
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hyperhalogen properties of early-transition-metal borates†
Jia-Yuan Liua,
Hai-Di Maa,
Yan-bo Suna,
Ying Lia,
Wei-Ming Sun b,
Di Wu*a and
Zhi-Ru Lia
b,
Di Wu*a and
Zhi-Ru Lia
aInstitute of Theoretical Chemistry, Laboratory of Theoretical and Computational Chemistry, Jilin University, Changchun 130023, China. E-mail: wud@jlu.edu.cn
bDepartment of Basic Chemistry, The School of Pharmacy, Fujian Medical University, Fuzhou 350108, China
First published on 5th October 2017
Abstract
The equilibrium structures, stability and magnetic properties of Sc(BO2)n−/0 (n = 1–4) clusters were investigated on the basis of density functional theory calculations. The BO2 ligands prefer to stretch out in the most stable Sc(BO2)n− anions but tend to come together in the lowest-lying Sc(BO2)4 structure. According to the MP2 results, the Sc(BO2)4− species could be classified as hyperhalogen anions since they have larger vertical electron detachment energies (VDEs, 5.44–8.85 eV) than that of the superhalogen anion BO2−. With titanium and vanadium playing the role of central atom, the Ti(BO2)n−/0 (n = 1–5) and V(BO2)n−/0 (n = 1–6) clusters were studied in a similar manner. In these cases, the central transition metal atoms are inclined to keep their intrinsic spin. In addition, the hyperhalogen identity of the Ti(BO2)n− (n = 4, 5) and V(BO2)n− (n = 3–6) species were also confirmed by the calculated VDE values.
1. Introduction
It is well-known that halogen atoms possess the highest electron affinities (EAs) in the periodic table, and the chlorine atom possesses the maximum value, namely 3.61 eV.1 Atoms and clusters with high EA usually show remarkable oxidation capacity in chemical reactions. Bartlett and co-workers found that PtF6 could even oxidize a Xe atom,2 and then confirmed that the PtF6 molecule possesses a very high EA of 6.8 eV.3 Such molecules that have higher EAs than the chlorine atom were termed superhalogen by Boldyrev and Gutsev in 1981.4 Besides, they recommended a simple formula MXk+1 for constructing superhalogens, where M is a main group or transition metal atom, X is a halogen atom, and k is the maximal formal valence of M.4 The first experimental evidence of such species, namely MX2− (M = Li, Na, and X = Cl, Br, I), was obtained by Wang's group in 1999.5 Afterwards, an increasing number of superhalogen anions have been theoretically predicted or experimentally detected, such as MX3− (M = Be, Mg, Ca; X = Cl, Br)6–8 and MCl4− (M = Sc, Y, La).9 Recently, many kinds of superhalogens that are beyond the MXk+1 formula have been proposed and characterized. To facilitate extra-electron delocalization, multinuclear superhalogen anions have been designed such as P2F11− and As2F11−,10 Mg2F5−,11 Al2F7−,10 and H12F13−.12 As a result, higher vertical electron detachment energy can be achieved. By introducing non-halogen ligands, the scope of superhalogen has been further extended to include the chalcogen-based MnO4,13 CrO4,14 BO2,15 AlO2,16 VO3,16 BrO3,17 IO3,17 IO4,17 BS2,18 and BSO18 molecules, and those involving electrophilic substituent,19 organic group,20 and acidic functional group21 as ligands.Superhalogens are strong electron-acceptors and can be used to oxidize systems with high ionization potential, such as benzene22 and small water clusters.23 They may combine with superalkalis to form ionic compounds that are predicted to possess excellent nonlinear optical response.24 Recent studies have also shown that MnCl3, one of the experimentally synthesized superhalogens, could be used to tune the electronic and magnetic properties of silicone,25 and that the AlF4 superhalogen can initiate a radical-substitution chain reaction as the trigger-compound.26 In 2015, Jena predicted that superhalogens could serve as a bridge between complex metal hydrides and electrolytes in Li-ion battery.27 Soon after, the suitability of superhalogen salt for Mg battery electrolyte was experimentally proved.28 The extensive application prospects of superhalogens make them promising agents in chemistry and material science and attract more and more attention.29,30
Hyperhalogens, another series of electronegative clusters whose EA values are even higher than those of their superhalogen ligands, were proposed by Jena and coworkers in 2010.31 Au(BO2)2 was reported as the first member of this type of molecules. Thereafter, the strategy of using superhalogen as ligands was employed to design a great many high EA species, e.g., Cun(BO2)m (n, m = 1, 2),32 Al(BO2)n,33 Ag(BO2)2,34 Na(BF4)2,35 Mg(BF4)3,36 Al(BF4)4,37 Al(BH4)4,37 and M(IO3)2− (M = H, Li, Na, K).38 Furthermore, Jena et al. proposed the concept of “magnetic hyperhalogens” by studying the Fe(BO2)n and Mn(BO2)n clusters.39 Recently, two series of hyperhalogen anions involving planar and cage-like superhalogen ligands were reported, respectively, and the concept of aromatic hyperhalogen was brought forward as well.40,41
In this work, theoretical investigation of borates of the first three subgroup elements, namely scandium, titanium, and vanadium, was presented. On the one hand, these transition metal (TM) elements have unfilled d orbitals, which may render the resulting compounds magnetism. On the other hand, they have multivalent properties, so it is interesting to make clear the minimum number of BO2 ligands they need to exhibit hyperhalogen character with. Besides, our study also aims to (1) reveal the geometrical feature of the resulting Sc(BO2)n−/0 (n = 1–4), Ti(BO2)n−/0 (n = 1–5), and V(BO2)n−/0 (n = 1–6) clusters; (2) detect their stability through examining their dissociation energies of predetermined dissociation pathways; (3) explore the spin state evolution of central atoms and VDE values of the TM(BO2)n− (TM = Sc, Ti, V) anions.
2. Computational details
With different spin multiplicity taken into account, the optimized geometries of the Sc(BO2)n−/0 (n = 1–4), Ti(BO2)n−/0 (n = 1–5), and V(BO2)n−/0 (n = 1–6) species were obtained by using the M06 functional42 of density functional theory. Frequency and natural bond orbital (NBO) analyses43 were performed at the same level. Single point energies were computed by using the second-order Møller–Plesset (MP2) method. For all calculations, the 6-311+G(3df) basis set was used for B and O atoms, while the Los Alamos set of the double-zeta type LANL2DZ basis set and effective core potential (ECP) were used for the Sc, Ti, and V atoms.The vertical detachment energies (VDEs) of the Sc(BO2)n− (n = 1–4), Ti(BO2)n− (n = 1–5), and V(BO2)n− (n = 1–6) anions were obtained by two methods. First, the VDE values were indirectly computed as the energy difference between the neutral and the anion both at the anion's geometry by using the MP2 method. Second, the restricted outer valence Green function (OVGF)44–46 method was used to estimate the VDE values. For all investigated anions, the pole strengths (PSs) are greater than 0.85, justifying the validity of OVGF approximation.47 For comparison, the adiabatic detachment energy (ADE) of each anion was also obtained by using the MP2 method, which was computed as the difference in total energy between the neutral and anion at their respective optimized geometries.
All calculations were performed using the GAUSSIAN 09 program package.48 Dimensional plots of the molecular structures were generated with the GaussView program.49
3. Results and discussion
3.1 Sc(BO2)n− and Sc(BO2)n (n = 1–4)
 | ||
| Fig. 1 Equilibrium structures and critical bond lengths (in Å) of the Sc(BO2)n− (n = 1–4) clusters. (Color legend: Sc, grey; O, red; B, light pink. Symmetry in parentheses.) | ||
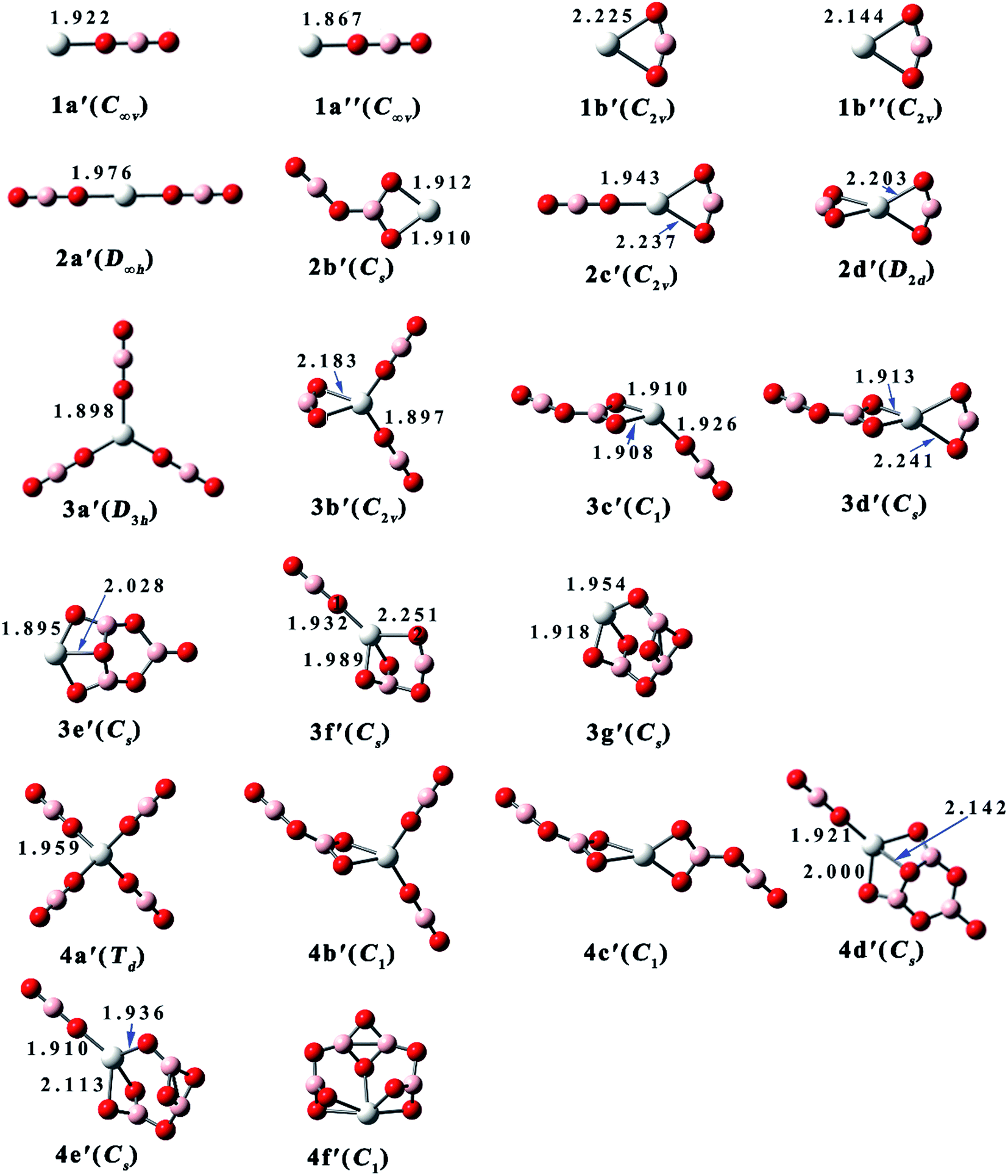 | ||
| Fig. 2 Equilibrium structures and critical bond lengths (in Å) of the Sc(BO2)n (n = 1–4) clusters. (Color legend: Sc, grey; O, red; B, light pink. Symmetry in parentheses.) | ||
| Cluster | Isomer | Erel | VDEOVGF | VDEMP2 | ADE (MP2) | D1 | D2 |
|---|---|---|---|---|---|---|---|
| Sc(BO2)− | 1a | 0.0 | 1.70 (0.888) | 1.41 | 1.23 | 53.6 | 163.9 |
| 1b | 17.6 | 1.21 (0.896) | 1.28 | 1.26 | 31.7 | 142.0 | |
| Sc(BO2)2− | 2a | 0.0 | 2.21 (0.882) | 1.94 | 1.91 | 82.0 | 149.7 |
| 2b | 15.6 | 1.68 (0.881) | 1.56 | 1.50 | 64.6 | 132.3 | |
| 2c | 21.7 | 1.44 (0.852) | 1.59 | 1.27 | 63.0 | 130.8 | |
| 2d | 44.1 | 0.80 (0.817) | 0.82 | 0.79 | 43.3 | 111.1 | |
| Sc(BO2)3− | 3a | 0.0 | 2.94 (0.940) | 2.59 | 2.45 | 94.3 | 134.8 |
| 3b | 9.0 | 3.87 (0.933) | 3.46 | 2.35 | 84.7 | 125.2 | |
| 3c | 13.2 | 2.36 (0.946) | 2.10 | 2.03 | 83.3 | 123.8 | |
| 3d | 20.2 | 3.40 (0.933) | 3.02 | 1.90 | 73.8 | 114.3 | |
| 3e | 27.1 | 3.19 (0.980) | 2.97 | 2.84 | 61.5 | 102.0 | |
| 3f | 27.3 | 3.57 (0.934) | 3.21 | 1.75 | 66.4 | 106.9 | |
| 3g | 36.5 | 2.00 (0.977) | 1.84 | 1.77 | 53.0 | 93.5 | |
| Sc(BO2)4− | 4a | 0.0 | 9.08 (0.917) | 8.69 | 8.67 | 103.0 | 142.7 |
| 4b | 6.9 | 8.40 (0.915) | 8.85 | 7.09 | 98.6 | 138.4 | |
| 4c | 17.5 | 7.87 (0.913) | 7.98 | 6.63 | 88.6 | 128.4 | |
| 4d | 23.5 | 5.82 (0.929) | 5.44 | 4.45 | 79.9 | 119.7 | |
| 4e | 30.2 | 6.93 (0.922) | 7.08 | 5.64 | 74.3 | 114.1 | |
| 4f | 49.2 | 6.02 (0.921) | 6.02 | 4.91 | 54.3 | 94.1 |
| Cluster | Isomer | Erel | D′1 | D′2 |
|---|---|---|---|---|
| Sc(BO2) | 1a′-t | 0.0 | 112.2 | — |
| 1a′-s | 10.2 | 121.3 | — | |
| 1b′-t | 19.8 | 99.4 | — | |
| 1b′-s | 28.4 | 108.8 | — | |
| Sc(BO2)2 | 2a′ | 0.0 | 122.5 | 206.8 |
| 2b′ | 6.1 | 124.0 | 208.3 | |
| 2c′ | 6.8 | 120.4 | 204.7 | |
| 2d′ | 18.1 | 111.7 | 196.1 | |
| Sc(BO2)3 | 3a′ | 0.0 | 130.7 | 207.7 |
| 3b′ | 6.8 | 125.3 | 202.2 | |
| 3c′ | 3.5 | 132.5 | 209.4 | |
| 3d′ | 7.5 | 127.6 | 204.5 | |
| 3e′ | 35.9 | 101.3 | 178.2 | |
| 3f′ | 11.2 | 122.4 | 199.3 | |
| 3g′ | 20.8 | 117.6 | 194.5 | |
| Sc(BO2)4 | 4a′ | 73.8 | 33.7 | 120.7 |
| 4b′ | 44.4 | 44.3 | 131.3 | |
| 4c′ | 44.3 | 46.1 | 133.1 | |
| 4d′ | 0.0 | 82.1 | 169.1 | |
| 4e′ | 34.2 | 50.6 | 137.6 | |
| 4f′ | 36.4 | 54.1 | 141.1 |
As shown in Fig. 1, isomer 1a of ScBO2− is linear and similar to the reported structures of FeBO2, MnBO2, and MnBO2−.39 The other isomer 1b has C2v symmetry, where the BO2 ligand bends to 156.6° and binds with the Sc atom via two O atoms. Both 1a and 1b have magnetic moment of 1 μB. However, the neutral ScBO2 (see Fig. 2) has variable magnetism. The most stable isomer 1a′ of ScBO2 has a magnetic moment of 2 μB, which is similar to the case of ScCl and ScF.50 Isomer 1a′′ also has a linear configuration, only it has a magnetic moment of 0 μB and slightly shorter Sc–O bond length of 1.867 Å. As for 1b′ and 1b′′, which share similar structural features with 1b, they possess different Sc–O bond lengths (2.225 Å and 2.114 Å, respectively) and magnetic moments (2 μB and 0 μB, respectively).
As one more BO2 ligand is introduced, four isomers have been obtained for both anionic Sc(BO2)2− and neutral Sc(BO2)2. The spin multiplicities of Sc(BO2)2− and Sc(BO2)2 are 1 and 2, respectively. 2a, the lowest-energy isomer of Sc(BO2)2−, has a linear structure, in which each BO2 ligand is attached to the middle Sc atom through a Sc–O bond. The same case was reported for the ground states of Mn(BO2)2− and Fe(BO2)2−.39 The Sc–O bond length (1.979 Å) of 2a is slightly shorter than that of 1a (2.006 Å). Isomer 2b has a planar structure, where the two BO2 ligands combine with each other, forming a B2O4 unit that shows a similar structure to the recently reported (BO2)2− anion.51 In the next isomer 2c, two BO2 ligands bind with the Sc atom via one and two Sc–O bonds, respectively. The ∠O1B1O2 angle of 2c is 147.5°, indicating a larger ligand distortion relative to structure 1b. As for the last isomer 2d, it has a D2d-symmetric structure where each BO2 unit is linked to the center Sc atom by two Sc–O bonds. The Sc atom is coplanar with each BO2 ligand in this structure and the two BO2 planes are perpendicular to each other. The ∠OBO angle in 2d is 148.5°, which is close to that of 2c. From Fig. 2 and Table 2, the neutral Sc(BO2)2 isomers share similar structures and the same total energy order with their corresponding anions.
Sc(BO2)3− and Sc(BO2)3 have magnetic moments of 1 μB and 0 μB, respectively. Seven structures were obtained for Sc(BO2)3−, and the D3h-symmetric structure 3a is the most stable one. In 3a, each BO2 ligand binds with the center Sc atom via one Sc–O bond of 1.990 Å. In contrast, a ScOBO quadrilateral forms in the rest isomers. The C2v-symmetric isomer 3b can be viewed as one more BO2 ligand attaching to structure 2c, which is supported by NBO analysis. Both isomers 3c and 3d can be obtained by adding a BO2 ligand to structure 2b. The difference is that the additional BO2 ligand bonds with the Sc atom via one O atom in the former but via two O atoms in the latter. In C2v-symmetric 3e, three BO2 ligands get together and form a B3O6 unit, which is similar to the structure of (BO2)3−.51 In the next isomer 3f, a B2O4 unit forms and is linked to Sc through three Sc–O bonds. The least favorable structure of Sc(BO2)3− is 3g with Cs symmetry, which contains a different B3O6 unit compared with 3e. As for neutral Sc(BO2)3, all isomers possess a similar structure to their corresponding anions but their stability follows a different order. The largest structural difference is found between 3c and its neutral counterpart, namely 3c′ where one of the BO2 ligands deviates 45.7° from the horizontal.
Six optimized configurations (μB = 0) were obtained for Sc(BO2)4− and all of them can be considered derived from the Sc(BO2)3− structures. The Td-symmetric 4a is the most stable isomer of Sc(BO2)4−, in which each BO2 ligand connects to the Sc atom via one O atom, forming four Sc–O bonds of 1.958 Å. The four BO2 ligands keep away from each other and retain their linear geometry in this isomer. In contrast, two or more BO2 units are combined together in the higher-lying isomers. Isomer 4b lies 6.9 kcal mol−1 higher in energy than 4a. It can be obtained by attaching one more BO2 ligand to the vertex boron atom of 3b. With two B2O4 units linked to the Sc atom separately, the C2-symmetric 4c is generated. A trimeric BO2 (B3O6) unit forms in both 4d and 4e isomers, and the energy difference between them is 6.7 kcal mol−1. From Fig. 1, 4d and 4e basically inherit the structures of 3e and 3g, respectively, apart from an additional BO2 ligand being attached to the Sc atom of their precursors. In the least stable isomer 4f, four BO2 ligands polymerize to form a tetrameric B4O8, which is bound to the Sc atom through five Sc–O bonds. Note that all anionic Sc(BO2)4− have corresponding neutral Sc(BO2)4 structures (μB = 1), only the latter has different total energy order from the former. For instance, the most stable isomer of Sc(BO2)4, namely 4d′, has a similar structure to 4d, a low-lying isomer of Sc(BO2)4−. Meanwhile, the lowest-energy isomer of Sc(BO2)4− is corresponding to the least favorable configuration (4a′) of Sc(BO2)4. This reflects the fact that the loss of extra electron only slightly affects geometry of the studied anions, but varies their relative stability.
For comparison, the previously reported VDE values of Al(BO2)n−,33 Sc(BH4)n−,52 and ScFn− (ref. 50) are shown in ESI.† Given that ScF4− is a superhalogen anion with halogen as ligands, it is not surprising that it possesses a smaller VDE value of 7.74 eV than that of Sc(BO2)4− (8.69 eV in the present work). Note that Sc(BO2)4− also outperforms other hyperhalogen anions. From Table S1,† the VDE value of Sc(BO2)4− is much larger than that of Sc(BH4)4− (6.47 eV), although BO2− has a smaller VDE value compared with BH4− (4.57 eV). Likewise, Sc(BO2)4− is probably a stronger oxidizing agent than Al(BO2)4− (whose VDE = 8.28 eV) where a trivalent main group atom plays the role of central core.
3.2 Ti(BO2)n− (n = 1–5) and V(BO2)n− (n = 1–6)
Next to Sc, the Ti and V atoms have electron configurations of [Ar]3d24s2 and [Ar]3d34s2 and possess maximum valence of +4 and +5, respectively. To reveal how many BO2 ligands Ti and V require to qualify their borates for being classified as hyperhalogens, the evolution of VDE values of the Ti(BO2)n− (n = 1–5) and V(BO2)n− (n = 1–6) systems were also studied in this work. Based on above analysis, all the lowest-lying Sc(BO2)n− isomers feature a structure where the BO2 ligands spread apart and each binds with the central Sc atom through a Sc–O bond. In view of this, only the isomers with separated BO2 ligands were considered for the Ti(BO2)n− (n = 1–5) and V(BO2)n− (n = 1–6) anions. These structures, together with their corresponding neutral ones, were optimized by the M06 method. Different spin multiplicities were taken into account during optimizations. Note that the neutral Ti(BO2)n (n = 1–5) and V(BO2)n (n = 1–6) configurations were obtained on the basis of their corresponding anionic structures instead of a thorough structure searching. They are possibly local, but not global minima on the potential energy surfaces.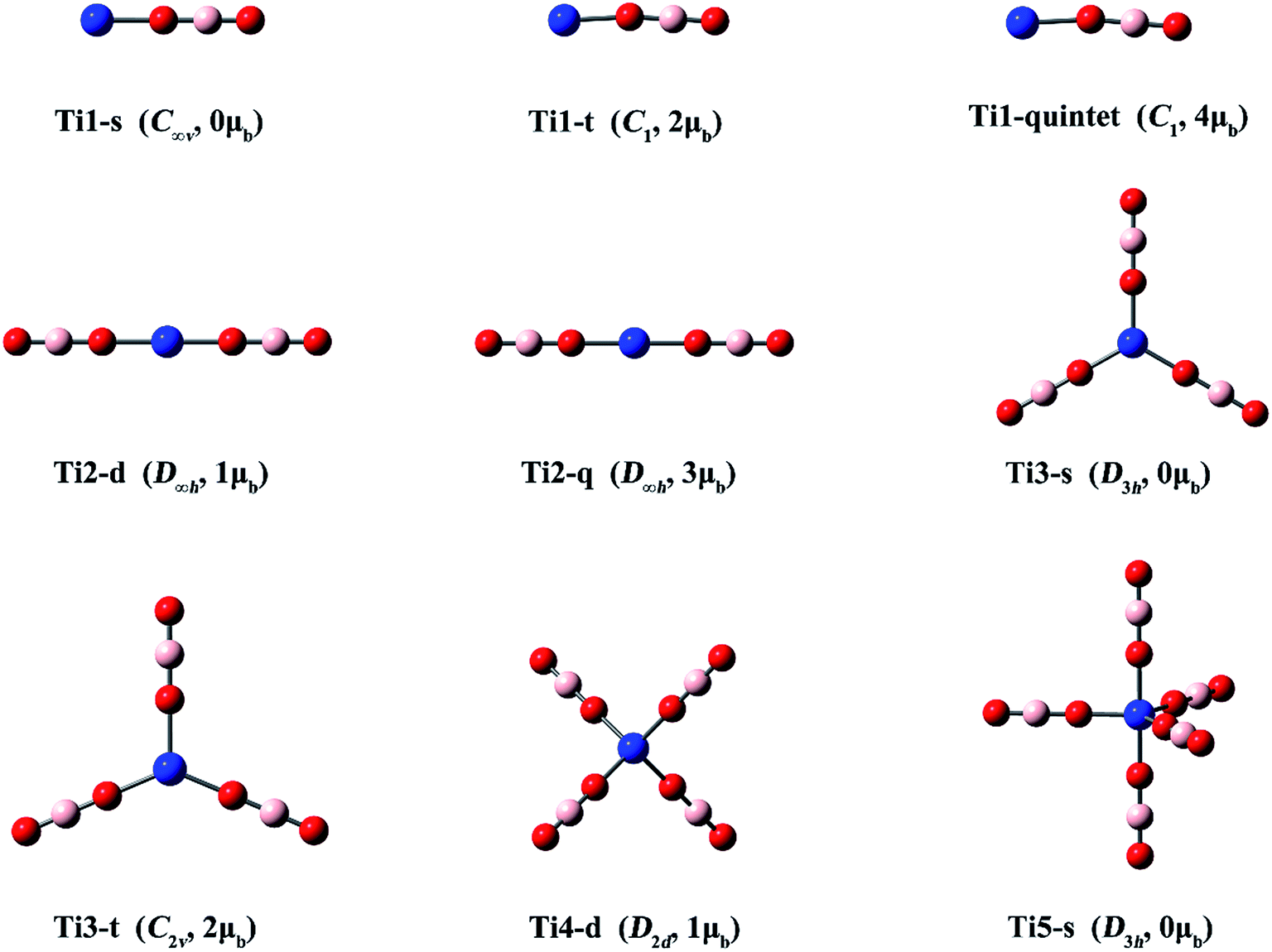 | ||
| Fig. 3 Optimized structures of the Ti(BO2)n− (n = 1–5) clusters. (Color legend: Ti, blue; O, red; B, light pink; symmetry and magnetic moment in parentheses.) | ||
 | ||
| Fig. 4 Optimized structures of the Ti(BO2)n (n = 1–5) clusters. (Color legend: Ti, blue; O, red; B, light pink; symmetry and magnetic moment in parentheses.) | ||
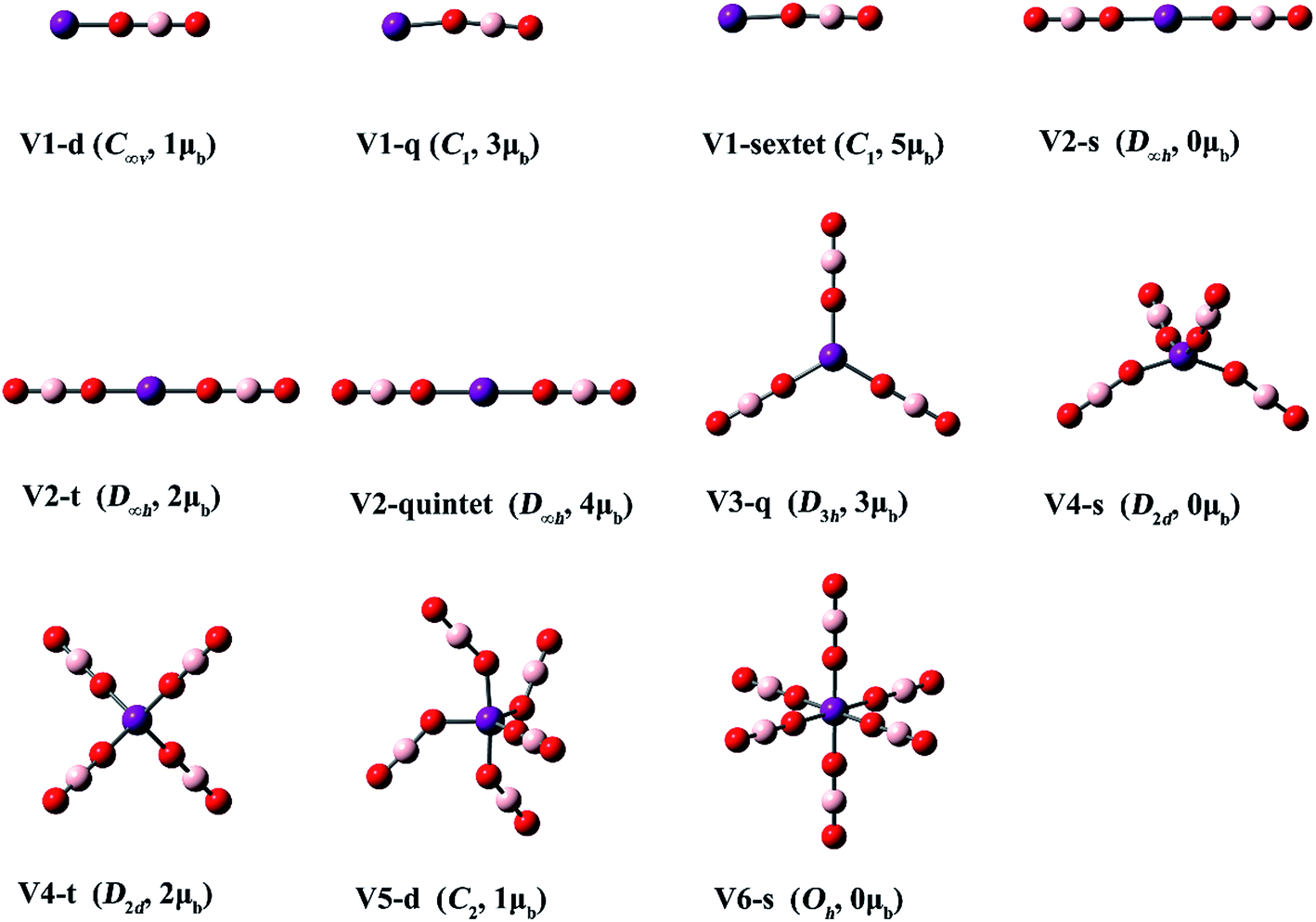 | ||
| Fig. 5 Optimized structures of the V(BO2)n− (n = 1–6) clusters. (Color legend: V, purple; O, red; B, light pink; symmetry and magnetic moment in parentheses.) | ||
 | ||
| Fig. 6 Optimized structures of the V(BO2)n (n = 1–6) clusters. (Color legend: V, purple; O, red; B, light pink; symmetry and magnetic moment in parentheses.) | ||
Three structures with different spin states (singlet, triplet, quintet) were found for Ti(BO2)−. The singlet isomer is linear, while the other two are slightly bent. It can be found from Fig. 3 and Table 3 that, the isomer where the Ti atom maintains its spin is the most stable, followed by that in a higher spin state, while the one with a total spin state of zero is the least favorable. For example, the stability order is Ti1-t > Ti1-quintet > Ti1-s. The same is valid for the Ti(BO2)2− and Ti(BO2)3− isomers. That is, the Ti2-q and Ti3-t isomers with two unpaired d electrons from Ti are 3.0 and 17.3 kcal mol−1 more stable than Ti2-d and Ti3-s, respectively. Due to Jahn–Teller effect, Ti3-t has C2v instead of D3h symmetry and the symmetry of Ti4-d is lowered to D2d compared with the lowest-lying structure of Sc(BO2)4−. In contrast, Ti3-s and its corresponding neutral Ti3′-d hold D3h symmetry. Likewise, Ti5-s possesses a trigonal bipyramidal geometry with D3h symmetry. The structures of neutral Ti(BO2)n basically resemble those of their corresponding anions. The only exception here is Ti5′-d, in which two BO2 ligands are combined together.
| Species | Erel | VDEOVGF | VDEMP2 | ADEMP2 | De |
|---|---|---|---|---|---|
| Ti1-s | 23.5 | 1.57 (0.885) | 1.25 | 0.66 | 28.5 |
| Ti1-t | 0.0 | 1.77 (0.864) | 1.76 | 1.68 | 53.4 |
| Ti1-quintet | 9.9 | 1.26 (0.922) | 0.62 | 0.61 | 26.3 |
| Ti2-d | 3.0 | 3.08 (0.815) | 3.67 | 3.58 | 141.6 |
| Ti2-q | 0.0 | 2.06 (0.960) | 1.65 | 1.61 | 136.1 |
| Ti3-s | 17.3 | 2.95 (0.878) | 2.58 | 2.07 | 126.9 |
| Ti3-t | 0.0 | 4.20 (0.922) | 3.71 | 2.82 | 130.1 |
| Ti4-d | — | 6.66 (0.889) | 4.86 | 4.11 | 126.3 |
| Ti5-s | — | 9.23 (0.911) | 9.30 | 6.29 | 94.1 |
| V1-d | 71.7 | 2.01 (0.854) | 0.93 | 0.91 | 14.6 |
| V1-q | 0.0 | 1.95 (0.858) | 3.03 | 2.98 | 59.5 |
| V1-sextet | 5.8 | 1.49 (0.983) | 0.97 | 0.86 | 45.6 |
| V2-s | 44.3 | 2.49 (0.849) | 2.54 | 2.44 | 107.5 |
| V2-t | 24.6 | 4.46 (0.811) | 3.62 | 3.30 | 139.5 |
| V2-quintet | 0.0 | 2.38 (0.957) | 2.11 | 2.06 | 134.2 |
| V3-q | — | 5.18 (0.908) | 5.10 | 4.02 | 127.9 |
| V4-s | 49.7 | 7.38 (0.882) | 7.23 | 3.68 | 68.1 |
| V4-t | 0.0 | 8.44 (0.906) | 8.72 | 5.84 | 95.5 |
| V5-d | — | 9.09 (0.909) | 9.27 | 5.62 | 93.6 |
| V6-s | — | 9.44 (0.910) | 8.23 | 5.87 | 46.2 |
From Fig. 5 and Table 3, the V atom maintains its spin in the lowest-lying isomer of V(BO2)−, namely V1-q. Consequently, it has a magnetic moment of 3 μB. High-spin isomer V1-sextet and low-spin isomer V1-d are 5.8 and 71.7 kcal mol−1 higher in energy than V1-q, respectively. All three structures of V(BO2)2− are linear with D∞h symmetry. The magnetic moment is 4 μB for the lowest-energy configuration V2-quintet. The electronic structure characteristics of both Ti(BO2)n− and V(BO2)n− species indicate that the central transition metal atoms are inclined to keep the intrinsic electronic state in their borates. For V(BO2)n− (n = 3–6), the BO2 ligands are distributed individually in each cluster. Obviously, Jahn–Teller distortion also appears in the V(BO2)n− series. The geometry of V3-q is still of D3h-symmetry. In contrast, both V4-t and V4-s are distorted to D2d-symmetry as the V atom possesses two d electrons therein. V5-d is lowered to C2-symmetry because the V atom has one d electron left. Similar to the case of 4a and Ti5-s, V6-s does not have d electron from the central metal atom, so it appears to be a regular octahedron with Oh-symmetry. As for the neutral V(BO2)n clusters, the BO2 ligands are also isolated in every structure except that two BO2 ligands dimerized in V6′-d.
From Table 3, the zero-point-corrected dissociation energies of Ti(BO2)n− and V(BO2)n− are in the range of 26.3–141.6 kcal mol−1 and 14.6–139.5 kcal mol−1, respectively, indicating the stability of Ti(BO2)n− and V(BO2)n− with respect to emission of a BO2 unit.
4. Conclusions
The structural, electronic and magnetic properties of three series of early-transition-metal borates, namely Sc(BO2)n−/0 (n = 1–4), Ti(BO2)n−/0 (n = 1–5), and V(BO2)n−/0 (n = 1–6), were studied by performing density functional theory and ab initio calculations. In view of the positive dissociation energies, all studied anionic clusters are stable against the loss of a BO2 ligand.The lowest-lying Sc(BO2)n− isomers feature a structure where the BO2 ligands spread apart, whereas the BO2 ligands begin to combine into a trimer in the lowest-energy Sc(BO2)4 structure. This can be related to the maximum valence state of +3 of the Sc atom. By the same token, there is a sharp increase in VDE value from Sc(BO2)3− to Sc(BO2)4−, and the latter species can be classified as hyperhalogens since they possess larger VDE values than that of its superhalogen ligand BO2−. As for Ti and V atoms, they require four and three BO2 ligands, respectively, to enable their borates to be termed hyperhalogen. Besides, the Ti4-d, V3-q, V4-t, and V5-d species can be considered as magnetic hyperhalogens owing to their nonzero magnetic moment.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21603032, 21375017), State Key Development Program for Basic Research of China (Grant No. 2013CB834801), Natural Science Foundation of Fujian Province (Grant No. 2016J05032), and MiaoPu Foundation of Fujian Medical University (Grant No. 2015MP034).References
- H. Hotop and W. C. Lineberger, J. Phys. Chem. Ref. Data, 1985, 14, 731 CrossRef CAS.
- N. Bartlett, Proc. Chem. Soc., London, 1962, 6, 218 Search PubMed.
- N. Bartlett and D. H. Lohmann, J. Chem. Soc., 1962, 5253 RSC.
- G. L. Gutsev and A. I. Boldyrev, Chem. Phys., 1981, 56, 277 CrossRef CAS.
- X.-B. Wang, C.-F. Ding, L.-S. Wang, A. I. Boldyrev and J. Simons, J. Chem. Phys., 1999, 110, 4763 CrossRef CAS.
- I. Anusiewicz and P. Skurski, Chem. Phys. Lett., 2002, 358, 426 CrossRef CAS.
- I. Anusiewicz, M. Sobczyk, I. Dąbkowska and P. Skurski, Chem. Phys., 2003, 291, 171 CrossRef CAS.
- B. M. Elliott, E. Koyle, A. I. Boldyrev, X.-B. Wang and L.-S. Wang, J. Phys. Chem. A, 2005, 109, 11560 CrossRef CAS PubMed.
- J. Yang, X.-B. Wang, X.-P. Xing and L.-S. Wang, J. Chem. Phys., 2008, 128, 201102 CrossRef PubMed.
- M. Sobczyk, A. Sawicka and P. Skurski, Eur. J. Inorg. Chem., 2003, 2003, 3790 CrossRef.
- I. Anusiewicz and P. Skurski, Chem. Phys. Lett., 2007, 440, 41 CrossRef CAS.
- S. Freza and P. Skurski, Chem. Phys. Lett., 2010, 487, 19 CrossRef CAS.
- G. L. Gutsev, B. K. Rao, P. Jena, X.-B. Wang and L.-S. Wang, Chem. Phys. Lett., 1999, 312, 598 CrossRef CAS.
- G. L. Gutsev, P. Jena, H.-J. Zhai and L.-S. Wang, J. Chem. Phys., 2001, 115, 7935 CrossRef CAS.
- H.-J. Zhai, L.-M. Wang, S.-D. Li and L.-S. Wang, J. Phys. Chem. A, 2007, 111, 1030 CrossRef CAS PubMed.
- K. Pradhan and P. Jena, J. Chem. Phys., 2011, 135, 144305 CrossRef PubMed.
- H. Wen, G.-L. Hou, W. Huang, N. Govind and X.-B. Wang, J. Chem. Phys., 2011, 135, 184309 CrossRef PubMed.
- L.-J. Zhao, H.-G. Xu, G. Feng, P. Wang, X.-L. Xu and W.-J. Zheng, Phys. Chem. Chem. Phys., 2016, 18, 6175 RSC.
- I. Anusiewicz, J. Phys. Chem. A, 2009, 113, 6511 CrossRef CAS PubMed.
- J. Zhang, P. Yang, Z.-R. Sun and X.-B. Wang, J. Phys. Chem. A, 2014, 118, 8074 CrossRef CAS PubMed.
- I. Anusiewicz, J. Phys. Chem. A, 2009, 113, 11429 CrossRef CAS PubMed.
- M. Czapla, S. Freza and P. Skurski, Chem. Phys. Lett., 2015, 619, 32 CrossRef CAS.
- M. Marchaj, S. Freza, O. Rybacka and P. Skurski, Chem. Phys. Lett., 2013, 574, 13 CrossRef CAS.
- Y. Li, D. Wu and Z.-R. Li, Inorg. Chem., 2008, 47, 9773 CrossRef CAS PubMed.
- T. Zhao, S. Zhang, Q. Wang, Y. Kawazoe and P. Jena, Phys. Chem. Chem. Phys., 2014, 16, 22979 RSC.
- M. Marchaj, S. Freza and P. Skurski, Chem. Phys. Lett., 2014, 612, 172 CrossRef CAS.
- S. Giri, S. Behera and P. Jena, Angew. Chem., Int. Ed., 2014, 126, 14136 CrossRef.
- O. Tutusaus, R. Mohtadi, T. S. Arthur, F. Mizuno, E. G. Nelson and Y. V. Sevryugina, Angew. Chem., Int. Ed., 2015, 54, 7900 CrossRef CAS PubMed.
- H. Zhao, J. Zhou, H. Fang and P. Jena, ChemPhysChem, 2016, 17, 184 CrossRef CAS PubMed.
- T.-s. Zhao, J. Zhou, Q. Wang and P. Jena, J. Phys. Chem. Lett., 2016, 7, 2689 CrossRef CAS PubMed.
- M. Willis, M. Götz, A. K. Kandalam, G. F. Ganteför and P. Jena, Angew. Chem., Int. Ed., 2010, 49, 8966 CrossRef CAS PubMed.
- Y. Feng, H.-G. Xu, W. Zheng, H. Zhao, A. K. Kandalam and P. Jena, J. Chem. Phys., 2011, 134, 094309 CrossRef PubMed.
- G. L. Gutsev, C. A. Weatherford, L. E. Johnson and P. Jena, J. Comput. Chem., 2012, 33, 416 CrossRef CAS PubMed.
- H. Chen, X.-Y. Kong, W. Zheng, J. Yao, A. K. Kandalam and P. Jena, ChemPhysChem, 2013, 14, 3303 CrossRef CAS PubMed.
- C. Paduani and P. Jena, J. Phys. Chem. A, 2012, 116, 1469 CrossRef CAS PubMed.
- C. Paduani and P. Jena, J. Nanopart. Res., 2012, 14, 1035 CrossRef.
- C. Paduani, M. M. Wu, M. Willis and P. Jena, J. Phys. Chem. A, 2011, 115, 10237 CrossRef CAS PubMed.
- G.-L. Hou, M. M. Wu, H. Wen, Q. Sun, X.-B. Wang and W.-J. Zheng, J. Chem. Phys., 2013, 139, 044312 CrossRef PubMed.
- P. Koirala, K. Pradhan, A. K. Kandalam and P. Jena, J. Phys. Chem. A, 2013, 117, 1310 CrossRef CAS PubMed.
- W.-M. Sun, X.-H. Li, Y. Li, D. Wu, C.-Y. Li, J.-H. Chen and Z.-R. Li, ChemPhysChem, 2016, 17, 1468 CrossRef CAS PubMed.
- W.-M. Sun, D. Hou, D. Wu, X.-H. Li, Y. Li, J.-H. Chen, C.-Y. Li and Z.-R. Li, Dalton Trans., 2015, 44, 19901 RSC.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2006, 110, 13126 CrossRef CAS PubMed.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS.
- L. S. Cederbaum, J. Phys. B: At. Mol. Phys., 1975, 8, 290 CrossRef CAS.
- J. V. Ortiz, J. Chem. Phys., 1988, 89, 6348 CrossRef CAS.
- V. G. Zakrzewski, W. v. Niessen, A. I. Boldyrev and P. v. R. Schleyer, Chem. Phys., 1993, 174, 167 CrossRef CAS.
- V. G. Zakrzewski, O. Dolgounitcheva and J. V. Ortiz, J. Chem. Phys., 1996, 105, 5872 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. B. G. Scalmani, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta Jr, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09, Revision D.01, Gaussian, Inc, Wallingford CT, 2009 Search PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView, Version 5.09, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- K. Pradhan, G. L. Gutsev and P. Jena, J. Chem. Phys., 2010, 133, 144301 CrossRef PubMed.
- A. K. Kandalam, B. Kiran, P. Jena, S. Pietsch and G. Gantefor, Phys. Chem. Chem. Phys., 2015, 17, 26589 RSC.
- Y.-Z. Liu, J. Zhou and P. Jena, J. Phys. Chem. C, 2015, 119, 11056 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Selected molecular orbitals of the studied borate anions, and VDE values of some related cluster anions. See DOI: 10.1039/c7ra10238k |
| This journal is © The Royal Society of Chemistry 2017 |