
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Prediction of supramolecular synthons and crystal packings of supramolecular HMX/solvent assemblies†
Zhichao Liua,
Yu Liub,
Jinshan Lib and
Weihua Zhu *a
*a
aInstitute for Computation in Molecular and Materials Science, Department of Chemistry, Nanjing University of Science and Technology, Nanjing 210094, China. E-mail: zhuwh@njust.edu.cn
bInstitute of Chemical Materials, China Academy of Engineering Physics, P. O. Box 919-327, Mianyang, Sichuan 621900, China
First published on 7th December 2017
Abstract
We report the most stable packings of five HMX/solvent supramolecular assemblies. A series of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 supramolecular synthons of both α-form and β-form HMX conformers with solvent molecules were investigated. Both α-form and β-form HMX conformers have similar stability when combining with solvent molecules into supramolecular synthons. AIM analysis was performed to evaluate the properties of the intermolecular hydrogen bonds between the HMX and solvent molecules. The most stable polymorphs among the 10 most common space groups for α-HMX/solvent supramolecular assemblies were predicted and compared with available experimental data.
:1 supramolecular synthons of both α-form and β-form HMX conformers with solvent molecules were investigated. Both α-form and β-form HMX conformers have similar stability when combining with solvent molecules into supramolecular synthons. AIM analysis was performed to evaluate the properties of the intermolecular hydrogen bonds between the HMX and solvent molecules. The most stable polymorphs among the 10 most common space groups for α-HMX/solvent supramolecular assemblies were predicted and compared with available experimental data.
1. Introduction
Cocrystallization has proven to be an effective method and has broad applications in many fields by generating novel materials with improved physicochemical properties from existing materials.1–4 In the energetics field, explosive cocrystals that are often composed of two or more explosive components or solvents possess distinct detonation properties and stability from the pure components or their physical mixture.5–19 Thus, cocrystallization has presented great opportunities to improve conventional energetic materials and reuse a large amount of candidate materials with less desirable performance. On the other hand, recent use of cocrystallization to increase sensitivity and produce primary explosives (high-sensitivity explosives) from secondary explosives (low-sensitivity explosives) is another aspect of interest.18By applying this strategy on the traditional explosives, such like 2,4,6,8,10,12-hexanitrohexaazaisowurtzitane (CL-20), 2,4,6-trinitrotoluene (TNT), octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX), and benzotrifuroxan (BTF), a series of energetic cocrystals with tailored sensitivity and well maintained detonation performance were prepared.8–15,17 The cocrystallization of HMX with various cocrystal formers remarkably reduced the material sensitivity and maintained most of the energy when compared to pure HMX.13 The cocrystals of CL-20 with TNT (1:1), HMX (2:1), and 1,3-dinitrobenzene (DNB, 1:1) have been prepared with improved stability. However, due to the performance criteria, only a few of the prepared energetic cocrystals meet these requirements, e.g. CL-20/HMX,11 CL-20/TNT,9 CL-20/BTF,10 BTF/TNT,15 and etc. On the other aspect, the prepared HMX/solvent cocrystals lead to better safety but tremendous reduction in detonation performance as compared to pure HMX. By removing the solvents, the HMX/solvent cocrystals can be selectively “activated”.13 However, for the solvates of high explosive HMX and TNT, only a few of them have been structurally characterized. This is mainly due to the weak interactions between the supramolecular synthons that feature the electron-withdraw NO2 groups. Therefore, to understand the alteration in the sensitivity of the cocrystals that stems from the pure components gathering into supramolecular assemblies, it is necessary to explore the intermolecular interactions between different supramolecular synthons.
In this work, five HMX/solvent supramolecular assemblies are examined in their intermolecular interactions and the self-assembling behaviors. The aims are to understand how the HMX/solvent supramolecular assemblies are defined by the relationships between noncovalent interactions and most stable molecular packings. The calculated results are useful for the design of a library of suitable energetic cocrystals with tailored properties.
2. Computational details
Quantum mechanical calculations were carried out B3LYP/6-311+G** level as implemented in Gaussian 09W package.20 For the structure optimization, the most stable synthons for the supramolecular assemblies of HMX with different solvents were proposed with respect to different conformations of HMX.To find the possible molecular packing for the HMX/solvent assemblies, 10 space groups that account for about 85.26% of the organic molecular solids were considered: P21/c (36.59%), P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (16.92%), P212121 (11.00%), C2/c (6.95%), P21 (6.35%), Pbca (4.24%), Pna21 (1.63%), R
(16.92%), P212121 (11.00%), C2/c (6.95%), P21 (6.35%), Pbca (4.24%), Pna21 (1.63%), R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c (0.11%), R (0.46%), and Pbcn (1.01%).21–23 The 10 space groups21 considered here are slightly different from those in ref. 22. But these most possible space groups are enough to find the most stable polymorphs for the organic compounds. The Monte Carlo sampling was performed for each of the space groups using fixed molecular structures. During the MC packing simulation, the MC trial consists of reorientation and movement of each rigid unit. The lattice vectors of the unit cell are then adjusted. The MC simulated annealing regime involves an initial heating, followed by cooling, and the whole process is repeated for each space group. The Metropolis criterion is used to determine whether the generated trial structures are accepted or rejected. The empirical Dreiding Force Field (FF) as implemented in polymorph code24–29 was used to minimize the most table structures while allowing the molecules and the lattice to relax. The most possible molecular packings were predicted according to both the total energy and the crystal density.
c (0.11%), R (0.46%), and Pbcn (1.01%).21–23 The 10 space groups21 considered here are slightly different from those in ref. 22. But these most possible space groups are enough to find the most stable polymorphs for the organic compounds. The Monte Carlo sampling was performed for each of the space groups using fixed molecular structures. During the MC packing simulation, the MC trial consists of reorientation and movement of each rigid unit. The lattice vectors of the unit cell are then adjusted. The MC simulated annealing regime involves an initial heating, followed by cooling, and the whole process is repeated for each space group. The Metropolis criterion is used to determine whether the generated trial structures are accepted or rejected. The empirical Dreiding Force Field (FF) as implemented in polymorph code24–29 was used to minimize the most table structures while allowing the molecules and the lattice to relax. The most possible molecular packings were predicted according to both the total energy and the crystal density.
During the optimization of the predicted crystal structures, we used the third-order corrected self-consistent charge density functional tight binding method30–33 (SCC-DFTB) with the first-principles London dispersion correction D3 (ref. 34) (DFTB-D) as implemented in CP2K package.35 The solid state calculation was performed with symmetry constrains in finding the crystal packing arrangements.
3. Results and discussion
3.1 Supramolecular synthons for HMX/solvent assemblies
To promote the rational design for new energetic cocrystals with tailored properties, it is necessary to understand the possible supramolecular synthons. Fig. 1 shows the chemical structures of N,N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), butyrolactone (GBL), hexamethylphosphoramide (HMPA), and N-methyl-2-pyrrolidinone (NMP). Landenberger et al. found that the HMX forms cocrystals in three motifs: chair–chair/layered, chair–chair/pocket, and chair/layered.13 For polar solvents, the experimentally prepared HMX/NMP, HMX/2-pyrrolidone, HMX/4-picoline-N-oxide, and HMX/DMF cocrystals possess the chair–chair/pocket motif, in which the solvents serve as interchangeable guest molecules (see Fig. 2).13 In this motif, the HMX molecules are the boat α-form (α-HMX) rather than the chair β-form (β-HMX) conformer. The two HMX conformers are found in different polymorphs of HMX (α-phase and β-phase).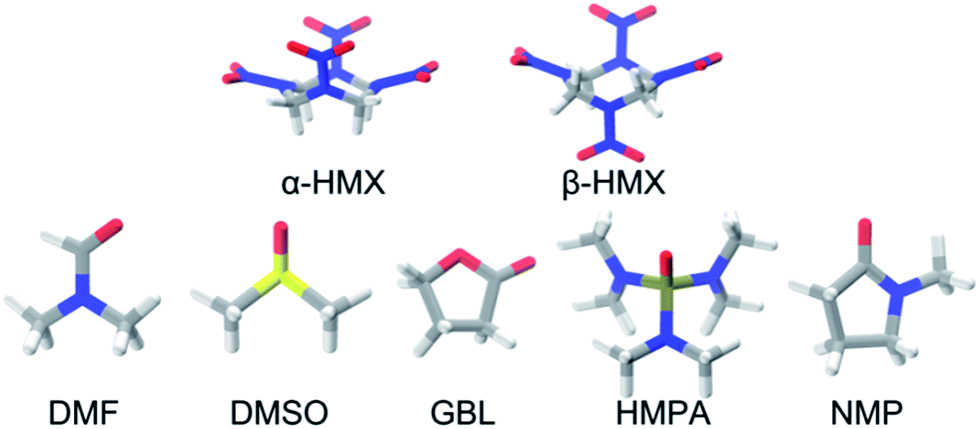 | ||
| Fig. 1 Structures of α-form and β-form HMX molecules (α-HMX and β-HMX) and five solvents (DMF, DMSO, GBL, HMPA, and NMP). The C, H, O, and N are represented by gray, white, red, and blue sticks, respectively. | ||
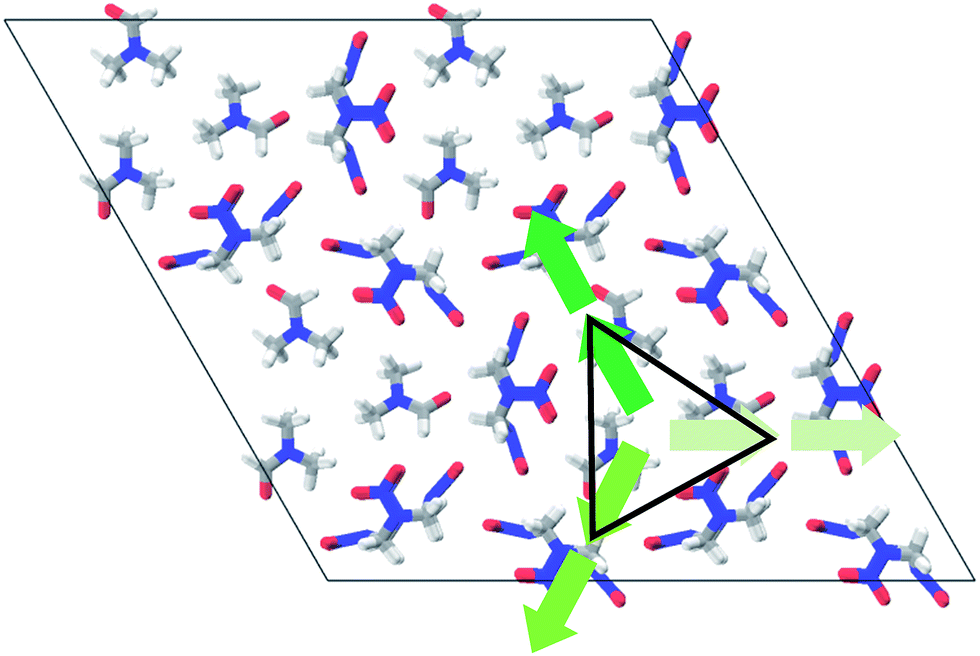 | ||
| Fig. 2 Spatial distribution of dipole moments (green arrows) for the chair–chair/pocket motif of HMX/solvent supramolecular assemblies (HMX/DMF taken as an example). The triangle area denotes the interchangeable guest solvent molecules. The C, H, O, and N are represented by gray, white, red, and blue sticks, respectively. | ||
To understand the driving force in this motif, it is prerequisite to confirm whether the supramolecular synthon exist or not and what the role it plays in the HMX/solvent assemblies. We note that some of the HMX and solvents molecules with hydrogen bonding between the O and H atoms have the same dipole direction (see Fig. 2). Driven by the dipole–dipole interaction, the HMX molecule always combines with one solvent molecule with four intermolecular hydrogen bonds between them, which leads to a 1:1 mole ratio of supramolecular assembly. This is common in other chair–chair/pocket motifs of other polar solvents.5–7,13
Based on these phenomena, we considered the 1:1 molecular pairs that may act as the supramolecular synthons. For the polar conformer of HMX (α-form), the initial structures of the HMX and solvent were set with same dipole direction. For the nonpolar conformer (β-form), the solvent molecule was set with dipole moment pointing to the HMX molecule. In Fig. 3a and b, the structures of the dimers of α-form and β-form HMX conformers with different solvents were relaxed without symmetry constraints at the B3LYP/6-311+G** level. The energetics is listed in Table 1. For different solvents, the calculated binding energies between the two HMX/solvent dimers are similar. The ΔE values for β-HMX/solvent dimers are slightly lower than those of the α-HMX/solvent dimers. This suggests that both two kinds of dimers have similar stability.
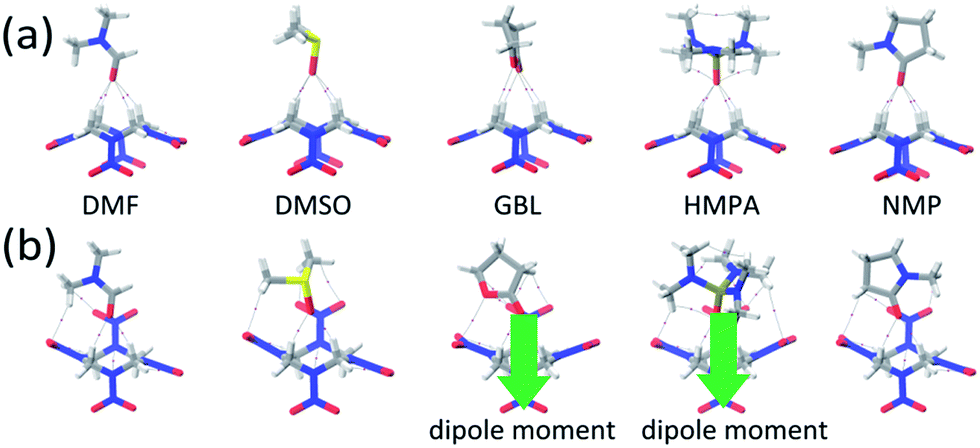 | ||
| Fig. 3 Relaxed structures of supramolecular synthons of (a) α-form and (b) β-form HMX conformers with five solvents. The lines denote the bond paths and the purple spheres correspond to the (3, −1) BCPs. | ||
During the relaxation, the configurations of HMX molecules maintained with exceptions for β-HMX/GBL and β-HMX/HMPA dimers, in which the HMX molecules are polarized with one NO2 group distorted by the presence of solvent molecules, leading to a net dipole moment of 3.30 debye for the HMX molecules. This suggests that the β-HMX/solvent dimers are unstable in these polar solvents (see Fig. 3b). Thus, we did not consider them further when packing them into crystals.
Next, we performed Bader's atoms in molecules (AIM) analysis for the 10 dimers to understand the interactions between molecules. The critical points (CPs) of the electron density (ρ) are the positions at which ∇ρ vanishes except at infinity. The CPs have four types: (3, −3), (3, −1), (3, +1), and (3, +3). The (3, −1) CP generally appears between attractive atom pairs and hence called bond critical points (BCP). The value of ρ and the sign of ∇2ρ at BCP are closely related to bonding strength and bonding type respectively. The topological parameters at the (3, −1) bond critical points (BCP) are summarized in Tables 2 and 3. The energy density Hb is the sum of the Lagrangian kinetic energy Gb and the potential energy density Vb. The hydrogen bond energies Eb is estimated by the Vb at corresponding BCP, which can be approximately described as Eb = 1/2Vb.36 It is found that both the AIM and the DFT calculations at the B3LYP/6-311+G** level yield consistent variation trend in predicting the total hydrogen bond energies between the HMX and solvent molecules.
| Solvent | Bond | d | ρ × 102 | ∇2ρ × 102 | Gb × 102 | Vb × 102 | Hb × 102 | |Vb|/Gb | Eb |
|---|---|---|---|---|---|---|---|---|---|
| DMF | H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C C |
2.51 | 0.887 | 0.274 | 0.602 | −0.517 | 0.0845 | 0.859 | 6.79 |
| H⋯OC |
2.51 | 0.888 | 0.275 | 0.602 | −0.518 | 0.0846 | 0.860 | 6.80 | |
| H⋯OC |
2.44 | 0.929 | 0.312 | 0.670 | −0.559 | 0.111 | 0.834 | 7.34 | |
| H⋯OC |
2.44 | 0.929 | 0.312 | 0.670 | −0.559 | 0.111 | 0.834 | 7.34 | |
| DMSO | H⋯OC |
2.43 | 1.05 | 0.332 | 0.729 | −0.629 | 0.100 | 0.863 | 8.26 |
| H⋯OC |
2.43 | 1.04 | 0.329 | 0.724 | −0.625 | 0.0992 | 0.863 | 8.20 | |
| H⋯OC |
2.34 | 1.18 | 0.395 | 0.857 | −0.726 | 0.132 | 0.847 | 9.53 | |
| H⋯OC |
2.34 | 1.18 | 0.394 | 0.854 | −0.723 | 0.130 | 0.847 | 9.49 | |
| GBL | H⋯OC |
2.55 | 0.83 | 0.253 | 0.556 | −0.479 | 0.0761 | 0.862 | 6.29 |
| H⋯OC |
2.5 | 0.796 | 0.277 | 0.588 | −0.483 | 0.106 | 0.821 | 6.34 | |
| H⋯COC | 2.54 | 0.863 | 0.258 | 0.570 | −0.495 | 0.0743 | 0.868 | 6.50 | |
| H⋯COC | 2.49 | 0.816 | 0.279 | 0.595 | −0.493 | 0.103 | 0.829 | 6.47 | |
| HMPA | H⋯OP |
2.39 | 1.16 | 0.365 | 0.803 | −0.693 | 0.110 | 0.863 | 9.10 |
| H⋯OP |
2.43 | 1.01 | 0.329 | 0.713 | −0.604 | 0.109 | 0.847 | 7.93 | |
| H⋯OP |
2.38 | 1.17 | 0.367 | 0.807 | −0.697 | 0.110 | 0.864 | 9.15 | |
| H⋯OP |
2.43 | 1.02 | 0.331 | 0.718 | −0.608 | 0.110 | 0.847 | 7.98 | |
| NMP | H⋯OC |
2.47 | 0.967 | 0.301 | 0.659 | −0.566 | 0.0933 | 0.859 | 7.43 |
| H⋯OC |
2.47 | 0.952 | 0.300 | 0.654 | −0.559 | 0.095 | 0.855 | 7.34 | |
| H⋯OC |
2.44 | 0.958 | 0.319 | 0.686 | −0.575 | 0.110 | 0.838 | 7.55 | |
| H⋯OC |
2.44 | 0.951 | 0.316 | 0.679 | −0.570 | 0.110 | 0.839 | 7.48 |
| Solvent | Bond | d | ρ × 102 | ∇2ρ × 102 | Gb × 102 | Vb × 102 | Hb × 102 | |Vb|/Gb | Eb |
|---|---|---|---|---|---|---|---|---|---|
| DMF | H⋯OC |
2.14 | 1.73 | 0.622 | 0.133 | −1.10 | 0.226 | 8.271 | 14.44 |
| H⋯OC |
2.24 | 1.37 | 0.517 | 1.08 | −0.868 | 0.213 | 0.804 | 11.39 | |
| NO2⋯H | 2.76 | 0.481 | 0.159 | 0.339 | −0.282 | 0.265 | 0.832 | 3.700 | |
| DMSO | H⋯SO |
2.08 | 1.83 | 0.736 | 1.55 | −1.26 | 0.289 | 0.813 | 16.54 |
| H⋯SO |
2.12 | 1.87 | 0.699 | 1.51 | −1.27 | 0.237 | 0.841 | 16.67 | |
| NO2⋯H | 2.51 | 0.768 | 0.257 | 0.547 | −0.454 | 0.0937 | 0.830 | 5.960 | |
| NO2⋯H | 2.72 | 0.551 | 0.180 | 0.387 | −0.325 | 0.062 | 0.840 | 4.270 | |
| GBL | H⋯OC |
2.25 | 1.30 | 0.508 | 1.05 | −0.828 | 0.221 | 0.789 | 10.87 |
| H⋯OC |
2.28 | 1.36 | 0.470 | 1.00 | −0.829 | 0.173 | 0.829 | 10.88 | |
| NO2⋯H | 2.65 | 0.661 | 0.213 | 0.464 | −0.396 | 0.0675 | 0.853 | 5.200 | |
| HMPA | H⋯PO |
2.07 | 1.92 | 0.745 | 1.58 | −1.31 | 0.277 | 0.829 | 17.20 |
| H⋯PO |
2.07 | 2.03 | 0.743 | 1.62 | −1.38 | 0.238 | 0.852 | 18.12 | |
| NO2⋯H | 2.74 | 0.319 | 0.109 | 0.227 | −0.181 | 0.0459 | 0.797 | 2.380 | |
| NO2⋯H | 2.97 | 0.304 | 0.122 | 0.238 | −0.170 | 0.0677 | 0.714 | 2.230 | |
| NO2⋯H | 3.10 | 0.478 | 0.157 | 0.338 | −0.283 | 0.0553 | 0.837 | 3.720 | |
| NMP | H⋯OC |
2.13 | 1.63 | 0.640 | 1.34 | −1.07 | 0.265 | 0.799 | 14.05 |
| H⋯OC |
2.20 | 1.62 | 0.564 | 1.21 | −1.02 | 0.198 | 0.843 | 13.39 | |
| NO2⋯H | 2.85 | 0.414 | 0.155 | 0.319 | −0.251 | 0.0677 | 0.787 | 3.300 | |
| NO2⋯H | 3.09 | 0.282 | 0.110 | 0.217 | −0.160 | 0.0578 | 0.737 | 2.100 | |
| NO2⋯H | 3.32 | 0.203 | 0.823 | 0.152 | −0.0985 | 0.0536 | 0.648 | 1.290 |
Both α-form and β-form HMX conformers combine the solvent molecules with weak hydrogen bonds with Eb values in the range 1.29–18.1 kJ mol−1 and bond lengths in the range 2.07–3.20 Å. For α-HMX/solvent dimers, the hydrogen bond strengths between two molecules are similar. While for β-HMX/solvent dimers, the strengths of CH2⋯OX (X = C, P, and S atoms) hydrogen bonds between the HMX and solvents vary in a wide range, with Eb values in the range 10.9–18.1 kJ mol−1 and bond lengths in the range 2.07–2.28 Å. What is more, the NO2⋯H hydrogen bonds are much weaker with Eb values in the range 1.29–5.96 kJ mol−1 and bond lengths in the range 2.51–3.32 Å. The net result is that the asymmetric structures for the β-HMX/solvent dimers result in the instability despite CH2⋯OX (X = C, P, and S atoms) hydrogen bonds being relatively stronger.
3.2 Molecular packings for HMX/solvent supramolecular assemblies
The 10 space groups that are commonly found in 85.26% of the organic molecular solids were investigated for the most stable molecular packings of the HMX/solvent synthons gathering into supramolecular assemblies. The FF-predicted energies and densities of the structures from each of the 10 best space groups are compared in Fig. S1.† The FF-predicted structures with lowest energy for the α-HMX/solvent synthons in the 10 most common space groups are depicted in Fig. S2.† The structures predicted by Dreiding FF were fully optimized using the DFTB-D method with symmetry constrains. The obtained energies and densities of the most stable packing structures in the 10 most common space groups are presented in Fig. 4. The DFTB-D-predicted most stable molecular packings for the α-HMX/solvent synthons in the 10 most common space groups are summarized in Table 4 with available experimental data.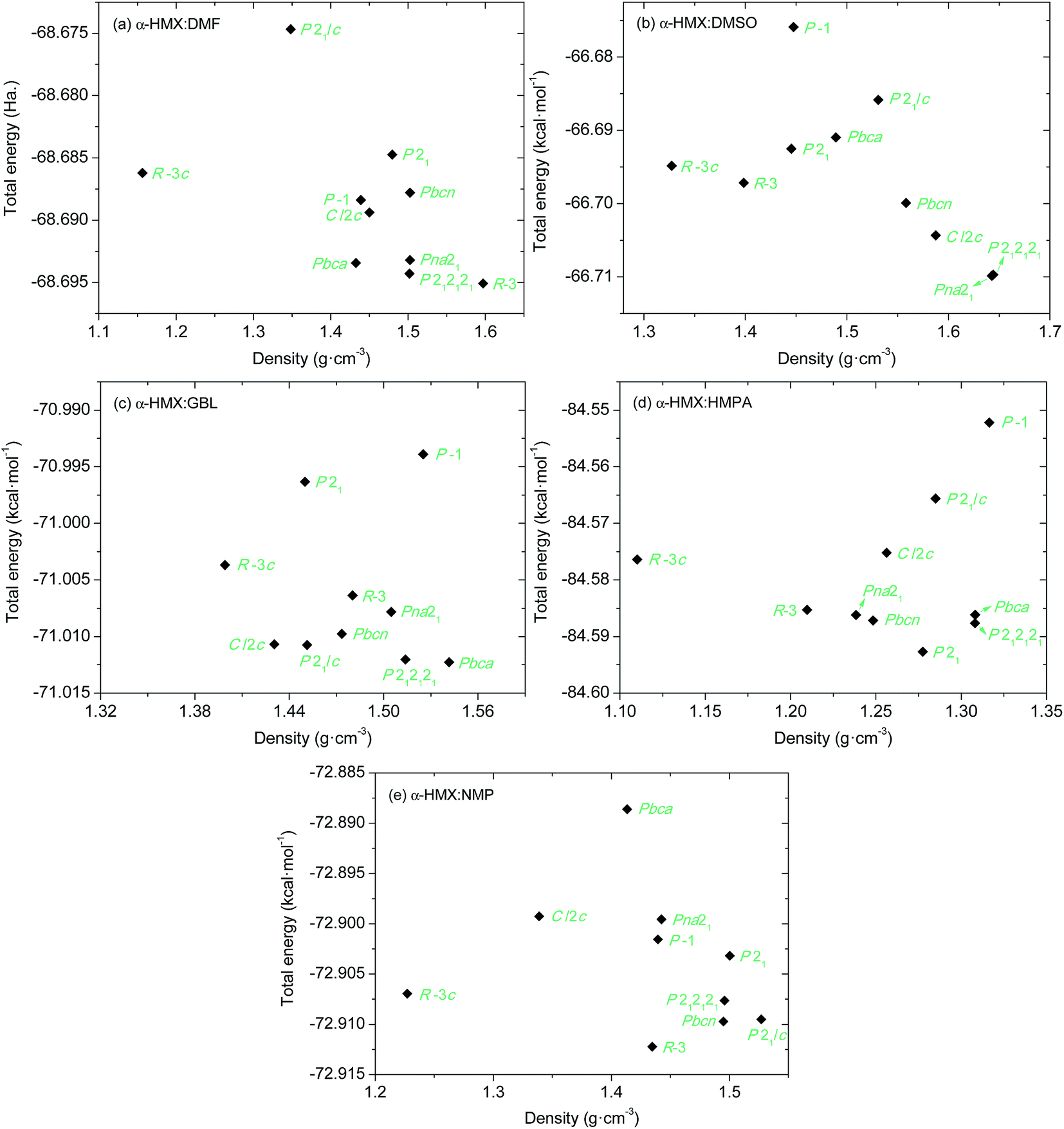 | ||
| Fig. 4 Relationship between crystal density and total energy of the packings for α-HMX/solvent supramolecular assemblies in different space groups from DFTB-D. | ||
| Solvent | Space group | a (Å) | b (Å) | c (Å) | β (Å) | V (Å3) | ρ (g cm−3) |
|---|---|---|---|---|---|---|---|
| a Experimental data in parenthesis were taken from ref. 5.b Experimental data in parenthesis were taken from ref. 7. | |||||||
| DMF | R |
16.10 (15.99)a | 16.10 (15.99) | 30.77 (30.92) | 90.00 (90.00) | 6911.6 (6845.6) | 1.597 (1.61) |
| DMSO | Pna21 | 12.67 | 11.18 | 10.69 | 90.00 | 1513.3 | 1.643 |
| GBL | Pbca | 10.71 | 18.86 | 16.31 | 90.00 | 3293.9 | 1.542 |
| HMPA | P21 | 7.703 | 14.65 | 12.25 | 63.39 | 1235.8 | 1.277 |
| NMP | R |
28.75 (16.61)b | 28.75 (16.61) | 11.51 (31.51) | 90.00 (90.00) | 8235.8 (7525.00) | 1.435 (1.51) |
In general, the FF-predicted structures from different space groups are maintained after DFTB-D optimization. But the DFTB-D results show large differentiations of both the energies and densities of the structures from the FF ones, leading to the variation of the most stable packings for different space groups. The DFTB-D-predicted lattice parameters of HMX/solvent supramolecular assemblies with 10 possible space groups were provided in Table S1.† We compared the DFTB-predicted most stable structures with the available experimental data to evaluate the differences between the experiment and theory (see Fig. 5). Experimentally, the HMX/DMF cocrystal has two polymorphs: Rc and C2/c.5,6 The Rc structure has higher crystal density of 1.612 g cm−3 than the C2/c one of 1.607 g cm−3.5,6 However, the DFTB-D method predicts the R structure to be the most possible packing (lowest energy and highest crystal density) within the global rank for HMX/DMF supramolecular assemblies. The predicted R structure has lower symmetry but similar crystal density 1.60 g cm−3 with the experiments. For HMX/NMP, the predicted lowest-energy packing also has R symmetry (crystal density of 1.435 g cm−3), which also has lower symmetry than the experimentally determined Rc symmetry (crystal density of 1.570 g cm−3).7
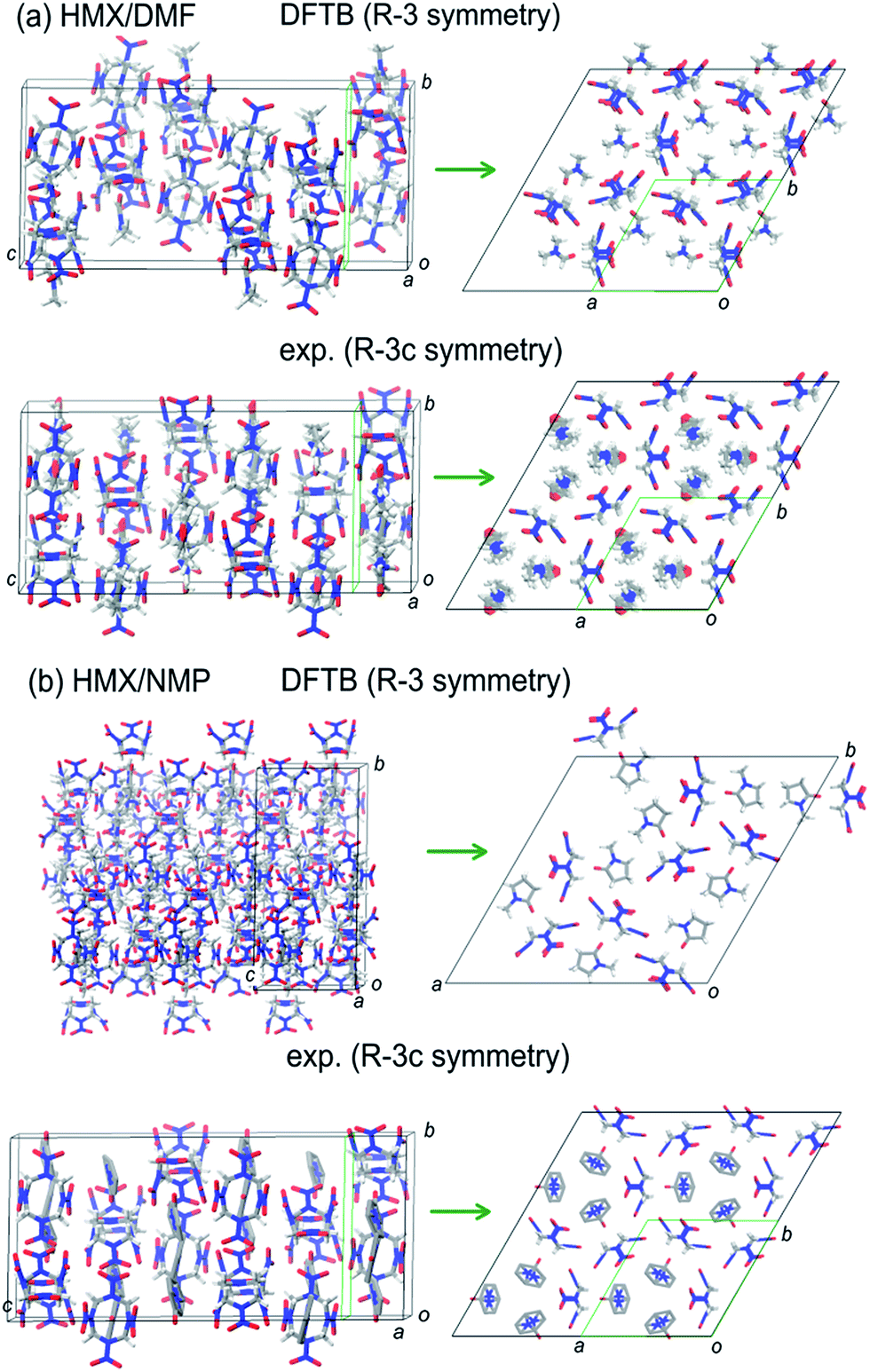 | ||
| Fig. 5 A comparison between the DFTB-predicted most stable structures and the experiments for two supramolecular assemblies (HMX/DMF and HMX/NMP). | ||
It is found that both the prepared DMF and NMP molecules are disordered in two sites. The coordinates of the disordered DMF and NMP molecules were not reported in the experimental data (CSD codes DADDOB and HMXDMF). But the atoms of the solvent molecules (C, N, and O atoms provided only) can be added artificially according to the literatures. For HMX/DMF, despite the disorder of the solvent molecules, the molecular orientation matches well with each other (see Fig. 5). But for HMX/NMP, the predicted R packing seems not match well the experiments.
For the other three assemblies, no experimental data are available. The most possible packing for HMX/GBL has Pbca symmetry. While for HMX/DMSO and HMX/HMPA, none of the packing possess the lowest-energy and the highest density, while their lowest-energy packing predicted by the DFTB-D method has Pna21 and P21 symmetry, respectively. Especially, the packing crystal structure for HMX/DMSO with Pna21 and P212121 symmetry are quite similar, making them to be the most possible packing at the same time (see Fig. 6).
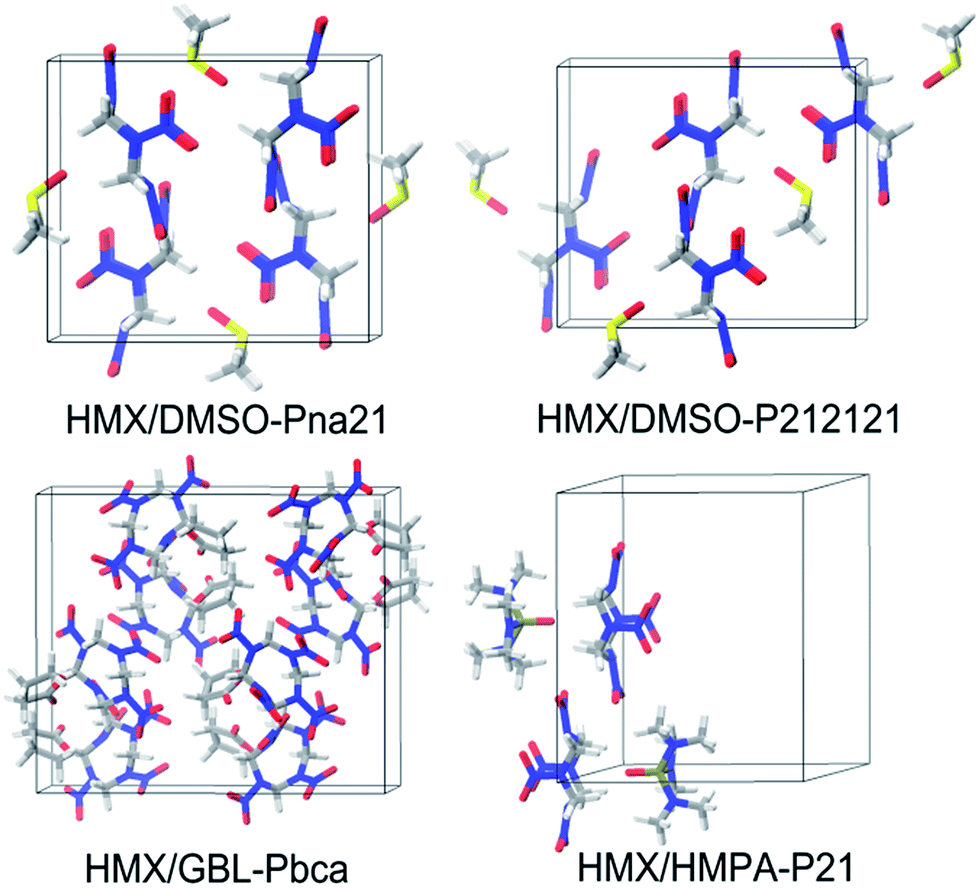 | ||
| Fig. 6 Most stable molecular packings for the HMX/DMSO, HMX/GBL, and HMX/HMPA by DFTB-D method. | ||
4. Conclusions
The most stable packings of supramolecular assemblies for HMX and five solvents were investigated. Both α-form and β-form HMX conformers have similar stability when combining with the solvent molecules into 1:1 supramolecular synthons. The AIM analysis shows that the intermolecular interactions between the HMX and solvent molecules are weak hydrogen bonds with Eb values in the range 1.29–18.1 kJ mol−1 and bond lengths in the range 2.07–3.20 Å. For α-HMX/solvent synthons, the hydrogen bond strengths between two molecules are similar. While for β-HMX/solvent synthons, the strengths of CH2⋯OX (X = C, P, and S atoms) are much stronger than the NO2⋯H hydrogen bonds.
The most stable polymorphs among 10 most common space groups for α-HMX/DMF and α-HMX/NMP assemblies have R symmetry, with relative lower symmetry but similar crystal densities as compared to experiments with Rc space groups. The most possible packing for α-HMX/DMSO and α-HMX/GBL has Pna21 and Pbca symmetry respectively. While for the α-HMX/HMPA, DFTB-D method predicts the lowest-energy packing has P21 symmetry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21773119), the NSAF Foundation of National Natural Science Foundation of China and China Academy of Engineering Physics (No. U1530104), and the Science Challenging Program (No. TZ2016001).References
- P. Sanphui, N. R. Goud, U. B. R. Khandavilli and A. Nangia, Cryst. Growth Des., 2011, 11, 4135–4145 CAS.
- A. Sun, J. W. Lauher and N. S. Goroff, Science, 2006, 312, 1030 CrossRef CAS PubMed.
- S. Horiuchi and Y. Tokura, Nat. Mater., 2008, 7, 357–366 CrossRef CAS PubMed.
- L. Zhu, Y. Yi, Y. Li, E. Kim, V. Coropceanu and J. Brédas, J. Am. Chem. Soc., 2012, 134, 2340–2347 CrossRef CAS PubMed.
- R. E. Cobbledick and R. W. H. Small, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 2805–2808 CrossRef.
- T. M. Haller, A. L. Rheingold and T. B. Brill, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1983, 39, 1559–1563 CrossRef.
- T. M. Haller, A. L. Rheingold and T. B. Brill, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1985, 41, 963–965 CrossRef.
- K. B. Landenberger and A. J. Matzger, Cryst. Growth Des., 2010, 10, 5341–5347 CAS.
- O. Bolton and A. J. Matzger, Angew. Chem., Int. Ed., 2011, 50, 8960–8963 CrossRef CAS PubMed.
- Z. Yang, H. Li, X. Zhou, C. Zhang, H. Huang, J. Li and F. Nie, Cryst. Growth Des., 2012, 12, 5155–5158 CAS.
- O. Bolton, L. R. Simke, P. F. Pagoria and A. J. Matzger, Cryst. Growth Des., 2012, 12, 4311–4314 CAS.
- D. I. A. Millar, H. E. Maynard-Casely, D. R. Allan, A. S. Cumming, A. R. Lennie, A. J. Mackay, I. D. H. Oswald, C. C. Tang and C. R. Pulhama, CrystEngComm, 2012, 14, 3742–3749 RSC.
- K. B. Landenberger and A. J. Matzger, Cryst. Growth Des., 2012, 12, 3603–3609 CAS.
- Y. Wang, Z. Yang, H. Li, X. Zhou, Q. Zhang, J. Wang and Y. Liu, Propellants, Explos., Pyrotech., 2013, 35, 1–7 Search PubMed.
- H. Zhang, C. Guo, X. Wang, J. Xu, X. He, Y. Liu, X. Liu, H. Huang and J. Sun, Cryst. Growth Des., 2013, 13, 679–687 CAS.
- K. B. Landenberger, O. Bolton and A. J. Matzger, Angew. Chem., Int. Ed., 2013, 52, 6468–6471 CrossRef CAS PubMed.
- C. Zhang, Z. Yang, X. Zhou, C. Zhang, Y. Ma, J. Xu, Q. Zhang, F. Nie and H. Li, Cryst. Growth Des., 2014, 14, 3923–3928 CAS.
- K. B. Landenberger, O. Bolton and A. J. Matzger, J. Am. Chem. Soc., 2015, 137, 5074–5079 CrossRef CAS PubMed.
- J. C. Bennion, A. McBain, S. F. Son and A. J. Matzger, Cryst. Growth Des., 2015, 15, 2545–2549 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, P. Iiskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 9 (Revision A.01), Gaussian, Inc, 2009 Search PubMed.
- W. H. Baur and D. Kassner, Acta Crystallogr., Sect. B: Struct. Sci., 1992, 48, 356–369 CrossRef.
- J. L. Mendoza-Cortes, Q. An, W. A. Goddard, C. Ye and S. Zybin, J. Comput. Chem., 2016, 37, 163–167 CrossRef CAS PubMed.
- C. P. Brock and J. D. Dunitz, Chem. Mater., 1994, 6, 1118–1127 CrossRef CAS.
- R. L. C. Akkermans, N. A. Spenley and S. H. Robertson, Mol. Simul., 2013, 39, 1153–1164 CrossRef CAS.
- R. J. Gdanitz, Chem. Phys. Lett., 1992, 190, 391–396 CrossRef CAS.
- R. J. Gdanitz, H. R. Karfunkel and F. J. J. Leusen, J. Mol. Graphics, 1993, 11, 275–276 CrossRef.
- H. R. Karfunkel and R. J. Gdanitz, J. Comput. Chem., 1992, 13, 1171–1183 CrossRef CAS.
- H. R. Karfunkel, B. Rohde, F. J. J. Leusen, R. J. Gdanitz and G. Rihs, J. Comput. Chem., 1993, 14, 1125–1135 CrossRef CAS.
- H. R. Karfunkel, F. J. J. Leusen and R. J. Gdanitz, J. Comput.-Aided Mater. Des., 1994, 1, 177–185 CrossRef CAS.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260–7268 CrossRef CAS.
- B. Aradi, B. Hourahine and T. Frauenheim, J. Phys. Chem. A, 2007, 111, 5678–5684 CrossRef CAS PubMed.
- M. Elstner, J. Phys. Chem. A, 2007, 111, 5614–5621 CrossRef CAS PubMed.
- M. Elstner, Theor. Chem. Acc., 2006, 116, 316–325 CrossRef CAS.
- J. G. Brandenburg and S. Grimme, J. Phys. Chem. Lett., 2014, 5, 1785–1789 CrossRef CAS PubMed.
- CP2K Developers Group, CP2K Code, 2000–2006, http://cpk2.berlios.de.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10043d |
| This journal is © The Royal Society of Chemistry 2017 |