DOI:
10.1039/C7RA09489B
(Paper)
RSC Adv., 2017,
7, 50027-50034
A ternary composite oxides S2O82−/ZrO2–TiO2–SiO2 as an efficient solid super acid catalyst for depolymerization of lignin†
Received
27th August 2017
, Accepted 22nd October 2017
First published on 26th October 2017
Abstract
The solid, super, acid catalyst S2O82−/ZrO2–TiO2–SiO2, which has both a strong Brønsted acid and Lewis acid, was prepared and applied in lignin depolymerization. The results indicate that S2O82−/ZrO2–TiO2–SiO2 is an effective catalyst for lignin depolymerisation since the liquid product was almost completely dissolved in ethyl acetate, indicating that it was comprised of mostly oligomeric lignin degradation products. The effect of hydrogenation co-catalysts (Pd/C, Pt/C and Ru/C, Rh/C) were also studied and found to improve the yield of aromatic compounds and the petroleum ether soluble fraction, which are mostly monomeric and dimeric products. The resulting yields of aromatic compounds and petroleum ether soluble products were 14.8% and 46%, respectively, when S2O82−/ZrO2–TiO2–SiO2 and Pt/C mixed catalyst was used at 310 °C for 24 hours. These results show that the use of a combination Brønsted and Lewis acid catalyst in combination with a hydrogenation catalyst (S2O82−/ZrO2–TiO2–SiO2 together with a noble metal catalyst) opens a practical route for efficient lignin depolymerization.
1 Introduction
Lignocellulosic biomass is one of the most abundant renewable sources in nature that can be used to produce high value-added chemicals and liquid fuel. Lignocellulosic biomass mainly contains lignin, cellulose, and hemicellulose. Lignin is an integral part of the secondary cell walls of lignocellulosic biomass such as plants and some algae.1,2 The proportion of lignin in lignocellulosic biomass is 25–35% by weight. Lignocellulosic biomass has a high energy density, causing lignin depolymerization to receive a lot of attention.3 In 2004, almost 100 million tons of technical lignin was produced as a by-product in the pulp and paper industry. More than 95% of the lignin was used as fuel in the chemical recovery process of alkaline pulping, while the remaining lignin was been used in other applications.4,5 Although the chemical recovery is essential for the economic viability of the alkaline pulping processes, approximately 10–15% of lignin can be isolated for valorization without negatively impacting the recovery processes. Therefore, there is roughly 10 million tons of technical lignin that could be available for use if a higher value-added application is found relative to its current value as a fuel.
So lignin depolymerization will be a key path for developing higher value-added applications, but currently there are limited research efforts on the topic.6,7 Lignin depolymerization faces great challenges due to its highly irregular polymeric structure. It is a complex three-dimensional amorphous polymer consisting of three basic phenyl-propane units (syrigyl-, guaiacyl, and p-hydroxyphenyl-propane).8 The different phenyl-propane units are linked together by C–O and C–C bonds. The most common linkages are the β-O-4, β-1, β-5, α-O-4, and 5–5′ linkages.9,10 To achieve the goal of lignin depolymerization, the primary task is to break these chemical bonds in order to decrease its molecular weight.
There are a lot of technologies including acid/base catalysed depolymerization, pyrolysis, catalytic hydrotreatment, chemical oxidation and biodegradation to produce valuable chemicals and bio-fuels.11 In term of acid catalysts, different types of mineral acids, zeolites, Lewis acids, and organic acids had been used in lignin depolymerization.12 For a long period of time, homogeneous acids had mainly been used for the separation and removal of lignin and to cleave the lignin into simple monomeric compounds. In 1924, hydrochloric acid was the first homogeneous mineral acid used in lignin depolymerization and conversion.13 The yield of aromatic monomers depended mainly on the concentration of the acid. Stronger acids can result in a higher conversion rate, but it also leads to serious corrosion of the reactor. The use of strong acid presents a very serious environment pollution problem as well.14 Consequently, there has been an increase in the number of researchers applying environmentally friendly, solid, acid catalysts for the transformation of lignocellulosic materials.15
Lignin contains a lot of ether bonds in the structure, and acid could effectively catalyst their cleavage.16 The acid-catalyst plays a significant role in hydrolytic cleavage of α- and β-aryl ether linkage.17 Williams and Lawton found that using a catalyst with coexisting Lewis and Brønsted acids can cause significant improvements in the overall performance of lignin depolymerization.18 Vuori and Ekerdt also have demonstrated that solid catalysts become more active for lignin depolymerization when Lewis acids were converted to the Brønsted acids form.19,20
Sulfated metal oxides are common strong solid acids, which include both a Lewis and Brønsted acid site. SO42−/SnO2, SO42−/TiO2, SO42−/ZrO2, and SO42−/ZrO2–SiO2 were employed in biomass conversion processes, and achieved promising results.21,22 Deepa demonstrated that the catalytic efficiency of one-component solid acid catalyst was lower than the efficiency of a multicomponent solid acid catalyst due to the difference of each component affecting the activity and stability of the catalyst.23 The strength and ratio of Lewis and Brønsted acid mainly rely on the concentration of the impregnation liquid and the type of oxide.24 TiO2 and ZrO2 contain Lewis acid sites and therefore they could be easily used to synthesize a solid super acid.25 Silica, a neutral support, can provide considerably high surface areas and stability, which can influence the performance of a heterogeneous catalyst.26 In a sulphated metal oxide, the inductive effect of S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O makes the solid super acid catalyst have a stronger Lewis acidity, whereas Brønsted acidity can form in the presence of water.27 Sulfuric acid is the most common acidifier, but it is dangerous in the process of impregnation. Ammonium persulfate is a safer acidifier and therefore, was used to improve the number of Brønsted and Lewis acid sites. In this paper, a ternary solid acid catalyst containing silica, zirconia, and titanium, was synthesized. It is a stable, environment-friendly catalyst that has strong Brønsted and Lewis acid sites. In any acid catalysed lignin depolymerisation, lignin condensation is inevitable, resulting in a limited yield of aromatic monomers during lignin degradation.28,29 Hydrogenation catalysts have commendable activity in hydrogenolysis and hydrogenation reaction, but it could alleviate self-condensation phenomenon in lignin degradation.30
O makes the solid super acid catalyst have a stronger Lewis acidity, whereas Brønsted acidity can form in the presence of water.27 Sulfuric acid is the most common acidifier, but it is dangerous in the process of impregnation. Ammonium persulfate is a safer acidifier and therefore, was used to improve the number of Brønsted and Lewis acid sites. In this paper, a ternary solid acid catalyst containing silica, zirconia, and titanium, was synthesized. It is a stable, environment-friendly catalyst that has strong Brønsted and Lewis acid sites. In any acid catalysed lignin depolymerisation, lignin condensation is inevitable, resulting in a limited yield of aromatic monomers during lignin degradation.28,29 Hydrogenation catalysts have commendable activity in hydrogenolysis and hydrogenation reaction, but it could alleviate self-condensation phenomenon in lignin degradation.30
In this study, a composite oxide solid super acid catalyst S2O82−/ZrO2–TiO2–SiO2 was prepared and characterized using various analytical test to investigate the catalyst properties. The effects of substrate concentration, reaction temperature, co-catalyst, and reaction time on lignin depolymerization yields using this novel catalyst system were investigated.
2 Material and methods
2.1 Materials
The lignin used in the experiment was Indulin AT™, which was purchased from Meadwestvaco (Shanghai, China). All organic solvents in this experiment were produced by Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). (NH4)2S2O8 and 5 wt% Ru/C, Rh/C, Pt/Cr, Pd/C were purchased from Aladdin (Shanghai, China). All Double distilled water was used in the experiments. All reagents in this study were analytic grade and did not require further purification.
2.2 Catalyst preparation
S2O82−/ZrO2–TiO2–SiO2 was prepared in laboratory. Tetraethyl orthosilicate was dissolved in 35 mL ethanol and 40 mL water mixture solvents. Then 1 mol L−1 hydrochloric acid was added dropwise with stirring at room temperature to a pH of 3. One hour later, titanium isopropoxide and zirconyl dichloride were slowly added into the solution at 80 °C with stirring until it gelled. Then the gel was dried at 80 °C for 24 hours. Finally, the obtained sample was calcined at 550 °C for 4 hours. The obtained powder was dispersed in 100 mL ammonium persulfate solution (1 g/4 mL) and maintained for 4 hours. After agitation/impregnation, the powder was dried at 80 °C for one day. The resulting material was calcined in tube furnace at 550 °C for 4 hours in ambient air to obtain the final S2O82−/ZrO2–TiO2–SiO2.
2.3 Lignin depolymerization
To study the effect of substrate concentration, different amounts of lignin (by weight) were dissolved in 30 mL of dioxane-water 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) and reacted at 270 °C for 1 h with 0.1 g S2O82−/ZrO2–TiO2–SiO2 as catalyst, a blank experiment was also conducted. After dissolving 0.5 g lignin completely in 25 mL of dioxane water solvent, the homogeneous solution and 0.1 g of the solid acid catalyst were loaded into the reactor to investigate the effect of reaction temperature on lignin depolymerisation in the temperature range of 250 °C to 310 °C. Considering the excellent hydrogenation capability of noble metals, the hydrogenolysis catalysts (Pd/C, Pt/C, Ru/C, Rh/C) were chosen as co-catalyst, which were mixed with a ratio of 1:1. The performance of solid super acid and co-catalyst at different temperature and reaction time was also studied.
:1 (v/v) and reacted at 270 °C for 1 h with 0.1 g S2O82−/ZrO2–TiO2–SiO2 as catalyst, a blank experiment was also conducted. After dissolving 0.5 g lignin completely in 25 mL of dioxane water solvent, the homogeneous solution and 0.1 g of the solid acid catalyst were loaded into the reactor to investigate the effect of reaction temperature on lignin depolymerisation in the temperature range of 250 °C to 310 °C. Considering the excellent hydrogenation capability of noble metals, the hydrogenolysis catalysts (Pd/C, Pt/C, Ru/C, Rh/C) were chosen as co-catalyst, which were mixed with a ratio of 1:1. The performance of solid super acid and co-catalyst at different temperature and reaction time was also studied.
A certain amount of lignin was dissolved in dioxane–water 5:1 (v/v) system and the resulting homogeneous solution and catalyst were charged in a 50 mL stainless reactor. Subsequently the reactor was repeatedly flashed with hydrogen three times to remove all atmosphere air from the reactor. The reactor was pressurized with hydrogen at 2 MPa. Ultimately the reactor was heated to the desired temperature with 700 rpm magnetic stirring. When the reaction ended, the reactor was rapidly cooled by immersion in a cold-water bath. Liquid product was separated from solid residual by filtration. The solid fraction was washed with dioxane three times and dried at 60 °C. The solid residual product includes catalyst and char from the lignin. The weight of char (WC) was obtained by subtracting the weight of catalyst (WCat) from the weight of the solid residual product (WR). GC-MS was used to identify the liquid products to obtain the most likely aromatic compounds (A), and the products were finally identified and quantified by GC. The yields of aromatic compounds were calculated using an internal standard (acetophenone). Due to the absence of standard sample of known aromatic compounds, the average correction factor of the identified aromatic monomers was used as the correction factor to estimate the yield of unknown aromatic compounds.31 The weight of aromatic compound was calculated as follows:
Mi: the weight of aromatic compound,
Ai: the peak area of aromatic compound,
Ms: the weight of acetophenone,
As: the peak area of acetophenone,
f: correction factor.
The liquid product was divided into two equal part and all the solvent in liquid product parts was removed by rotary evaporator to obtain the weight of the liquid product (WLP). One of the liquid products was dissolved in 1 mL of acetone, and the solution was added dropwise into 200 mL of petroleum ether. The soluble petroleum product was separated from the insoluble petroleum ether product by centrifugation. Finally, rotary evaporator was used to remove the petroleum ether to obtain petroleum ether soluble product (PES). The other part was extracted with 100 mL ethyl acetate and the ethyl acetate soluble product (EAS) was separated from ethyl acetate insoluble product by centrifugation. The ethyl acetate was evaporated under reduced pressure at 35 °C to obtain the weight of ethyl acetate soluble (WEAS). The yield of product was calculated by the following equations (eqn (1)–(6)).
| | |
YA (%) = yield of aromatic compounds = WA/WL × 100%
| (1) |
| | |
YPES (%) = yield of petroleum ether soluble = 2 × WPES/WL × 100%
| (2) |
| | |
YC (%) = yield of char = (WR − WCat)/WL × 100%
| (3) |
| | |
YLP (%) = yield of liquid product = 2 × WLP/WL × 100%
| (4) |
| | |
YV (%) = yield of volatile product = 1 − YLP – YR
| (5) |
| | |
YEAS (%) = yield of ethyl acetate soluble = 2 × WEAS/WL × 100%
| (6) |
WA: the weight of aromatic compounds,
WPES: the weight of petroleum ether soluble,
WR: the weight of residue,
WV: the weight of volatile product,
WL: the weight of lignin (Indulin AT),
WLP: the weight of liquid product,
WEAS: the weight of ethyl acetate soluble,
WCat: the weight of catalyst.
2.4 Analysis methods
2.4.1 Catalyst characterization. EDS was used to analyse elements in the solid super acid. The surface of the catalyst was observed by the scanning electron microscope (SEM; SIRION200, USA). The detailed information of the chemical bond in the catalyst was provided by Fourier transform infrared spectrometry (FT-IR; Nicolet 8700, USA).
2.4.2 Product analysis. Gas chromatography/mass spectrometry (GC-MS, QP2010S, Shimadzu, Japan) equipped with Rtx-5MS column (30 m × 25 mm × 0.25 μm) was used for qualitative analysis of the volatile products. The oven temperature was programmed to hold for 3 min at 40 °C, ramp up to 180 °C at 4 °C min−1, then ramped up to 260 °C at 10 °C min−1, and held for another 10 min. Helium was used as the carrier gas. Quantitative analysis of the volatile products was detected by SHIMADZU GC-2010 gas chromatograph with a FID and a WondaCAP5 column. The oven temperature was programmed to hold for 3 min at 50 °C, ramp up to 250 °C at 10 °C min−1, and hold for 10 min. The injector was kept at 280 °C in spit mode (5:1) with helium as the carrier gas.
3 Results and discussion
3.1 Catalysts characterization
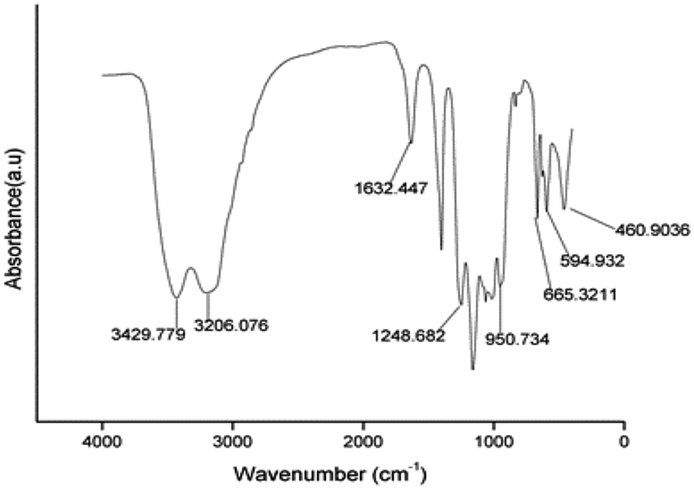
3.1.1 FT-IR analysis. Fig. 1 shows the FT-IR spectrum of S2O82−/ZrO2–TiO2–SiO2. The stretching band detected in the region of 3200–3600 cm−1 indicates the presence of coordinated water (–OH bond) in the catalyst.22 The broad band in the range of 790 cm−1 to 800 cm−1 are the characteristic bands of SiO2. The peak at 1632 cm−1 is the absorption of ZrO2, and in the range of 500 to 700 cm−1 are the absorption of TiO2. In the region of 1300–800 cm−1, asymmetric stretching absorptions of partially ionized SO double bands and S–O bonds are found.26
 |
| | Fig. 1 FT-IR spectra of S2O82−/ZrO2–TiO2–SiO2 composites. | |
3.2 Influence of reaction conditions
3.2.1 Effect of substrate concentration. Yields of the petroleum ether soluble and the ethyl acetate soluble were used as bench marks for the efficiency of lignin depolymerisation, as Indulin AT is insoluble in petroleum ether and only partially soluble in ethyl acetate.33 It has been demonstrated that lignin degradation product soluble in petroleum ether consist mainly of monomeric and dimeric products whereas those soluble in ethyl acetate contains mainly of oligomeric degradation products.31–34 As shown in Table 1 (entries 1–4), the substrate concentration had little influence on the yields of aromatic compounds, petroleum ether soluble, and ethyl acetate soluble. Furthermore, by comparing entry 3 with entry 5 in Table 1, the yield of ethyl acetate soluble increased from 52% to 91% when the solid super acid was used. These results indicate that solid super acid is an effective catalyst that can break lignin down to oligomeric compounds. In addition, the char was twice higher without the addition of the solid acid catalyst as shown in Table 2 (entry 3 vs. entry 5). Since softwood kraft lignin is completely soluble in the solvent system, the amount of char formation indicates the extent of condensation of lignin and its degradation products.
Table 1 Effect of substrate concentrationa
| Entry |
Lignin/g |
Yield of aromatic monomers/% |
Yield of petroleum ether soluble/% |
Yield of ethyl acetate soluble/% |
| Reaction conditions: 0.1 g catalyst (S2O82−/ZrO2–TiO2–SiO2), 30 mL dioxane-water (5:1, v/v), 2 MPa H2, 270 °C, 1 h. Blank test: reaction without catalyst. |
| 1 |
0.1 |
5.6 |
9 |
93 |
| 2 |
0.3 |
5.7 |
8 |
94 |
| 3 |
0.5 |
5.9 |
8 |
91 |
| 4 |
1.0 |
5.8 |
10 |
92 |
| 5b |
0.5 |
3.7 |
5 |
52 |
Table 2 Material balance under various conditionsa
| Entry |
Lignin/g |
Char/% |
Liquid product/% |
Volatile product/% |
| Reaction conditions: 0.1 g catalyst (S2O82−/ZrO2–TiO2–SiO2), 30 mL dioxane–water (5:1, v/v), 2 MPa H2, 270 °C, 1 h. Blank test: reaction without catalyst. |
| 1 |
0.1 |
2 |
96 |
2 |
| 2 |
0.3 |
2 |
95 |
3 |
| 3 |
0.5 |
4 |
95 |
1 |
| 4 |
1.0 |
5 |
93 |
2 |
| 5b |
0.5 |
8 |
91 |
1 |
3.2.2 Influence of temperature. The product results are shown in Fig. 2 and 3. It can be seen in Fig. 2, the yield of aromatic compounds and petroleum ether soluble increased with increasing temperature. However, the yield of ethyl acetate soluble increased initially with increasing temperature and reached the maximum value (91%) around 270–280 °C. Thereafter, it decreased gradually as the temperature increased from 280 °C to 310 °C. Effect of temperature on product distribution is shown in Fig. 3. The yield of liquid product reached 98% and the yield of char was about 1% at 250 °C. As the temperature increased from 250 °C to 310 °C, the yields of both char and volatile product increased to 7% and 2%, respectively, whereas liquid product gradually decreased to 91% at 310 °C. The decrease in both ethyl acetate soluble and liquid product in addition to the presence of char are clear indications of the occurrence of condensation reactions between lignin and its degradation products.
 |
| | Fig. 2 Effect of temperature on the product yield. | |
 |
| | Fig. 3 Effect of temperature on the distribution of liquid product, char and volatile distribution. | |
Considering the data presented in Fig. 2 and 3, it is obvious that both degradation and condensation of lignin occur simultaneously during the reactions. High temperature favours both degradation and condensation reactions. We consider 270 °C as the optimal temperature as it gives maximum yield of ethyl acetate soluble (oligomeric lignin products, ∼91%) and minimum amounts of residue (∼3%) and volatiles (∼1%).
3.2.3 Influence of co-catalyst. Although the solid super acid catalyst is effective in lignin depolymerization, it also causes lignin condensation. Catalytic hydrogenation is known to minimize condensation reaction.33,35 Carbon supported metal catalysts, Pd/C, Pt/C, Ru/C, and Rh/C, were investigated individually as a co-catalyst to the solid acid catalyst, which were mixed with a ratio of 1:1. The reaction was carried out at 270 °C for 1 hour, keeping other conditions the same as before. Control experiments, one without co-catalyst and one with Pt/C as the only catalyst, were also carried out to explore the effect of co-catalyst on lignin depolymerization under the same conditions. The distribution of liquid product, char and volatile products are shown in Fig. 4. As expected, the amount of char formed decreased when any of the hydrogenation catalysts were used as co-catalyst. The yields of degraded lignin products are summarized in Table 3. Except for Pt/C used alone, the yield of ethyl acetate soluble was high, approximately 90%, regardless of whether the solid acid catalyst was used with or without the co-catalyst, indicating that the solid acid catalyst, used alone or with co-catalyst, is efficient in depolymerizing lignin to oligomeric products. The hydrogenation catalyst Pt/C is apparently not effective in depolymerisation of lignin, as the yield of ethyl acetate soluble was only 58% as opposed to 91% with solid acid catalyst (entry 6 vs. entry 1). The yield of petroleum ether soluble more than doubled when the co-catalyst was used with the solid acid catalyst suggesting that suppression of condensation may be a factor in the yield of monomeric and dimeric products.
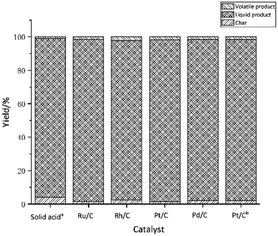 |
| | Fig. 4 Effect of co-catalyst on the distribution of liquid product, char and volatile. aSolid acid catalyst as the only catalyst. bWithout solid acid catalyst. | |
Table 3 Effect of co-catalyst on the yields of degradation productsa
| Entry |
co-catalyst |
Yield of aromatic compounds/% |
Yield of petroleum ether soluble/% |
Yield of ethyl acetate soluble/% |
| Reaction conditions: 0.5 g lignin, 0.1 g catalyst (S2O82−/ZrO2–TiO2–SiO2), 0.1 g co-catalyst, 25 mL dioxane and 5 mL water solvent, 270 °C, 2 MPa H2, 1 h, 700 rpm. Solid acid catalyst as the only catalyst. Without solid acid catalyst. |
| 1b |
Solid acid |
5.86 |
8 |
91 |
| 2 |
Ru/C |
5.92 |
16 |
89 |
| 3 |
Rh/C |
5.94 |
26 |
87 |
| 4 |
Pt/C |
6.81 |
20 |
92 |
| 5 |
Pd/C |
6.30 |
18 |
90 |
| 6c |
Pt/C |
4.80 |
14 |
58 |
3.2.4 Influence of reaction time. The positive effect of the addition of co-catalyst in suppressing condensation prompts us to investigate if longer reaction times can increase the yields of degradation products. Pt/C was selected as co-catalyst for further research. The effect of reaction time on the degradation products was investigated under the same conditions as shown in Table 3 except the reaction temperature was increased to 310 °C as more char and volatile products were formed at this temperature (Fig. 3). Blank control experiments were also carried out for comparison. The effect of reaction time on the distribution of liquid product, char and volatile product is shown in Fig. 5. With or without the co-catalyst, the solid acid catalyst can supress char formation, presumably due to their ability to promote lignin degradation. The effect of the co-catalyst in suppression of char formation becomes obvious only after long reaction time (18 and 24 hours). Fig. 5 also shows that the solid acid catalyst with the co-catalyst appears to increase the amount of volatile product. However, the yields of volatile product are relatively small, increasing from 3% to 8% when the reaction time was extended from 1 to 24 hours. In the meantime, the yield of liquid product decreased from 91% to 84% with the remaining char. It is noteworthy that, at 12 hours, the yield of char reached the maximum (11%), but it is reduction with the extension of time, because the residue have further reaction in the experiment. Without the co-catalyst, the amount of char increased from 7–15% as the reaction time was extended from 1 to 24 hours. Without either catalysts, the char formation increases to 25% in a 24 hour period. The effects of reaction time on the yields of aromatic compounds and petroleum ether soluble are shown in Fig. 6 and 7, respectively. While both increase steadily with increasing time, the effect of the catalysts is apparent, indicating the ability of the solid acid catalyst to catalyse the depolymerisation of lignin to monomeric and dimeric products. As shown in Fig. 7, the yield of petroleum ether soluble increased from 30% to 46% when the reaction time was prolonged from 1 hour to 24 hours using both the solid acid and Pt/C as catalysts. The yield of petroleum ether soluble increased from 18% to 42% without the co-catalyst and increased from 8% to 25% without any catalyst.
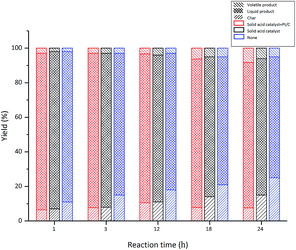 |
| | Fig. 5 Effect of reaction time on the distribution of liquid product, char and volatile. | |
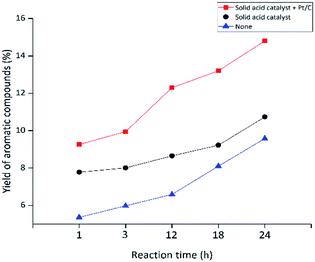 |
| | Fig. 6 Effect of reaction time on the yield of aromatic compounds. | |
 |
| | Fig. 7 Effect reaction time on the yield of petroleum ether soluble. | |
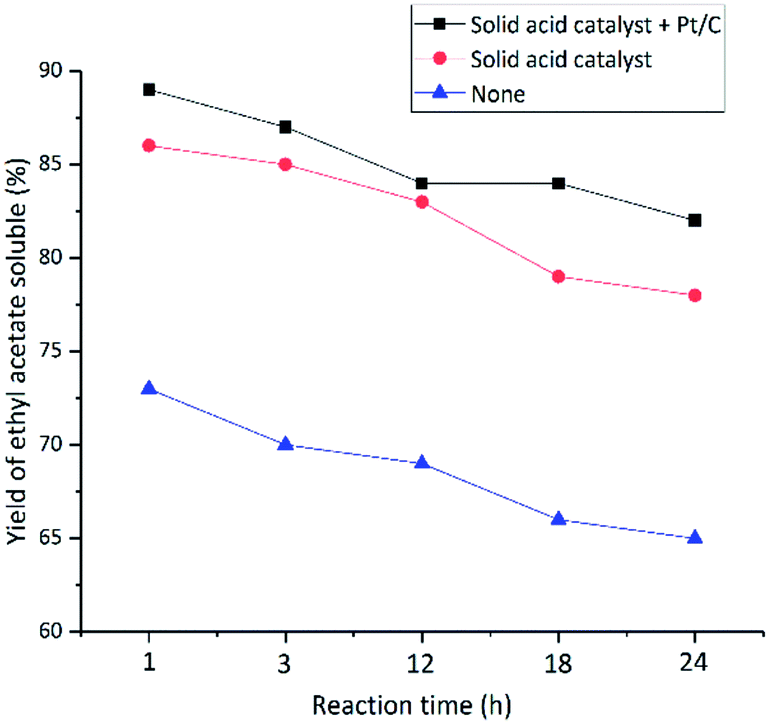
Surprisingly, the yield of ethyl acetate soluble fraction (which represents oligomeric lignin degradation products) as a function of reaction time, behaved differently as compared to the yield of aromatic products and the petroleum ether soluble fraction (which represents the monomeric and dimeric products). As shown in Fig. 8, the yield of ethyl acetate soluble declined steadily, as reaction time was extended from 1 to 24 hours. The results also showed the ability of the solid acid and the co-catalyst to catalyze the depolymerization of lignin. When the reaction time was extended from 1 hour to 24 hours and catalyzed by solid acid catalyst and co-catalyst Pt/C, the yield of liquid product decreased from 91% to 84% (Fig. 5). Meanwhile, the ethyl acetate soluble decreased from 89% to 82% (Fig. 8), indicating that the liquid product produced in the reaction with the acid catalyst with Pt/C as co-catalyst is almost exclusively ethyl acetate soluble. The results shown in Fig. 5–8 also give support to the earlier conclusion that both degradation and condensation occur simultaneously during the reaction and that the acid catalyst, without co-catalyst, can promote degradation reactions and suppress the condensation reactions, what's more, the acid catalyst used with co-catalyst have more excellent effect in suppress the condensation reactions in the reaction.
 |
| | Fig. 8 Effect of reaction time on the yield of ethyl acetate soluble. | |
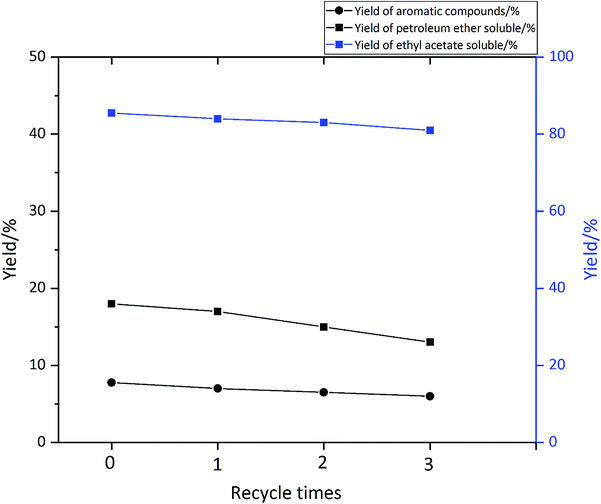
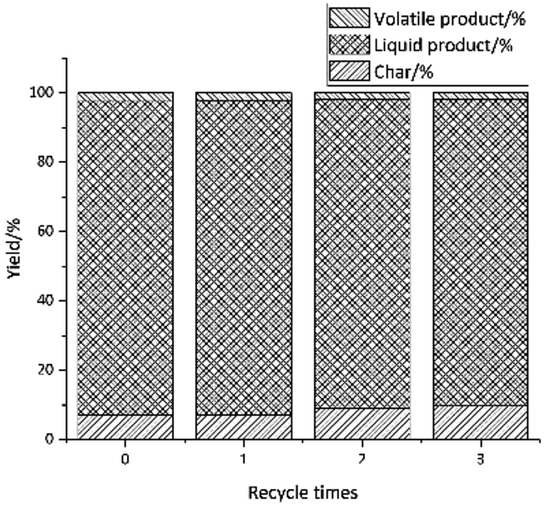
3.2.5 Investigation of the catalyst lifetime. In this experiment, a 3-cycle experiment was carried out to examine the recycle of the catalyst. The reaction condition was at 310 °C, 1 hour and with 0.1 g catalyst. After the reaction, the catalyst was filtered, then washed with 1.4-dioxane and acetone, and lastly dried in oven at 80 °C. The effect of the reaction cycles on the yield is shown in Fig. 9. As can been seen from Fig. 9, at the first cycle the yield of petroleum ether soluble was 18%, after 1 cycle, the yield decreased to 16% and the yield on the 3rd cycle was 13% which is very similar to the yield of the 1st cycle. The yield of aromatic compound and ethyl acetate soluble only decrease by 1.3% and 5% after 3-time cycles. Considering the data presented in Fig. 10, with an increasing number of reuse cycles, the yield of volatile product have little change. And the yield of char increased from 7% to 10%. This result indicates that the catalyst is partly deactivated but most of the activity is preserved after 3 cycles.
 |
| | Fig. 9 Effect of catalyst circulation cycles on product yield. | |
 |
| | Fig. 10 Effect of recycle time on the distribution of liquid product, char and volatile distribution. | |
3.2.6 The leaching of Brønsted acid. The NH3-TPD was performed to obtain the concentrations of the acid sites and the TPD profiles was displayed in Fig. 11. The fresh catalyst has two temperature regions, located at 100–700 °C and 700–1000 °C. The high temperature acid sites correspond to the Brønsted acid sites, whereas the low temperature acid site are Lewis acid sites due to the generally weak basicity.36–38
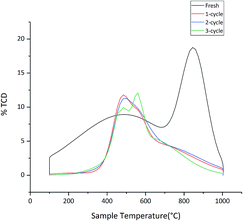 |
| | Fig. 11 NH3-TPD desorption profiles of catalysts. | |
The amount of NH3 absorbed was shown in Table 4. The amount of NH3 absorbed significantly decreases after 1-cycle. However, the catalyst which was cycled 3 times has similar ammonia desorption patterns with 1-cycle. The peak at about 800–900 °C characterizes stronger acid sites, and after three cycles of the catalyst, the peak intensity significantly decreases. The area of peak, in the low temperature region, is slightly decreased after 1-cycles. The result indicated that leaching of Brønsted acid actually occurred in the first recycle reactions. But leaching of Brønsted acid is nearly invariable with an increasing number of reuse cycles. The result shows that the product yield only slightly decreased after the catalyst is re-used up to 3 reaction cycles. As can been seen from Table 4, leaching of Brønsted acid only occurred during the first cycle. The result indicates that leaching of Brønsted acid, during the first cycle, is useless for lignin depolymerization. However, as seen from the recycle result, the product yield only slightly decreases after the catalyst is re-used up to 3 reaction cycles. It indicates that the leaching part of Brønsted acid is ineffective or inefficient to lignin depolymerization. Previous research has shown that the sulfate group binding with metal oxide surface in a chelating bidentate fashion can show the catalytic activity.39
Table 4 Amount of NH3 desorbed of catalysts
| |
Fresh |
1-Cycle |
2-Cycle |
3-Cycle |
| Total (μmol g−1) |
1634.1 |
877.0 |
755.9 |
730.3 |
| Peak 1 (μmol g−1) |
1062.2 |
788.4 |
687.9 |
656.5 |
| Peak 2 (μmol g−1) |
571.9 |
78.9 |
68.0 |
65.7 |
4 Conclusions
Solid super acid catalyst (S2O82−/ZrO2–TiO2–SiO2) which both have Brønsted acid and Lewis acid functionality was prepared and its performance for degradation of lignin was investigated in this study. In the control experiment with no catalyst, the yield of ethyl acetate soluble fraction was 52% as compared a yield of 90+% with the solid acid catalyst. This demonstrated that solid acid catalyst was an effective catalyst for lignin depolymerization. When S2O82−/ZrO2–TiO2–SiO2 and Pt/C was used in a ratio of 1:1, the yield of petroleum ether soluble fraction increased to 46% at 310 °C for 24 hours. The yield of char reached its maximum value (11%) after 12 hours and decreased when the reaction time was extended to 24 hours, because residue in this experiment undergoes further reaction at the prolonged reaction times. These results show that the use of a combination Brønsted and Lewis acid catalyst in combination with a hydrogenation catalyst opens a practical route for efficient lignin depolymerization.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was financially supported by the Program of National Natural Science Foundation of China (51676178), the National Key Technology R&D Program of China (NO. 2015BAD15B06), Science and Technological Fund of Anhui Province for Outstanding Youth (1508085J01) and the international technology cooperation plan of Anhui (No. 1503062030).
Notes and references
- M. G. Adsul, M. S. Singhvi, S. A. Gaikaiwari and D. V. Gokhale, Bioresour. Technol., 2011, 102, 4304–4312 CrossRef CAS PubMed.
- B.-C. Zhao, B.-Y. Chen, S. Yang, T.-Q. Yuan, A. Charlton and R.-C. Sun, ACS Sustainable Chem. Eng., 2017, 5, 1113–1122 CrossRef CAS.
- J. Yu, N. Paterson, J. Blamey and M. Millan, Fuel, 2017, 191, 140–149 CrossRef CAS.
- W. O. S. Doherty, P. Mousavioun and C. M. Fellows, Ind. Crops Prod., 2011, 33, 259–276 CrossRef CAS.
- R. J. A. Gosselink, E. de Jong, B. Guran and A. Abacherli, Ind. Crops Prod., 2004, 20, 121–129 CrossRef CAS.
- C. Crestini, M. Crucianelli, M. Orlandi and R. Saladino, Catal. Today, 2010, 156, 8–22 CrossRef CAS.
- L. A. Pfaltzgraff, M. De Bruyn, E. C. Cooper, V. Budarin and J. H. Clark, Green Chem., 2013, 15, 307–314 RSC.
- S. Stiefel, C. Marks, T. Schmidt, S. Hanisch, G. Spalding and M. Wessling, Green Chem., 2016, 18, 531–540 RSC.
- Z. C. Jiang, H. Zhang, T. He, X. Y. Lv, J. Yi, J. M. Li and C. W. Hu, Green Chem., 2016, 18, 4109–4115 RSC.
- F. X. Yue, F. C. Lu, R. C. Sun and J. Ralph, Chem.–Eur. J., 2012, 18, 16402–16410 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- C. Z. Li, X. C. Zhao, A. Q. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- A. A. Geronikaki and K. A. Abduazimov, Khim. Prir. Soedin., 1976, 646–648 CAS.
- T. Klamrassamee, T. Tana, N. Laosiripojana, L. Moghaddam, Z. Y. Zhang, J. Rencoret, A. Gutierrez, J. C. del Rio and W. O. S. Doherty, RSC Adv., 2016, 6, 92638–92647 RSC.
- D. K. Shen, J. Zhao and R. Xiao, Energy Convers. Manage., 2016, 124, 61–72 CrossRef CAS.
- B. Guvenatam, E. H. J. Heeres, E. A. Pidko and E. J. M. Hensen, Catal. Today, 2016, 269, 9–20 CrossRef.
- Z. P. Xu, W. Z. Li, Z. J. Du, H. Wu, H. Jameel, H. M. Chang and L. L. Ma, Bioresour. Technol., 2015, 198, 764–771 CrossRef CAS PubMed.
- D. Bradley, G. Williams and M. Lawton, J. Mol. Catal. A: Chem., 2010, 317, 68–71 CrossRef.
- S. Y. Jia, B. J. Cox, X. W. Guo, Z. C. Zhang and J. G. Ekerdt, Ind. Eng. Chem. Res., 2011, 50, 849–855 CrossRef CAS.
- A. Vuori and M. Niemela, Holzforschung, 1988, 42, 327–334 CrossRef CAS.
- R. Akkari, A. Ghorbel, N. Essayem and F. Figueras, Appl. Catal., A, 2007, 328, 43–51 CrossRef CAS.
- F. Yang, Y. Li, Q. Zhang, X. Sun, H. Fan, N. Xu and G. Li, Carbohydr. Polym., 2015, 131, 9–14 CrossRef CAS PubMed.
- A. K. Deepa and P. L. Dhepe, RSC Adv., 2014, 4, 12625–12629 RSC.
- H. Z. Chen, B. Yu and S. Y. Jin, Bioresour. Technol., 2011, 102, 3568–3570 CrossRef CAS PubMed.
- S. Uchiyama, T. Isobe, S. Matsushita, K. Nakajima, M. Hara and A. Nakajima, J. Mater. Sci., 2011, 47, 341–349 CrossRef.
- Y. Wang, J. H. Ma, D. Liang, M. M. Zhou, F. X. Li and R. F. Li, J. Mater. Sci., 2009, 44, 6736–6740 CrossRef CAS.
- M. M. Hepditch and R. W. Thring, Can. J. Chem. Eng., 2000, 78, 226–231 CrossRef CAS.
- J. Papadopoulos, C. L. Chen and I. S. Goldstein, Holzforschung, 1981, 35, 283–286 CrossRef CAS.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Nature, 2014, 515, 249–252 CrossRef CAS PubMed.
- L. Yan, X. X. Liu and Y. Fu, RSC Adv., 2016, 6, 109702–109705 RSC.
- X. Jiang, D. Savithri, X. Du, S. Pawar, H. Jameel, H.-m. Chang and X. Zhou, ACS Sustainable Chem. Eng., 2017, 5, 835–842 CrossRef CAS.
- J. M. Lavoie, W. Bare and M. Bilodeau, Bioresour. Technol., 2011, 102, 4917–4920 CrossRef CAS PubMed.
- J. Wang, W. Li, H. Wang, Q. Ma, S. Li, H.-m. Chang and H. Jameel, Bioresour. Technol., 2017, 243, 100–106 CrossRef CAS PubMed.
- Z. S. Yuan, S. N. Cheng, M. Leitch and C. B. Xu, Bioresour. Technol., 2010, 101, 9308–9313 CrossRef CAS PubMed.
- D. Meier, R. Ante and O. Faix, Bioresour. Technol., 1992, 40, 171–177 CrossRef CAS.
- R.-t. Guo, Y. Zhou, W.-g. Pan, J.-n. Hong, W.-l. Zhen, Q. Jin, C.-g. Ding and S.-y. Guo, J. Ind. Eng. Chem., 2013, 19, 2022–2025 CrossRef CAS.
- J. Lif, I. Odenbrand and M. Skoglundh, Appl. Catal., A, 2007, 317, 62–69 CrossRef CAS.
- Y. Shen, S. Zhu, T. Qiu and S. Shen, Catal. Commun., 2009, 11, 20–23 CrossRef CAS.
- C. Zhang, J. Zhang, Y. Zhao, J. Sun and G. Wu, Catal. Lett., 2016, 146, 1256–1263 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09489b |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Jindong Wanga,
Hou-min Changb,
Hasan Jameelb,
Qi Zhangc,
Song Lib and
Lele Jina
*a,
Jindong Wanga,
Hou-min Changb,
Hasan Jameelb,
Qi Zhangc,
Song Lib and
Lele Jina