
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid and sensitive detection of trace malachite green and its metabolite in aquatic products using molecularly imprinted polymer-coated wooden-tip electrospray ionization mass spectrometry
Yanying Huang†
a,
Yanfang Ma†b,
Huawen Hu† a,
Pengran Guob,
Lei Miaoa,
Yunyun Yang*b and
Min Zhang*a
a,
Pengran Guob,
Lei Miaoa,
Yunyun Yang*b and
Min Zhang*a
aSchool of Materials Science and Energy Engineering, Foshan University, Foshan 528000, China. E-mail: zhangmin@gic.ac.cn
bGuangdong Engineering and Technology Research Center for Ambient Mass Spectrometry, Guangdong Provincial Key Laboratory of Emergency Test for Dangerous Chemicals, Guangdong Institute of Analysis (China National Analytical Center Guangzhou), 100 Xianlie Middle Road, Guangzhou 510070, China. E-mail: yy_yang@vip.126.com
First published on 9th November 2017
Abstract
In this study, a molecularly imprinted polymer-coated wooden-tip (MIPCWT) electrospray ionization mass spectrometry (ESI-MS) method was developed for rapid and sensitive detection of trace malachite green (MG) and its metabolite in aquatic products. Such a method was realized by applying a silicone-modified acrylate molecularly imprinted emulsion (SMAMIE) onto the surface of wooden tips to specially design a MIPCWT solid-phase micro-extraction (SPME) probe for selective enrichment of MG and its metabolite from aquatic products. Subsequently, a high voltage and some spray solvent were applied to the MIPCWT SPME probe, and ESI was induced for direct MS analysis under ambient and open-air conditions. The MIPCWT-SPME probe exhibits a high enriching capacity of approximately 1500–2000 fold toward MG and leucomalachite green (LMG), with detection limit reaching 0.01 μg L−1. In addition, a good linearity is obtained for both MG and LMG, with correlation coefficient values (R2) of no less than 0.998. The present method was successfully applied to analyze MG and LMG in real-life tap water, river water and fish samples, and good recoveries in the range of 93–103%, 92–108% and 106–113%, respectively, were found. All of these demonstrated that our developed MIPCWT-ESI-MS method holds great potential for rapid, direct, sensitive, and reliable detection and analysis of trace veterinary drug residues in aquatic products.
Introduction
Various industries relating to chemical synthesis (such as textiles, ceramics, printing, leather and plastics) are discharging an increasing quantity of effluents containing dye wastes. The widespread use of synthetic dyes, as one of the most important and concerning categories of contaminants nowadays, poses a serious threat to the aquaculture environment, due to their hazardous nature, high stability and complex aromatic structures.1,2 Malachite Green (MG), also known as aniline green and basic green 4, etc., is a cationic azo-compound with three aryl groups. MG is classified as a Class II Health Hazard by reason of its toxicity to human cells, induction of liver tumor formation, and potential risk to aquatic life.3,4 When injected into an organism, MG can be readily converted to leucomalachite green (LMG), which is most likely to accumulate in the serum, liver, kidney, muscle, fertilized eggs and other tissues of the organism, causing teratogenic, carcinogenic and mutagenic potential to humans.5–7 On the other hand, owing to its high efficiency and low cost, MG has been widely used as a biological repellent and insecticide in aquaculture industries because it is effective at treating aquatic mildew, gill mildew and parasitic diseases in aquatic products.8,9 Considering the growing importance and significance of environmental protection, exploration of rapid and sensitive methods for sensing and detection of various contaminants, including MG, has become a hot topic over recent decades.10,11 However, it remains a great challenge to develop a novel, sensitive, and rapid method for MG detection, with cost-effective and portable probes developed as the key component. Herein, we report the very first example of a novel, rapid, sensitive, and low-cost method for detection of trace malachite green and its metabolite in aquatic products using molecularly imprinted polymer-coated wooden-tip (MIPCWT) based electrospray ionization mass spectrometry (ESI-MS).Liquid chromatography (LC) coupled with mass spectrometry (MS) is one of the most frequently used approaches to detect and quantify MG and its metabolite in different matrices, such as water, aquatic products, and fish feedstuffs. However, conventional LC-MS methods are generally labor-intensive and time-consuming, since multi-step sample pretreatments are needed for enrichment of trace analytes to a detectable level.8 Moreover, matrix interference needs to be eliminated to obtain better-resolved detection results, requiring additional steps and hence consuming more time. A protracted chromatographic separation is also required, rendering this traditional method highly labor-intensive and time-consuming. It is therefore highly desirable to explore a method for rapid, direct, sensitive, and reliable analysis of MG and its metabolite in complex matrices.
Ambient MS gives such an opportunity. During the past two decades, a series of ambient ionization techniques, such as desorption electrospray ionization (DESI),12 extractive electrospray ionization (EESI),13–15 direct analysis in real time (DART),16–18 probe electrospray ionization (PESI),19,20 low-temperature plasma (LTP),21,22 desorption corona beam ionization (DCBI),23,24 laser ablation electrospray ionization (LAESI),25–27 paper spray,28–32 and wooden-tip ESI have been developed,33–40 which facilitate rapid, direct and straightforward analysis of various samples under ambient and open-air conditions with minimal or no sample pretreatment. However, MG and its metabolite usually present in the environment or aquatic products with a trace level of concentrations (around the μg L−1 level), and the compositions of matrices are also unknown with complexities. In this regard, it is rather difficult to directly detect MG and its metabolite using the existing ambient MS methods.
Solid-phase micro-extraction (SPME) coupled with ambient MS has been developed to directly detect complex samples with high sensitivity and low matrix interference.41–44 To date, SPME has been successfully coupled to several ambient ionization techniques, such as DESI,45 DART,46–48 LTP,49,50 and DCBI.23,24 In addition, direct ionization of SPME fibers,51 a SPME probe coupled with nanoelectrospray ionization (nanoESI),52–54 coated blade spray (CBS),55–57 and surface-coated wooden-tip electrospray ionization (SCWT-ESI)58,59 have been explored as novel strategies for rapid extraction of trace analytes for direct MS analysis of complex samples. Direct desorption/ionization can also be realized by MS analysis. Considering the complexity of aquatic products, SPME with higher selectivity and better stability should be developed.
Molecularly imprinted polymers (MIPs) have drawn significant attention recently owing to their specific recognition and enrichment of MG molecules, even in trace analysis.60–63 MIPs have been successfully used as cartridge column fillers for realizing solid-phase extraction (SPE) of MG in aquatic products in several previous studies.64–67 However, before the analytical process based on the high selectivity of MIPs and high sensitivity of modern instruments (e.g., HPLC or HPLC-MS), the initial sample preparation is generally complex and tedious, and involves multiple solvent extractions, centrifugation, rotary evaporation, solid phase extraction and elution.65,68 Even though advances have recently also been made in the fabrication of various kinds of MIPs for determination of MG in aquatic products, such as magnetic MIPs,68–71 CdTe quantum dots,8,72 carbon nanotubes73 or polymeric dopamine hydrochloride film,74 it remains time-consuming and labor-intensive to employ these MIPs in the detection of trace MG in aquatic products.
In this article, we report the very first example of exploring a molecularly imprinted polymer-coated wooden-tip electrospray ionization mass spectrometry (MIPCWT-ESI-MS) method (with cost-effective and portable probes developed as the key component) for rapid and sensitive analysis of MG and its metabolite in water and fish samples. We simplify the extraction and enrichment processing of MG by using a pinprick, in this case a wooden tip, through fish muscles for sampling. A MIPCWT-SPME probe that possesses imprinting cavities of MIPs with specific identification to MG is then specially designed for selective enriching and rapid detection of trace MG. The detection of trace MG from water and fish samples using a real-time direct analysis MS method is finally realized. In this study, a comparison is also made between the present method and many reported state-of-the-art approaches. The main content of this study is schematically illustrated in Fig. 1.
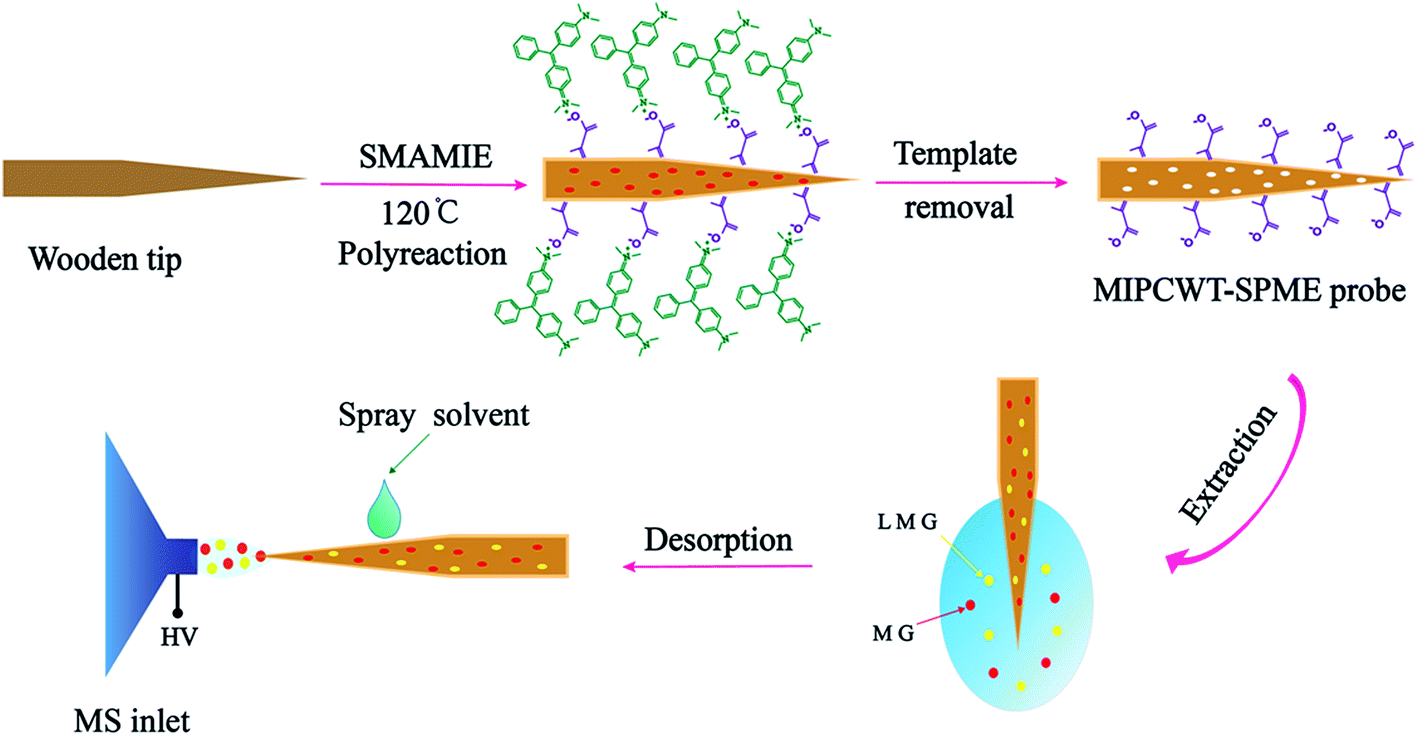 | ||
| Fig. 1 Schematic diagram showing the detection of MG and LMG in water by the present MIPCWT-ESI-MS method. | ||
Experimental
Materials and reagents
Wooden toothpicks (birch wood) were purchased from a PARKnSHOP supermarket in Hong Kong. Reference standards of MG, LMG and d5-MG were purchased from Witega (Berlin, Magnusstrasse 11, Germany). Thioflavin T (ThT) and rosolic acid were obtained from Shanghai Macklin Biochemical Co., Ltd (Shanghai, China). Tris(2-methoxyethoxy) vinylsilane (A-172) was provided by Fisher Scientific (Geel, Belgium). 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone (DDQ, 98% purity) was purchased from Sigma-Aldrich (St. Louis, MO, USA). High-purity chromatographic grade acetonitrile and methanol (MeOH) were obtained from Fisher Scientific (Geel, Belgium). Analytical grade methacrylic acid (AA), styrene (St), butyl acrylate (BA), methyl methacrylate (MMA), hydroxyropyl acrylate (HPA), sodium dodecyl sulfate (SDS), 2-ethylhexyl acrylate (2-EHA), polyoxyethlene octyphenol ether (OP-10), p-toluenesulfonic acid (p-TSA) and potassium persulfate (K2S2O8) were obtained from Aladdin (Southern California, L.A, USA). Sodium bicarbonate (NaHCO3), ammonium acetate (NH4Ac), ammonia, glacial acetic acid (HAc), L(+)-ascorbic acid, and sodium acetate anhydrous were supplied by Guangzhou Chemical Reagent Factory (Guangzhou, China). Deionized water (DI water) was purified on a Milli-Q water-purification system (Milford, MA, USA). Tap water and river water were collected from our laboratory and the Pearl River (Guangzhou, China), respectively. The butterfish sample was purchased from Wal-Mart Stores (Guangzhou, China).Standard solutions
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :47.5:5. A DDQ solution (0.001 M) was also adopted to assist in the mixing. Calibrants were used to generate a MG standard curve as for the fish sample. Calibration standards were prepared each time the experiment was carried out.
:47.5:5. A DDQ solution (0.001 M) was also adopted to assist in the mixing. Calibrants were used to generate a MG standard curve as for the fish sample. Calibration standards were prepared each time the experiment was carried out.Preparation of MIPCWT-SPME probes
000 rpm vigorous stirring for 15 min to form a pre-emulsion. In a four-necked round-bottom flask equipped with a mechanical stirrer, a reflux condenser, a temperature controller and an additional funnel, the 7.5 g of pre-emulsion, 13 mL of 1.2 mmol L−1 NaHCO3 solution and 2.5 mL of 0.11 mol L−1 K2S2O8 solution were added in the reactor and refluxed at 80 °C for 1 h to obtain a seed latex. Then, the remaining pre-emulsion and 2.5 mL of a 0.11 mol L−1 K2S2O8 solution were fed through an addition funnel over the course of 3–4 h. Afterwards, A-172 was added dropwise to the mixture over 30 min. After addition of all the monomers, the reaction mixture was maintained at 80 °C for an additional 1 h to complete the polymerization. Then, a stoichiometric amount of MG was added to the resulting aqueous emulsion after the mixture was cooled to room temperature, finally producing SMAMIE.:5, v/v), followed by MeOH, and then drying.Characterization of the prepared MIPCWT-SPME probe
Scanning electron microscope (SEM) and elemental mapping images analyses were performed on a S4800 field emission SEM instrument (Hitachi, Japan). Fourier transform-infrared spectroscopy (FTIR) analysis was conducted on a Cary 630 Fourier transform infrared spectrometer (Agilent Technologies, USA).Extraction
Detection
The loaded MIPCWT-SPME probe was mounted onto a three-dimensional moving stage, with the probe tip pointed to the MS inlet and adjusted to a position 10 mm away from the MS inlet.34 Afterwards, a high voltage of 4.5 kV released from the MS was applied to the probe. Subsequently, as a spray solvent, 10 μL of a mixture of MeOH and glacial acetic acid (99:1, v/v) was added onto the probe surface by a micropipette, for electrospray ionization mass spectrometric analysis.
Mass spectrometry
The mass spectrometric analysis was performed on a 6540 UHD Accurate-Mass Quadrupole Time-of-Flight mass spectrometer (Agilent Technologies, Santa Clara, USA). The ionization source was set up using the corresponding ESI configuration. The quadrupole time-of-flight mass spectrometer was operated in total transmission ion mode, with the experimental conditions shown in Table 1. Experimental control and MS data acquisition were conducted via Qualitative Analysis B.06.00 software (Agilent Technologies, Santa Clara, USA).| Analyte | Molecular formula | Parent ion (m/z) | Product ion (m/z) | Fragmentor (V) | Vcap (V) |
|---|---|---|---|---|---|
| a Internal standard. | |||||
| MG | C23H25ClN2 | 364.1706 | 329.2012 | 180 | 4500 |
| LMG | C23H26N2 | 330.2096 | 331.2168 | 180 | 4500 |
| ThT | C17H19ClN2S | 318.0957 | 283.1263 | 190 | 4500 |
| Rosolic acid | C19H14O3 | 290.0942 | 291.1015 | 200 | 4500 |
| d6-MGa | C29H22D5N5O7 | 562.6012 | 334.2326 | 180 | 4500 |
Quantitative calculation
Quantitative analysis was performed using the internal standard (IS) calibration curve method. Blank DI water was spiked with a series of concentrations of MG and LMG standards in order to establish the calibration curves in aqueous solution. For establishing the calibration curves in the fish sample, the homogenized fish matrixes were added to the MG standards at a series of concentrations. d5-MG was used as the IS for the analysis of MG, LMG, CV, LCV, ThT and rosolic acid. The IS compound was added into the spray solvent at a concentration of 50 ng mL−1. The peak area ratios of fragment ions (A) generated from the target analyte to those from the corresponding IS compounds (AIS) were used for quantitative calculation.Results and discussion
Optimization of the preparation conditions of MIPCWT-SPME probes
The preparation conditions of the MIPCWT-SPME probes include the eluent and elution time of the template (MG), and the dosage of the template. The eluent and elution time for processing the probe were first confirmed by immersing the probes with the template into MeOH, ACN, a mixture of MeOH and NH4Ac (95:5, v/v), and a mixture of ACN and NH4Ac (95:5, v/v) to wash the template for 12 h, 24 h, 36 h and 48 h at 100 rpm. The eluent was replaced every 12 h. The eluent of each cycle was monitored by an ultraviolet spectrophotometer at 618 nm to investigate the elution of the template for the probes. These probes were washed for four cycles, and for each cycle, mass spectrometry was used for the measurement. As shown in Fig. 2b, the absorbance value of the mixture of MeOH and NH4Ac (95:5, v/v) decreased steeply with elution time. It can also be seen that the signal intensities increased with increasing sample volume, and equilibrium was reached at 100 mL for most of the MG and LMG. Note that the extraction time and sample dosage were optimized by means of analyzing water samples spiked with 50 ng mL−1 of MG and LMG. Different extraction times (5, 10, 20, 30, 40, 50, and 60 min) were also studied because the distribution of analytes between the sample solution and the MIPCWT-SPME probe is a dynamic equilibrium process. For MG and LMG, the results demonstrate that the signal intensities become steady after the highest peak emerged at 40 min. Then, different sample volumes of 5, 10, 50, 100, 150, and 200 mL were investigated. It can be noted that the signal intensities increased with increasing sample volume, with an equilibrium reached at 100 mL for most of the MG and LMG.
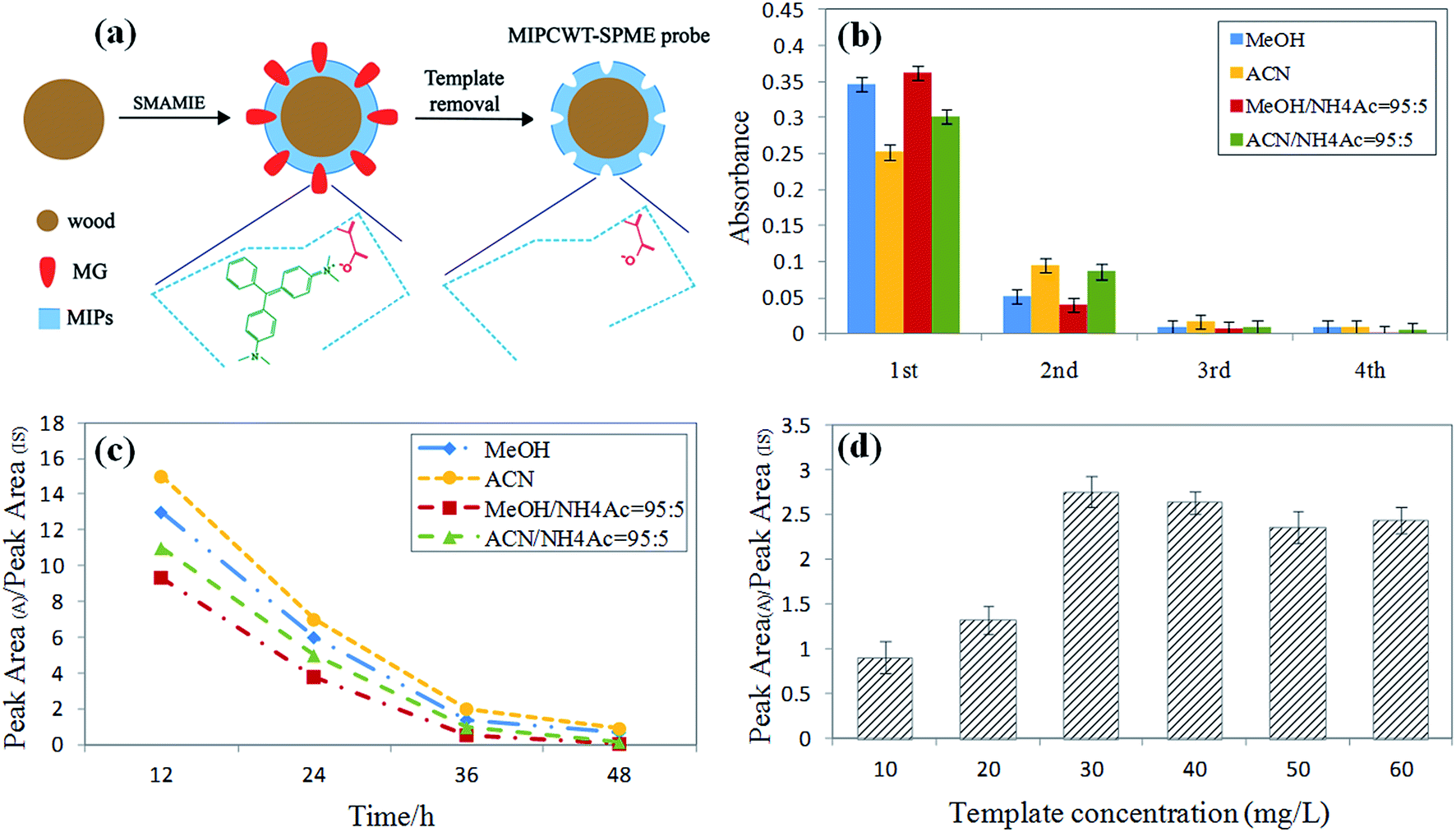 | ||
| Fig. 2 Optimization of the preparation conditions of the MIPCWT-SPME probe. (a) A schematic diagram showing the preparation of the MIPCWT-SPME probe. (b) Histograms of the UV/Vis absorbance of eluents vs. elusion time for different elution systems with various elution solvents; each cycle has a 12 h interval. (c) Plots of the ratio of the characteristic MS peak area (A) to the peak area (IS) as a function of elution time using different elution solvents. (d) Study of the impact of the MIT template concentration on the ratio of the characteristic MS peak area (A) to the IS peak area. | ||
Characterization of the MIPCWT-SPME probe
A layer of MIM can be observed on the MIPCWT-SPME probe surface by SEM (Fig. 3), which completely covers the porous structure of the wooden fiber. The selective extraction of MIM affords a high specific surface area for conjunction with MG analytes, as well as microchannels that facilitate transportation of the solvent to achieve ambient ionization for mass spectrometric analysis. | ||
| Fig. 3 SEM images at different magnifications of 100× (a) and 500× (b) of the pristine wooden tip, and of 100× (c), 500× (d) of the MIPCWT-SPME probe. | ||
FTIR experiments were applied to confirm the success of the modification, as shown in Fig. 4. In contrast to the original wooden tip, the MIPCWT-SPME probe surface of our modified wooden tip can be evidenced through several relatively strong characteristic peaks indexed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group at about 1730 cm−1, a C–O–C group at about 1160 and 1118 cm−1 (symmetric stretching vibration), and a C–O–C group at 1241 cm−1 (asymmetric stretching vibration). The characteristic peak at 1062 cm−1 can be ascribed to the stretching vibration of the Si–O bond. These FTIR absorptions are attributed to the existence of the SMAMIE, illuminating the successful synthesis of MIPs by the SMAMIE. All these results reveal the effective incorporation of adsorbents onto the MIPCWT-SPME probe surface.
O group at about 1730 cm−1, a C–O–C group at about 1160 and 1118 cm−1 (symmetric stretching vibration), and a C–O–C group at 1241 cm−1 (asymmetric stretching vibration). The characteristic peak at 1062 cm−1 can be ascribed to the stretching vibration of the Si–O bond. These FTIR absorptions are attributed to the existence of the SMAMIE, illuminating the successful synthesis of MIPs by the SMAMIE. All these results reveal the effective incorporation of adsorbents onto the MIPCWT-SPME probe surface.
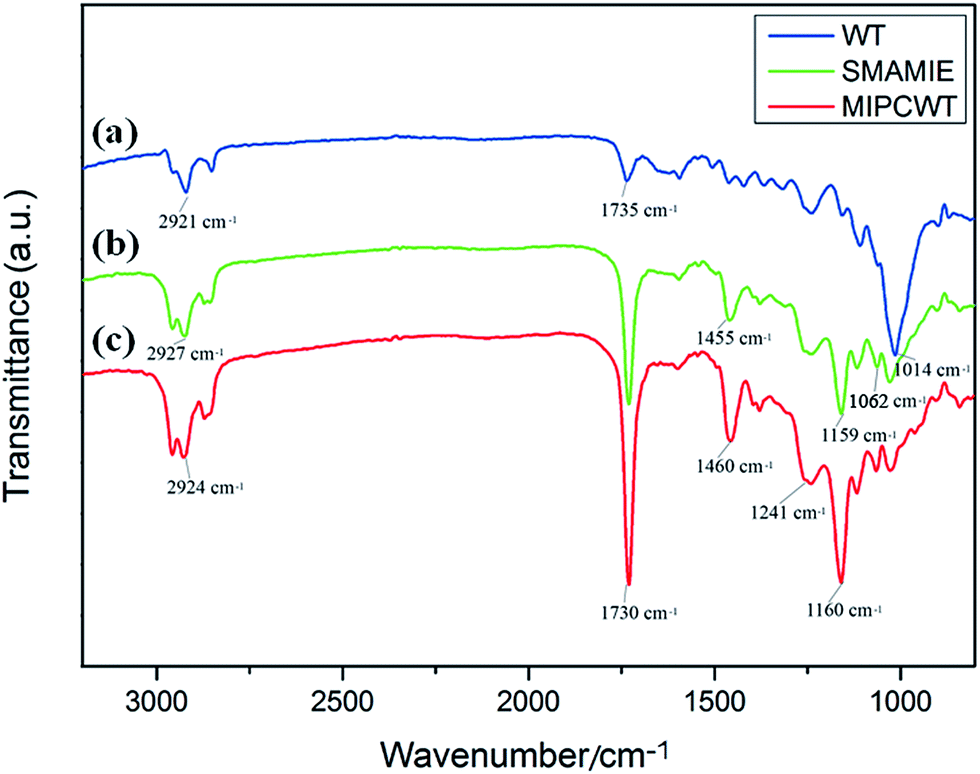 | ||
| Fig. 4 FTIR spectra of the pristine wooden-tip surface (a), and SMAMIE (b) and MIPCWT-SPME (c) probe surfaces. | ||
To further analyze the chemical structure of our fabricated probe, the elemental distribution of MIM on the MIPCWT-SPME probe surface was obtained by elemental mapping (Fig. 5). It can be seen that the MIPCWT-SPME probe surface is composed of a large amount of carbon (C), and a portion of oxygen (O) and silicon (Si) elements, of which C occupies the highest atomic percentage. It can also be seen that the MIM possesses Si element stemming from SMAMIE, and C and O elements from acrylics. Acrylics consist of short carbon chains, where C occupies the greatest atomic percentage.
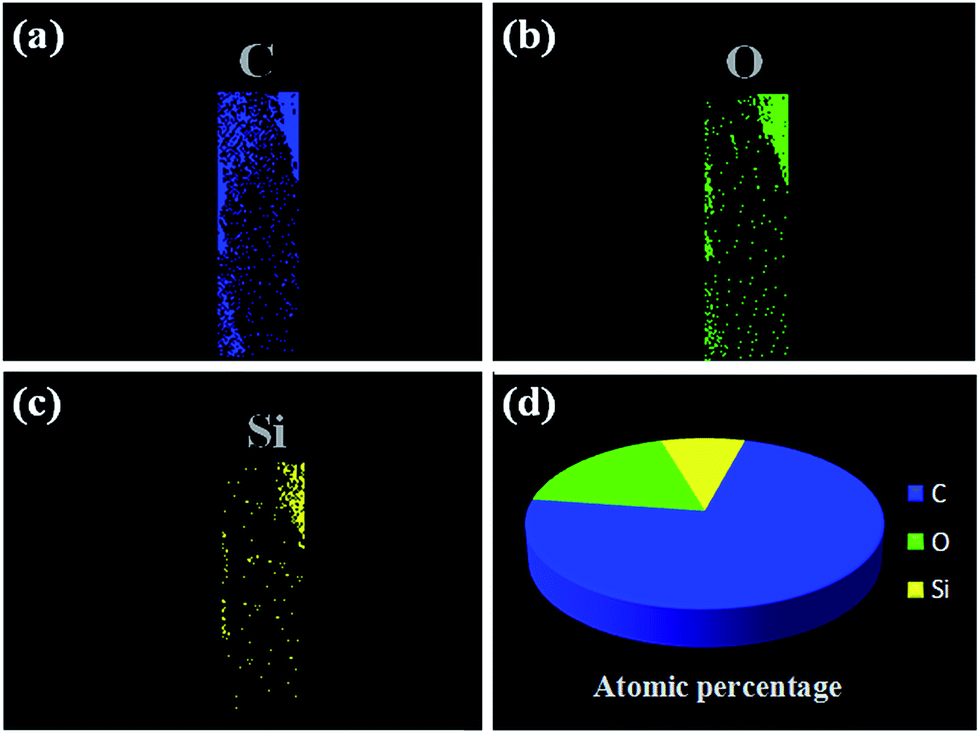 | ||
| Fig. 5 Elemental mapping images of the MIPCWT-SPME probe. Elemental distribution of C (a), O (b), and Si (c). (d) The atomic percentages of C, O, and Si. | ||
Optimization of extraction and desorption conditions
The extraction conditions, including extraction time and sample volume, were optimized by analyzing DI water samples spiked with 50 ng mL−1 of MG and LMG. First, different extraction times (5, 10, 20, 30, 40, 50, and 60 min) were studied because the distribution of analytes between the sample solution and the MIPCWT-SPME probe is a dynamic equilibrium process. As shown in Fig. 6a, the results demonstrate that the signal intensities, for MG and LMG, become steady after the highest peak appeared at 40 min. Then, different sample volumes of 5, 10, 50, 100, 150, and 200 mL were investigated. It was found that the signal intensities increased with increasing sample volume, and equilibrium was reached at 100 mL for most of the MG and LMG (Fig. 6b). | ||
| Fig. 6 Optimization of the extraction and desorption conditions under which the MIPCWT-SPME probe was prepared. (a) Plots of the ratio of the characteristic MS peak area (A) to the IS peak area as a function of sample extraction volume. (b) Plots of the ratio of the characteristic MS peak area (A) to the IS peak area as a function of extraction time. (c) The impact of the different spray solvents on the ratio of the characteristic MS peak area (A) to the IS peak area. | ||
The desorption conditions are vital to the direct analysis of MG and LMG for ambient ionization for mass spectrometric analysis. The signals of the spray solvent can be observed, including MeOH, ACN, a mixture of MeOH and HAc (9:1, v/v), a mixture of ACN and HAc (9:1, v/v), a mixture of MeOH and HAc (99:1, v/v), and a mixture of ACN and HAc (99:1, v/v). The obtained results show that the signal intensities increased with decreasing solution pH values. Because MG and LMG are polar compounds, the solution pH value can be lowered to some extent, causing the MG and LMG to ionize easily. Furthermore, MeOH with acid can shift the equilibrium towards the molecular form, thereby increasing their affinity for the sorbent. Compared to ACN, MeOH can be ionized with a lower ionization energy, which endows the spray solvent, namely the mixture of MeOH and HAc (99:1, v/v), with a remarkable extraction and ionization efficiency (Fig. 6c).
Selective extraction properties
Considering that malachite green and its metabolite are triphenylmethane compounds containing triphenylmethane and amidogen structures, the selective extraction of MIPs on the surface of wooden tips is based on the specific recognition sites matching either triphenylmethane or amidogen groups. The structural formulae of verified compounds are shown in Fig. 7a. The selective extraction properties of MIPCWT-SPME probes were investigated by extracting 50 ng mL−1 of hybrid intermediate solutions including MG, LMG, CV, LCV, ThT and rosolic acid for 40 min. The extracted MIPCWT-SPME probe tip was placed pointing to the MS inlet, with 4.5 kV applied to the probe. An aliquot of 10 μL of the mixture of MeOH and HAc (99:1, v/v), together with 50 ng mL−1 IS compound (d5-MG), was added onto the probe surface for mass spectrometric analysis. The experimental results demonstrate that the adsorption of the amidogen groups (CV, LCV and ThT) is much higher as compared to those with only triphenylmethane groups (rosolic acid), as shown in Fig. 7b. The data thus indicate that the MIPs of the MIPCWT-SPME probe are primarily extracted by the compounds containing amidogen groups, leading us to improve the selective and specific extraction of trace level compounds.
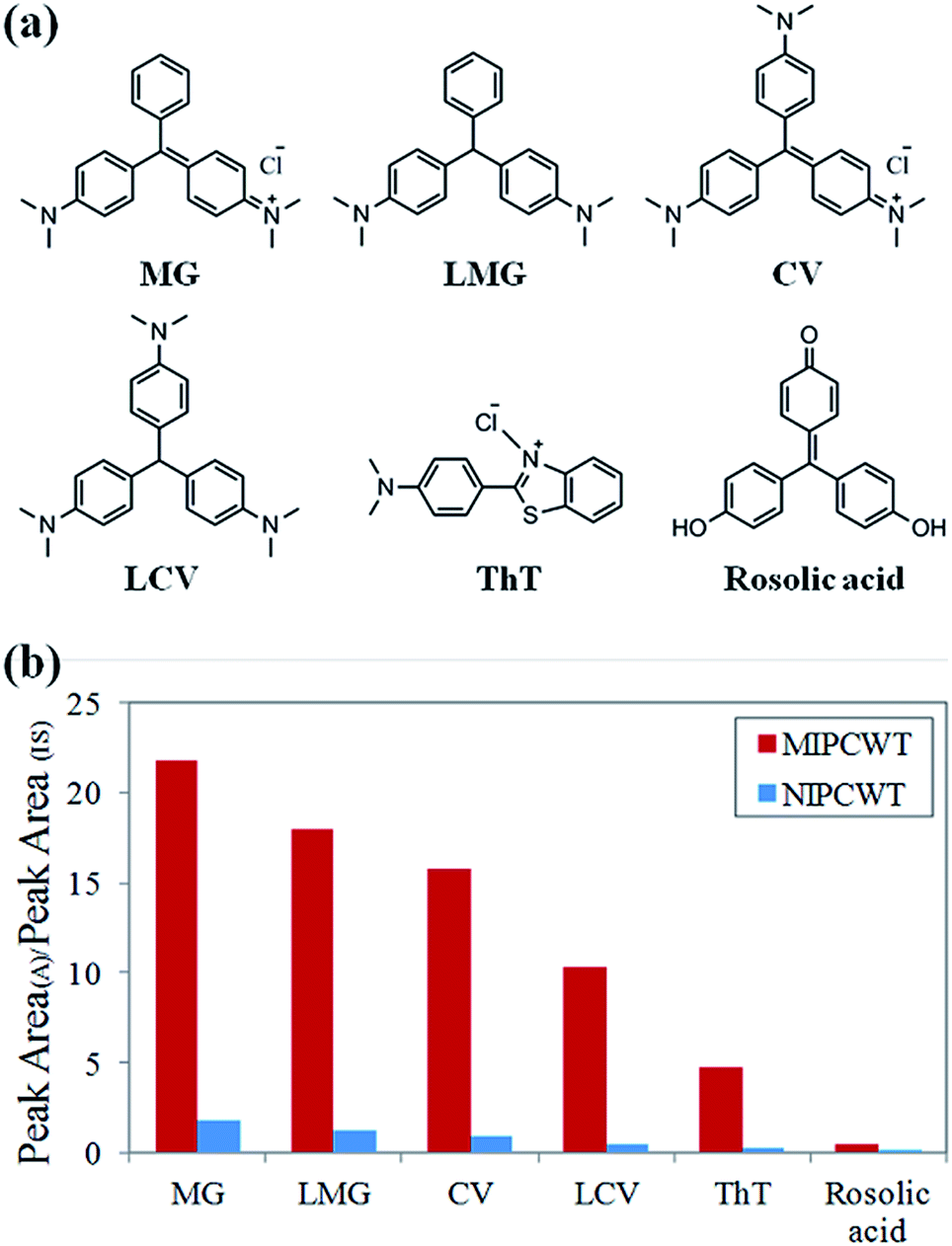 | ||
| Fig. 7 Study of the selective extraction conditions of the MIPCWT-SPME probe and the NIPCWT-SPME probe. (a) Chemical structures of the selectively extracted compounds. (b) Plots of the ratio of the characteristic MS peak area (A) to the IS peak area as a function of selective extraction between the MIPCWT-SPME probe and the NIPCWT-SPME probe. | ||
Extraction properties
The extraction properties were investigated by determining the enrichment factor (EF, defined as the ratio of the A/AIS obtained by MIPCWT-SPME probe to that of the original wooden tip) of each analyte in DI water. A 100 mL aliquot of 50 ng mL−1 spiked DI water sample was used for the study. The results show that the developed SPME probe possessed excellent extraction capacity for analysis of aqueous samples, with EF values of 1877 ± 241 and 1502 ± 232 for MG and LMG, respectively. All these results demonstrate that the proposed MIPCWT-ESI-MS method is suitable for direct quantitative determination of trace levels of malachite green and its metabolite.Quantitative analysis
The quantitative analysis is based on the quantitative calculation of the DI water sample and fish sample through optimization via analyzing the DI water sample spiked with MG and LMG at a series of concentrations, as well as the fish sample with MG at a series of concentrations. Quantitative calculation was performed using the IS calibration curve method. Into the spray solvent, d5-MG was added at a concentration of 50 ng mL−1 and used as the isotope IS compound. After MIPCWT-ESI-MS analysis, satisfactory linearity was obtained with impressive correlation coefficient values (R2) of no less than 0.9988 for MG and LMG in the concentration range of 0.1–100 ng mL−1 (Table 2). Under the optimal conditions, the latter protocol gives limits of detection (LOD) of 0.01 ng L−1 for MG and LMG in the DI water sample, and 0.05 ng L−1 for MG in the fish sample. Notably, the performance of our method in the detection of MG is superior to most of the state-of-the-art approaches recently reported, as displayed in Table 3.| Matrix | Analyte | Linear range (ng mL−1) | Regression equation | R2 | LODa (ng mL−1) | Repeatabilityb (RSD, %, n = 6) |
|---|---|---|---|---|---|---|
| a LOD was determined as the concentration producing a signal-to-noise ratio of 3.b Analysis of DI water and fish samples spiked with 50 ng mL−1 analytes. | ||||||
| DI water | MG | 0.1–100 | y = 0.0608x + 0.1128 | 0.9990 | 0.01 | 4.6 |
| LMG | 0.1–100 | y = 0.0054x + 0.0549 | 0.9988 | 0.01 | 5.1 | |
| Fish | MG | 0.1–100 | y = 0.4991x + 1.0385 | 0.9989 | 0.05 | 10.6 |
| Imprinted polymer | Application | Analytical method | Detection limits | Linear range | Ref. |
|---|---|---|---|---|---|
| a Methacrylic acid–ethylene glycol dimethacrylate.b Solid-phase extraction.c Electrochemiluminescence-molecularly imprinted solid phase extraction.d Liquid chromatography mass spectrometry.e 3-Methacryloxypropyltrimethoxy-silane.f Enzyme-linked immunosorbent assay.g Quantum dots.h Acrylamide.i (3-Aminopropyl)triethoxysilane.j Tetraethyl orthosilicate. | |||||
| MAA-EGDMAa bulk polymerization in acetonitrile | SPEb | ECL-MISPEc | 5 ng kg−1 | 0.02–5 μg L−1 | 64 |
| Commercial products obtained from MIP technologies AB | SPE | LC-MSd | 3 ng kg−1 | 0.02–5 μg L−1 | 65 |
| MAA–EGDMA in acetonitrile and water | SPE | HPLC | 0.14 μg kg−1 | — | 66 |
| MAA–EGDMA in methanol | SPE | HPLC | 0.05 μg kg−1 | 10–200 μg L−1 | 67 |
| Fe3O4, MAA, EGDMA in chloroform | Extraction of magnetic MIP | HPLC | 0.27 μg kg−1 | 0.5–40 μg L−1 | 68 |
| Fe3O4@SiO2–MPSe, MAA, EGDMA in acetonitrile | Extraction of magnetic MIP | ECL | 7.3 ng kg−1 | 0.29–290 μg L−1 | 69 |
| Fe3O4, MAA, EGDMA in chloroform | Extraction of magnetic MIP | HPLC | 0.71 μg kg−1 | 2–1000 μg L−1 | 70 |
| Fe3O4, MAA, EGDMA in chloroform | Extraction of magnetic MIP | ELISAf | 0.1 μg kg−1 | 0.1–10000 μg L−1 |
71 |
| CdTe QDsg, AMh, EGDMA in acetonitrile | Adsorbent (mixed with extraction solution) | Synchronous fluorescence quenching method | 30 nmol L−1 | 0.1–20 μmol L−1 | 72 |
| CdTe QDs, APTESi, TEOSj | Adsorbent (mixed with extraction solution) | Synchronous fluorescence quenching method | 12 μg kg−1 | 0.08–20 μg L−1 | 8 |
| Carbon nanotube, MAA, EGDMA in methylene chloride | Adsorbent (mixed with fish sample) | HPLC | 0.7 μg kg−1 | 3–300 μg L−1 | 73 |
| Dopamine hydrochloride film | Adsorbent on microplate | ELISA | 0.3 μg L−1 | — | 74 |
| MMA, acrylate, and silicone-modified acrylate | SMPE | ESI-MS | 0.05 ng kg−1 | 0.1–100 μg L−1 | This work |
To examine the repeatability, one SPME probe was used for six repeated extractions of the DI water samples spiked with 50 ng mL−1 of MG and LMG under the same conditions. Before reuse, the samples were washed with 10 mL of a mixture of MeOH and HAc (99:1, v/v) for 5 min. It should be noted that the relative standard deviations (RSD) of the repeated test results are less than 4.5 and 5.1% regarding MG and LMG, respectively. It is worth pointing out that, although the wooden tip-based probes are disposable after use, they can be reused without significant loss of the excellent initial detection properties.
Analysis of real-life samples
The optimized surface-coated wooden-tip ESI-MS method was applied for analysis of real-life tap water, river water and fish samples, with the results shown in Table 4. No MG and LMG could be detected in tap water, but it is well demonstrated that the recoveries ranged from 93.7 to 102.8% in tap water. In one river water sample, MG and LMG were determined to be 0.8 and 0.3 ng L−1, respectively, with the recoveries ranging from 95.2 to 108.3%. Considering that LMG could be oxidized to MG by DDQ in the fish sample, a large concentration of 3.4 ng mL−1 could be detected for MG, with the recoveries obtained in the range of 106.7 to 113.6%. The satisfactory results of the recovery measurements confirm that the aqueous matrices do not affect the sorbent materials, which is of technological importance for trace analysis.| Matrix | Spiked (ng mL−1) | MG | LMG | ||
|---|---|---|---|---|---|
| Measured (ng mL−1) | Recovery (%) | Measured (ng mL−1) | Recovery (%) | ||
| a N.D. = not detected. | |||||
| Tap water | 0 | N.D.a | — | N.D.a | — |
| 10 | 10.3 | 103 | 43.4 | 97 | |
| 50 | 48 | 96 | 424.5 | 93 | |
| River water | 0 | N.D.a | — | N.D.a | — |
| 10 | 10.8 | 108 | 43.3 | 98 | |
| 50 | 47.5 | 95 | 438.0 | 92 | |
| Fish | 0 | N.D.a | — | — | — |
| 10 | 11.3 | 113 | — | — | |
| 50 | 55.1 | 106 | — | — | |
Conclusions
A molecularly imprinted polymer-coated wooden-tip solid-phase micro-extraction (MIPCWT-SPME) probe has been fabricated by applying a silicone-modified acrylate molecularly imprinted emulsion (SMAMIE) onto the surface of wooden tips. Such a novel probe can be used for efficient extraction of malachite green, realizing sensitive detection and analysis. Using the cost-effective and portable MIPCWT-SPME probe as the key component, the MIPCWT-ESI-MS method has been developed for rapid and sensitive analysis of trace malachite green and its metabolite in real-life water and fish samples. The method proposed here possesses the advantages of simplicity, quantitativeness, reproducibility, and high throughput, showing promising prospects for rapid and sensitive detection of trace malachite green and its metabolite in aquatic products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the Science and Technology Project of Guangdong Province, China (2015A030401106, 2017A070702017, 2016A040403057, 2016A040403061, 2015A020218001 and 2013B031500003), the Innovation Project of Department of Education of Guangdong Province (2015KTSCX150), the Science and Technology Project of Foshan City (2015AG10011 and 2016GA10162), GDAS' Special Project of Science and Technology Development (No. 2017GDASCX-0104 and 2016GDASPT-0203), and Natural Science Foundation of Guangdong Province, China (No. 2017A030310233) are greatly acknowledged. We also thank Dr Jiewei Deng at Sun Yat-sen University for guidance of the paper.Notes and references
- L. Zhang, H. Zhang, W. Guo and Y. Tian, Appl. Clay Sci., 2014, 93, 85–93 CrossRef.
- E. A. Dil, M. Ghaedi, A. Asfaram, S. Hajati, F. Mehrabi and A. Goudarzi, Ultrason. Sonochem., 2017, 34, 677–691 CrossRef CAS PubMed.
- S. Srivastava, R. Sinha and D. Roy, Aquat. Toxicol., 2004, 66, 319–329 CrossRef CAS PubMed.
- A. Stolker, T. Zuidema and M. Nielen, TrAC, Trends Anal. Chem., 2007, 26, 967–979 CrossRef CAS.
- F. Jia, X. Yang and Z. Li, RSC Adv., 2016, 6, 92723–92728 RSC.
- Y. N. Rao, D. Banerjee, A. Datta, S. K. Das and A. Saha, RSC Adv., 2016, 6, 49083–49090 RSC.
- X. Jiang, S. Wang, L. Ge, F. Lin, Q. Lu, T. Wang, B. Huang and B. Lu, RSC Adv., 2017, 7, 38965–38972 RSC.
- L. Wu, Z.-z. Lin, H.-p. Zhong, A.-h. Peng, X.-m. Chen and Z.-y. Huang, Food Chem., 2017, 229, 847–853 CrossRef CAS PubMed.
- H. Zhang, F. Zhang and Q. Huang, RSC Adv., 2017, 7, 5790–5799 RSC.
- F. Jing, R. Liang, J. Xiong, R. Chen, S. Zhang, Y. Li and L. Wu, Appl. Catal., B, 2017, 206, 9–15 CrossRef CAS.
- Z. Shi, L. Li, Y. Xiao, Y. Wang, K. Sun, H. Wang and L. Liu, RSC Adv., 2017, 7, 30904–30910 RSC.
- R. G. Hemalatha, H. R. Naik, V. Mariappa and T. Pradeep, RSC Adv., 2015, 5, 50512–50522 RSC.
- H. Zhang, H. Lu, H. Huang, J. Liu, X. Fang, B.-F. Yuan, Y.-Q. Feng and H. Chen, Anal. Chim. Acta, 2016, 926, 72–78 CrossRef CAS PubMed.
- L. Song, J. Xu, K. Chingin, T. Zhu, Y. Zhang, Y. Tian, H. Chen and X. Chen, J. Agric. Food Chem., 2017, 65, 7006–7011 CrossRef CAS PubMed.
- L. A. Doezema, T. Longin, W. Cody, V. Perraud, M. L. Dawson, M. J. Ezell, J. Greaves, K. R. Johnson and B. J. Finlayson-Pitts, RSC Adv., 2012, 2, 2930–2938 RSC.
- F. Zhou, S. Liu, J. Xing, F. Song, Z. Liu and S. Liu, RSC Adv., 2016, 6, 98927–98934 RSC.
- X. Fan, C.-F. Wang, C.-Y. You, X.-Y. Wei, L. Chen, J.-P. Cao, Y.-P. Zhao, W. Zhao, Y.-G. Wang and J.-L. Lu, RSC Adv., 2016, 6, 105780–105785 RSC.
- H.-Y. Hsieh, L.-H. Li, R.-Y. Hsu, W.-F. Kao, Y.-C. Huang and C.-C. Hsu, Anal. Chem., 2017, 89, 6147–6153 Search PubMed.
- K. Hiraoka, K. Nishidate, K. Mori, D. Asakawa and S. Suzuki, Rapid Commun. Mass Spectrom., 2007, 21, 3139–3144 CrossRef CAS PubMed.
- L. C. Chen, K. Nishidate, Y. Saito, K. Mori, D. Asakawa, S. Takeda, T. Kubota, H. Hori and K. Hiraoka, J. Phys. Chem. B, 2008, 112, 11164–11170 CrossRef CAS PubMed.
- B. B. Wang, D. Gao, I. Levchenko, K. Ostrikov, M. Keidar, M. K. Zhu, K. Zheng and B. Gao, RSC Adv., 2016, 6, 87607–87615 RSC.
- H. Puliyalil, G. Filipic, J. Kovac, M. Mozetic, S. Thomas and U. Cvelbar, RSC Adv., 2016, 6, 95120–95128 RSC.
- D. Chen, Y.-Q. Huang, X.-M. He, Z.-G. Shi and Y.-Q. Feng, Analyst, 2015, 140, 1731–1738 RSC.
- D. Chen, H.-B. Zheng, Y.-Q. Huang, Y.-N. Hu, Q.-W. Yu, B.-F. Yuan and Y.-Q. Feng, Analyst, 2015, 140, 5662–5670 RSC.
- J. A. Fincher, A. R. Korte, B. Reschke, N. J. Morris, M. J. Powell and A. Vertes, Analyst, 2017, 142, 3157–3164 RSC.
- H. Li and A. Vertes, Analyst, 2017, 142, 2921–2927 RSC.
- F. A. M. G. van Geenen, M. C. R. Franssen, A. H. M. Schotman, H. Zuilhof and M. W. F. Nielen, Anal. Chem., 2017, 89, 4031–4037 CrossRef CAS PubMed.
- S. F. Resende, J. A. R. Teodoro, I. Binatti, R. L. Gouveia, B. S. Oliveira and R. Augusti, Int. J. Mass Spectrom., 2016, 418, 107–111 CrossRef.
- X. Wang, Y. Chen, Y. Zheng and Z. Zhang, Materials, 2017, 10, 769 CrossRef PubMed.
- T. Wang, Y. Zheng, X. Wang, D. E. Austin and Z. Zhang, Anal. Chem., 2017, 89, 7988–7995 CrossRef CAS PubMed.
- L. Di Donna, D. Taverna, S. Indelicato, A. Napoli, G. Sindona and F. Mazzotti, Food Chem., 2017, 229, 354–357 CrossRef CAS PubMed.
- A. K. Meher and Y.-C. Chen, RSC Adv., 2015, 5, 94315–94320 RSC.
- B. Hu, Y. Huang, G. Yin, G. Zhang, L. Zhang, T. Wang and Z.-P. Yao, Anal. Methods, 2016, 8, 6840–6846 RSC.
- Y. Yang, J. Deng and Z.-P. Yao, Anal. Chim. Acta, 2015, 887, 127–137 CrossRef CAS PubMed.
- B.-c. Yang, F. Wang, W. Deng, Y. Zou, F.-y. Liu, X.-d. Wan, X. Yang, H. Liu and O.-p. Huang, Anal. Methods, 2015, 7, 5886–5890 RSC.
- B.-c. Yang, F.-y. Liu, L.-q. Wang, Y. Zou, F. Wang, W. Deng, X.-d. Wan, X. Yang, M. He and O.-p. Huang, Anal. Methods, 2015, 7, 6125–6132 RSC.
- B.-c. Yang, F.-y. Liu, J.-b. Guo, L. Wan, J. Wu, F. Wang, H. Liu and O.-p. Huang, Anal. Methods, 2015, 7, 2913–2916 RSC.
- Q. Du, J. Deng, Y. Liu, X. Zhang, Y. Yang and J. Chen, Anal. Methods, 2015, 7, 4803–4810 RSC.
- Y. Yang and J. Deng, Anal. Chim. Acta, 2014, 837, 83–92 CrossRef CAS PubMed.
- G.-Z. Xin, B. Hu, Z.-Q. Shi, Y. C. Lam, T. T.-X. Dong, P. Li, Z.-P. Yao and K. W. K. Tsim, Anal. Chim. Acta, 2014, 820, 84–91 CrossRef CAS PubMed.
- R. Parchami, M. Kamalabadi and N. Alizadeh, J. Chromatogr. A, 2017, 1481, 37–43 CrossRef CAS PubMed.
- G. A. Gomez-Rios, C. Liu, M. Tascon, N. Reyes-Garces, D. W. Arnold, T. R. Covey and J. Pawliszyn, Anal. Chem., 2017, 89, 3805–3809 CrossRef CAS PubMed.
- M. F. Mirabelli, J.-C. Wolf and R. Zenobi, Anal. Chem., 2016, 88, 7252–7258 CrossRef CAS PubMed.
- N. R. Choi, Y. P. Kim, W. H. Ji, G.-S. Hwang and Y. G. Ahn, Talanta, 2016, 148, 69–74 CrossRef CAS PubMed.
- F. D. S. Araujo, R. L. Vieira, E. P. L. Molano, H. J. Maximo, R. J. D. Dalio, P. H. Vendramini, W. L. Araujo and M. N. Eberlin, RSC Adv., 2017, 7, 29953–29958 RSC.
- J. E. Giffen, J. Y. Rosati, C. M. Longo and R. A. Musah, Anal. Chem., 2017, 89, 7719–7726 CrossRef CAS PubMed.
- G. A. Gomez-Rios, E. Gionfriddo, J. Poole and J. Pawliszyn, Anal. Chem., 2017, 89, 7240–7248 CrossRef CAS PubMed.
- G. A. Gomez-Rios, T. Vasiljevic, E. Gionfriddo, M. Yu and J. Pawliszyn, Analyst, 2017, 142, 2928–2935 RSC.
- A. Nicolas Filippin, J. Ramon Sanchez-Valencia, J. Idigoras, M. Macias-Montero, M. Alcaire, F. Javier Aparicio, J. Pedro Espinos, C. Lopez-Santos, F. Frutos, A. Barranco, J. Antonio Anta and A. Borras, Adv. Mater. Interfaces, 2017, 4, 1601233 CrossRef.
- J. Chauvin, F. Judee, M. Yousfi, P. Vicendo and N. Merbahi, Sci. Rep., 2017, 7, 4562 CrossRef PubMed.
- S. Ahmad, M. Tucker, N. Spooner, D. Murnane and U. Gerhard, Anal. Chem., 2015, 87, 754–759 CrossRef CAS PubMed.
- B.-C. Yang, S.-F. Fang, X.-J. Wan, Y. Luo, J.-Y. Zhou, Y. Li, Y.-J. Li, F. Wang and O.-P. Huang, Anal. Chim. Acta, 2017, 973, 68–74 CrossRef CAS PubMed.
- G. A. Gomez-Rios, N. Reyes-Garces, B. Bojko and J. Pawliszyn, Anal. Chem., 2016, 88, 1259–1265 CrossRef CAS PubMed.
- H. Amanzadeh, Y. Yamini, M. Moradi and Y. A. Asl, J. Chromatogr. A, 2016, 1465, 38–46 CrossRef CAS PubMed.
- M. Tascon, G. A. Gomez-Rios, N. Reyes-Garces, J. Poole, E. Boyaci and J. Pawliszyn, Anal. Chem., 2017, 89, 8421–8428 CrossRef CAS PubMed.
- M. Tascon, G. A. Gomez-Rios, N. Reyes-Garces, J. Poole, E. Boyaci and J. Pawliszyn, Sci. Rep., 2017, 144, 106–111 CAS.
- G. A. Gomez-Rios and J. Pawliszyn, Angew. Chem., Int. Ed., 2014, 53, 14503–14507 CrossRef CAS PubMed.
- J. Deng, T. Yu, Y. Yao, Q. Peng, L. Luo, B. Chen, X. Wang, Y. Yang and T. Luan, Anal. Chim. Acta, 2017, 954, 52–59 CrossRef CAS PubMed.
- J. Deng, Y. Yang, L. Fang, L. Lin, H. Zhou and T. Luan, Anal. Chem., 2014, 86, 11159–11166 CrossRef CAS PubMed.
- P. Regal, M. Díaz-Bao, R. Barreiro, A. Cepeda and C. Fente, Cent. Eur. J. Chem., 2012, 10, 766–784 Search PubMed.
- A. Speltini, A. Scalabrini, F. Maraschi, M. Sturini and A. Profumo, Anal. Chim. Acta, 2017, 974, 1–26 CrossRef CAS PubMed.
- R. Mirzajani and M. Bagheban, Int. J. Environ. Anal. Chem., 2016, 96, 576–594 CrossRef CAS.
- S. Ansari, TrAC, Trends Anal. Chem., 2017, 90, 89–106 CrossRef CAS.
- Z. Guo, P. Gai, T. Hao, J. Duan and S. Wang, J. Agric. Food Chem., 2011, 59, 5257–5262 CrossRef CAS PubMed.
- M. J. Martinez Bueno, S. Herrera, A. Ucles, A. Aguera, M. D. Hernando, O. Shimelis, M. Rudolfsson and A. R. Fernandez-Alba, Anal. Chim. Acta, 2010, 665, 47–54 CrossRef CAS PubMed.
- C. Long, Z. Mai, Y. Yang, B. Zhu, X. Xu, L. Lu and X. Zou, J. Chromatogr. A, 2009, 1216, 2275–2281 CrossRef CAS PubMed.
- Z. Lian and J. Wang, Mar. Pollut. Bull., 2012, 64, 2656–2662 CrossRef CAS PubMed.
- Z.-z. Lin, H.-y. Zhang, A.-h. Peng, Y.-d. Lin, L. Li and Z.-y. Huang, Food Chem., 2016, 200, 32–37 CrossRef CAS PubMed.
- B. Huang, X. Zhou, J. Chen, G. Wu and X. Lu, Talanta, 2015, 142, 228–234 CrossRef CAS PubMed.
- Z.-z. Lin, H.-y. Zhang, L. Li and Z.-y. Huang, React. Funct. Polym., 2016, 98, 24–30 CrossRef CAS.
- L. Li, Z. Z. Lin, A. H. Peng, H. P. Zhong, X. M. Chen and Z. Y. Huang, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1035, 25–30 CrossRef CAS PubMed.
- J. Yang, Z.-Z. Lin, H.-P. Zhong, X.-M. Chen and Z.-Y. Huang, Sens. Actuators, B, 2017, 252, 561–567 CrossRef CAS.
- Y. Wang and L. Chen, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 1002, 98–106 CrossRef CAS PubMed.
- L. Li, A.-h. Peng, Z.-z. Lin, H.-p. Zhong, X.-m. Chen and Z.-y. Huang, Food Chem., 2017, 229, 403–408 CrossRef CAS PubMed.
- J. Ou, Y. Yang, J. Gan, C. Ha and M. Zhang, J. Appl. Polym. Sci., 2014, 131, 2540 Search PubMed.
- W. C. Andersen, S. B. Turnipseed and J. E. Roybal, J. Agric. Food Chem., 2006, 54, 4517–4523 CrossRef CAS PubMed.
- A. A. Fallah and A. Barani, Food Control, 2014, 40, 100–105 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |