
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multifunctional Prussian blue analogous@polyaniline core–shell nanocubes for lithium storage and overall water splitting†
Lu Zhang,
Tao Meng,
Baoguang Mao,
Donglei Guo,
Jinwen Qin* and
Minhua Cao *
*
Key Laboratory of Cluster Science, Ministry of Education of China, Beijing Key Laboratory of Photoelectronic/Electrophotonic Conversion Materials, School of Chemistry and Chemical Engineering, Beijing Institute of Technology, Beijing 100081, P. R. China. E-mail: caomh@bit.edu.cn
First published on 1st November 2017
Abstract
Developing efficient multifunctional electrode materials is highly effective to dramatically reduce the overall cost of the electrochemical devices. In this work, we for the first time demonstrate a facile strategy for preparing Prussian blue analogous@polyaniline core–shell nanocubes (denoted as PBAs@PANI) as multifunctional electrode materials for lithium storage and overall water splitting. By finely controlling the reaction parameters, PANI was uniformly coated on the surface of PBAs nanocubes and that the thickness of the PANI shell can be adjusted. The as-synthesized PBAs@PANI nanocubes yield improved electrochemical performance due to significantly enhanced charge transport, elastic buffer, and corrosion protection afforded by the PANI coating. More specifically, PBAs@PANI nanocubes show excellent lithium storage behavior with a reversible capacity of 626 mA h g−1 compared with the uncoated PBAs (203 mA h g−1) after 500 cycles at a high current density of 1 A g−1. Furthermore, PBAs@PANI nanocubes can also efficiently catalyze the hydrogen evolution reaction (HER) and oxygen evolution reaction (OER), and we therefore investigated their applications as bifunctional electrocatalysts (as both the anode and cathode) for overall water splitting. A current density of 10 mA cm−2 can be gained at a low cell voltage of 1.73 V over a long-term operation in base, representing a combined overpotential of 500 mV for full water splitting. These superior performances manifest that PBAs@PANI nanocubes can serve as high-performance anode for LIBs as well as promising bifunctional electrocatalysts for overall water splitting.
Introduction
Recently, metal–organic frameworks (MOFs), consisting of metal ions or clusters that are connected by organic ligands, feature well-defined pore structures, large specific surface areas, and structural flexibility. These properties make MOFs suitable as direct functional materials, efficient templates and/or precursors to develop new functional materials for a wide variety of applications, such as heterogeneous catalysis, gas adsorption, sensing and molecular release.1–6 Very recently, MOF-derived composites have been extensively studied, due to their great potential in energy conversion and storage like water electrolysis and rechargeable metal (Li, Na, K, Ca) ion batteries.7–10 Indeed, due to the redox of metal ions providing a pathway for electrons and open framework allowing highly reversible insertion/extraction of ions either in aqueous or organic electrolytes, MOFs also can directly act as outstanding candidates for use in energy conversion and storage systems, such as electrocatalysis,11–13 lithium-ion batteries (LIBs),14 sodium-ion batteries (NIBs),15 Li–S batteries,16 supercapacitors,17–19 and so on.20In particular, Prussian blue (PB) and its analogous (PBAs), as the old coordination compounds, are well known and have a face-centered cubic structures with the general chemical formula (AaMxM′y(CN)6·nH2O (where A is alkali metal cation, M and M′ are transition metal cations).21,22 The open-framework nature of PB and PBAs, containing open 〈100〉 channels and interstitial sites, enables rapid solid-state diffusion of a wide variety of ions, such as Li+, Na+, K+, NH4+, Rb+ and alkaline earth divalent ions (Scheme 1).23 Previous studies have reported that PB and PBAs are not suitable to be used for Li insertion/extraction in aqueous electrolyte because lithium ions are highly hydrated and become trapped within the channels of the lattices, resulting in the irreversible lithium storage.24,25 However, Nie et al., for the first time, have investigated PBAs as anode materials for LIBs in nonaqueous electrolyte, suggesting that Li+ can be reversibly intercalated from aprotic solvent.14,29 The minimal radius of lithium ion (0.69 Å) among above cations leads to the high capacity utilization of anodes due to comparatively fast electrochemical insertion/extraction kinetics. Therefore, diverse PB and PBAs are worthy of further study for high-performance LIBs because of their open frameworks for Li+ insertion/extraction, low cost, and environmental friendliness.
 | ||
| Scheme 1 Schematic illustration of the formation process of PBAs@PANI core–shell structure, starting from the open-framework of Fe–Co PBAs. The PANI shell was coated on the surface of PBAs nanocubes via in situ chemical oxidative polymerization. The resultant PBAs@PANI was applied as anode material for LIBs and bifunctional electrocatalysts for overall water electrolysis. | ||
Recently, the applications of MOFs have also been expanded to the electrocatalysts for the two core processes of water splitting, i.e. hydrogen evolution reaction (HER) and oxygen evolution reaction (OER).26 On the one hand, metal active species (Pt, Ru, Ir, Mn, Cu, Fe, Co and Ni) could be introduced into the structures of MOFs and directly act as catalysts for HER and/or OER.27 On the other hand, MOFs can serve as supports to uniformly disperse active species to increase the metal utilization, and accelerate mass transfer due to their porosity. In respect to these advances of MOFs, researchers inevitably turn to low-cost Fe, Co and Ni-based PBAs with abundant intrinsic molecular metal sites.9 However, the main drawback of PB and PBAs for LIBs or electrocatalysis is their poor electrical conductivity. In addition, PB and PBAs still suffer from structural degradation due to their structural un-stability in acidic or basic media and.28,29 Therefore, the structure optimization and surface engineering, especially hybridization with conductive nanomaterials, have been considered as the effective ways to improve their poor electronic conductivity and enhance the structural stability. As an ideal candidate, polyaniline (PANI) has become the desired material due to its simple synthetic method, low expense, nontoxicity, desirable chemical stability, and excellent electrical conductivity.30–34 As a consequence, the combination of conductive polymer PANI and PBAs is expected to exhibit notable synergetic effect with enhanced electrocatalytic performance and energy storage.
Herein, we demonstrate a strategy for facile and scalable synthesis of PBAs@PANI core–shell nanocubes through in situ polymerization of aniline monomer on the surface of Fe–Co PBAs nanocubes. In our case, besides serving as a good electron transfer mediator, PANI also serves as an elastic buffer to accommodate the volume variation during the insertion/extraction processes, as well as a protective layer to prevent etching of PBAs in basic media for HER and OER. As expected, the as-synthesized PBAs@PANI nanocubes achieve an enhanced reversible capacity as high as 865 mA h g−1 at 0.1 A g−1 after 100 cycles, and an outstanding cycling stability of 626 mA h g−1 at 1 A g−1 after 500 cycles, which is threefold higher than that of uncoated PBAs. Moreover, we also achieve 10 mA cm−2 water-splitting current at 1.73 V for over 10 h in 1.0 M KOH. In this work, we, for the first time, investigated the structural design of PBAs@PANI nanocubes as well as their multifunctional electrochemical performances for lithium storage and overall water splitting.
Experimental section
Synthesis of Fe–Co PBAs nanocubes
Fe–Co PBAs nanocubes were synthesized by a room-temperature solution method. In a typical procedure, cobalt chloride (CoCl2·6H2O, 2.5 mmol) and sodium citrate (Na3C6H5O7·2H2O, 6 mmol) were dissolved in 80 mL of deionized water to form a clear solution A. In the meantime, K3[Fe(CN)6] (1.5 mmol) was dissolved into 80 mL of deionized water to form a clear solution B. Then, A and B solutions were mixed under magnetic stirring. After being stirred for 24 h, the dark purple precipitate was collected by centrifugation and subsequently further washed with deionized water and ethanol for several times. Then the target product was obtained after drying under vacuum condition at 60 °C for 24 h.Synthesis of PBAs@PANI nanocubes
The PANI shell was coated on the surface of PBAs nanocubes via in situ chemical oxidative polymerization. In a typical synthesis, the as-prepared Fe–Co PBAs were dispersed in HCl solution (0.01 M, 50 mL) with continuous sonication for 1 h. Then, a certain amount of aniline (C6H7N, 0.5 mmol) was added slowly into the above solution and stirred for 1 h to form solution C. Ammonium persulfate ((NH4)2S2O8, 0.5 mmol) was dissolved in HCl solution (0.01 M, 50 mL) to obtain solution D. Subsequently, solution D was added dropwise into the above solution C with a speed of 2 mL min−1 under vigorous stirring in an ice bath. The mixed solution was further stirred for different times in an ice bath and thus-obtained products were denoted as PBAs@PANI-x (x = 6, 12, 18 and 24 h). Finally, the PANI-coated PBAs were collected by centrifugation and further washed with deionized water and ethanol for several times. In order to remove surface-adsorbed and interstitial water in PBAs, the as-prepared PBAs@PANI was dried under vacuum condition at 150 °C for 12 h for LIBs. If no otherwise specified, PBAs@PANI in the following discuss is referred as PBAs@PANI-18.Synthesis of PANI
Pure PANI was prepared in the same way as PBAs@PANI nanocubes in the absence of Fe–Co PBAs.Synthesis of PBAs&PANI nanocubes
The bare PBAs and pure PANI were physically mixed together for comparison and this sample was referred as PBAs&PANI (PANI: 14.56 wt%).Materials characterizations
The morphologies of as-obtained products were characterized by field emission scanning electron microscopy (FE-SEM, HITACHI S-4800). Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) were performed on a JEOL JEM-2010 microscope. Energy dispersive spectroscopy (EDS) elemental mapping images were taken on a scanning transmission electron microscope (STEM) (FEI Technai G2 F20). Powder X-ray diffraction (XRD) patterns of the as-prepared products were analyzed by a Bruker D8 Advance diffractometer with Cu–Kα radiation (λ ≈ 0.154 nm) at 40 kV and 40 mA in a scanning range of 10–80° (2θ). Fourier transform infrared (FT-IR) spectra were obtained using a Bruker Alpha FT-IR spectrometer (ATR-Ge, 400–4000 cm−1). Thermogravimetric analysis (TGA) was carried out on a Q50 thermoanalyzer with air as the carrier gas at a heating rate of 10 °C min−1. Raman spectra were recorded on an Invia Raman spectrometer, with an excitation laser wavelength of 532 nm. Inductively coupled plasma mass spectrometry (ICP-MS) measurements were recorded on a JY Ultima (French).LIB measurements
The working electrode for LIBs was prepared via coating the copper foil with a slurry, consisting of active material (70 wt%), acetylene black (20 wt%) as the conductive additive and sodium carboxymethyl cellulose (CMC, 10 wt%) dissolved in deionized water as the binder. The as-resultant slurry was dried at 150 °C for 24 h under vacuum before being assembled into a coin cell in an argon-filled glovebox. The mass loading of the active material was 1.0–1.2 mg cm−2. Coin-type cells (CR2025) were assembled within an argon-filled glovebox using Celgard 2400 as the separator, lithium foil as the counter and reference electrodes, and 1 M LiPF6 in ethylene carbonate (EC)/dimethyl carbonate (DMC)/diethyl carbonate (DEC) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, vol%) as the electrolyte. Galvanostatic cycling experiments of the cells were performed on a LAND CT2001A battery test system in the voltage range of 0.01–3.0 V (vs. Li+/Li) at room temperature. Cyclic voltammetry (CV) measurements were carried out on a CHI-760E electrochemical workstation at a scanning rate of 0.5 mV s−1 with a potential window between 0.01 and 3.0 V. Moreover, electrochemical impendence spectroscopy (EIS) was measured on an electrochemical workstation (CHI-604B) in a frequency range of 0.01 Hz to 100 kHz.
:1:1, vol%) as the electrolyte. Galvanostatic cycling experiments of the cells were performed on a LAND CT2001A battery test system in the voltage range of 0.01–3.0 V (vs. Li+/Li) at room temperature. Cyclic voltammetry (CV) measurements were carried out on a CHI-760E electrochemical workstation at a scanning rate of 0.5 mV s−1 with a potential window between 0.01 and 3.0 V. Moreover, electrochemical impendence spectroscopy (EIS) was measured on an electrochemical workstation (CHI-604B) in a frequency range of 0.01 Hz to 100 kHz.
OER and HER measurements
The working electrodes for OER and HER were prepared by the following processes. 2 mg of the catalyst was dispersed into a mixed solvent containing 450 μL of ethanol and 50 μL of 5 wt% Nafion (Dupont) followed by ultrasonication for 30 min. Then, 20 μL of above slurry was uniformly coated onto a freshly polished glassy carbon electrode (3 mm in diameter) and dried under ambient conditions.All electrochemical measurements of OER and HER were performed with a three-electrode system using CHI-760E electrochemical workstation. For the electrocatalytic HER and OER in alkaline media (1.0 KOH), an Ag/AgCl electrode in a 4 M KCl solution and a Pt foil (1 × 1 cm2) were used as the reference and counter electrodes, respectively. The current densities were normalized to the geometrical surface area and the measured potentials vs. Ag/AgCl were converted to potentials vs. the reversible hydrogen electrode (RHE) according to the Nernst equation [ERHE = EAg/AgCl + (0.059 × pH + 0.197)V]. Linear sweep voltammetry (LSV) was carried out in 1.0 M KOH electrolyte with a scan rate of 5 mV s−1 to obtain the polarization curves. The long-term stability test was performed by current density vs. time (I–t) curve at a certain potential for 10 h using a rotating speed of 1500 rpm. All of the data presented were not iR corrected.
Overall water splitting measurements
Overall water splitting was tested in a two-electrode system using CHI-760E electrochemical workstation. The PBAs@PANI catalyst coated onto nickel foam substrate was used as both the anode and cathode. Prior to the measurements, the iR compensation was not applied, which is similar with OER and HER measurements. The electrocatalytic activities of PBAs@PANI towards the overall water splitting were examined by polarization curves using LSV in 1.0 M KOH with scan rate of 5 mV s−1.Results and discussion
The synthesis procedure of PBAs@PANI nanocubes is schematically illustrated in Scheme 1. In this work, PBAs@PANI nanocubes were fabricated by a simple two-step method, including the formation of Fe–Co PBAs nanocubes and the polymerization of the PANI shell on the surface of the prepared PBAs (details about this process are given in experimental section). PBAs nanocubes were first prepared by a room-temperature wet-chemical method and then PANI was in situ polymerized on the surface of PBAs nanocubes. The morphologies of the obtained bare PBAs, PBAs@PANI and pure PANI (Fig. S1†) were systematically investigated by field emission scanning electron microscopy (FE-SEM) technique. As shown in Fig. 1, the as-prepared pristine PBAs consist of nanocubes with side lengths in the range of 140–200 nm and the surface of the nanocubes is relatively rough. It should be noted that the rough surface of PBAs can provide abundant nucleation sites for the uniform growth of the PANI shell. The thickness of the PANI shell can be simply controlled by varying the polymerization time from 6 to 12, 18 and 24 h (Fig. 1c–j). With increasing the reaction time, it can be clearly seen that the size of PBAs also increases accordingly, indicating PANI shell thickens progressively. Regardless of shorter reaction time or longer, the nanocubes are completely covered by the PANI shell, and that the cube shape is well inherited. With a further increase of the reaction time (24 h), PBAs nanocubes become very dense due to that thicker PANI shell was formed. Furthermore, transmission electron microscopy (TEM) images (Fig. 2a and b) reveal that the thickness of PANI after the reaction time of 18 h is around 5–10 nm and the surface of the PANI is fairly rough, indicating that the shell results from the self-assembly of small-sized PANI particles. As we all know, organic compounds and metal–organic frameworks both are unable to withstand electron beam bombardment for a relatively long duration when TEM measurements were carried out. As expected, we did not obtain high-resolution TEM images of PBAs@PANI nanocubes. Moreover, energy dispersive spectroscopy (EDS) elemental analysis (Fig. 2c and d) further confirm that PBAs@PANI nanocubes are composed of Co, Fe, K, C, N, and O elements and that these elements are uniformly distributed from the EDS element mappings (Fig. 2e). | ||
| Fig. 1 FE-SEM images of bare PBAs (a, b) and PBAs@PANI nanocubes for different polymerization times: 6 h (c, d), 12 h (e, f), 18 h (g, h) and 24 h (i, j). | ||
 | ||
| Fig. 2 (a, b) TEM images of PBAs@PANI after 18 h polymerization. (c) EDS spectrum, (d) FE-SEM image and (e) corresponding elemental mappings of PBAs@PANI. | ||
The crystallinity and phase composition of the as-prepared PBAs@PANI nanocubes were further characterized by X-ray diffraction (XRD) measurements. For comparison, bare PBAs and pure PANI were also measured. As shown in Fig. 3a, PBAs and PBAs@PANI nanocubes have same crystalline phase, which can be assigned to KCo[Fe(CN)6]·3H2O according to the ref. 34. This phase is further confirmed by combining inductively coupled plasma mass spectrometry (ICP-MS) result with thermogravimetric analysis (TGA) (Fig. 3d). In addition, the broad diffraction peak in the 2θ range of 20–30° can be assigned to amorphous PANI.35 To further verify the presence of PANI in PBAs@PANI, fourier transform infrared (FT-IR) and Raman spectra of PBAs, PANI and PBAs@PANI nanocubes were also collected. As shown in Fig. 3b, in the range of 400–4000 cm−1, the typical vibrations peaks of PBAs were detected at around 2098 and 597 cm−1, which are attributed to the stretching mode of cyanide functional group (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) and Fe–CN bending mode in the [Fe–CN] skeleton.32,36 Clearly, the above two peaks are also observed in the case of PBAs@PANI, indicating the presence of PBAs in our PBAs@PANI nanocubes. Meanwhile, the broad peak at about 3430 cm−1 (antisymmetric and symmetric O–H stretching modes) and the obvious peak at 1639 cm−1 (H–O–H bending mode) mainly come from the water incorporated in the framework of PBAs. For pure PANI, the adsorption bands at 1144, 1307, 1496 and 1581 cm−1 can be assigned to the C–H plane-bending vibration, C–N stretching vibration, C
N) and Fe–CN bending mode in the [Fe–CN] skeleton.32,36 Clearly, the above two peaks are also observed in the case of PBAs@PANI, indicating the presence of PBAs in our PBAs@PANI nanocubes. Meanwhile, the broad peak at about 3430 cm−1 (antisymmetric and symmetric O–H stretching modes) and the obvious peak at 1639 cm−1 (H–O–H bending mode) mainly come from the water incorporated in the framework of PBAs. For pure PANI, the adsorption bands at 1144, 1307, 1496 and 1581 cm−1 can be assigned to the C–H plane-bending vibration, C–N stretching vibration, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching of benzenoid ring, and CC stretching of quinoid rings of PANI, respectively.37–41 By comparison, it is clearly seen that the FT-IR spectrum of PBAs@PANI nanocubes also exhibits these adsorption bands, indicating the presence of PANI in as-prepared PBAs@PANI nanocubes.42 As for Raman spectrum of pure PANI (Fig. 3c), the distinctive and broad peaks appear in the 1000–1700 cm−1 region, which can be assigned to be C–C stretching, CC stretching, CN stretching, C–N stretching, N–H bending, etc. The peak broadening may result from the overlap of the Raman bands of PANI.43 Although the Raman spectrum of PBAs@PANI is not as prominent as that of the pure PANI, we can still distinguish the characteristic peaks of PANI. Moreover, there is an obvious peak located at about 2109 cm−1 in PBAs@PANI, which can be attributed to the stretching vibration of CN of PBAs. As a result, all the characteristic data can prove that the PANI shell is successfully formed on the surface of PBAs. TGA was applied to determine the amounts of PANI in a series of PBAs@PANI samples obtained with different polymerization times. As shown in Fig. 3d, the typical TGA curve for PBAs shows a multi-step mass loss. The first two weight losses correspond to the removal of interstitial water (below 150 °C) and lattice water (between 250 and 350 °C), respectively. The last weight loss occurring at >600 °C can be assigned to the decomposition of PBAs and liberation of CN groups.44 Based on the decomposition of bare PBAs, the PANI contents in a series of PBAs@PANI nanocubes are thus calculated to be about 5.45 wt% for PBAs@PANI-6, 8.15 wt% for PBAs@PANI-12, 14.56 wt% for PBAs@PANI-18, and 16.04 wt% for PBAs@PANI-24, respectively.45 It is clear that with increasing the polymerization time, the PANI content increases gradually, in good agreement with the FE-SEM observations.
C stretching of benzenoid ring, and CC stretching of quinoid rings of PANI, respectively.37–41 By comparison, it is clearly seen that the FT-IR spectrum of PBAs@PANI nanocubes also exhibits these adsorption bands, indicating the presence of PANI in as-prepared PBAs@PANI nanocubes.42 As for Raman spectrum of pure PANI (Fig. 3c), the distinctive and broad peaks appear in the 1000–1700 cm−1 region, which can be assigned to be C–C stretching, CC stretching, CN stretching, C–N stretching, N–H bending, etc. The peak broadening may result from the overlap of the Raman bands of PANI.43 Although the Raman spectrum of PBAs@PANI is not as prominent as that of the pure PANI, we can still distinguish the characteristic peaks of PANI. Moreover, there is an obvious peak located at about 2109 cm−1 in PBAs@PANI, which can be attributed to the stretching vibration of CN of PBAs. As a result, all the characteristic data can prove that the PANI shell is successfully formed on the surface of PBAs. TGA was applied to determine the amounts of PANI in a series of PBAs@PANI samples obtained with different polymerization times. As shown in Fig. 3d, the typical TGA curve for PBAs shows a multi-step mass loss. The first two weight losses correspond to the removal of interstitial water (below 150 °C) and lattice water (between 250 and 350 °C), respectively. The last weight loss occurring at >600 °C can be assigned to the decomposition of PBAs and liberation of CN groups.44 Based on the decomposition of bare PBAs, the PANI contents in a series of PBAs@PANI nanocubes are thus calculated to be about 5.45 wt% for PBAs@PANI-6, 8.15 wt% for PBAs@PANI-12, 14.56 wt% for PBAs@PANI-18, and 16.04 wt% for PBAs@PANI-24, respectively.45 It is clear that with increasing the polymerization time, the PANI content increases gradually, in good agreement with the FE-SEM observations.
 | ||
| Fig. 3 (a) XRD patterns, (b) FITR and (c) Raman spectra of bare PBAs, PBAs@PANI and pure PANI. (d) TGA curves of bare PBAs and a series of PBAs@PANI with different polymerization times. | ||
To confirm the potential application of the as-prepared PBAs@PANI nanocubes, we first evaluated them as an anode material for LIBs. However, it has been addressed that for vacancy-containing PB and PBAs, their poor cycling performance and low coulombic efficiency as electrode materials for LIBs mainly result from water molecules existing in their structures.46 In views of this fact, the as-prepared PBAs@PANI in our work was dried to remove water molecules under vacuum condition at 150 °C for 12 h before being assembled into coin cell, during which the cubic morphology of PBAs@PANI is not destroyed (Fig. S2†). Even after being soaked in 1.0 M KOH for 12 h, PBAs@PANI still maintains its cubic structure due to the PANI coating, revealing its excellent structure stability. In contrast, bare PBAs have completely transformed into nanosheets in similar alkaline medium (Fig. S3†).
Fig. 4a shows the galvanostatic charge/discharge voltage profiles of the typical PBAs@PANI at a current density of 100 mA g−1 in a voltage window of 0.01–3.0 V. The initial discharge and charge capacities are 1001 and 730 mA h g−1, which offer an initial coulombic efficiency of 72.9%. The low coulombic efficiency during the first cycle may result from irreversible capacity loss, including inevitable formation of SEI film, side reactions associated with residual water, and decomposition of the electrolyte. The discharge capacity maintains about 700 mA h g−1 after the 2nd cycle and gradually increases to 865 mA h g−1 after the 100th cycle. Fig. 4b shows the cycling performances of the PBAs@PANI, bare PBAs, pure PANI as well as the physically mixed PBAs and PANI (denoted as PBAs&PANI) electrodes at 100 mA g−1. Clearly, PBAs@PANI nanocubes possess the highest specific capacity and the best stability. Even after 100 charge/discharge cycles, the specific capacity of PBAs@PANI remains 1046 mA h g−1, while those of the bare PBAs, pure PANI and PBAs&PANI are only 713, 243 and 690 mA h g−1, respectively. More importantly, compared with bare PBAs (Fig. S4†), PBAs@PANI can still remain the cubic structure after 100 cycles at the current density of 100 mA g−1 (Fig. 4c), suggesting that the PANI shell can provide mechanical/structural integrity to effectively buffer the volume change during charging/discharging processes. Impressively, PBAs@PANI electrode exhibits remarkable rate capability (Fig. 4d). All the rate capabilities were tested at step-wise current densities from 0.1 to 1 A g−1 and then reversed to 0.1 A g−1. The average discharge capacities of PBAs@PANI electrode are 863, 742, 644, 577, 530 and 500 mA h g−1, at the current density from 0.1, 0.2, 0.4, 0.6, 0.8, 1.0 A g−1, all of which are higher than those of the uncoated PBAs, pure PANI and physically mixed sample. Remarkably, a stable capacity of PBAs@PANI can be recovered as high as 891 mA h g−1 when the current density goes back to 0.1 A g−1.
 | ||
| Fig. 4 (a) Galvanostatic charge/discharge profiles of PBAs@PANI at a current density of 100 mA g−1 between 0.01 and 3.0 V. (b) Cycling performances of PBAs@PANI, bare PBAs, pure PANI and PBAs&PANI at a current density of 100 mA g−1. (c) FE-SEM images of PBAs@PANI electrode after being tested for 100 cycles at a current density of 100 mA g−1. (d) Rate performances of PBAs@PANI, bare PBAs, and PBAs&PANI at different current densities. | ||
To reveal the excellent cycling performance of PBAs@PANI nanocubes at high current density, PBAs@PANI electrode is tested at 1 A g−1 for 500 cycles. As shown in Fig. 5a, the PBAs@PANI electrode delivers a discharge capacity as high as 626 mA h g−1 after 500 cycles. However, the bare PBAs without PANI coating, pure PANI (220 mA h g−1) and mechanical mixed mixture (246 mA h g−1) show poor cycling stability with severe capacity decay under the same test conditions and only 203, 220, and 246 mA h g−1 can be retained after 500 cycles. To further understand the reaction kinetic of PBAs@PANI nanocubes, electrochemical impendence spectroscopy (EIS) measurements are performed from 100 kHz to 0.01 Hz. The measured Nyquist plots of PBAs@PANI and bare PBAs, pure PANI and PBAs&PANI electrodes are compared in Fig. 5b. Each plot consists of a semicircle in the high-frequency region that can be attributed to the charge transfer process, and a sloping line in the low-frequency region that is related to the mass transfer of lithium ions. Clearly, compared with the bare PBAs, PBAs@PANI nanocubes display relatively smaller diameter of the semicircle in EIS spectrum, indicating a lower impedance value. Therefore, the PBAs@PANI electrode shows smaller charge transfer resistance, implying its good conductivity, and faster Li+ ion diffusion behavior. Fig. 5c shows schematic illustration of the insertion of lithium ions into the Fe–Co PBAs. Based on reported investigations on the PB and PBAs as electrode materials,47 this face-centered framework of Fe–Co PBAs could offer channels for rapid Li+ insertion into eight subunit cells throughout the lattice. The flexible open-framework structure enables structural integrity while accommodating multiple Li+ via phase-transition behavior during Li+ insertion. Meanwhile, the conductive PANI shell not only maintains the structural stability of PBAs during lithium insertion/extraction processes, but also offers effective transport pathways for electron.58
 | ||
| Fig. 5 (a) Long cycling performances of PBAs@PANI, bare PBAs, pure PANI and PBAs&PANI at 1 A g−1 after activation at 0.1 mA g−1 for initial ten cycles. (b) Nyquist plots of PBAs@PANI, bare PBAs, pure PANI and PBAs&PANI. (c) Schematic illustration of the insertion of lithium ions into the Fe–Co PBAs. (d) Rate and (e) long cycling performances for a series of PBAs@PANI at 1 A g−1 after activation at 0.1 A g−1 for initial ten cycles. | ||
Consequently, our elastic PANI could significantly prevent the fracture of the Li+ host, and thus the PBAs@PANI nanocubes exhibit an excellent cycling stability with almost 100% capacity retention after 500 cycles. Furthermore, rate and long cycling performances of a series of PBAs@PANI nanocubes with different polymerization times are also tested under the same conditions (Fig. 5d, e and S5a, b†). Clearly, the sample obtained with 18 h polymerization time exhibits the best lithium storage performance among all the as-synthesized samples in terms of capacity, rate capability.
The explanation is not far to seek. In our case, inadequate coverage of PBAs by the PANI shell definitely hinders electron transfer. However, excessive PANI coating on PBAs shields open framework of PBAs and thus impedes Li+ diffusion and limits their lithium storage capacity.
Next, the PBAs@PANI nanocubes were evaluated as an electrocatalyst towards OER by using a typical three-electrode setup in O2-saturated 1.0 M KOH solution at a scanning rate of 5 mV s−1. For comparison, bare PBAs, pure PANI, PBAs&PANI and IrO2/C were also investigated under the same conditions. From linear sweep voltammetry (LSV) curves (Fig. 6a), it can be seen that PBAs@PANI electrode exhibits a remarkably high activity with an onset potential of ∼1.52 V, which is evidently smaller than those of bare PBAs, pure PANI and PBAs&PANI. Furthermore, PBAs@PANI reaches 10 mA cm−2 in 1.0 M KOH solution at a potential of 1.56 V corresponding to an overpotential of 330 mV, which not only is much lower than those of PBAs&PANI (1.62 V), bare PBAs (1.66 V), pure PANI, and many other reported OER catalysts (Table 1), but also is comparable to that of the state-of-the-art IrO2/C catalyst (1.54 V). The OER kinetics of above catalysts are further examined by Tafel plots in Fig. 6b. The Tafel slopes are evaluated to be 70, 165, 148 and 43 mV dec−1 for PBAs@PANI, PBAs&PANI, bare PBAs and IrO2/C, respectively. Clearly, the Tafel slope of PBAs@PANI is only higher than that of IrO2/C catalyst and lower than those of PBAs&PANI and bare PBAs, suggesting the favorable kinetic of our PBAs@PANI. Furthermore, EIS analysis under operating condition is employed to study the OER kinetics occurring at the electrode/electrolyte interface. The diameter of the semicircle in high-frequency field can be assigned to the charge transfer resistance related to the electrocatalytic kinetics. As shown in Fig. 6c, compared with the bare PBAs, our PBAs@PANI nanocubes reveal smaller contact impedance and faster charge transport along with favorable reaction kinetics. It should be noted that the PANI shell coated on PBAs nanocubes can effectively improve electron transfer from PANI to PBAs due to the intimate adhesion. Electrochemical double-layer capacitance (Cdl) is measured to estimate the active surface area using typical CVs (Fig. S6†) in the region of 1.37–1.42 V (vs. RHE) at different scan rates (10–50 mV s−1). As depicted in Fig. 6d, PBAs@PANI possesses a significantly high Cdl value (29 mF cm−2), which is higher than 27 mF cm−2 for bare PBAs, 3 mF cm−2 for PBAs&PANI, and 2 mF cm−2 for pure PANI. We further investigated the durability of PBAs@PANI. The long-term electrochemical stability of PBAs@PANI is examined by measuring the I–t curve of OER electrolysis (Fig. 6e). The time-dependent current density curve at the fixed potential suggests that PBAs@PANI catalyst has superior durability over 10 h without any structure change (Fig. S7†). After continuous CV scanning for 1000 cycles at a scan rate of 100 mV s−1 in the voltage of 1.12–1.67 V, the polarization curve of PBAs@PANI (Fig. 6f) shows a negligible difference compared to the initial one. However, the same testing leads to a large activity loss for pure PBAs.
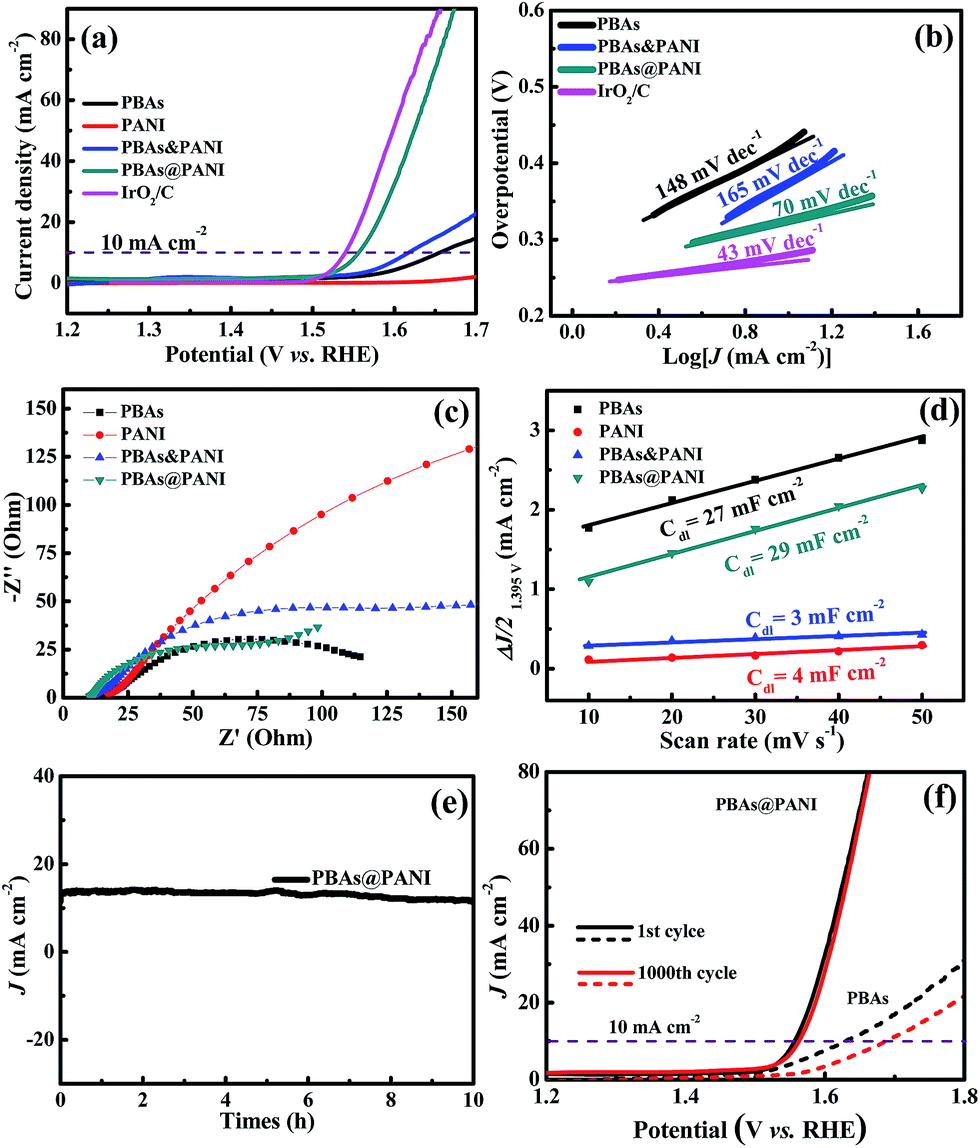 | ||
| Fig. 6 (a) Polarization curves of bare PBAs, pure PANI, PBAs&PANI, PBAs@PANI and IrO2/C in an O2-saturated 1.0 M KOH solution (scan rate of 5 mV s−1). (b) Tafel plots of bare PBAs, PBAs&PANI, PBAs@PANI and IrO2/C. (c) Nyquist plots of bare PBAs, pure PANI, PBAs&PANI and PBAs@PANI. (d) The capacitive currents at 1.395 V vs. RHE as a function of the scan rate for bare PBAs, pure PANI, PBAs&PANI and PBAs@PANI. (e) I–t curve obtained over PBAs@PANI at an overpotential of 330 mV over 10 h long electrocatalytic OER. (f) Polarization curves of the bare PBAs and PBAs@PANI before and after 1000 CV cycles. | ||
| Catalysts | Overpotential (V vs. RHE) at J = 10 mA cm−2 | Ref. | ||
|---|---|---|---|---|
| HER | OER | Full water splitting | ||
| a N.A. stands for not given. | ||||
| PBAs@PANI | 0.17 | 0.33 | 1.71 | This work |
| Mo–N/C@MoS2 | 0.117 | 0.39 | aN.A. | 48 |
| MoS2/Ni3S2 | 0.11 | 0.218 | 1.56 | 49 |
| Co–CoO/N-rGO | 0.31 | 0.39 | N.A. | 50 |
| Co–Ni–Se/C/NF | 0.09 | 0.275 | 1.60 | 51 |
| CP/CTs/Co–S | 0.19 | 0.31 | 1.743 | 52 |
| CoP2/rGO | 0.088 | 0.30 | 1.56 | 53 |
| np(Co0.52Fe0.48)2P | 0.064 | 0.27 | 1.53 | 54 |
| Co2P nanowires | 0.095 | 0.26 | 1.50 | 55 |
| Co-doped NiSe2 | 0.185 | 0.32 | 1.62 | 56 |
| PCPTF | 0.38 | 0.31 | N.A. | 57 |
The opposite reaction that can availably catalyze the corresponding hydrogen evolution process as well is an import merit to pursue bifunctional water electrolysis electrode and simplify the installation. Hence, the HER performances of bare PBAs, pure PANI, PBAs@PANI, PBAs&PANI and Pt/C are evaluated in N2-saturated 1.0 M KOH solution. As shown in Fig. 7a, PBAs@PANI holds a small overpotential of around 170 mV at the current density of 10 mA cm−2. Although the PBAs@PANI performance is inferior to that of the Pt/C electrode, it outperforms the bare PBAs (200 mV), PBAs&PANI (390 mV), pure PANI (623 mV), and some reported HER catalysts (Table 1). The corresponding Tafel plots are given in Fig. 7b, PBAs@PANI nanocubes also possess a small Tafel value of 173 mV dec−1, which is much smaller than those of bare PBAs (203 mV dec−1) and PBAs&PANI (182 mV dec−1), clearly indicating a favorable HER reaction kinetics for PBAs@PANI. Based on the above results, the good bifunctional electrocatalytic activities of PBAs@PANI observed in OER and HER prompt us to construct an integrated water-electrolysis unit. A current density of 10 mA cm−2 can be gained at a cell voltage of 1.73 V, representing a combined overpotential of 500 mV for full water splitting (Fig. 7c). The overall water splitting performance of PBAs@PANI outperforms that of the Ni foam substrate and some of the reported materials, e.g. CP/CTs/Co–S (1.743 V).62 Furthermore, when used as both the cathode and anode, PBAs@PANI nanocubes also exhibit no detectable current decay during the long-term testing with remarkable durability for >12 h (Fig. 7d). This outstanding performance manifests that PBAs@PANI can be served as a low-cost bifunctional electrocatalyst with high efficiency and durability for overall water electrolysis. OER and HER performances for a series of PBAs@PANI nanocubes with different polymerization times are also tested under the same conditions (Fig. 8). According to the LSV curves and Tafel slopes for OER, PBAs@PANI-18 with an optimal loading of PANI (14.56 wt%), exhibits the best electrocatalytic activity among the samples tested, giving the lowest overpotential (330 mV) and smallest Tafel slope (70 mV dec−1) at the same current density (10 mA cm−2). In addition, Nyquist plots for a series of PBAs@PANI in O2-saturated 1.0 M KOH solution (scan rate of 5 mV s−1) are also tested under the same conditions (Fig. S8†), revealing the favorable reaction kinetics of PBAs@PANI-18 as well. Furthermore, the LSV curves and kinetics of PBAs@PANI electrodes with different PANI loadings for HER are presented in Fig. 8c and d. The finding is consistent with the result from OER. Therefore, by tuning the mass ratio of PANI to PBAs, optimum overpotentials of 330 mV (OER) and 170 mV (HER) for PBAs@PANI-18 to reach 10 mA cm−2 are achieved.
 | ||
| Fig. 7 (a) Polarization curves of bare PBAs, pure PANI, PBAs@PANI, PBAs&PANI and Pt/C in a N2-saturated 1.0 M KOH solution with a scan rate of 5 mV s−1. (b) Tafel plots of bare PBAs, PBAs&PANI, PBAs@PANI and Pt/C. (c) Polarization curves of PBAs@PANI//PBAs@PANI, and Ni foam//Ni foam for overall water splitting in a two-electrode configuration at a scan rate of 5 mV s−1 in 1.0 M N2-saturated KOH. (d) I–t curve of PBAs@PANI//PBAs@PANI at an overpotential of 500 mV over 12 h for overall water splitting (inset shows a digital photograph of an electrolyzer, whose anode and cathode both are comprised of PBAs@PANI). | ||
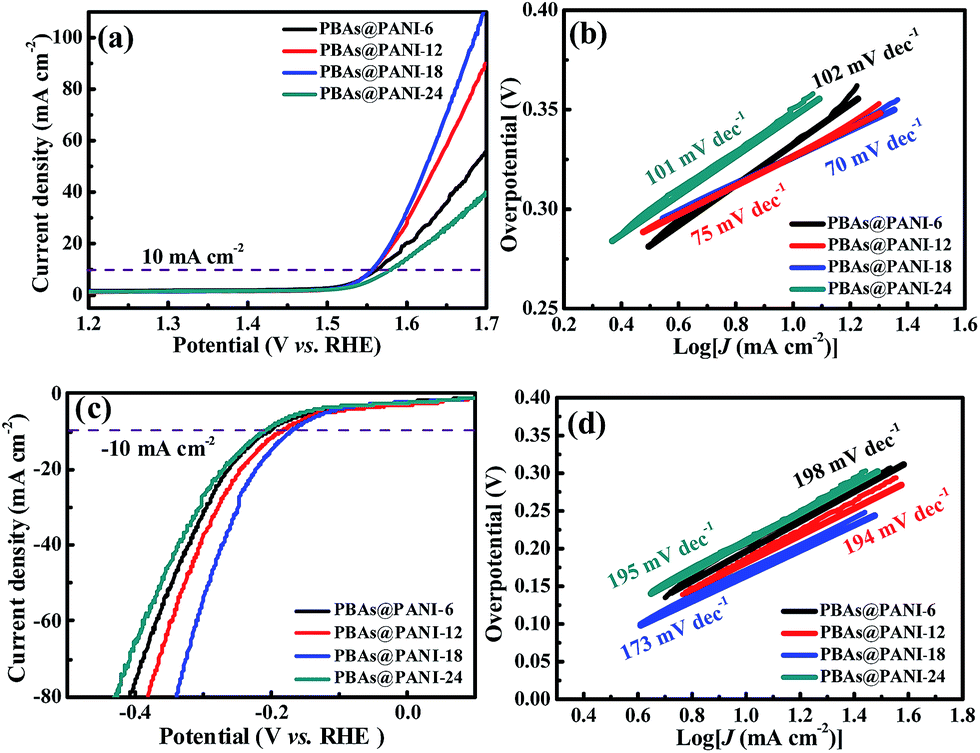 | ||
| Fig. 8 (a) Polarization curves and (b) Tafel plots of a series of PBAs@PANI for OER in an O2-saturated 1.0 M KOH solution (scan rate of 5 mV s−1). (c) Polarization curves and (d) Tafel plots of a series of PBAs@PANI for HER in an N2-saturated 1.0 M KOH solution (scan rate of 5 mV s−1). | ||
As reported,59–64 those materials with metal–nitrogen–carbon (Me–N–C) structure, i.e. the metal cations coordinated with nitrogen functional groups provided by organic ligands or N-doped carbon, have been demonstrated to be high-performance electrocatalysts for OER, HER, and ORR. The PBAs possess abundant Me–N–C, which may act as the active sites for OER and HER. This result is also confirmed by XRD and FE-SEM measurements of PBAs@PANI after OER tests, which keep well the original structure (Fig. S7†). As a consequence, we speculate that the Me–N species in PBAs manifest the material with the overall properties. Moreover, the excellent electrocatalytic activities of our PBAs@PANI nanocubes towards OER and HER are closely related to the PANI coating, which has been widely studied for a variety of applications in anticorrosion protection of metals, showing high resistance against the corrosion in both acidic and basic conditions.65–67 In our work, the availability of abundant N-species in PANI allows to couple with PBAs, thus leading to the formation of the chemically bonded interface between PBAs and PANI, which can effectively protect PBAs from structural degradation in basic media up to pH 14.0. Therefore, the excellent durability of PBAs@PANI nanocubes for OER and HER is due to the stable PANI shell, which can avoid PBAs from direct exposure to the corrosive environment.
Conclusion
We have investigated PBAs@PANI nanocubes as multifunctional electrode materials for lithium storage and overall water splitting. The as-prepared PBAs@PANI nanocubes exhibit higher lithium storage capacity, smaller overpotentials, and better stability for OER and HER in basic media compared to pure PBAs and physical mixture PBAs&PANI. In overall water splitting testing, PBAs@PANI nanocubes, used as electrodes for both cathode and anode, realize a current density of 10 mA cm−2 at an overpotential of 500 mV with good durability. This work will pave an alternative and low-cost pathway for designing multifunctional nanocomposites, aiming at developing regenerative energy storage and conversion system.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21601014, 21471016 and 21271023) and the 111 Project (B07012). The authors would like to thank the Analysis & Testing Center of Beijing Institute of Technology for performing FESEM and TEM measurements.References
- X. B. Xu, Z. C. Zhang and X. Wang, Adv. Mater., 2015, 27, 5365–5371 CrossRef CAS PubMed.
- B. Kong, C. Selomulya, G. F. Zheng and D. Y. Zhao, Chem. Soc. Rev., 2015, 44, 7997–8018 RSC.
- D. M. Wu, Z. D. Xu, T. Zhang, Y. B. Shao, P. X. Xi, H. Li and C. L. Xu, RSC Adv., 2016, 52, 103116 RSC.
- M. Hu, A. A. Belik, M. Imura and Y. Yamauchi, J. Am. Chem. Soc., 2013, 135, 384 CrossRef CAS PubMed.
- M. Hu, S. Furukawa, R. Ohtani, H. Sukegawa, Y. Nemoto, J. Reboul, S. Kitagawa and Y. Yamauchi, Angew. Chem., Int. Ed., 2012, 124, 1008 CrossRef.
- M. Hu, S. Ishihara and Y. Yamauchi, Angew. Chem., Int. Ed., 2013, 125, 1273 CrossRef.
- W. Xia, A. Mahmood, R. Q. Zou and Q. Xu, Energy Environ. Sci., 2015, 8, 1837–1866 CAS.
- S. L. Li and Q. Xu, Energy Environ. Sci., 2013, 6, 1656–1683 CAS.
- L. Zhang, H. B. Wu, S. Madhavi, H. H. Hng and X. W. Lou, J. Am. Chem. Soc., 2012, 134, 17388–17391 CrossRef CAS PubMed.
- X. Z. Li, Y. Y. Fang, X. Q. Lin, M. Tian, X. C. An, Y. Fu, R. Li, J. Jin and J. T. Ma, J. Mater. Chem. A, 2015, 3, 17392–17402 CAS.
- A. Carné Sánchez, I. Imaz, M. Cano Sarabia and D. Maspoch, Nat. Chem., 2013, 5, 203–211 CrossRef PubMed.
- L. J. Han, P. Y. Tang, Á. Reyes Carmona, B. Rodríguez García, M. Torréns, J. R. Morante, J. Arbiol and J. R. Galan Mascaros, J. Am. Chem. Soc., 2016, 138, 16037–16045 CrossRef CAS PubMed.
- M. Jahan, Z. L. Liu and K. P. Loh, Adv. Funct. Mater., 2013, 23, 5363–5372 CrossRef CAS.
- P. Nie, L. Shen, H. F. Luo, B. Ding, G. Y. Xu, J. Wang and X. G. Zhang, J. Mater. Chem. A, 2014, 2, 5852–5857 CAS.
- H. W. Lee, R. Y. Wang, M. Pasta, S. Woo Lee, N. Liu and Y. Cui, Nat. Commun., 2014, 5, 5280 CrossRef CAS PubMed.
- R. Demir Cakan, M. Morcrette, F. Nouar, C. Davoisne, T. Devic, D. Gonbeau, R. Dominko, C. Serre, G. Férey and J. M. Tarascon, J. Am. Chem. Soc., 2011, 133, 16154–16160 CrossRef CAS PubMed.
- Y. Z. Zhang, T. Cheng, Y. Wang, W. Y. Lai, H. Pang and W. Huang, Adv. Mater., 2016, 28, 5242–5248 CrossRef CAS PubMed.
- R. Díaz, M. G. Orcajo, J. A. Botas, G. Calleja and J. Palma, Mater. Lett., 2012, 68, 126–128 CrossRef.
- C. Qu, Y. Jiao, B. Zhao, D. C. Chen, R. Q. Zou, K. S. Walton and M. L. Liu, Nano Energy, 2016, 26, 66–73 CrossRef CAS.
- A. Morozan and F. Jaouen, Energy Environ. Sci., 2012, 5, 9269–9290 CAS.
- D. Asakura, C. H. Li, Y. Mizuno, M. Okubo, H. S. Zhou and D. R. Talham, J. Am. Chem. Soc., 2013, 135, 2793–2799 CrossRef CAS PubMed.
- M. Pasta, C. D. Wessells, N. Liu, J. Nelson, M. T. McDowell, R. A. Huggins, M. F. Toney and Y. Cui, Nat. Commun., 2014, 5, 3007 Search PubMed.
- R. Y. Wang, C. D. Wessells, R. A. Huggins and Y. Cui, Nano Lett., 2013, 13, 5748–5752 CrossRef CAS PubMed.
- A. L. Crumbliss, P. S. Lugg and N. Morosoff, Inorg. Chem., 1984, 23, 4701–4708 CrossRef CAS.
- N. Imanishi, T. Morikawa, J. Kondo, Y. Takeda, O. Yamamoto, N. Kinugasa and T. Yamagishi, J. Power Sources, 1999, 79, 215–219 CrossRef CAS.
- D. J. Jing, C. Sheng and Z. Chuan, Nat. Commun., 2017, 8, 15341 CrossRef PubMed.
- S. B. Wang, Y. D. Hou, S. Lin and X. C. Wang, Nanoscale, 2014, 6, 9930–9934 RSC.
- S. D. Wang, H. C. Lan, H. J. Liu and J. H. Qu, Phys. Chem. Chem. Phys., 2016, 18, 9437–9445 RSC.
- M. Hu, N. L. Torad and Y. Yamauchi, Eur. J. Inorg. Chem., 2012, 2012, 4795–4799 CrossRef CAS.
- Q. Wang, J. Yan, Z. J. Fan, T. Wei, M. L. Zhang and X. Y. Jing, J. Power Sources, 2014, 247, 197–203 CrossRef CAS.
- M. Kotal, A. K. Thakur and A. K. Bhowmick, ACS. Appl. Mater. Interfaces, 2013, 5, 8374–8386 CAS.
- F. N. Maluin, M. Sharifah, P. Rattanarat, W. Siangproh, O. Chailapakul, A. M. Issam and N. S. A. Manan, Anal. Methods, 2016, 8, 8049–8058 RSC.
- L. Wang, X. Feng, L. T. Ren, Q. H. Piao, J. Q. Zhong, Y. B. Wang, H. W. Li, Y. F. Chen and B. Wang, J. Am. Chem. Soc., 2015, 137, 4920–4923 CrossRef CAS PubMed.
- M. Hu, S. Ishihara, K. Ariga, M. Imura and Y. Yamauchi, Chem.–Eur. J., 2013, 19, 1882–1885 CrossRef CAS PubMed.
- D. Chaudhuri, A. Kumar, D. D. Sarma, M. G. A. Hernández, J. P. Joshi and S. V. Bhat, Appl. Phys. Lett., 2003, 82, 1733–1735 CrossRef CAS.
- R. J. Chen, H. Y. Xin, X. Man, Z. Q. Yun, Z. X. Xiao, L. Li and W. Feng, ACS Appl. Mater. Interfaces, 2016, 8, 16078–16086 CAS.
- G. S. Nan, Z. Yong, Y. Y. Yan, Y. L. Min, J. C. Fan, Q. J. Xu and H. Yun, J. Power Sources, 2016, 316, 176–182 CrossRef.
- S. S. Ding, P. He, W. R. Feng, L. Li, G. L. Zhang, J. C. Chen, F. Q. Dong and H. C. He, J. Phys. Chem. Solids, 2016, 91, 41–47 CrossRef CAS.
- W. D. Zhou, Y. C. Yu, H. Chen, F. J. DiSalvo and H. D. Abruña, J. Am. Chem. Soc., 2013, 135, 16736–16743 CrossRef CAS PubMed.
- C. Lai, H. Z. Zhang, G. R. Li and X. P. Gao, J. Power Sources, 2011, 196, 4735–4740 CrossRef CAS.
- C. W. Hu, T. Kawamoto, H. Tanaka, A. Takahashi, K. M. Lee, S. Y. Kao, Y. C. Liao and K. C. Ho, J. Mater. Chem. C, 2016, 4, 10293–10300 RSC.
- M. Cochet, G. Louarn, S. Quillard, J. P. Buisson and S. Lefrant, J. Raman Spectrosc., 2000, 31, 1041–1049 CrossRef CAS.
- Y. You, X. L. Wu, Y. X. Yin and Y. G. Guo, J. Mater. Chem. A, 2013, 1, 14061–14065 CAS.
- X. H. Zhao, H. J. Ahn, K. W. Kim, K. K. Cho and J. H. Ahn, J. Phys. Chem. C, 2015, 119, 7996–8003 CAS.
- D. Z. Yang, J. Xu, X. Z. Liao, H. Wang, Y. S. He and Z. F. Ma, Chem. Commun., 2015, 51, 8181–8184 RSC.
- X. K. Huang, S. M. Cui, J. B. Chang, P. B. Hallac, C. R. Fell, Y. T. Luo, B. Metz, J. Jiang, P. T. Hurley and J. H. Chen, Angew. Chem., Int. Ed., 2015, 54, 1490–1493 CrossRef CAS PubMed.
- J. M. Jeong, B. G. Choi, S. C. Lee, K. G. Lee, S. J. Chang, Y. K. Han, Y. B. Lee, H. U. Lee, S. Kwon, G. Lee, C. S. Lee and Y. S. Huh, Adv. Mater., 2013, 25, 6250–6255 CrossRef CAS PubMed.
- I. S. Amiinu, Z. H. Pu, X. B. Liu, K. A. Owusu, H. G. R. Monestel, F. O. Boakye, H. N. Zhang and S. Mu, Adv. Funct. Mater., 2017, 1702300 CrossRef.
- J. Zhang, T. Wang, D. Pohl, B. Rellinghaus, R. H. Dong, S. H. Liu, X. D. Zhuang and X. L. Feng, Angew. Chem., Int. Ed., 2016, 128, 6814–6819 CrossRef.
- X. E. Liu, W. Liu, M. Ko, M. Park, M. G. Kim, P. Oh, S. Chae, S. Park, A. Casimir, G. Wu and J. Cho, Adv. Funct. Mater., 2015, 25, 5799–5808 CrossRef CAS.
- F. W. Ming, H. F. Liang, H. H. Shi, X. Xu, G. Meia and Z. C. Wang, J. Mater. Chem. A, 2016, 4, 15148–15155 CAS.
- J. Wang, H. X. Zhong, Z. L. Wang, F. L. Meng and X. B. Zhang, ACS Nano, 2016, 10, 2342–2348 CrossRef CAS PubMed.
- J. M. Wang, W. R. Yangac and J. Q. Liu, J. Mater. Chem. A, 2016, 4, 4686–4690 CAS.
- Y. W. Tan, H. Wang, P. Liu, Y. H. Shen, C. Cheng, A. Hirata, T. Fujita, Z. Tangc and M. W. Chen, Energy Environ. Sci., 2016, 9, 2257–2261 CAS.
- Z. Y. Jin, P. P. Lia and D. Xiao, Green Chem., 2016, 18, 1459–1464 RSC.
- T. T. Liu, A. M. Asirib and X. P. Sun, Nanoscale, 2016, 8, 3911–3915 RSC.
- Y. Yang, H. L. Fei, G. D. Ruan and J. M. Tour, Adv. Mater., 2015, 27, 3175–3180 CrossRef CAS PubMed.
- W. W. Xu, K. N. Zhao, C. J. Niu, L. Zhang, Z. Y. Cai, C. H. Han, L. He, T. Shen, M. Y. Yan, L. B. Qu and L. Q. Mai, Nano Energy, 2014, 8, 196–204 CrossRef CAS.
- J. Wang, K. Li, H. X. Zhong, D. Xu, Z. L. Wang, Z. Jiang, Z. J. Wu and X. B. Zhang, Angew. Chem., Int. Ed., 2015, 54, 10530–10534 CrossRef CAS PubMed.
- S. Dou, L. Tao, J. Huo, S. Wang and L. Dai, Energy Environ. Sci., 2016, 9, 1320–1326 CAS.
- T. Y. Ma, J. L. Cao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2016, 55, 1138–1142 CrossRef CAS PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- Y. Zhao, K. Watanabe and K. Hashimoto, J. Am. Chem. Soc., 2012, 134, 19528–19531 CrossRef CAS PubMed.
- M. Zeng, Y. L. Liu, F. P. Zhao, K. Q. Nie, N. Han, X. X. Wang, W. J. Huang, X. N. Song, J. Zhong and Y. G. Li, Adv. Funct. Mater., 2016, 26, 4397–4404 CrossRef CAS.
- R. Ansari and A. H. Alikhani, J. Coat. Technol. Res., 2009, 6, 221–227 CrossRef CAS.
- S. J. Tian, J. Y. Liu, T. Zhu and W. Knoll, Chem. Commun., 2003, 21, 2738–2739 RSC.
- S. G. Chen, Z. D. Wei, X. Q. Qi, L. C. Dong, Y. G. Guo, L. J. Wan, Z. G. Shao and L. Li, J. Am. Chem. Soc., 2012, 134, 13252–13255 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10292e |
| This journal is © The Royal Society of Chemistry 2017 |