DOI:
10.1039/C7RA10057D
(Paper)
RSC Adv., 2017,
7, 50017-50026
Sinomenine inhibits ovarian cancer cell growth and metastasis by mediating the Wnt/β-catenin pathway via targeting MCM2
Received
9th September 2017
, Accepted 25th September 2017
First published on 26th October 2017
Abstract
Sinomenine (SIN), an isoquinoline isolated from the Chinese medicinal plant Sinomenium acutum, is well known for its curative effect on rheumatic and arthritic diseases. Recently, SIN has been reported to possess potent anti-tumor effects in various types of cancers. However, the role and the underlying mechanism of the inhibitory action of SIN on ovarian cancer remain unknown. In this study, we preliminarily found that SIN might suppress tumor proliferation and metastasis by modulating the oncogene MCM2 and the Wnt/β-catenin signaling pathway. The results demonstrated that SIN could inhibit ovarian cancer cell proliferation in a dose-dependent manner. IGROV1 or HeyA8 cells treated with 0.25 mM, 0.5 mM, and 1 mM SIN exhibited gradually reduced growth capacity. Correspondingly, the protein expression levels of c-myc and cyclinD1 were also obviously decreased. In addition, reduction in migration and invasion rates was found in IGROV1 or HeyA8 cells treated with 0.25 mM and 0.5 mM SIN. Western blot results revealed that SIN could upregulate the protein expression levels of epithelial markers such as ZO-1 and E-cadherin; it also downregulated the protein expression levels of mesenchymal markers, i.e., vimentin, N-cadherin, ZEB1, and Snail. Furthermore, Wnt/β-catenin signaling attenuation was followed by the suppression of total β-catenin, cytoplasmic β-catenin, and nuclear β-catenin due to SIN treatment. MCM2 was verified to exhibit a positive relation with ovarian cancer malignancy in 75 ovarian cancer tissue and 15 paracancer tissue samples. Furthermore, MCM2 could be reduced by SIN, and its overexpression could rescue the loss of β-catenin, ZEB1, c-myc, and cyclinD1 mRNA levels together with β-catenin and ZEB1 protein expression levels, which could markedly be inhibited by MCM2 knockdown again. In summary, SIN might play anti-proliferation and anti-metastasis functions in ovarian cancer via targeting MCM2 and thereby regulating the Wnt/β-catenin signaling pathway. Thus, it could be considered to be a promising chemopreventive compound to treat ovarian carcinoma.
1. Introduction
Ovarian cancer (OC) is the most lethal gynecologic malignancy mainly because in most patients, this disease is diagnosed at the advanced stage.1 Its aggressiveness is based on the rapid dissemination of OC cells to the peritoneum, omentum, and organs located in the peritoneal cavity. Researchers have estimated that 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 440 new cases of ovarian cancer would be diagnosed and 14080 patients would die of OC in the United States in 2017.2 Although the disease is sensitive to platinum- and taxane-based chemotherapy following debulking surgery, most patients still experience recurrence and die of progressive chemotherapy-resistant diseases.3 Therefore, it is urgent to explore more effective reagents to suppress OC progression.
440 new cases of ovarian cancer would be diagnosed and 14080 patients would die of OC in the United States in 2017.2 Although the disease is sensitive to platinum- and taxane-based chemotherapy following debulking surgery, most patients still experience recurrence and die of progressive chemotherapy-resistant diseases.3 Therefore, it is urgent to explore more effective reagents to suppress OC progression.
Plants are one of the most important sources of compounds for chemo-prevention, and a number of carcinoma therapeutics in preclinical trials are based on natural products.4 SIN (7,8-didehydro-4-hydroxy-3,7-dimethoxy-17-methyl-α, 13α, 14α-mor-phinan-6-one) was extracted by Ishiwari from a medicinal plant in the 1920s5 (Fig. 1A). Numerous reports have demonstrated that SIN has been widely used to clinically treat various rheumatic diseases6 and has a number of pharmacological effects such as anti-inflammatory, anti-rheumatic, and antiangiogenic functions.7 Recently, accumulating data have suggested that SIN has potential anti-tumor functions against various cancers such as hepatocellular cancer, lung cancer, and breast cancer.8 SIN could inhibit breast cancer metastasis and hepatocellular carcinoma cell growth.9,10 However, the anti-tumor effects of SIN against OC and anti-tumor molecular mechanism have not been elucidated to date.
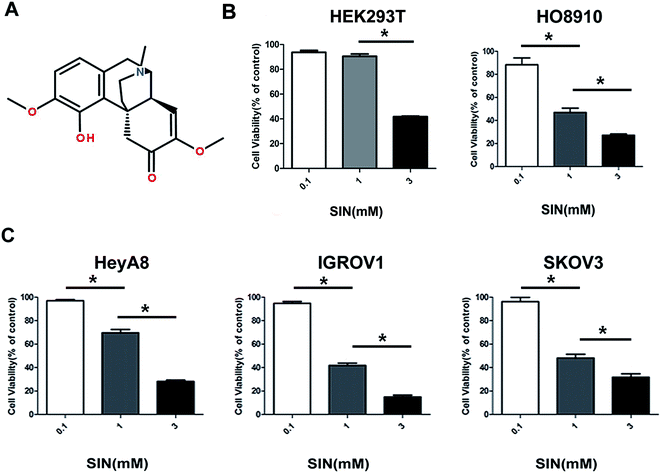 |
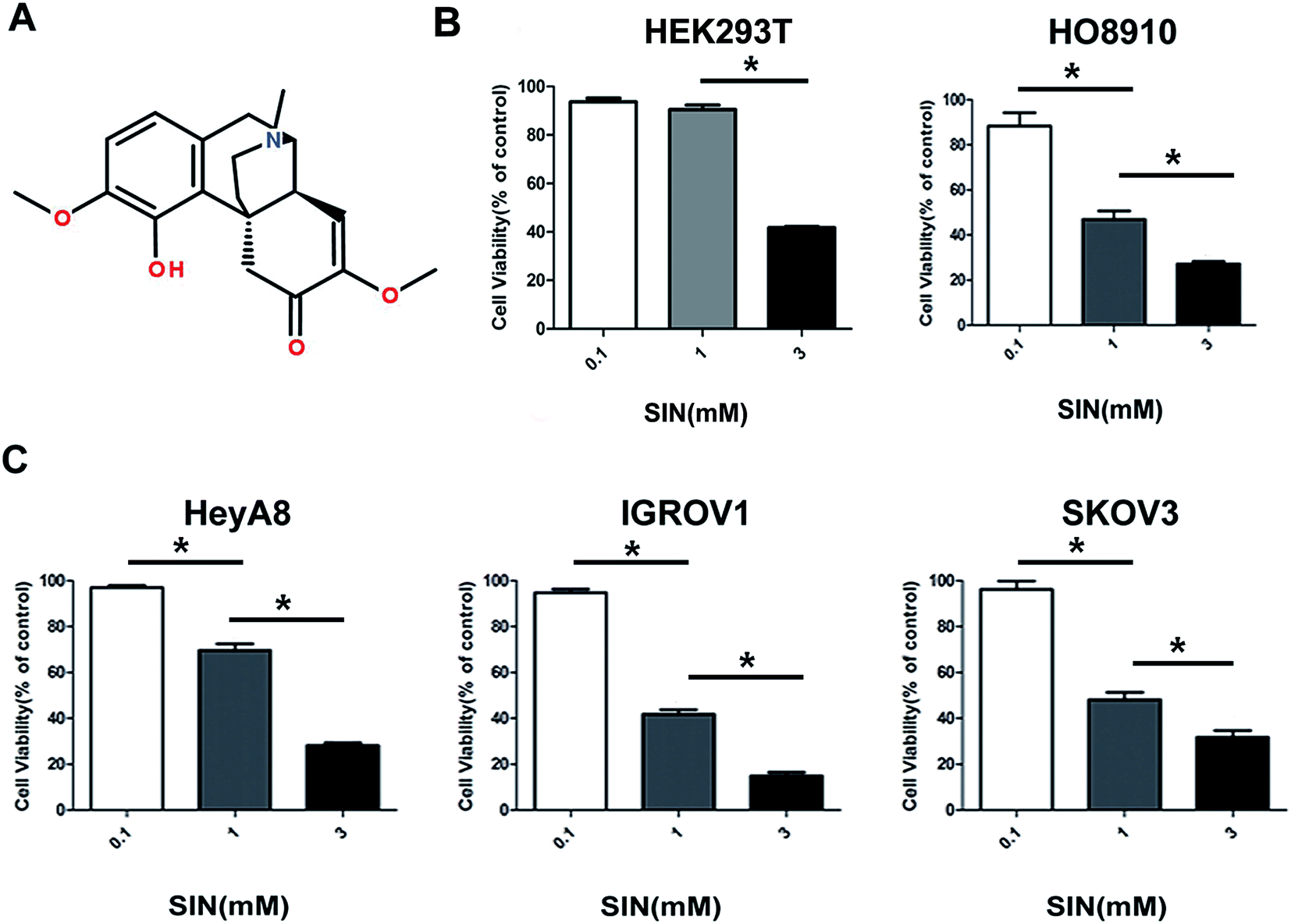
| | Fig. 1 The structure and anti-growth feature of SIN. The structure of SIN is shown in Fig. A. (B and C) HEK293T cells and human ovarian cancer cell lines HO8910, HeyA8, IGROV1, and SKOV3 were treated with SIN (0, 0.1, and 3.0 mM) for 48 h. The dose-dependent inhibition effect on cell viability was measured by the CCK8 assay; *P < 0.05. Data were represented as mean ± S.D of three independent experiments of the cells. | |
Metastasis is a multistage process that comprises EMT (epithelial–mesenchymal transition), collective invasion, reorganization of cytoskeleton, and remodeling.11 Studies have demonstrated that EMT, accompanied by loss of polarity and gain of motility characteristics, plays an important role in the process of metastasis.12 EMT is an essential biological process that allows polarized epithelial cells to show a loss of cell–cell contact and polarity to acquire a mesenchymal cell phenotype.13 Cells undergoing EMT exhibit downregulation of epithelial markers (e.g. E-cadherin, ZO-1, and occludin)14,15 and upregulation of mesenchymal markers such as Vimentin, Snail1, N-cadherin, β-catenin, Twist, ZEB1, and Fibronectin.16 In recent years, accumulating evidence suggest that activation of Wnt/β-catenin signaling pathway plays a pivotal role in EMT.17 Aberrant activation of this signaling pathway is associated with ovarian cancer and other cancers,18 which leads to the accumulation of β-catenin in the cytoplasm as well as translocation of β-catenin to the nucleus.19 The Wnt/β-catenin signaling pathway is critical in promoting EMT during kidney development, and nuclear β-catenin induces ZEB1 expression.20 EMT could be regulated by the transcriptional regulator Snail and ZEB1.21 Wnt-induced transcriptional regulation causes the acquisition of target genes, such as c-myc and ZEB1, which, as transcriptional factors, may promote proliferation22 and metastasis,23 respectively. MCM2 (minichromosome maintenance protein 2) is the main regulator of eukaryotic DNA replication, which is related to the initiation and extension of DNA replication; reports have demonstrated that it acts as a significant proliferation marker.24 In addition, it is associated with the regulation of cell migration and invasion.25
Data acquired from this study demonstrated that SIN might inhibit OC proliferation and metastasis along with EMT reversion and the loss of c-myc and cyclinD1. In addition, SIN could suppress Wnt/β-catenin expression and its nuclear accumulation. Furthermore, MCM2 rescued Wnt/β-catenin signaling attenuation caused by SIN. The application of post-expression and siRNA, wound healing assay, transwell assay, quantitative PCR, and western blot analysis assists us to further investigate the SIN functions and underlying molecular mechanism.
2. Materials and methods
2.1 Cell culture
Typically, four established OC cell lines IGROV1, HeyA8, HO8910, and SKOV3 and HEK293T were used in this study. SKOV3 cell line (ATCC) was cultured in McCoy's 5A medium. HeyA8 (obtained from Peter's laboratory), HO8910, and HEK293T cell lines were maintained in the HyClone DMEM/F12 1:1 medium. IGROV1 cell lines were cultured in the RPMI-1640 medium. All cell lines were incubated in the medium containing 10% FBS (fetal bovine serum, Excell), 100 U mL−1 penicillin, and 100 μg mL−1 streptomycin at 37 °C in a humidified atmosphere of 5% CO2. When OC cells reached the logarithmic growth phase, we performed the treatment with SIN.
2.2 Transient transfection
The expression plasmids of pcDNA3.1-MCM2 expression plasmid and its empty vector pcDNA3.1 were stored in our group. MCM2 siRNA and the negative control (NC) were designed and synthesized in Genepharma Company (Suzhou, China). SiRNA sequences targeting MCM2 and the negative sequence are listed in Table 1. HeyA8 or IGROV1 cells were incubated in 6-well culture plates when they grew upto 70–80% confluence, and transient transfections were performed in HeyA8 or IGROV1 cells with Lipofectamine™ 2000 (Invitrogen, USA) according to the manufacturer's suggestion. Then, 4 μg plasmids or 4 μL siRNA were seeded in each well. After 5 hours, the transfection system was replaced with 2 mL culture medium with 10% FBS, and the cells were continually cultured at 37 °C for 48 h.
Table 1 Sense and antisense sequences of MCM2 siRNA and negative control
| SiRNA |
|
Sequence |
| MCM2 |
Sense |
5′-CCAUCUAUCAGAACUACCATT-3′ |
| Antisense |
5′-UGGUAGUUCUGAUAGAUGGTT-3′ |
| Negative control |
Sense |
5′-UUCUCCGAACGUGUCACGUTT-3′ |
| Antisense |
5′-ACGUGACACGUUCGGAGAATT-3′ |
2.3 Cell viability assay
The cell viability assay was performed using the Cell Counting Kit (CCK8, Dojindo Laboratories, Kumamoto) according to the manufacturer's instructions. Herein, four kinds of cell lines were seeded into 96-well plates (1 × 103 cells per well). After culturing for 24 hours, cells were incubated in the media with or without SIN. After 24 h, 48 h, and 72 h, the media were removed, and 100 μL medium containing 10 μL CCK8 was added to each well followed by incubation for other 2 h. The absorbance was measured at 450 nm using a microplate reader (Thermo, USA).
2.4 Clone formation assay
The clone ability of cancer cells is closely related to tumor recurrence. To assess the impact of SIN on the monoclonal ability of the OC cells, clone formation assay was performed. Herein, five hundred HeyA8 or IGROV1 cells were seeded into 6-well plates and then treated with different concentrations of SIN on the second day. The process was continued for 14 days until visible colonies were obtained. The cells were fixed in 4% paraformaldehyde for 15 min and washed two times with PBS before staining with 0.1% crystal violet for 10 min. The plates were imaged using the ECL ChemiDoc imaging system (Bio-Rad, USA). The results were assessed by counting the colonies using the IPP software (Image Pro Plus).
2.5 Wound-healing assay
Wound-healing assay was performed to indicate the migration ability of the cells exposed to SIN. When HeyA8 and IGROV1 cells grew up to 80–90% confluence in 6-well plates, artificial wounds were created by a 200 μl pipette tip. A serum-free medium containing SIN (0, 0.25, and 0.5 mM) was added to the cells. Images were obtained using an inverted microscope (Leica, Germany) at 0, 12, and 24 h.
2.6 Transwell migration and invasion assay
HeyA8 or IGROV1 cells in a 200 μL serum-free medium with SIN (0, 0.25, and 0.5 mM) were added to the upper chambers (24-well insert; 8 mm; Corning) spread with or without matrigel. The lower chambers were added with a 700 μL medium containing 10% FBS. After 24 h and 30 h, the non-migrated cells were, respectively, removed by a cotton swab. The migratory cells on lower surface of the chambers were fixed in 4% paraformaldehyde and stained with 2% crystal violet. Moreover, five random field images were obtained using a microscope (100; Olympus IX70; Olympus Corporation, Osaka, Japan).
2.7 Western blotting assay
After being incubated with various concentrations (0, 0.25, and 0.5 mM) of SIN for 48 h, cells were obtained and homogenized in RIPA lysis buffer (Beyotime Biotechnology) containing 1% PMSF (phenylmethylsulfonyl fluoride) and 1% protease inhibitor cocktail (Sigma), and the lysates were incubated on ice for 30 min, followed by 15 min centrifugation at 12000 rpm at 4 °C. The supernatant was obtained for further use. The nuclear protein samples were extracted using the Nuclear and Cytoplasmic Protein Extraction Kit (KeyGEN) according to the manufacturer's instructions. Protein samples mixed with 5× loading buffer were separated by 10% SDS–PAGE gel depending on the target protein size followed by transfer to NC membranes. After being blocked in 5% skim milk at room temperature for one hour, the membranes were incubated with primary antibodies including E-cadherin(proteintech), vimentin (proteintech), ZO-1(proteintech), N-cadherin (CST), Snail (proteintech), ZEB1 (proteintech), β-catenin (proteintech), MCM2 (Abcam), c-myc (proteintech), and cyclinD1 (proteintech) overnight at 4 °C. Then, the membranes were washed with TBST and incubated with HRP-conjugated secondary antibodies for one hour. The enhanced chemiluminescence ECL system (advanstar) was employed in immunodetection.
2.8 Tissue microassay
Tissue chip of OC was supplied by SHANGHAI OUTDO BIOTECH. The tissue microarray (TMA) analysis was carried out in small representative tissue samples obtained from 75 patients evaluated herein. Briefly, the slides were incubated in the MCM2 antibody (1:80) overnight. The sections were then incubated with biotinylated anti-rabbit antibody for 20 min followed by treatment with 3,3′-diaminobenzidine working solution and counterstaining with hematoxylin. MCM2 staining results were scored based on staining intensity (0 = negative, 1 = weak, 2 = moderate, and 3 = strong) and the percentage of positive cells (0 = 0–5%, 1 = 6–25%, 2 = 26–50%, 3 = 51–75%, and 4 = 76–100% positive OC cells).
2.9 Immunofluorescence assay
Cells grown on 15 mm diameter glass coverslips were washed with 1XPBS for 5 min and then fixed with 4% paraformaldehyde for 20 min, followed by permeabilization with 0.1% Triton X-100 and blocking with bovine serum albumin (BSA) for 30 min at 37 °C. Cells were incubated with the primary antibodies p65 for overnight at 4 °C. After being washed with 1XPBS, the cells were incubated with a FITC-labelled secondary antibody for 90 min, and then, propidium iodide (PI) was used to counterstain the nuclei. Slides mounted with coverslips containing an anti-quenching solution were observed using a fluorescence microscope (Nikon).
2.10 Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from the IGROV1 cells with TRIzol reagent (Invirtrogen, USA) followed by chloroform-isopropanol extraction and ethanol precipitation. Then, total RNA acting as the first template strand was converted into cDNA using the reverse transcription reagent kit (Takara, Japan). After this, samples were tested with a PCR array (Takara, Japan). Subsequently, real-time PCR assay was performed to quantify ZEB1, β-catenin, and β-actin gene transcripts with SYBR Green II dye (Takara, Japan). The forward and reverse primer sequences of the gene were designed by the Primer Premier 5.0 software and are listed in Table 2. The β-actin gene was used as an internal standard. Data were analyzed by the ΔΔCt method.
Table 2 Upstream and downstream sequences of the qRT-PCR primers
| Gene |
|
Sequence |
| ZEB1 |
Upstream: |
5′-TTACACCTTTGCATACAGAACCC-3′ |
| Downstream: |
5′-TTTACGATTACACCCAGACTGC-3′ |
| β-Catenin |
Upstream: |
5′-CCCAGCGTCGTCTGCTTTA-3′ |
| Downstream: |
5′-CGATTCGCTCTCCCCGTAAC-3′ |
| c-Myc |
Upstream: |
5′-CACGCTAGCCTGGATTTTTTTCGGGTAGTGG-3′ |
| Downstream: |
5′-CACGCGGCCGCTTACGCACAAGAGTTCCGTAG-3′ |
| CyclinD1 |
Upstream: |
5′-CCGTCCATGCGGAAGATC-3′ |
| Downstream: |
5′-GAAGACCTCCTCCTCGCACT-3′ |
| β-Actin |
Upstream: |
5′-CCTCTCCCAAGTCCACACAG-3′ |
| Downstream: |
5′-GGGCACGAAGGCTCATCATT-3′ |
2.11 Statistical analysis
Statistical analysis was performed with IBM SPSS Statistics 19.0. The results are expressed as mean ± standard deviation (S.D) and evaluated by the X2 test and Student's t-test. ANOVA and SNK test were employed to evaluate IC50. Each experiment was performed in triplicate. P < 0.05 was defined as statistically significant.
3. Results
3.1 SIN inhibits ovarian cancer cell line proliferation
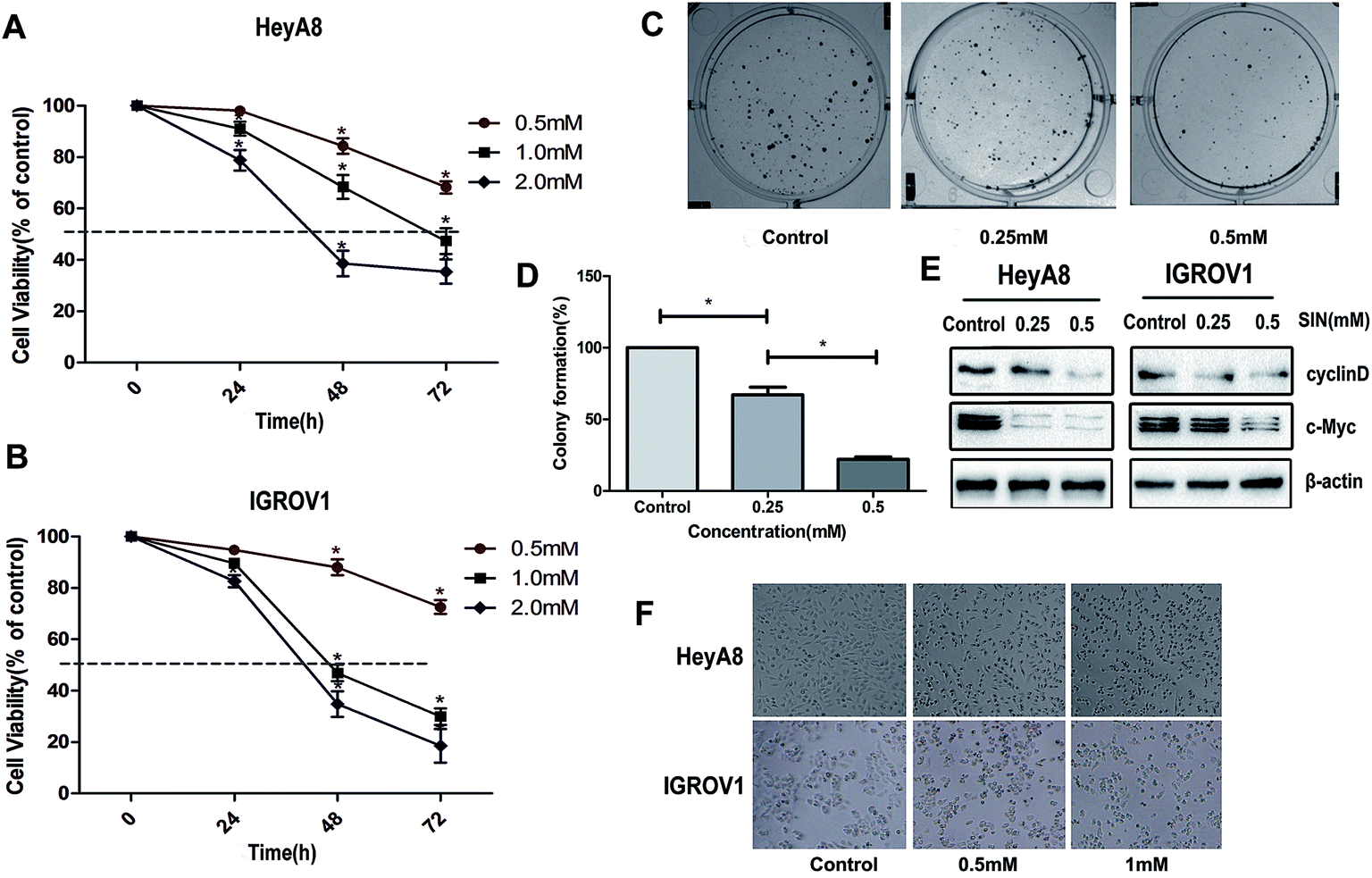
To extensively explore the effects of SIN on OC cells, HEK293T cell line and four kinds of OC cells lines, i.e. HO8910, HeyA8, IGROV1, and SKOV3, were incubated with SIN at the concentrations of 0, 0.1 mM, 1 mM, and 3 mM for 48 h. Cell viability was detected with CCK8. The results indicated that SIN inhibited OC cell proliferation at both concentrations of 1 mM and 3 mM to different extents in the four kinds of OC cells; however, its toxicity to HEK293T was feeble at a concentration of 1 mM (Fig. 1B and C). HeyA8 and IGROV1 cell lines were chosen to further detect the suppression effect of SIN in OC. They were treated with SIN (0, 0.5, 1, and 2 mM), and the OD was measured at 24 h, 48 h, and 72 h after treatment. The results demonstrated that cell viability decreased with an increase in time and dose (Fig. 2A and B). Cell proliferation was inhibited by SIN in a time- and dose-dependent manner. Clone-formation assay was performed to assess the long-term effects of SIN in the OC cells. The clone number and size gradually decreased with the increasing concentrations (0, 0.25, and 0.5 mM) (Fig. 2C), and a significant difference was revealed in the chart (P < 0.05, Fig. 2D). Results have demonstrated that the SIN may possess the potential ability to inhibit the OC cell, i.e. IGROV1, growth. To further detect the effects of SIN on OC cell proliferation, c-myc and cyclinD1 as representative proliferation markers in OC were investigated by western blot and clarified to be obviously suppressed by SIN (Fig. 2E). Moreover, changes were observed in the morphology of cells, and the results indicated that the shape of the OC cells changed to round and the number of cells was decreased (Fig. 1F); both these changes occurred in a dose-dependent manner. Data showed that SIN could inhibit the OC cell proliferation.
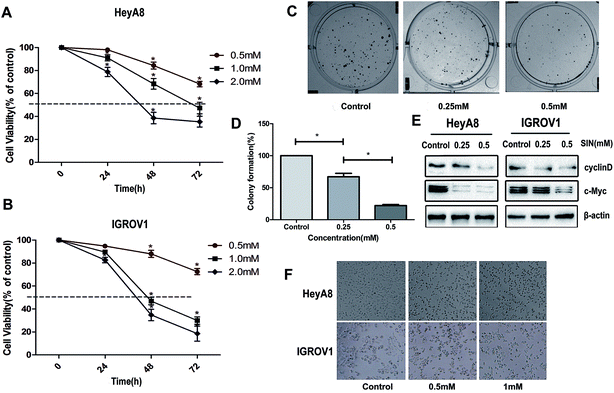 |
| | Fig. 2 SIN inhibited ovarian cancer cell viability. The dose-dependent inhibition effect on cell viability was measured by the CCK8 assay; (A and B) time- and dose-dependent inhibition of SIN was further investigated at indicated concentrations (0, 0.25, and 0.5 mM). Data were represented as mean ± S.D of three independent experiments. (C and D) Cell clone formation assay and its statistic analysis exhibited OC cell viability reduction by SIN. (E) The protein expression suppression of c-myc and cyclinD1 was verified by western blot. (F) Microscopic observations were performed to detect morphological changes in the OC cells treated with SIN. c-Myc, MYC proto-oncogene, BHLH transcription factor; cyclinD1, G1/S-specific cyclin D1; and β-actin, Actin Beta; *P < 0.05. Data are represented as mean ± S.D of three independent experiments of the cells. | |
3.2 SIN inhibits the OC cell migration and invasion
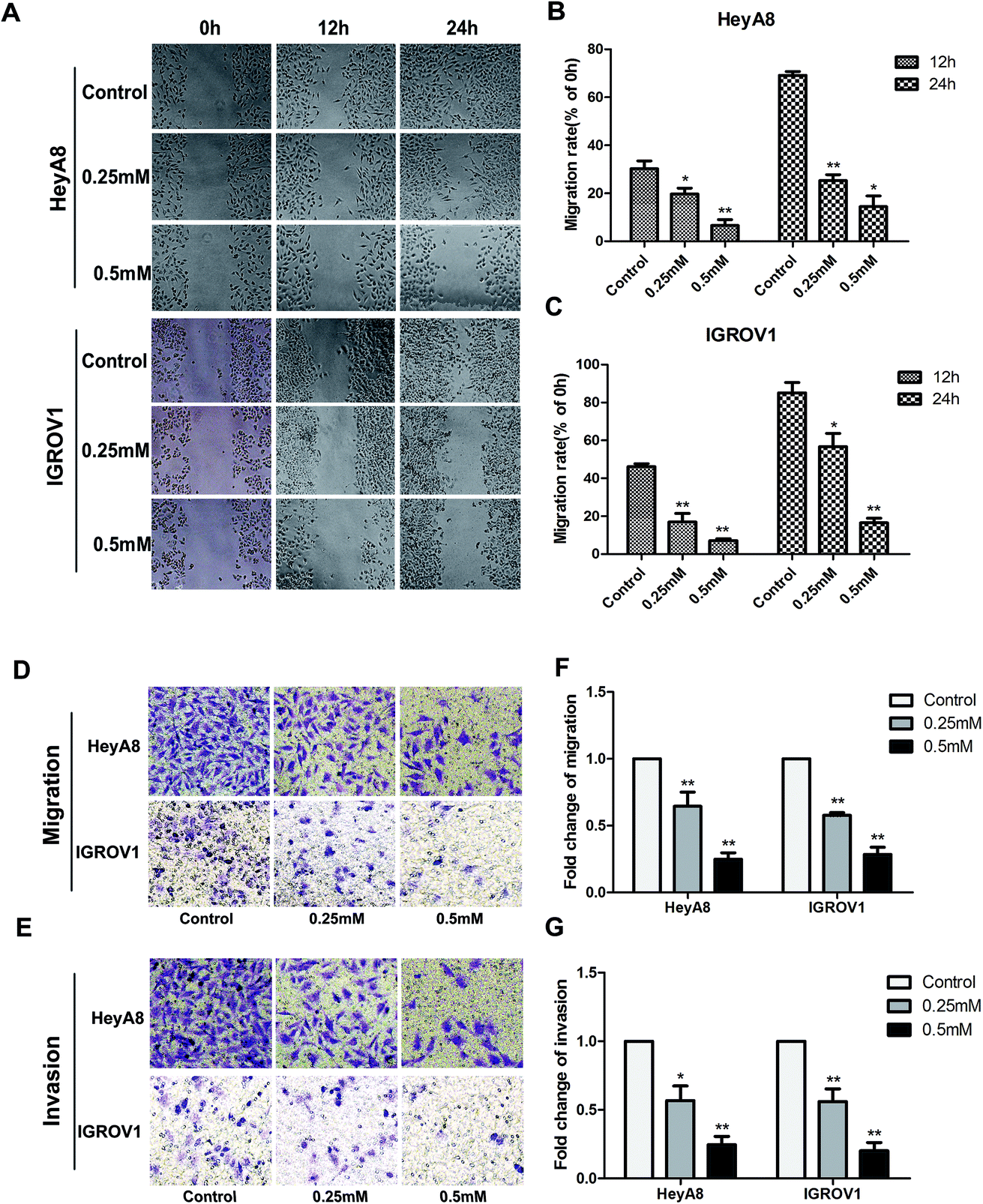
Accumulating studies exhibited that SIN could suppress some types of cancer cell metastasis and invasion. Wound-healing assay was performed in this study to detect the anti-metastasis effects of SIN on the OC cells. HeyA8 or IGROV1 cells treated with SIN at indicated concentrations displayed markedly different wound-healing abilities. SIN could obviously interfere with wound healing (Fig. 3A). The migration rate of HeyA8 and IGROV1 cells treated with SIN was gradually decreased with the increasing dosages, which was changed from 0.25 mM to 0.5 mM. Furthermore, HeyA8 cells treated with 0.25 mM SIN showed nearly 30% migration rate, whereas for the cells treated with 0.5 mM SIN, the migration rate decreased to almost 20%. In addition, the migration rate was changed from 60% to almost 20% when the SIN concentration was changed from 0.25 mM to 0.5 mM in the IGROV1 cells (P < 0.05, Fig. 3B and C). The inhibition of migration ability by 0.25 mM and 0.5 mM SIN was consistent with the result of the wound-healing assay. The transwell migration and invasion assay results indicated that the fold of migration change in HeyA8 and IGROV1 cells was gradually decreased by the treatment with SIN at different concentrations (0, 0.25, and 0.5 mM) (Fig. 3D and E). Furthermore, histogram according to the migrated cells number was shown to illustrate the inhibition of the OC metastasis by SIN (P < 0.01, Fig. 3F and G). Results suggest that SIN possesses potential anti-migration ability in the OC cells.
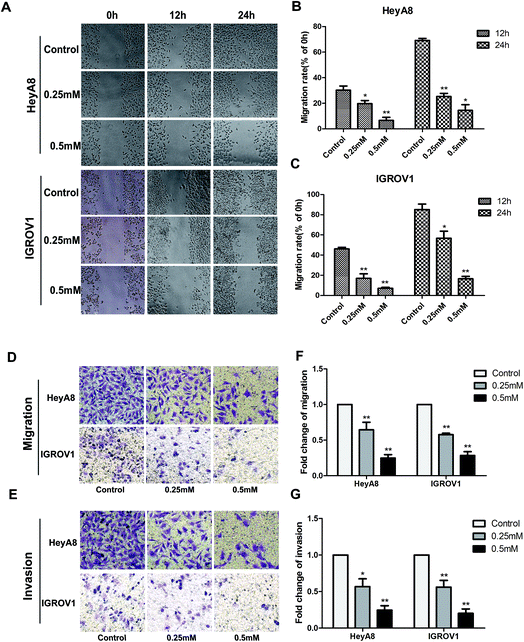 |
| | Fig. 3 Inhibition effects of SIN in metastasis and invasion were investigated in HeyA8 or IGROV1 cells. (A) Wound-healing assay was conducted in HeyA8 or IGROV1 cells to evaluate cell migration. An artificial wound was created after treatment with 0, 0.25, and 0.5 mM SIN, and images were obtained at 0, 12, and 24 h. (B and C) Statistical analysis results illustrated significant differences between SIN-treated group and the control group. Data are represented as mean ± S.D of three independent experiments. (D and E) The reduction in migrated rates was observed through transwell migration and invasion assay. (F and G) In the bar chart, significant differences of migrated rates were evaluated between SIN-treated groups and the control group. *P < 0.05; **P < 0.01. Data are represented as mean ± S.D of three independent experiments of the cells. | |
3.3 SIN inhibits OC cell EMT-related protein expression
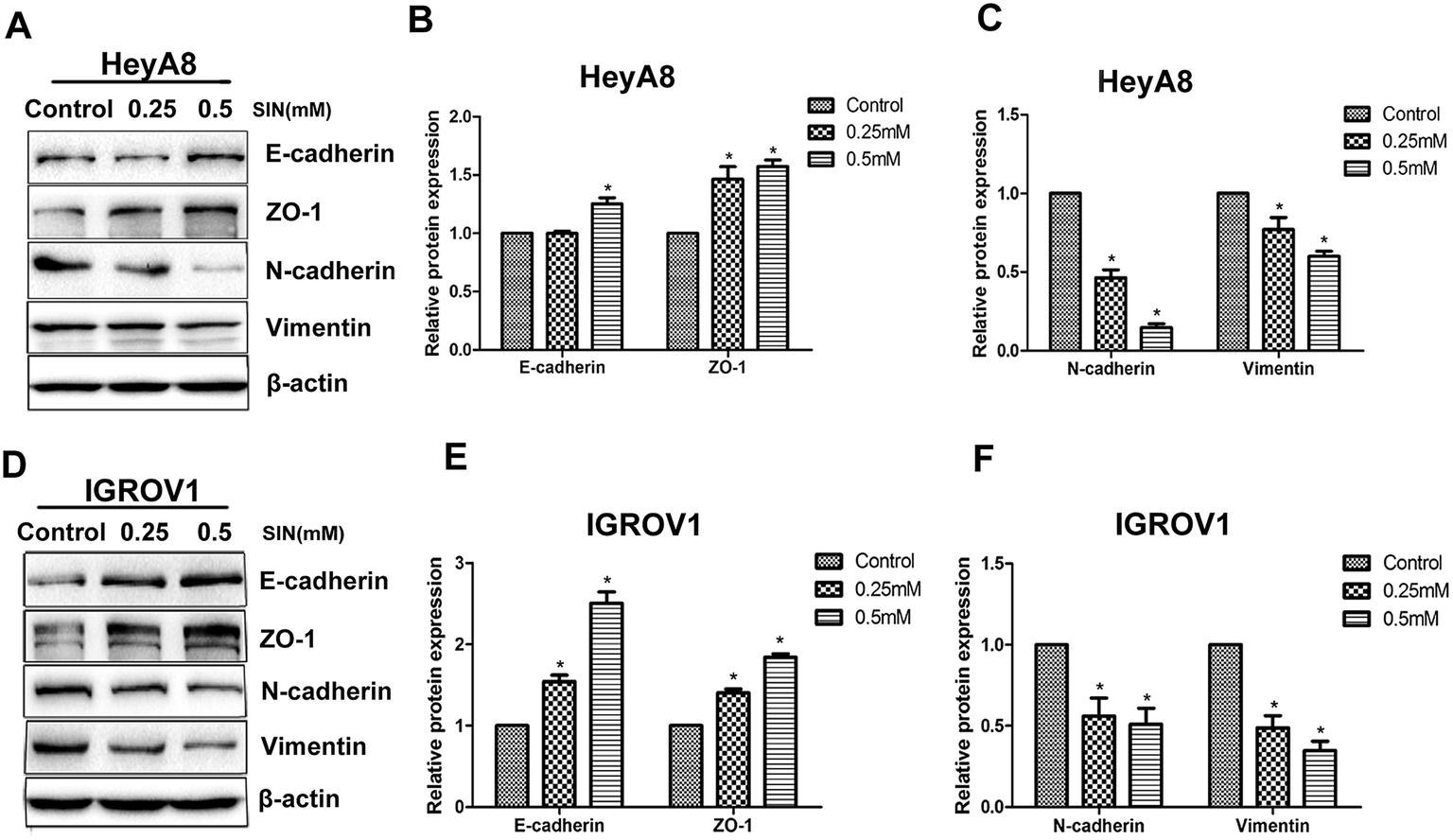
Reports have indicated that EMT can be reversed when the ability of migration is inhibited in various types of cancers. EMT is typically characterized by the attenuation of the epithelial markers, such as E-cadherin, β-catenin, and ZO-1, and the increase in mesenchymal markers such as N-cadherin, ZEB1, and vimentin. The results clarified the significant increase in the mesenchymal markers E-cadherin and ZO-1, accompanied by the reduction in vimentin and N-cadherin (Fig. 4A and D). E-cadherin protein expression level showed significant differences between 0.25 mM and 0.5 mM SIN (P < 0.05), whereas there was no significant difference between the control group and 0.25 mM SIN-treated HeyA8 cells (Fig. 4B). The gradual acquisition of ZO-1 occurred along with the gradual loss of N-cadherin and vimentin (P < 0.05, Fig. 4B and C). In IGROV1 cells, the EMT marker proteins exhibited different significances between any two groups (P < 0.05, Fig. 4E and F). Data indicated that SIN might inhibit the OC cell EMT.
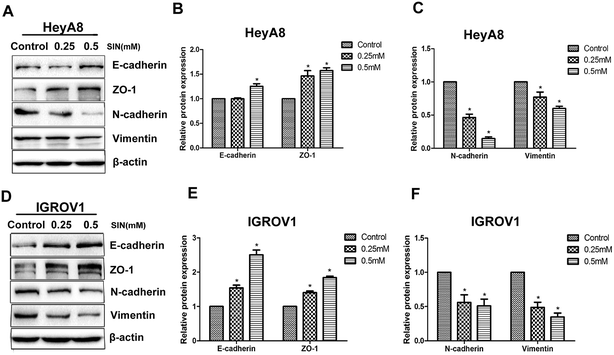 |
| | Fig. 4 SIN decreased the protein expression levels of EMT-related proteins. (A) Western blot results indicated that the expression of E-cadherin and ZO-1 were up-regulated together with the down-regulation of N-cadherin and vimentin in HeyA8 cells; (B and C) histogram exhibited statistically significant differences between SIN-treated groups and the control group; (D) the same western blot detection of the protein expression levels of E-cadherin, ZO-1, N-cadherin and vimentin was conducted in IGROV1 cells; (E and F) histogram exhibited statistically significant differences between SIN-treated groups and the control group. E-cadherin, epithelial cadherin; ZO-1, zona occludens 1; N-cadherin, neural cadherin; *P < 0.05. Data was represent in three independent experiments. | |
3.4 SIN suppresses the Wnt/β-catenin signaling pathway and MCM2
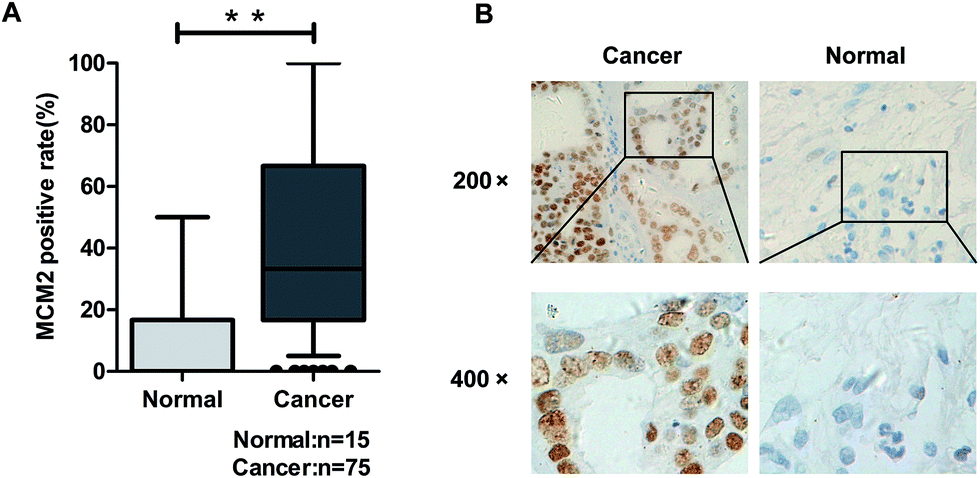
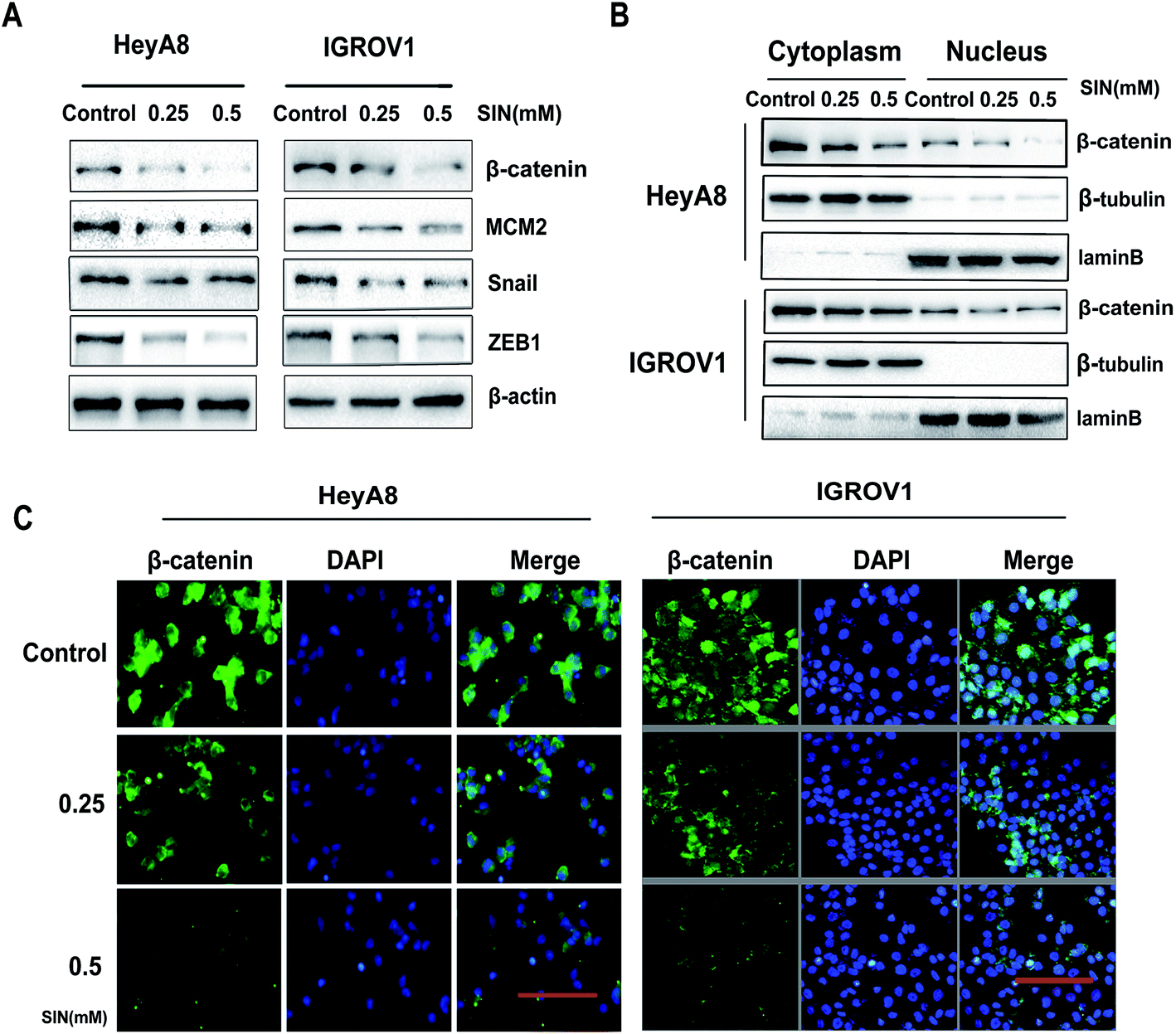
MCM2 clinical expression was measured in the samples obtained from 75 patients, and the results in this study clarified the significant difference of the MCM2 positive rate between ovarian cancer and the corresponding normal tissues (P < 0.001, Table 3, and Fig. 5A). We also extensively verified MCM2 nuclear location in ovarian tissues (Fig. 5B). To further explore the molecular mechanism of SIN in inhibiting metastasis, nuclear and cytoplasmic proteins were respectively extracted and detected, and the results indicated that total β-catenin, cytoplasmic β-catenin, and nuclear β-catenin, attenuated in a SIN dose-dependent manner (Fig. 6A and B). Snail, the downstream target gene of the Wnt/β-catenin pathway, was significantly downregulated accompanied by the evident reduction of ZEB1 (Fig. 6A). Moreover, MCM2 acting as a pivotal regulatory factor in DNA replication exhibited a protein expression loss caused by SIN (Fig. 6A). β-Catenin was further examined by immunofluorescence in HeyA8 and IGROV1 cells. It was observed that nuclear β-catenin staining together with cytoplasm β-catenin was visibly weakened by SIN in a concentration-dependent manner (Fig. 6C). Therefore, it was indicated that SIN treatment led to a decrease in the protein expression level of total, nuclear, and cytoplasmic β-catenin. Total β-catenin contains active and inactive β-catenin, and the reduction in the accumulation of nuclear β-catenin, derived from active β-catenin undergoing nuclear translocation, forbade the Wnt/β-catenin signaling attenuation; this suggested that SIN might suppress the Wnt/β-catenin signaling pathway in OC cells.
Table 3 Statistic analysis of MCM2 in normal and cancer tissues
| Variable |
MCM2 |
Number |
Positive rate (%) |
X2 |
P |
| − |
+ |
| P < 0.001, P value was acquired by Pearson's chi-square test. |
| Normal |
13 |
2 |
15 |
13.3 |
14.575 |
0.000a |
| Cancer |
25 |
50 |
75 |
66.7 |
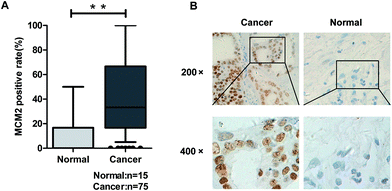 |
| | Fig. 5 MCM2 expression was increased in OC tissues than that in normal tissues. (A) Statistic analysis clarified the significant difference between OC tissues and normal ovarian tissues; (B) immunohistochemical staining of MCM2 showed the visible difference of positive expression between OC tissues and normal ovarian tissues. MCM2 nuclear location was also present. **P < 0.01 (Pearson's chi-square test). | |
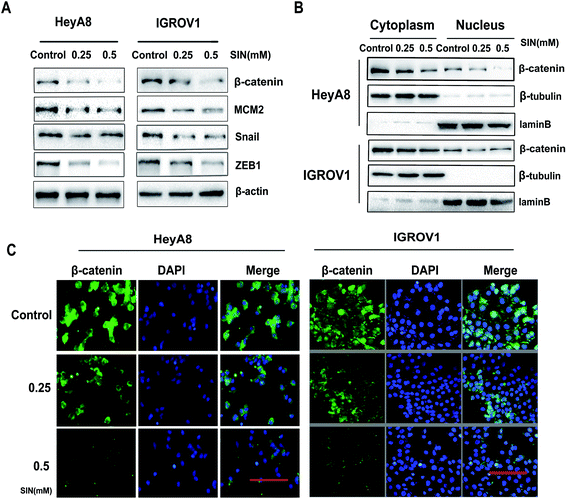 |
| | Fig. 6 SIN attenuated MCM2 and blocked the Wnt/β-catenin signaling pathway. (A) After cells were treated with SIN (0, 0.25, 0.5 mM), western blot was performed to detect the decrease in β-catenin, MCM2, Snail and ZEB1. (B) The reduction in cytoplasm β-catenin together with nuclear β-catenin was verified by western blot. (C) In HeyA8 or IGROV1 ovarian cancer cells, immunofluorescence assay exhibited gradually weakened staining of cytoplasm β-catenin and nuclear β-catenin in SIN-treated groups compared with that in the control group. ZEB1, zinc finger E-box binding homeobox1; Snail, zinc finger protein SNAI1; β-catenin, catenin beta 1 β-tubulin, tubulin beta class I; MCM2, minichromosome maintenance complex component 2; DAPI, 4′,6-diamidino-2-phenylindole; scale bars: 100 μm. | |
3.5 SIN inhibits the Wnt/β-catenin signaling pathway via targeting MCM2
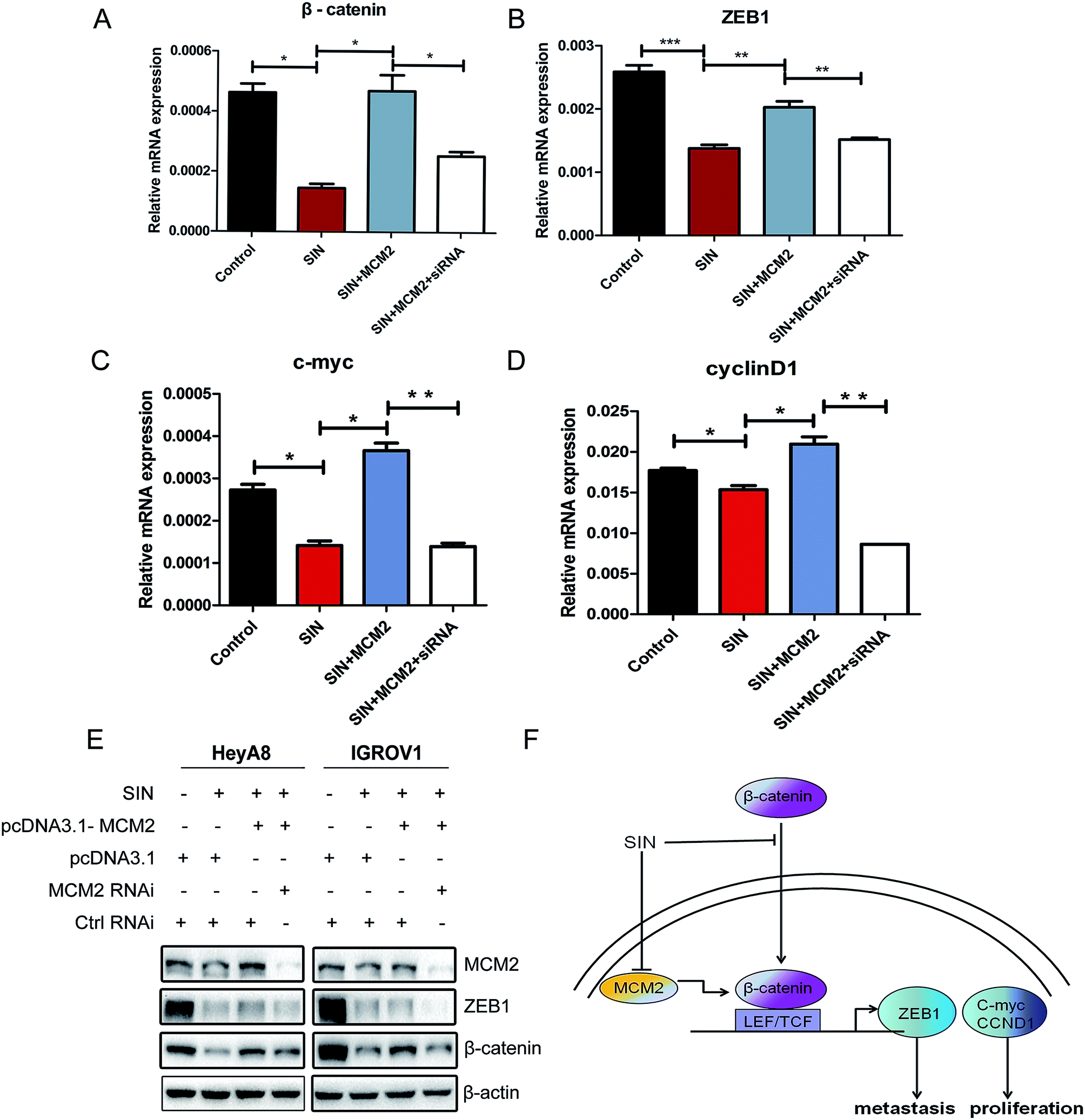
To further investigate the role of MCM2 in OC cell progression, MCM2 expression plasmids and MCM2 siRNA were used to modulate the MCM2 protein expression level in OC cells treated with SIN. QPCR assay was performed to explore whether MCM2 affected the transcription of β-catenin and its downstream factors in the Wnt/β-catenin signaling pathway. Results conveyed that β-catenin together with ZEB1 mRNA was regulated by MCM2 and showed positive correlation with MCM2 in the OC cells. The increase in the amount of MCM2 was followed by evident rescue of β-catenin mRNA (P < 0.05) and ZEB1 mRNA levels (P < 0.01), as expected. Furthermore, MCM2 knockdown using siRNA led to a significant degradation of β-catenin (P < 0.05) and ZEB1 (P < 0.01) mRNA levels again (Fig. 7A and B). c-Myc and cyclinD1 mRNA levels were also clarified to be regulated by MCM2 according to the regulatory custom of MCM2 to β-catenin and ZEB1 (Fig. 7C and D). In accordance with the phenomena that occurred in the mRNA levels of β-catenin, ZEB1, western blot assay results showed that MCM2 overexpression and knockdown were respectively followed by the rescue and further loss of these protein expression levels (Fig. 7E). The involvement of MCM2 further hinted that SIN might suppress the Wnt/β-catenin signaling pathway via regulation of MCM2. Thus, it was inferred that SIN might inhibit the Wnt/β-catenin pathway through targeting MCM2, thereby performing anti-metastasis and anti-proliferation functions in the OC cells (Fig. 7F). The involvement of MCM2 in the Wnt/β-catenin signaling pathway has been explored in our study for the first time.
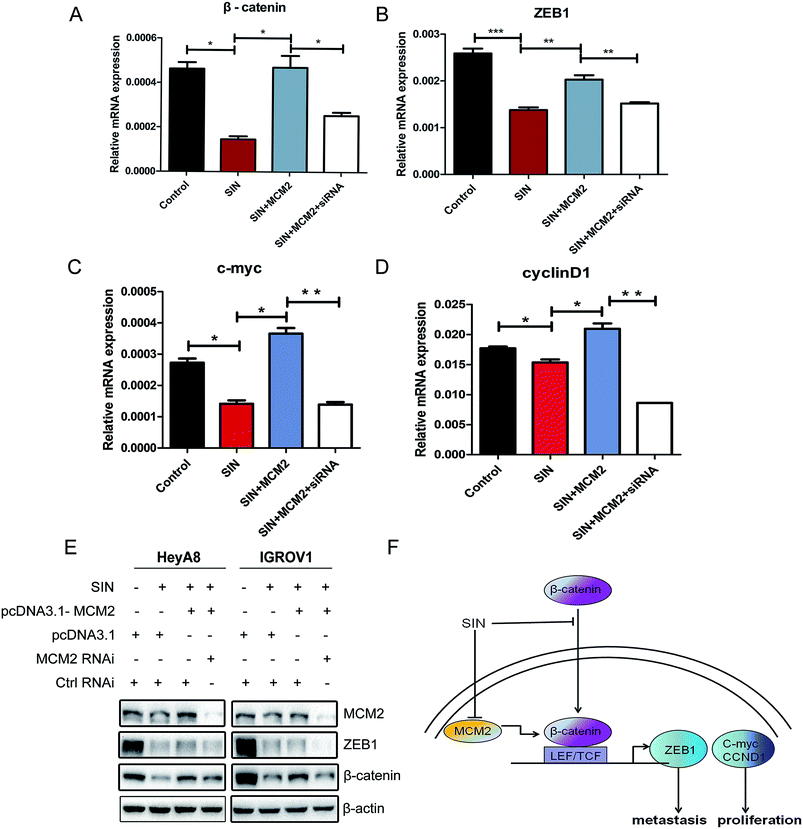 |
| | Fig. 7 SIN inhibited the Wnt/β-catenin signaling pathway via targeting MCM2. HeyA8 or IGROV1 cells treated with SIN were transfected with pcDNA3.1-MCM2 or its corresponding control, MCM2 siRNA and its negative control. (A and B) QPCR was performed to measure the mRNA levels of β-catenin and ZEB1. (C and D) QPCR was performed to measure the mRNA levels of c-myc and cyclinD1. (E) Western blot was carried out to detect the protein expression levels of MCM2, β-catenin and ZEB1. (F) It was exhibited that SIN inhibits OC proliferation and metastasis through the MCM2-mediated Wnt/β-catenin signaling pathway. *P < 0.05; **P < 0.01; ***P < 0.001. Data were obtained from three independent experiments. | |
4. Discussion
In this study, we have found that SIN plays anti-tumor functions by inhibiting proliferation and metastasis in a time- and concentration-dependent manner in the OC cells. In addition, SIN might suppress metastasis together with reversing EMT and reducing β-catenin nuclear accumulation. ZEB1, acting as a significant EMT regulator, and β-catenin are modulated by MCM2. Results may provide a vital clue to further detect the anti-tumor molecular mechanism of SIN in OC.
In recent years, studies on medicines for the prevention or treatment against various types of cancers have significantly increased. SIN, as an alkaloid, has been used to clinically treat mesangial proliferative nephritis, neuropathic pain hypersensitivity, and various rheumatoid diseases.26 In addition, evidence have indicated that SIN has potential neuroprotective,27 immunoregulatory, and anti-inflammatory activities.28 SIN was discovered to have an inhibitory impact on cell proliferation.29 The anti-cancer effects of SIN gradually gained attention. Recently, studies have clarified that SIN can inhibit proliferation and metastasis and induce apoptosis in multiple cancers including synovia, esophagus, colon, gastric, liver, breast, and lung cancers.30 Via applying two OC cell lines, namely HeyA8 and IGROV1, in the present study, it has been proven that SIN can notably inhibit OC proliferation and metastasis in a time- and concentration-dependent manner.
Cancer metastasis is a highly complex and multi-stage biological process, and a number of studies have demonstrated that EMT is involved in tumor migration and invasion. SIN inhibited breast cancer metastasis by attenuating the inflammation-related EMT.9 In the current studies, it was observed that SIN could reduce migration and invasion and reverse EMT by regulating the protein expression of EMT markers including vimentin, N-cadherin, and E-cadherin.31 Downregulation of E-cadherin, which releases free β-catenin, correlates with EMT.32 Transcriptional repression of tight junction molecules, including occludin, ZO-1/2, and E-cadherin, is a pivotal phenomenon during EMT. Our results indicated EMT reversion occurrence after SIN treatment in OC, as shown by the elevation of ZO-1 and E-cadherin as well as the loss N-cadherin and vimentin levels. Accumulating studies have associated the metastasis and EMT induction with Wnt/β-catenin signaling pathway activation.33 Aberrant activation of Wnt/β-catenin, which acts as a transcription factor, has been detected in a large proportion of human malignancies.34 β-Catenin accumulation and nuclear translocation lead to aggressive tumor formation and metastasis by promoting the disruption of adheren junctions, dedifferentiation, and EMT transcriptional program.35 Snail1, ZEB1, and ZEB2 can bind directly to E-boxes and repress the E-cadherin transcription.36 The observation of the loss of β-catenin, ZEB1, and Snail in our study has provided a clue that SIN may inhibit metastasis evolution by inactivating Wnt/β-catenin-mediated EMT. Carcinoma cells with nuclear β-catenin accumulation appear to trigger the activity of transcriptional factors, such as ZEB1, Snail, c-myc, and cyclinD1, that undergo EMT-mediated metastasis and proliferation. Following the deregulation of c-myc expression by gene amplification or abnormal signaling, abnormal cell proliferation might occur visibly.37 Data in our study illustrated that c-myc and cyclinD1 were obviously decreased by SIN treatment of the OC cells; moreover, it was indicated that SIN might suppress OC cell proliferation through Wnt/β-catenin-mediated cyclinD1 and c-myc activity. MCM2 and its homologues form a functional complex that comprises an important part of the pre-replicative complex (PRE-RC) of replication proteins found at the replication origins during the G1 phase.38 Deregulated MCM2 in various types of cancers is involved in carcinoma proliferation and metastasis. We demonstrated the association between MCM2 and rescue of β-catenin as well as ZEB1, the association between the MCM2 knockdown, and the reduction in β-catenin and ZEB1 again in HeyA8 or IGROV1 cells with SIN treatment.
In summary, this was the first study that highlighted the anti-tumor effect and the underlying molecular mechanism of SIN in OC. Our study preliminarily unveiled that SIN inhibited the proliferation and metastasis of OC cells via the Wnt/β-catenin signaling pathway. Our results also preliminarily illustrated a novel regulatory effect of MCM2 on the Wnt/β-catenin signaling pathway. These results may open up new possibilities for future therapeutic interventions. The reduction function of SIN in OC progression, especially in cancer metastasis, suggests that SIN may act as a promising reagent in anti-tumor therapies and deserves further exploration.
Conflicts of interest
None.
Acknowledgements
There was no funding to support the research. All authors are likely to show their appreciation to reviewers for their helpful comments.
References
- X. Liu, R. Dong, Z. Jiang, Y. Wei, Y. Li, L. Wei, H. Sun, Y. Li, N. Yang, Q. Yang, Z. Liu and B. Kong, Tumor Biol., 2015, 36, 4261–4269 CrossRef CAS PubMed.
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2017, 67, 7–30 CrossRef PubMed.
- C. S. Leung, T. L. Yeung, K. P. Yip, S. Pradeep, L. Balasubramanian, J. Liu, K. K. Wong, L. S. Mangala, G. N. Armaiz-Pena, G. Lopez-Berestein, A. K. Sood, M. J. Birrer and S. C. Mok, Nat. Commun., 2014, 5, 5092 CrossRef CAS PubMed.
- V. Brower, JNCI, J. Natl. Cancer Inst., 2008, 12, 838–839 CrossRef PubMed.
- Q. Wang and X.-K. Li, Int. Immunopharmacol., 2011, 11, 373–376 CrossRef CAS PubMed.
- Q. Zhu, Y. Sun, J. Zhu, T. Fang, W. Zhang and J.-X. Li, Sci. Rep., 2014, 4, 7270 CrossRef CAS PubMed.
- L. Qian, Z. Xu, W. Zhang, B. Wilson, J. S. Hong and P. M. Flood, J. Neuroinflammation, 2007, 4, 23 CrossRef PubMed.
- J. Wang, Z. R. Yang, W. G. Dong, J. X. Zhang, X. F. Guo, J. Song and S. Qiu, World J. Gastroenterol., 2013, 19, 8292–8300 CrossRef CAS PubMed.
- X. Li, P. Li, C. Liu, Y. Ren, X. Tang, K. Wang and J. He, Oncotarget, 2017, 8, 13560–13574 Search PubMed.
- X. L. Lu, J. Zeng, Y. L. Chen, P. M. He, M. X. Wen, M. D. Ren, Y. N. Hu, G. F. Lu and S. X. He, Int. J. Oncol., 2013, 42, 229–238 CrossRef CAS PubMed.
- H. Y. Niu, W. Zhou, Y. X. Xu, Z. Q. Yin, W. Z. Shen, Z. Ye, Y. H. Liu, Y. A. Chen, S. Yang, R. Xiang, L. N. Wang and P. P. Qu, Oncotarget, 2017, 8, 76266–76278 Search PubMed.
- S. Hori, K. Wadhwa, V. Pisupati, V. Zecchini, A. Ramos-Montoya, A. Y. Warren, D. E. Neal and V. J. Gnanapragasam, Int. J. Cancer, 2017, 140, 1881–1887 CrossRef CAS PubMed.
- S. Lin, X. Lyu, J. Yu, L. Sun, D. Du, Y. Lai, H. Li, Y. Wang, L. Zhang, H. Yin and S. Yuan, Phytomedicine, 2016, 23, 1053–1063 CrossRef CAS PubMed.
- C. W. Lin, L. K. Wang, S. P. Wang, Y. L. Chang, Y. Y. Wu, H. Y. Chen, T. H. Hsiao, W. Y. Lai, H. H. Lu, Y. H. Chang, S. C. Yang, M. W. Lin, C. Y. Chen, T. M. Hong and P. C. Yang, Nat. Commun., 2016, 7, 13867 CrossRef CAS PubMed.
- J. Tang, Y. Li, J. Wang, Z. Wen, M. Lai and H. Zhang, Cancer Lett., 2016, 371, 301–313 CrossRef CAS PubMed.
- M. Zeisberg and E. G. Neilson, J. Clin. Invest., 2009, 1429–1437 CrossRef CAS PubMed.
- J. Sun, X. Yang, R. Zhang, S. Liu, X. Gan, X. Xi, Z. Zhang, Y. Feng and Y. Sun, Cancer Med., 2017, 6, 834–844 CrossRef CAS PubMed.
- R. Nusse and H. Clevers, Cell, 2017, 169, 985–999 CrossRef CAS PubMed.
- S. Shin, H. J. Im, Y. J. Kwon, D. J. Ye, H. S. Baek, D. Kim, H. K. Choi and Y. J. Chun, Oncotarget, 2017, 8, 61604–61617 Search PubMed.
- Y. Wang, F. Bu, C. Royer, S. Serres, J. R. Larkin, M. S. Soto, N. R. Sibson, V. Salter, F. Fritzsche, C. Turnquist, S. Koch, J. Zak, S. Zhong, G. Wu, A. Liang, P. A. Olofsen, H. Moch, D. C. Hancock, J. Downward, R. D. Goldin, J. Zhao, X. Tong, Y. Guo and X. Lu, Nat. Cell Biol., 2014, 16, 1092–1104 CrossRef CAS PubMed.
- C. Min, S. F. Eddy, D. H. Sherr and G. E. Sonenshein, J. Cell. Biochem., 2008, 104, 733–744 CrossRef CAS PubMed.
- J. D. Gordan, C. B. Thompson and M. C. Simon, Cancer Cell Int., 2007, 12, 108–113 CrossRef CAS PubMed.
- P. Zhang, Y. Sun and L. Ma, Cell Cycle, 2015, 14, 481–487 CrossRef CAS PubMed.
- M. D. Ramer, E. S. Suman, H. Richter, K. Stanger, M. Spranger, N. Bieberstein and B. P. Duncker, J. Biol. Chem., 2013, 288, 14926–14935 CrossRef CAS PubMed.
- K. M. Lau, Q. K. Chan, J. C. Pang, K. K. Li, W. W. Yeung, N. Y. Chung, P. C. Lui, Y. S. Tam, H. M. Li, L. Zhou, Y. Wang, Y. Mao and H. K. Ng, Oncogene, 2010, 29, 5475–5489 CrossRef CAS PubMed.
- H. Shi, K. Zheng, Z. Su, H. Su, M. Zhong, X. He, C. Zhou, H. Chen, Q. Xiong and Y. Zhang, J. Neuroimmunol., 2016, 299, 28–34 CrossRef CAS PubMed.
- Y. Yang, H. Wang, L. Li, X. Li, Q. Wang, H. Ding, X. Wang, Z. Ye, L. Wu, X. Zhang, M. Zhou and H. Pan, Front. Neurosci., 2016, 10, 580 Search PubMed.
- Z.-J. Zhao, C. Zhao, J. Xiao and J.-C. Wang, Molecules, 2016, 21, 1520 CrossRef PubMed.
- Y. Lv, C. Li, S. Li and Z. Hao, Oncol. Lett., 2011, 4, 741–745 Search PubMed.
- H. Zhang, Y. Ren, X. Tang, K. Wang, Y. Liu, L. Zhang, X. Li, P. Liu, C. Zhao and J. He, Sci. Rep., 2015, 5, 8888 CrossRef CAS PubMed.
- X. Li, P. Li, C. Liu, Y. Ren, X. Tang, K. Wang and J. He, Oncotarget, 2017, 8, 13560–13574 Search PubMed.
- E. Scarpa, A. Szabo, A. Bibonne, E. Theveneau, M. Parsons and R. Mayor, Dev. Cell, 2015, 34, 421–434 CrossRef CAS PubMed.
- M. Luo, L. Hou, J. Li, S. Shao, S. Huang, D. Meng, L. Liu, L. Feng, P. Xia, T. Qin and X. Zhao, Cancer Lett., 2016, 373, 1–11 CrossRef CAS PubMed.
- J. Deka, N. Wiedemann, P. Anderle, F. Murphy-Seiler, J. Bultinck, S. Eyckerman, J. C. Stehle, S. Andre, N. Vilain, O. Zilian, S. Robine, M. Delorenzi, K. Basler and M. Aguet, Cancer Res., 2010, 70, 6619–6628 CrossRef CAS PubMed.
- H. Clevers, Cell, 2006, 127, 469–480 CrossRef CAS PubMed.
- J. Y. Shih, M. F. Tsai, T. H. Chang, Y. L. Chang, A. Yuan, C. J. Yu, S. B. Lin, G. Y. Liou, M. L. Lee, J. J. Chen, T. M. Hong, S. C. Yang, J. L. Su, Y. C. Lee and P. C. Yang, Clin. Cancer Res., 2005, 11, 8070–8078 CrossRef CAS PubMed.
- M. D. Cole and V. H. Cowling, Nat. Rev. Mol. Cell Biol., 2008, 9, 810–815 CrossRef CAS PubMed.
- J. J. Blow and B. Hodgson, Trends Cell Biol., 2002, 12, 72–80 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Zhikun Linb,
Yuxin Baia,
Xinming Chid,
Hailu Fua,
Rui Sunc,
Meizi Liub,
Xuan Liua,
Liying Chena and
Shujuan Shao*d
a,
Zhikun Linb,
Yuxin Baia,
Xinming Chid,
Hailu Fua,
Rui Sunc,
Meizi Liub,
Xuan Liua,
Liying Chena and
Shujuan Shao*d