancer therapy against colon cancer.
1. Introduction
Colorectal cancer is considered the third most common malignant disease and remains a significant cause of morbidity and mortality worldwide.1,2 Factors promoting colon cancer are low physical activity, cigarette smoking, alcohol consumption, diabetes mellitus, ulcerative colitis, some hereditary cases and so on.3Reactive oxygen species is a product of normal metabolism and xenobiotic exposure, and plays a crucial role in normal biochemical functions and abnormal pathological processes.4 ROS-induced oxidative stress is involved in a number of physiological and pathological processes, including cancer. Accumulating evidence has suggested that cancer cells exhibited higher ROS levels than that in normal cells; and excessive ROS can induce apoptosis and necrosis.5,6 Tumor cells have limited capacity to deal with the elevated ROS level, which makes them vulnerable to oxidative stress.7 Thus oxidative stress is linked to the onset of colon carcinogenesis. It has been demonstrated that ROS plays a key role in microbiota-linked colon cancer via inflammation pathways, oxidative stress induction, and anti-oxidative defense regulation.8 Apoptotic cell death induced by ROS is mediated by the activation of MAPK family members, including p38 and JNK.9
Conventional chemotherapy and radiotherapy are commonly used in the treatment of colorectal cancer, which are often accompanied with serious side effects. Therefore, it is urgent to identify alternative drugs to chemotherapeutics with lower toxicity and substantial antitumor effect. Garlic, used traditionally as a common food additive, has been suggested as an anticancer agent for several decades. Epidemiological investigations have provided evidence that regular consumption of garlic and garlic products could reduce the risk of colon cancer.10 The beneficial effects of garlic are attributed to its organosulfur compounds. SAMC is one of the water-soluble sulfur compounds present in aged garlic extract.11 Previous studies have shown that SAMC is effective in inhibiting cell growth and inducing apoptosis of various cancer cells.12,13 SAMC is also shown to have anti-oxidative and anti-inflammatory properties.14,15
It has been reported that the anti-cancer effects of organosulfur compounds were attributed to interactions with ROS.16 However, the role of ROS in SAMC-induced apoptosis has not been investigated. Thus, in the present study, we aimed to investigate the inhibitory effect of SAMC on human colon cancer growth both in vitro and in vivo and to determine the underlying mechanisms.
2. Materials and methods
2.1. Materials
SAMC was synthesized and purified in our laboratory with a modified procedure as previously reported.17 The purity is 96% and was determined by HPLC.18 SAMC was freshly prepared as a stock solution in phosphate-buffered saline (PBS) for the in vitro assay and was suspended in 10% (w/v) L-dextrose, 1% (w/v) gum Arabic (Sigma-Aldrich, St. Louis, MO) for application in mice. Cisplatin (DDP) was purchased from QILU Pharmaceutical Co., LTD. (Jinan, China) and was suspended in normal saline for application in mice. Propidium iodide (PI), 4′,6-diamidino-2-phenylindole (DAPI) and sulforhodamine B (SRB) were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). Dichlorofluorescein diacetate (DCFH-DA) was purchased from Invitrogen Biotechnology (California, USA). NAC was purchased from the Beyotime Institute of Biotechnology (Beijing, China).2.2. Cell culture
Human colon cancer cell line HCT116 was obtained from ATCC (Manassas, VA USA) and maintained in RPMI-1640 supplemented with 10% fetal bovine serum, 100 U ml−1 of penicillin and 100 μg ml−1 of streptomycin and maintained in a humidified incubator of 5% CO2 at 37 °C. Cells were harvested by brief incubation in 0.02% ethylenediamine tetraacetic acid-phosphate-buffered saline.2.3. Cell viability assay
Cell viability was measured by SRB method.19 Cancer cells (5 × 103 per well) seeded in 96-well plates were exposed to increasing concentrations of SAMC for the indicated time. The treated cells were then fixed with 10% trichloroacetic acid for 1 h at 4 °C; the 96-well plates were washed three times with distilled water and allowed to dry in the air. Each well received 100 μl of SRB solution and the staining was completed at room temperature for 15 min. The SRB stain solution was removed by washing the plates quickly with 1% (v/v) acetic acid three times, and the plates were dried in the air. The dried materials in each well were solubilized by adding 200 μl of 10 mM unbuffered Tris base (pH 10.5). The cell viability was detected by measuring the absorbance at 540 nm on a plate reader (Safire2, TECAN, France). All experiments were repeated at least three times.2.4. DAPI staining
DNA fragmentation during cell apoptosis can be detected by DAPI staining. Treated HCT116 cells cultured on cover glasses in 12-well plates were washed with PBS and then fixed with cold methanol/acetone (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, store at −20 °C) for 5 min at room temperature. The solution was removed and washed with PBS, and then incubated with the DAPI solution for 10 min. Fluorescent cells were observed under a fluorescence microscope (Olympus, Tokyo, Japan).
:1, store at −20 °C) for 5 min at room temperature. The solution was removed and washed with PBS, and then incubated with the DAPI solution for 10 min. Fluorescent cells were observed under a fluorescence microscope (Olympus, Tokyo, Japan).
2.5. Cell cycle analysis
HCT116 cells were pretreated with or without 5 mM NAC for 1 h, and followed by 400 μM SAMC for 24 h. Then cells were harvested and fixed in cold 70% ethanol at −20 °C overnight. Next, cells were incubated with RNase and stained with PI (50 μg ml−1) for 30 min. Cell cycle distribution was determined using a Cytomics FC500 Flow Cytometer (Beckman Coulter, USA). The percentages of cells in G1, G2, and S phases were analyzed by using ModFit LT software 3.0 (Varity Software House).2.6. Annexin V and PI staining assay
HCT116 cells seeded in six-well plates (1 × 105 per well) were with or without NAC (5 mM) for 1 h and then cultured in the presence or absence of 400 μM SAMC for 24 h. Cells were harvested and washed with cold PBS. Cell surface phosphatidylserine in apoptotic cells was quantitatively estimated using annexin-V FITC along with PI based on the manufacturer's instructions (Invitrogen, Carlsbad, USA). FITC fluorescence was analyzed using the Cytomics FC500 Flow Cytometer (Beckman Coulter, USA).2.7. Measurement of reactive oxygen species generation
Generation of ROS was measured by flow cytometry as described previously.20 Briefly, cells were pretreated with or without 5 mM NAC for 1 h and then cultured in the presence or absence of 400 μM SAMC for 4 h. After the incubation, the cells were harvested, washed, and suspended in serum-free medium containing 10 μM of DCFH-DA at 37 °C for 20 min in the dark. Cells were washed and resuspended in PBS, and the induction of ROS was examined by flow cytometry.2.8. Western blot analysis
Western blotting was performed to analyze the contents of apoptotic and mitogenic molecules in cancer cells. In the in vitro assay, cells were harvested and the lysates were fractionated using 10% SDS-PAGE. For the HCT116 xenografts, cancer tissues were dispersed in PBS mechanically. The supernatant liquids were then collected and the total proteins were determined. Cell lysates were separated using 10% SDS-PAGE and electro-transferred onto the polyvinylidene difluoride membrane. After incubation in blocking buffer (1× TBS, 0.1% Tween-20 and 5% w/v dry nonfat milk) at 4 °C, the membranes were incubated with the desired primary antibody, followed by incubation with the appropriate peroxidase-conjugated secondary antibody. The primary antibodies included those to cleaved PARP (5625), cleaved caspase-9 (7237), phospho-p38 (4511), p38 (8690), ERK1/2 (4695), phospho-ERK1/2 (4370), JNK (9252), phospho-JNK (4668, cell signaling), Akt (sc-8312), phospho-Akt/Sre-474 (sc-135651), cyclin B1 (1495-1, epitomics), Bcl-2 (ab32124), Bax (ab32503), p53 (ab32389), PCNA (ab92552), GAPDH (ab9482, Abcam) and β-actin (TA-09, ZSGB-BIO).2.9. Animal tumor models and treatment
Female BALB/c nude mice (16 g, aged 5–6 weeks) were purchased from Institute of Laboratory Animal Sciences, Cams&Pumc (Beijing, China, SCXK 2014-0004), and housed under standard conditions (12:12 h light/dark cycle at 25 ± 2 °C and 40–70% humidity) in a specific pathogen free environment. The mice had free access to standard commercial diet and water. All animal study procedures were complied with the institutional guidelines of the Animal Care and Use Committee of Shandong University and approved by the same Committee. The xenografts were established via the subcutaneous inoculation of HCT116 cells (1 × 107 cells per mouse) into the armpit of one mouse. After two weeks of growth, the cancer tissues were cut into pieces with the dimensions of 1.5 × 1.5 × 1.5 mm3 and inoculated subcutaneously into the right armpit of the mice with a puncture needle. When tumor volume reached approximately 100 mm3, mice were randomly divided into five groups (n = 6): vehicle control, SAMC (40 mg kg−1), SAMC (60 mg kg−1), SAMC (80 mg kg−1), and cisplatin (5 mg kg−1). Cisplatin was used as the positive control and was injected intraperitoneally once a week. SAMC and drug vehicle were given by intraperitoneal injection daily for 30 days. Tumor volume and body weight were measured every four days. The volume was calculated using the formula: V = 1/2 × L × W2, where length (L) and width (W) were determined in millimeters. The tumor inhibition rate was calculated using the following formula:| (%) = (1 − tumor volume of treatment group/tumor volume of control group) × 100%. |
After whole blood was collected from eye canthus in each animal under general anesthesia, all animals were euthanized by cervical dislocation. The weight of tumor tissue were measured and then frozen in −80 °C for Western blotting.
The blood was centrifuged and serum was obtained for analysis of AST, ALT, creatinine and BUN. This analysis was performed using commercial kits (Nanjing Jiancheng Bioengineering Institute, China).
2.10. Statistical analysis
The data were described as the mean ± SEM and analyzed using one-way ANOVA. Differences with p values <0.05 were considered statistically significant. Statistical analysis was performed with SPSS/Win13.0 software (SPSS, Inc., Chicago, IL).3. Results
3.1. SAMC inhibits cell proliferation and induces cell cycle arrest in HCT116 cells
To assess the anti-proliferative activity of SAMC, HCT116 cells were exposed to increasing concentrations of SAMC for 24 h and cell survival was detected by SRB assay. As shown in Fig. 1A, the survival of HCT116 cells was markedly suppressed by SAMC in a dose-dependent manner. Statistical analysis indicated that the IC50 value was 314.7 μM. It is well documented that cancer-related activities of organic sulfur-containing compounds from Allium species are associated with ROS generation.16 Therefore, we explored whether ROS was associated with SAMC-induced growth inhibition. Cells were treated with SAMC in the presence or absence of NAC, a ROS scavenger. As a result, anti-proliferative effect of SAMC was almost completely abolished by NAC in HCT116 cells (Fig. 1B).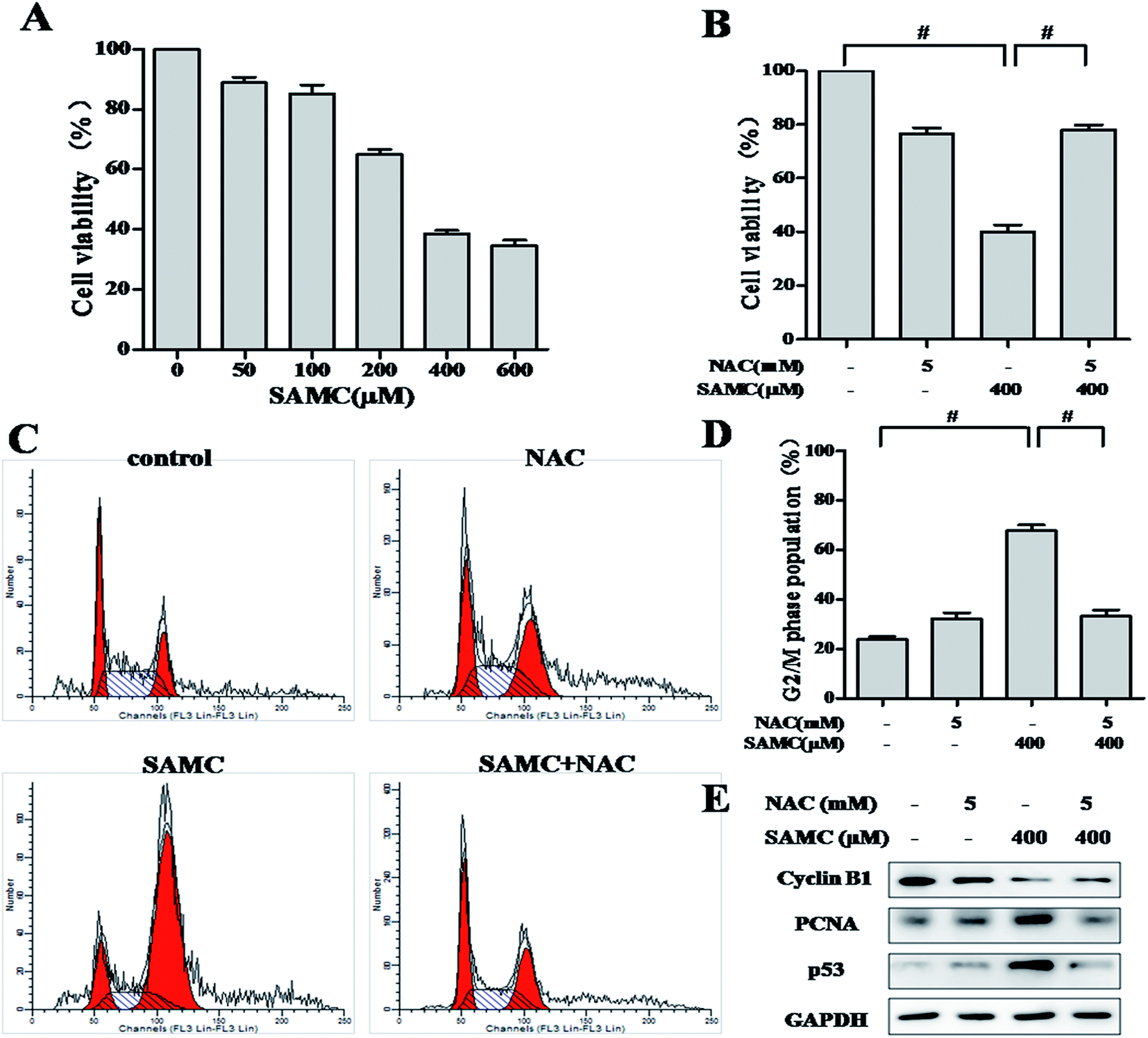 | ||
| Fig. 1 Effect of SAMC on cell growth and cell cycle distribution in human colon cancer cells. (A) The inhibition of SAMC on HCT116 cell growth was estimated by the SRB assay. Cells were exposed to various concentrations of SAMC for 24 h, followed by SRB assay. (B–E) HCT116 cells were pretreated with or without 5 mM NAC for 1 h, and then followed by 400 μM SAMC for 24 h. (B) Cell proliferation was measured by SRB assay in the absence or presence of NAC. (C) Arrest of cell cycle in G2 phase in HCT116 cells. Cancer cells were exposed to SAMC with or without NAC for 24 h and subjected to flow cytometry analysis. (D) Bar diagram shows the percentage of cells in G2 phase of cell cycle. (E) Effects of SAMC on the expression of various cell cycle-related proteins in human HCT116 colon cancer cells. Western blotting was performed for cyclin B1, PCNA and p53. GAPDH was used as a loading control. Data represent the mean ± SEM of three independent experiments. #p < 0.01, compared with the indicated group. | ||
Given that growth inhibition of cancer cell is usually associated with cell cycle arrest, we thus examined the effect of SAMC on cell cycle in HCT116 cells. As shown in Fig. 1C, treatment with 400 μM SAMC resulted in a distinct accumulation of cells in G2/M phase as compared with controls. The percentage of G2/M phase was increased from 23.8 ± 1.3% to 67.8 ± 3.4%. As expected, 5 mM NAC dramatically blocked cell cycle arrest induced by SAMC (Fig. 1D). To explore the mechanism underlying SAMC-mediated G2/M arrest, we investigated the effect of SFN on the expression of cell cycle-related proteins, including cyclin B1, PCNA and p53. As shown in Fig. 1E, SAMC treatment at the indicated concentration caused a decreased expression of cyclin B1, whereas PCNA expression and p53 expression were increased by SAMC treatment. Furthermore, we found that blocking ROS generation prevented SAMC-induced G2/M cell cycle arrest and down-regulation of cell cycle-related protein cyclin B1 and decreased the expression of PCNA and p53. These results indicate that ROS plays an important role in SAMC-induced cell growth inhibition and cell cycle arrest in HCT116 cells.
3.2. SAMC induces ROS production and apoptosis in HCT116 cells
To investigate whether SAMC could trigger ROS generation in HCT116 cells, we performed DCFH-DA staining assays to analyze the change of intracellular ROS. As compared with controls, ROS was significantly induced in HCT116 cells when treated with 400 μM SAMC for 4 h. The mean fluorescence intensity was increased from 56.6 ± 3.5 to 112.7 ± 4.3 (p < 0.05). In addition, we found that pretreatment with NAC caused a notable decrease of ROS production induced by SAMC (Fig. 2A).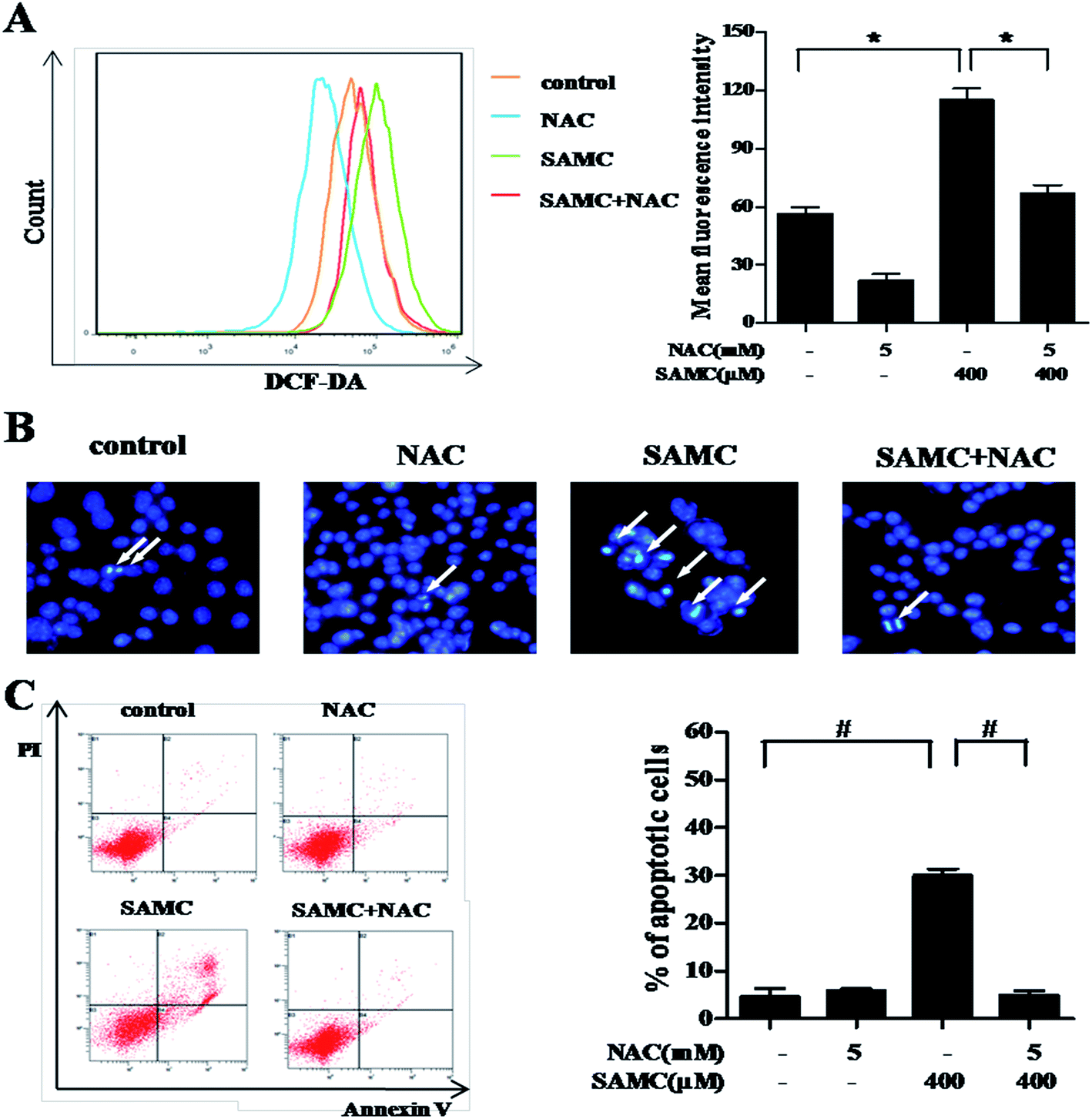 | ||
| Fig. 2 Effect of SAMC on ROS production and apoptosis in human colon cancer cells. (A) Cells were pretreated with or without NAC (5 mM) for 1 h and then cultured in the presence or absence of 400 μM SAMC for 4 h. DCFH-DA staining was used to evaluate ROS production. The fluorescence intensity of ROS-positive cells is shown as indicated in HCT116 cells. Pretreatment with NAC attenuates SAMC-induced apoptosis. Cells were pretreated with NAC (5 mM) for 1 h, followed by 400 μM SAMC for 24 h. (B) Cells were stained with DAPI to investigate apoptotic-like nuclear morphology. White arrows indicate condensed nuclei. (C) Flow cytometric analysis was carried out by double-staining with annexin V-FITC and PI. All assays were performed in triplicate. *p < 0.05 and #p < 0.01 compared with the indicated group. | ||
Next, we attempted to test whether ROS was involved in SAMC-induced apoptosis. Cells were co-treated with SAMC for 24 h with or without 5 mM NAC pretreatment for 1 h. The cell apoptosis was detected by DAPI staining assay and flow cytometry with annexin V/PI staining. After treatment with 400 μM SAMC, HCT116 cells exhibited typical apoptotic features, such as nuclear fragmentation, as evidenced by the white arrows in Fig. 2B. However, co-treatment with NAC effectively prevented these morphological changes. Furthermore, flow cytometric analysis showed that apoptosis was attenuated by NAC pretreatment compared with the effects on apoptosis in response to SAMC treatment alone (Fig. 2C). Collectively, these data further support the role of ROS in SAMC-induced apoptosis in HCT116 cells.
3.3. SAMC modulates the activities of major signaling pathways in HCT116 cells through a ROS-dependent manner
To explore the mechanisms underlying the antitumor activity of SAMC and determine the role of ROS, we analyzed apoptosis-related proteins and MAPK pathways with SAMC treatment. HCT116 cells were treated with SAMC in the presence or absence of NAC. As shown in Fig. 3A, the expression level of Bax was notably up-regulated, while the expression of Bcl-2 was down-regulated after SAMC treatment. Further analysis showed increased levels of cleaved PARP and cleaved caspase 9 following SAMC treatment. Moreover, we found that SAMC enhanced phosphorylation of JNK and p38 in HCT116 cells (Fig. 3B). However, we did not find a notable effect of SAMC on phosphorylation of Erk (data not shown). Importantly, blocking ROS generation by NAC recovered these changes. These data suggest that ROS generation is involved in SAMC-induced JNK MAPK and p38 MAPK activation.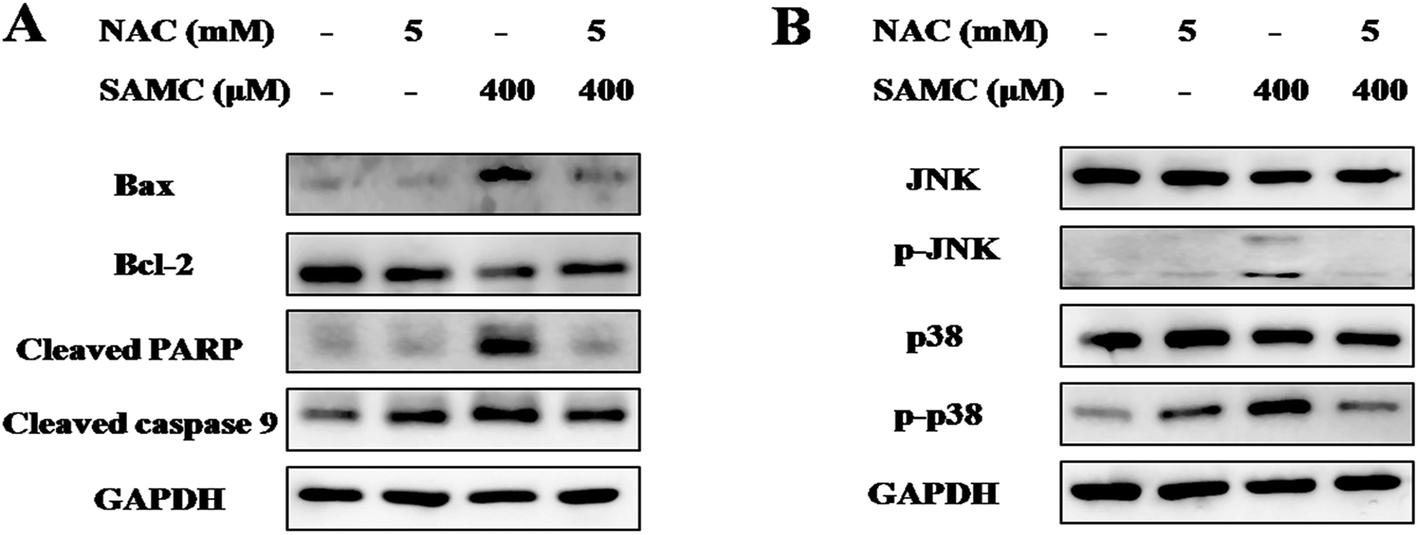 | ||
| Fig. 3 Effect of SAMC on the activities of major signaling pathways in colon cancer cells. HCT116 cells were pretreated with or without 5 mM NAC for 1 h, and then followed by 400 μM SAMC for 24 h. Cell lysates were collected and subjected to Western blotting for apoptosis related proteins (A) and MAPK signals (B). | ||
3.4. SAMC inhibits xenograft tumor growth
Given in vitro inhibitory effect of SAMC on colon cancer cell growth, we next determined the effect of SAMC on the growth of xenograft colon tumors in nude mice. As shown in Fig. 4A, mice treated with SAMC or DDP slowdown the tumor growth compared with control mice. At a dose of 80 mg kg−1, SAMC suppressed HCT116 xenograft tumor growth by 58.1% (p < 0.01) on day 30. At doses of 60 and 40 mg kg−1, SAMC inhibited tumor growth by 54.2% and 50.7%, respectively (p < 0.05). Tumor volume was lower in DDP-treated mice, which inhibited HCT116 tumor volume by 67.7%. However, DDP at this dosage was found to possess toxicity to mice, showing loss of body weight, an indicator of the health of animals. Instead, there is no significant difference in body weight change among the control group and SAMC-treated groups (Fig. 4B). At the end of experiments, the tumors were isolated and weighted. The mean tumor weight was significantly lower in SAMC treated mice as compared with control mice (p < 0.05) (Fig. 4C and D). To quantitatively evaluate the anti-proliferation effect of tumors treated with SAMC, we examined the level of PCNA, a proliferation marker, in tumor sections. As shown in Fig. 4E, treatment with SAMC dramatically reduced the expression of PCNA.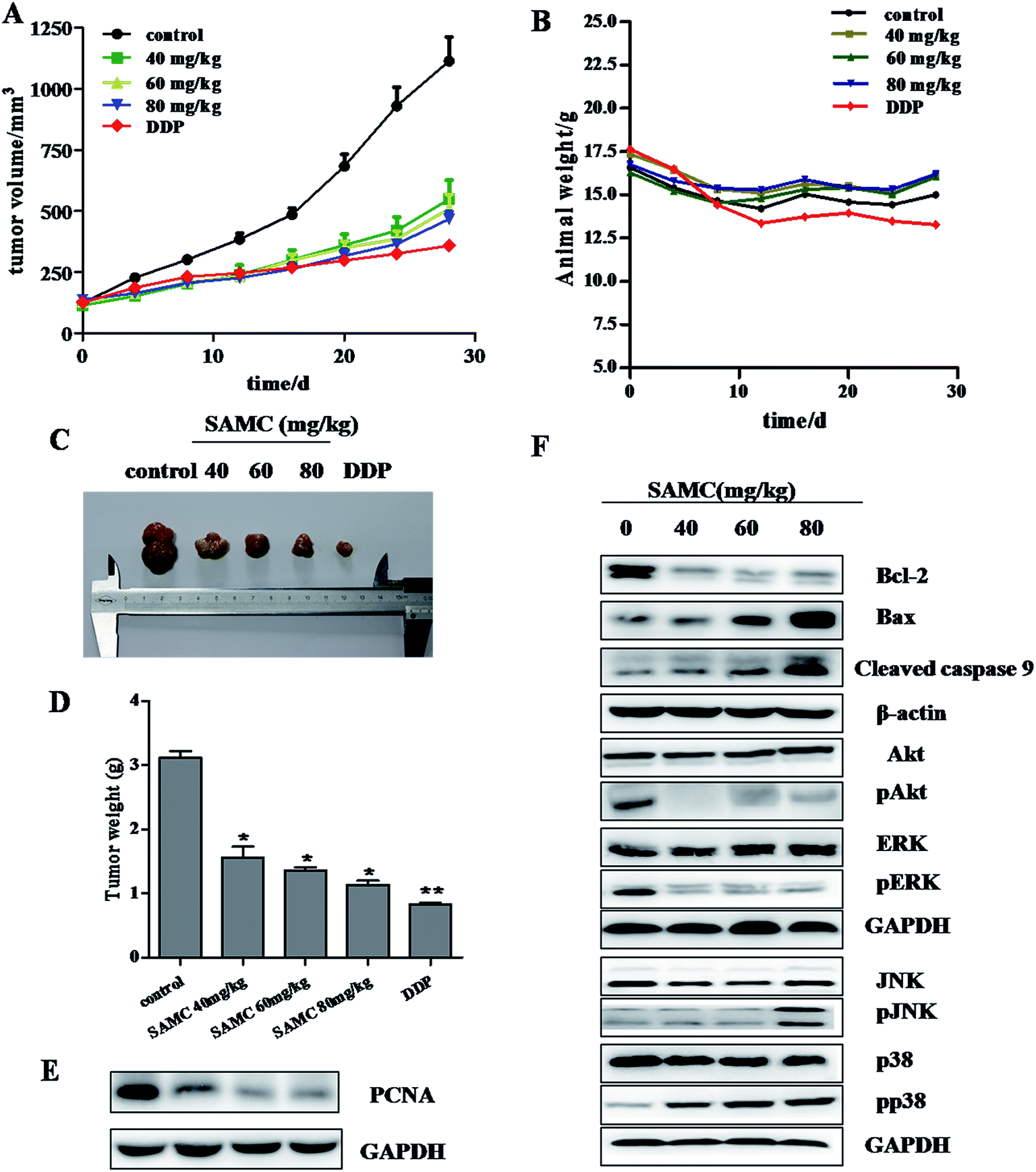 | ||
| Fig. 4 Inhibition of xenograft tumor growth by SAMC. Time courses of tumor growth (A) and animal weight (B) were measured in each group at the indicated time point of various treatments. (C) A representative picture of tumor growth in mice is shown. (D) Bar graphs represent mean of the tumor weight. Data were presented as mean ± SEM. (n = 6/group). *p < 0.05 and **p < 0.01. (E) Western blotting assay determined the expression of PCNA in cancer tissues. (F) SAMC induces apoptosis in vivo. Tumors tissues from representative mice were homogenated, lysed, and then subjected to Western blotting using specific primary antibodies. | ||
To further confirm the regulation of major pathways by SAMC, tumors tissues from mice were homogenated, lysed, and then subjected to Western blotting using specific primary antibodies, including Bcl-2, Bax, cleaved caspase 9, p-Akt, Akt, pERK, ERK, pJNK, JNK, pp38 and p38. Similar to in vitro study, SAMC reduced Bcl-2 expression, increase the expression of Bax and cleaved caspase 9 and activated the MAPK/JNK and MAPK/p38 pathways. Furthermore, we found that SAMC inhibited the activity of PI3K/Akt and MAPK/ERK pathway (Fig. 4F).
The examination of the serum BUN, Cr, ALT and AST was performed to evaluate the toxicity. In DDP-treated mice, high concentrations of serum BUN, Cr, ALT and AST were measured (p < 0.05), indicating that DDP exhibited notable kidney and liver toxicity. However, there was no significant difference in serum BUN, Cr, ALT and AST levels between SAMC-treated and control mice. These observations implicate that the indicated dose of SAMC did not cause significant kidney and liver injury in mice, supporting that it may be a safe and effective agent for colon cancer therapy at least in xenograft tumor models (Fig. 5).
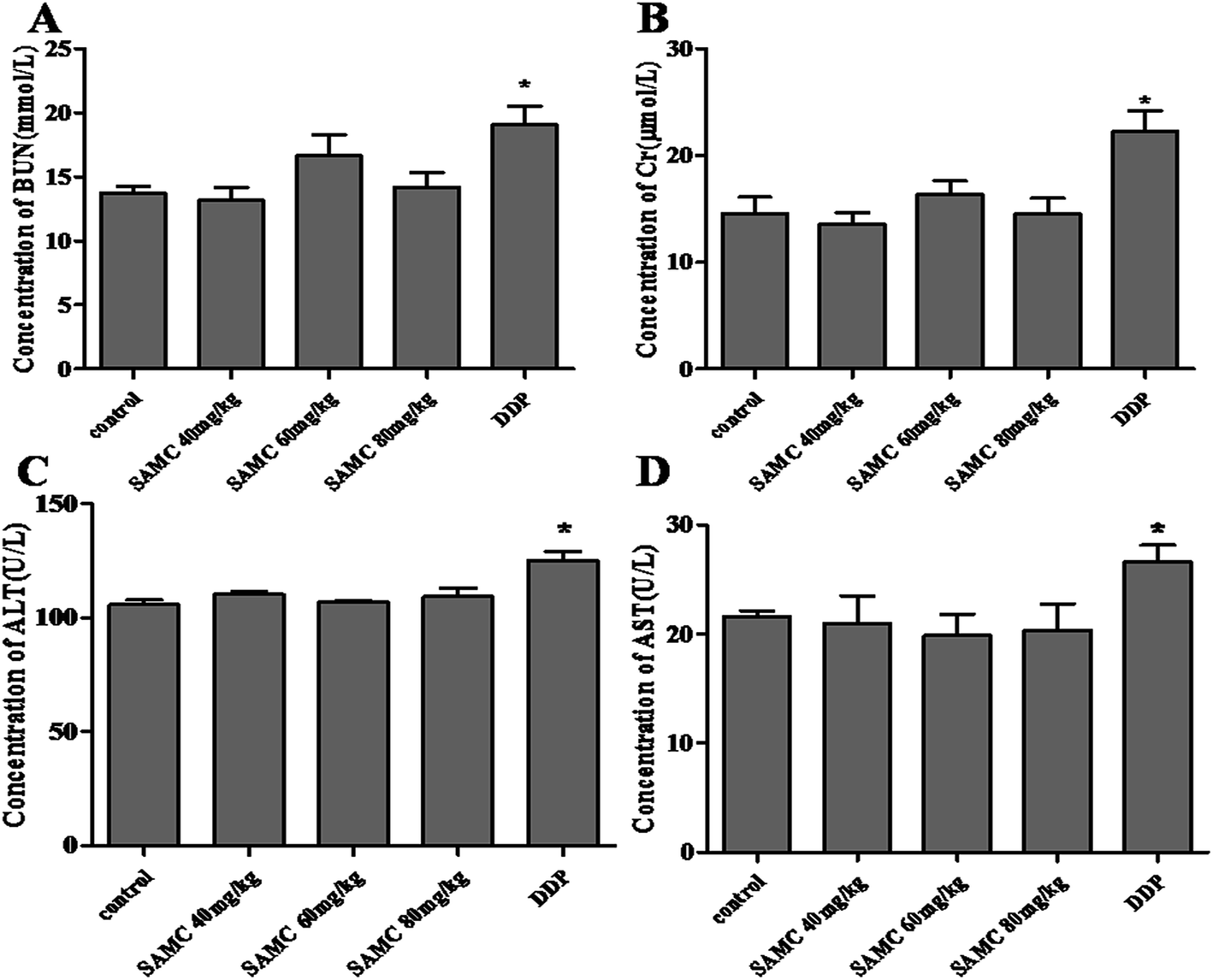 | ||
| Fig. 5 Biochemical serum analysis of SAMC treated mice. Concentration of (A) BUN (mmol L−1), (B) Cr (μmol L−1), (C) ALT (U L−1) and (D) AST (U L−1) in serum of the mice shown as mean ± SEM. *p < 0.05 compared with control. | ||
4. Discussion
A considerable amount of epidemiological studies have indicated that a garlic-rich diet decreases risk of some cancers such as gastric, stomach, and colon.12 Some in vitro and in vivo studies using garlic-derived organosulfur compound, SAMC, have presented encouraging outcome of experimental therapy for colon cancer.21–23 However, the anticancer effects on HCT116 colon cancer cells of SAMC have not been revealed upon our knowledge. In the present study, we undertook a comprehensive analysis of the effect of SAMC on colon cancer using both in vitro and in vivo models. Since generation of ROS plays a vital role in the anticancer activity, we investigated the ROS level in SAMC-treated HCT116 colon cancer cells. Our data showed that SAMC could suppress cell proliferation, cause G2/M phase arrest and induce apoptosis via ROS-mediated p38 and JNK signaling pathway in human HCT116 cells. Moreover, we found that SAMC inhibited the growth of colon cancer in vivo and administration of SAMC in the doses used had no apparent toxic side effects on the experimental mice, through functional studies of liver and kidney.In the cytotoxicity test, SAMC shows a dose-dependent inhibitory effect on the proliferation of HCT116 cells, while pretreatment with NAC resulted in partial rescue of some cells with an increased survival rate. The G2 checkpoint prevents cells from entering mitosis when DNA is damaged, which offers an opportunity for repair and stopping the proliferation of damage cells.24 Our data showed that SAMC-treated colon cancer cells were arrested in G2/M phase, which was accompanied by a decline in the levels of cyclin B1 and an increase in the levels of PCNA and p53. ROS scavenger NAC efficiently abolished SAMC induced G2/M phase cell arrest and inhibited p53 expression in HCT116 cells. It is well-documented that p53 is a vital molecular target in cancer biology because it plays a crucial role in cell cycle arrest and apoptosis.25,26 Collectively, these observations suggest that SAMC may cause cell growth inhibition and G2/M phase arrest by a ROS-dependent pathway.
Apoptosis is crucially involved in killing tumor cells and mitochondria is thought to be the major pathway for apoptosis.27 Mitochondria mediated-apoptosis is mainly regulated by the Bcl-2 family proteins including anti-apoptotic member Bcl-2 and pro-apoptotic member Bax. The balance between the expression levels of pro- and anti-apoptotic proteins is critical for cell survival or cell death. Overexpression of Bax results in mitochondrial cytochrome c release, which triggers the activation of caspase-9 and caspase-3 and subsequently the cleavage of PARP, eventually leading to apoptosis.28 In our studies, cells treated with SAMC caused down-regulation of Bcl-2, up-regulation of Bax as well as activation of PARP and caspase 9, suggesting that SAMC treatment facilitated apoptosis. ROS is considered as a signal mediator for cell death. Accumulating evidence suggests that high levels of ROS can damage cellular components such as DNA and protein, leading to cell apoptosis.29 Previous studies have implicated ROS generation in apoptosis induction by garlic derivatives. For instance, the DADS-induced apoptosis in HL-60 cells was associated with ROS generation.30 One study has implicated ROS production in apoptotic response to DATS in the LNCaP cell line.31 In another study, the ajoene-induced apoptosis in human promyeloleukemic cells was accompanied by generation of ROS and activation of nuclear factor-κB.32 In the present study, SAMC induced a significant increase in ROS generation and pretreatment with ROS inhibitor NAC remarkably reversed SAMC-induced apoptosis. Our results indicated that the induction of ROS might contribute to SAMC-induced apoptosis in human colon cancer cells.
Growing evidence demonstrates that activation of p38 MAPK and JNK plays a critical role in cell growth, survival and death.33,34 Consistent with these reports, SAMC induced significant elevation in the phosphorylation of p38 MAPK and JNK in colon cancer cells. Studies have reported that increased ROS levels promote apoptosis by activating pro-apoptotic signaling molecules, including MAPKs.33 Indeed, we found that apoptosis and p38 MAPK and JNK activation induced by SAMC were markedly attenuated by NAC. These findings suggest that ROS generation is involved in SAMC-induced MAPK activation.
In conclusion, our study identifies antitumor effects of SAMC on colon cancer in vitro and in vivo and the potential molecular mechanisms. We found that SAMC significantly induced G2/M cell cycle arrest and cell apoptosis regulated via ROS-mediated JNK and p38 signaling pathway. We also demonstrated that SAMC significantly decreased tumor growth in mice bearing xenografts without obvious toxicity. This study indicates that SAMC is a novel antitumor drug candidate that has great potential as a promising agent for anticancer therapy in colon cancer.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the funds from National “Major Science and Technology Project–Prevention and Treatment of AIDS, Viral Hepatitis, and Other Major Infectious Diseases” (Grant #2013ZX10005004), Major Project of Science and Technology of Shandong Province (Grant #2015ZDJS04001), Science & Technology Enterprise Technology Innovation Fund of Jiangsu Province (Grant #BC2014172), Small & Medium Enterprise Technology Innovation Project of Lianyungang City (Grant #CK1333).References
- J. Ferlay, I. Soerjomataram, R. Dikshit, S. Eser, C. Mathers, M. Rebelo, D. M. Parkin, D. Forman and F. Bray, Int. J. Cancer, 2015, 136, E359–E386 CrossRef CAS PubMed.
- R. L. Siegel, K. D. Miller, S. A. Fedewa, D. J. Ahnen, R. G. Meester, A. Barzi and A. Jemal, Ca-Cancer J. Clin., 2017, 67, 177–193 CrossRef PubMed.
- C. M. Johnson, C. Wei, J. E. Ensor, D. J. Smolenski, C. I. Amos, B. Levin and D. A. Berry, Canc. Causes Contr., 2013, 24, 1207–1222 CrossRef PubMed.
- C. Zhang, S. Cao, B. P. Toole and Y. Xu, Int. J. Cancer, 2015, 136, 2001–2011 CrossRef CAS PubMed.
- A. D. Kim, K. A. Kang, H. S. Kim, D. H. Kim, Y. H. Choi, S. J. Lee, H. S. Kim and J. W. Hyun, Cell Death Dis., 2013, 4, e750 CrossRef CAS PubMed.
- K. Larocque, P. Ovadje, S. Djurdjevic, M. Mehdi, J. Green and S. Pandey, PLoS One, 2014, 9, e87064 Search PubMed.
- H. Pelicano, D. Carney and P. Huang, Drug Resist. Updates, 2004, 7, 97–110 CrossRef CAS PubMed.
- J. Gagniere, J. Raisch, J. Veziant, N. Barnich, R. Bonnet, E. Buc, M. A. Bringer, D. Pezet and M. Bonnet, World J. Gastroenterol., 2016, 22, 501–518 CrossRef CAS PubMed.
- H. M. Shen and Z. G. Liu, Free Radical Biol. Med., 2006, 40, 928–939 CrossRef CAS PubMed.
- J. T. Pinto, S. Lapsia, A. Shah, H. Santiago and G. Kim, in Nutrition and Cancer Prevention, Springer, 2001, pp. 83–106 Search PubMed.
- J. Imai, N. Ide, S. Nagae, T. Moriguchi, H. Matsuura and Y. Itakura, Planta Med., 1994, 60, 417–420 CrossRef CAS PubMed.
- H. M. Yun, J. O. Ban, K. R. Park, C. K. Lee, H. S. Jeong, S. B. Han and J. T. Hong, Pharmacol. Ther., 2014, 142, 183–195 CrossRef CAS PubMed.
- W. Zhang, H. Xiao and K. L. Parkin, Food Chem. Toxicol., 2014, 68, 1–10 CrossRef PubMed.
- K. Wang, Y. Wang, Q. Qi, F. Zhang, Y. Zhang, X. Zhu, G. Liu, Y. Luan, Z. Zhao, J. Cai, J. Cao and S. Li, Int. Immunopharmacol., 2016, 34, 37–43 CrossRef CAS PubMed.
- J. Pedraza-Chaverri, D. Barrera, P. D. Maldonado, Y. I. Chirino, N. A. Macias-Ruvalcaba, O. N. Medina-Campos, L. Castro, M. I. Salcedo and R. Hernandez-Pando, BMC Clin. Pharmacol., 2004, 4, 5 CrossRef PubMed.
- C. Scherer, C. Jacob, M. Dicato and M. Diederich, Phytochem. Rev., 2009, 8, 349 CrossRef CAS.
- C. Starkenmann, Y. Niclass and M. Troccaz, J. Agric. Food Chem., 2011, 59, 9457–9465 CrossRef CAS PubMed.
- X. Zhu, X. Jiang, A. Li, Y. Sun, Y. Liu, X. Sun, X. Feng, S. Li and Z. Zhao, Biochem. Biophys. Res. Commun., 2017, 491, 821–826 CrossRef CAS PubMed.
- P. Houghton, R. Fang, I. Techatanawat, G. Steventon, P. J. Hylands and C. C. Lee, Methods, 2007, 42, 377–387 CrossRef CAS PubMed.
- A. Cossarizza, R. Ferraresi, L. Troiano, E. Roat, L. Gibellini, L. Bertoncelli, M. Nasi and M. Pinti, Nat. Protoc., 2009, 4, 1790–1797 CrossRef CAS PubMed.
- Y. Zhang, H. Y. Li, Z. H. Zhang, H. L. Bian and G. Lin, Oncol. Lett., 2014, 8, 2591–2596 Search PubMed.
- D. Liang, Y. Qin, W. Zhao, X. Zhai, Z. Guo, R. Wang, L. Tong, L. Lin, H. Chen, Y. C. Wong and Z. Zhong, Cancer Lett., 2011, 310, 69–76 CrossRef CAS PubMed.
- D. Xiao, J. T. Pinto, J.-W. Soh, A. Deguchi, G. G. Gundersen, A. F. Palazzo, J.-T. Yoon, H. Shirin and I. B. Weinstein, Cancer Res., 2003, 63, 6825–6837 CAS.
- G. R. Stark and W. R. Taylor, Methods Mol. Biol., 2004, 280, 51–82 CAS.
- X. Wang, E. R. Simpson and K. A. Brown, Cancer Res., 2015, 75, 5001–5007 CrossRef CAS PubMed.
- M. L. Antony, S. H. Kim and S. V. Singh, PLoS One, 2012, 7, e32267 CAS.
- S. Fulda, L. Galluzzi and G. Kroemer, Nat. Rev. Drug Discovery, 2010, 9, 447 CrossRef CAS PubMed.
- I. M. Ghobrial, T. E. Witzig and A. A. Adjei, Ca-Cancer J. Clin., 2005, 55, 178–194 CrossRef PubMed.
- M. C. Maiuri, E. Zalckvar, A. Kimchi and G. Kroemer, Nat. Rev. Mol. Cell Biol., 2007, 8, 741–752 CrossRef CAS PubMed.
- K. B. Kwon, S. J. Yoo, D. G. Ryu, J. Y. Yang, H. W. Rho, J. S. Kim, J. W. Park, H. R. Kim and B. H. Park, Biochem. Pharmacol., 2002, 63, 41–47 CrossRef CAS PubMed.
- Y. A. Kim, D. Xiao, H. Xiao, A. A. Powolny, K. L. Lew, M. L. Reilly, Y. Zeng, Z. Wang and S. V. Singh, Mol. Cancer Ther., 2007, 6, 1599–1609 CrossRef CAS PubMed.
- V. M. Dirsch, A. L. Gerbes and A. M. Vollmar, Mol. Pharmacol., 1998, 53, 402–407 CAS.
- P. D. Ray, B. W. Huang and Y. Tsuji, Cell. Signalling, 2012, 24, 981–990 CrossRef CAS PubMed.
- M. L. Circu and T. Y. Aw, Free Radical Biol. Med., 2010, 48, 749–762 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10346h |
| This journal is © The Royal Society of Chemistry 2017 |