
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic catalysis of nano-Pd and nano rare-earth oxide/AC: complex nanostructured catalysts fabricated by a photochemical route for selective hydrogenation of phenol†
Yanji Zhang,
Jicheng Zhou * and
Jiaqi Si
* and
Jiaqi Si
Key Laboratory of Green Catalysis and Chemical Reaction Engineering of Hunan Province, School of Chemical Engineering, Xiangtan University, Xiangtan 411105, Hunan Province, China. E-mail: zhoujicheng2012@126.com
First published on 30th November 2017
Abstract
Cyclohexanone is an important industrial intermediate in the manufacture of polyamides in the chemical industry, but direct selective hydrogenation of phenol to cyclohexanone under mild conditions to achieve both high conversion and selectivity is a challenge. Here we report novel complex nanostructured catalysts Pd/@-CeO2/AC and Pd/@-La2O3/AC prepared by a photochemical route. These catalysts were characterized by XRD, BET, TEM, H2-TPR and XPS. The results showed that Pd nanoparticles were well-dispersed on CeO2/AC or La2O3/AC, and the complex nanostructured catalysts exhibit a synergistic effect between nano-Pd and nano-La2O3/AC or nano-CeO2/AC. These catalysts had an excellent catalytic performance for the hydrogenation of phenol, achieved 100% conversion and a selectivity higher than 96% within 3 h at 70 °C and 0.7 MPa H2 pressure. The results indicated that the remarkable performance may result from the synergistic effect between active component Pd and rare-earth oxide, their complex nanostructured properties and the high dispersion of Pd particles on the large surface area. Furthermore, this work revealed synergistic catalysis of nano-metal Pd and nano rare-earth oxide and opened a new direction of synergistic catalysis of nanostructured catalysts.
1. Introduction
Cyclohexanone is an important intermediate in the production of caprolactam, which is used in the manufacture of nylon 6 and nylon 66.1 In general, cyclohexanone can be obtained from cyclohexane oxidation or phenol hydrogenation.2 The former route requires high temperatures and high pressures and generates a large number of byproducts, but low conversion and high energy consumption.3 Therefore, selective hydrogenation of phenol to cyclohexanone is an important chemical process that has been extensively studied to date.4,5 In the hydrogenation of phenol, cyclohexanone is generally obtained through a one-step or a two-step process. In the two-step procedure, phenol is first hydrogenated to cyclohexanol and then cyclohexanol is dehydrogenated to cyclohexanone at high temperature.6 In one-step, the selective hydrogenation of phenol directly into cyclohexanone,7 however, cyclohexanone can be further hydrogenated to cyclohexanol easily, so it is extremely necessary to prepare high selectivity and conversion catalysts. In recent years, supported Pd-based catalysts have been widely used for the selective hydrogenation of phenol to cyclohexanone,8 such as Pd/C,9 Pd/MCM-41,10 Pd–Al2O3,11 Pd/ZrO2,12 Pd/Ce–AlOx (ref. 13) and so on. All of those catalysts indicated that Pd is an excellent metal catalyst for phenol hydrogenation. However, to achieving both excellent conversion (≥80%) and high selectivity (≥90%) under mild conditions is still a challenging problem.14Notably, Han Buxing and his co-worker15 reported that a complete phenol conversion with >99.9% selectivity to cyclohexanone could be achieved on a dual-supported Pd-Lewis acid catalyst in sc-CO2, while the conditions is so sophisticated (supercritical carbon dioxide as solvent, which requires high H2 and CO2 pressures >7.0 MPa). Most recently, Wang Yong and co-works designed a kind of carbon nitride material as support to prepare Pd-based catalysts,16 which achieved both excellent conversion and selectivity in the hydrogenation of phenol in aqueous media. Nevertheless, the preparation of the mpg-C3N4 and/or CN-x was complicated and involved the use of NH4HF2 or HF which are hazardous. Zhaoyin Hou17 prepared a Amberlyst-45 palladium catalyst (Pd/A-45), achieved 89% cyclohexanone selectivity and 100% phenol conversion at 100 °C and 1 MPa H2 pressure in water. They think the good performance of Pd/A-45 can be attributed to its strong acidity enhance the desorption of phenoxy species. On the other hand, Chen and co-works18–21 utilized polymer-functionalized CNF, hydroxyapatite-bound and/or ionic liquid-like copolymer to stabilize Pd, and the as-prepared catalysts have been proven to be efficient for phenol hydrogenation. However, the preparation of these catalysts required many expensive polymers and had the disadvantage of high preparation cost. In conclusion, although a few catalysts show a decent activity under an appropriate conditions, either the harsh reaction conditions, the low cyclohexanone selectivity (<90%) or the complicated method to prepare catalysts. Therefore, it is necessary to use a facile method to prepare catalyst for high efficient selective hydrogenation of phenol.
Recently, rare earth oxide has been great interest in the use of catalytic reaction due to its unique characteristics. Especially, the CeO2 and La2O3 are the most widely used.22,23 CeO2 has a good performance of stockpile and release oxygen, it can not only enhance the catalytic activity but also improve thermostability.24,25 La2O3 has a lot of attractive properties for applications such as catalysts, high k gate dielectric material and optical filters.26 Even so, they are low activity for used as catalyst directly. But when they combined with noble metal, it could influence the catalytic performance. Salvatore Scirè27 studied the gas-phase phenol hydrogenation reaction over Pd/CeO2 (40% conversion, 92% selectivity) and Pd/La2O3 (35% conversion, 95% selectivity) at 160 °C. The groups of Inagaki28 studied the reaction in the vapor phase over Pd/CeO2 at 180 °C (80% conversion, 50% selectivity). However, the impact of the interaction between the metal oxide and noble metal on the catalytic performance of these catalysts has not been discussed. In addition, these catalysts has not achieve both excellent conversion and high selectivity simultaneously.
The utilization efficiency of noble metals (Pt, Au, Pd) in conventional supported catalysts is far less than satisfactory. Recently, research has been reported that the metal with higher dispersity, many of the metal atoms are present at a surface, available for catalysis and offer the efficiency. Flytzani-Stephanopoulos et al.29–31 showed that atomically dispersed Au or Pt cations exhibited excellent catalytic activity for water–gas shift reaction, while Au or Pt particles had no chemical action. Another solution of the significantly reducing the use of precious metal, preparation of support single-atom catalysts (SAC), was proposed by Zheng and coworkers.32–34 We proposed a novel strategy to utilize noble metal efficiently, and prepared a novel complex nanostructured catalyst Au@TiO2/MCM-41 and Au@TiO2/MCM-22 by photochemical route, these catalysts exhibited excellent activity and chemical stability for cyclohexane oxidation.35–37 Herein, we used a novel strategy to prepare a complex nanostructured catalyst, which combined nano-metals with nano-semiconductor to fabricate a novel nano-Pd complex nanostructured catalyst Pd/@-MexOy/AC (MexOy: CeO2 or La2O3) by photochemical route. These complex catalysts were characterized by XRD, BET, TEM and XPS, and employed in the selective hydrogenation of phenol to cyclohexanone at low temperature and H2 pressure in the liquid phase. These catalysts exhibited a higher activity and selectivity to cyclohexanone, the remarkable catalytic performance could be ascribed to the synergistic effect between Pd nanoparticles and nano-semiconductor (CeO2 or La2O3), their complex nano-structured properties and the large contact surface.
2. Experimental
2.1 Catalyst preparation
The obtained CeO2/AC and La2O3/AC composites were denoted as xCeO2/AC and xLa2O3/AC, where x represents the CeO2/AC and La2O3/AC loading by weight.
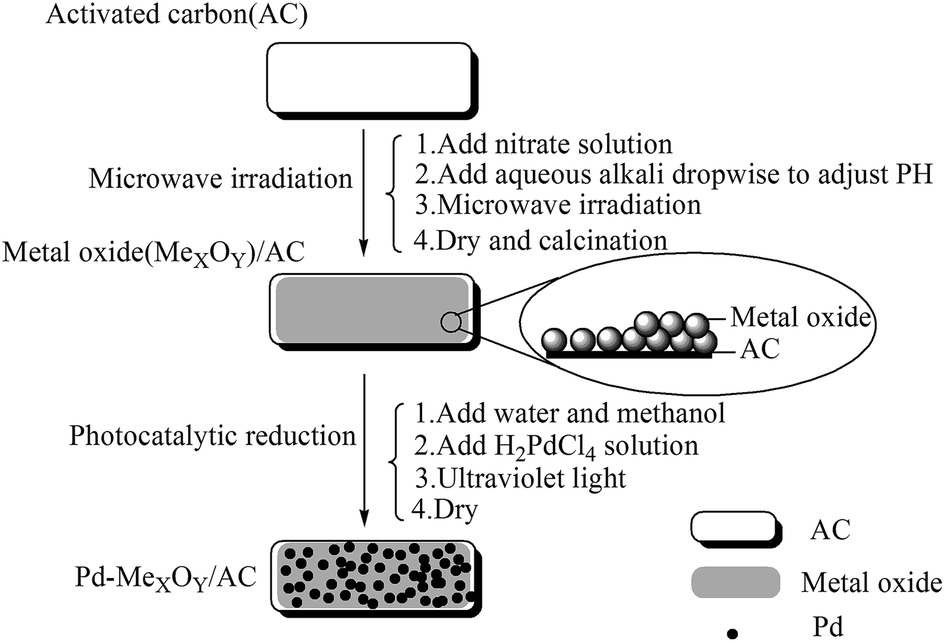 | ||
| Scheme 1 Preparation of the Pd/@-CeO2/AC and Pd/@-La2O3/AC catalysts. | ||
Our previous work involving photocatalytic reduction of noble Au,35–37 the preparation process and condition was simple and mild, and the noble nanoparticles was uniform dispersed, this method is also applicable to reduction Pd. Photochemical deposition of Pd films or small particles on oxide substrates such as TiO2,39,40 or graphene sheets/ZnO.41 Similarly, CeO2 is also an interesting material due to its unique redox properties as well as strong UV absorption at ∼400 nm.42 La2O3 is also a rare earth metal oxide and has similar chemical properties to CeO2. It is well known that a semiconductor can be excited and then generate electrons (e−) and holes (h+) in the conduction band (CB) and valence band (VB) if the energy of the photons of the incident light is larger than that of the band gap of the semiconductor.39 The reaction mechanism of photochemical route is in Scheme 2. When composite support (CeO2/AC or La2O3/AC) absorbed ultraviolet, metal oxide semiconductor can be excited and then generate electrons (e−) and holes (h+), forming electron–hole pairs. In the presence of methanol, the holes were scavenged, thus leaving the electrons to accumulate on the surface of the metal oxide semiconductor, the stored electrons were then transferred to the metal ions Pd2+ and reduced Pd2+ to form Pd on the surface of composite support.
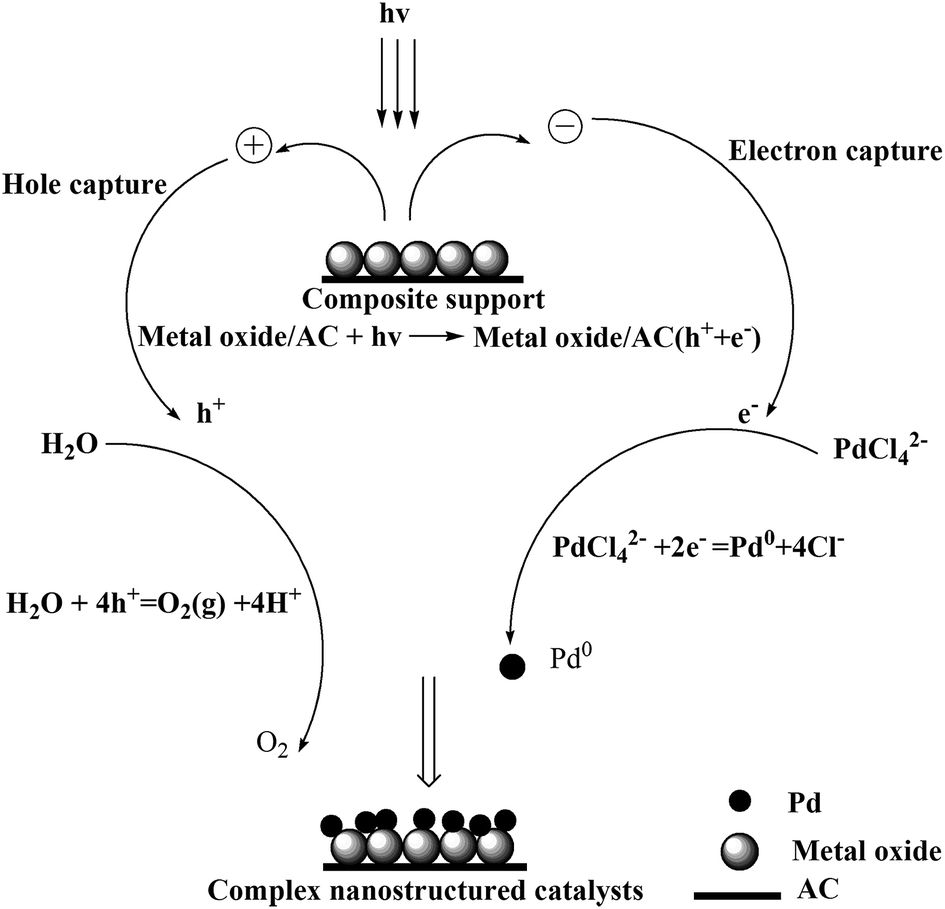 | ||
| Scheme 2 Reaction mechanism of photocatalytic reduction method. | ||
2.2 Catalyst characterization
X-ray diffraction (XRD) of samples was obtained on a Rigaku D/max-II/2500 X-ray powder diffractometer, Cu Kα radiation was employed and the working voltage and current were 40 kV and 30 mA, respectively. Transmission electron microscope (TEM) images were obtained with a JEOL JEM-2100F at an acceleration voltage of 200 kV. X-ray photoelectron spectroscopy (XPS) was performing using ESCALAB 250Xi (Thermo) with Al Kα radiation. Inductively coupled plasma-atomic emission spectrometry (ICP-AES) data were obtained using an Intrepid XSP (IRIS). The specific surface areas of the samples were calculated by the BET method by N2 adsorption–desorption with a NOVA-2200e volumetric desorption analyzer. Temperature-programmed reduction by hydrogen (H2-TPR) was carried out on Chem Bet 3000 analyzer. All samples (0.1 g) were pretreated in the flow of Ar at 250 °C for 1 hour.2.3 Catalytic tests
The typical procedure for hydrogenation of phenol was as follows: 0.1 g phenol, 0.353 g catalyst (0.75 mol% Pd relative to phenol), and 20 mL solvent were placed in a autoclave. The autoclave was purged with H2 to remove the air 3 times. Then the reaction was stirred at 800 rpm under 0.7 MPa H2 pressure until the temperature reached 70 °C. After the end of the reaction, the autoclave was cooled to room temperature, then the products were analyzed using an Agilent 6890N GC (Fig. S1†) with an HP-5 (30 m × 0.32 mm × 0.25 μm) capillary column and FID detector. The temperature of gasify room and detector were 250 °C and 280 °C respectively. The air at a constant flow of 400 mL min−1, the mixed gas H2 and N2 flow of 30 mL min−1. The initial column temperature was fixed at 50 °C for 7 min and then increased to 180 °C at a rate of 60 °C min−1. The final temperature was maintained for 2 min.3. Results and discussion
3.1 Characterization
The XRD patterns of AC, CeO2/AC, La2O3/AC, Pd/@-CeO2/AC and Pd/@-La2O3/AC catalysts were shown in Fig. 1. All samples present broadened peaks at about 2θ = 25° and 42°, which could be attributed to the AC support. Due to the high dispersion and low loading of CeO2 on the AC, just a very weak peaks at 2θ = 56.1°. The diffraction peak at 2θ = 31.3° was attributed to La2O3. Four characteristic diffraction peaks of Pd were detected at 2θ = 40.2°, 46.6°, 68.2° and 82.2°, which were attributed to the (111), (200), (220) and (311) crystalline planes. Comparison with La2O3/AC, the peaks attributed to La2O3 in XRD patterns of Pd/@-La2O3/AC became very weak or disappeared, which result from the uniform mixture of AC with La2O3, because the strong mechanical and chemical effects of an ultrasound wave and ultraviolet lamp dispersed the precipitates immediately.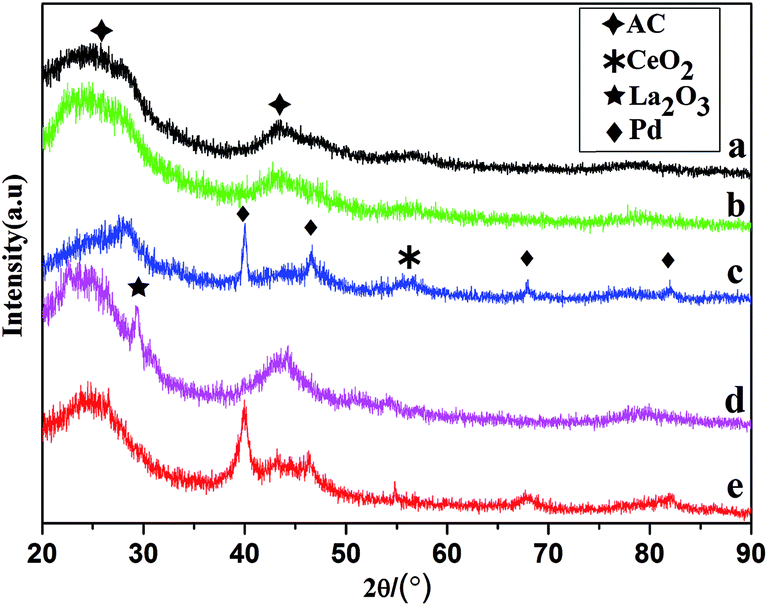 | ||
| Fig. 1 XRD patterns of samples: (a) AC (b) 10% CeO2/AC, (c) 3% Pd/@-10% CeO2/AC, (d) 10% La2O3/AC (e) 3% Pd/@-10% La2O3/AC. | ||
TEM and HRTEM images of 3% Pd/@-10% CeO2/AC and 3% Pd/@-10% La2O3/AC were recorded and shown in Fig. 2. For 3% Pd/@-10% CeO2/AC (Fig. 2a), the Pd nanoparticles are evident as larger dark spots, while the CeO2 substrate is smaller, it was very clear that the Pd particles were dispersed mainly on the composite support. Particles are distribution analysis in Fig. 2b, which reveals that the particles are in the range of 5–30 nm with 13 nm as an average diameter. The result was consistent with the average particle size 13.77 nm that obtained from XRD by the Scherrer equation. Fig. 2c represents the HRTEM image of 3% Pd/@-10% CeO2/AC, the lattice fringe with a d-spacing of 0.312 nm corresponds to the (111) plane of CeO2, and the distance of 0.223 nm (measured crystal DigitalMicrograph) corresponds to the (111) plane of Pd. Fig. 2d show the EDX data of 3% Pd/@-10% CeO2/AC, the data indicate the presence of elemental constituents within the as-synthesized samples. For 3% Pd/@-10% La2O3/AC (Fig. 2e), the situation is different, the Pd nanoparticles was more smaller than 3% Pd/@-10% CeO2/AC, which resulted from the interaction between Pd and La2O3, because the Pd nanoparticles could be smaller when the sample contains La2O3.43 The image revealed highly dispersed Pd with a mean size of 5.5 nm (Fig. 2f). Fig. 2g represents the HRTEM image of 3% Pd/@-10% La2O3/AC, the lattice fringe with a d-spacing of 0.297 nm corresponds to the (111) plane of La2O3, and the distance of 0.225 nm corresponds to the (111) plane of Pd. In Fig. 2h, we can also observe that the presence of elemental constituents within the as-synthesized samples.
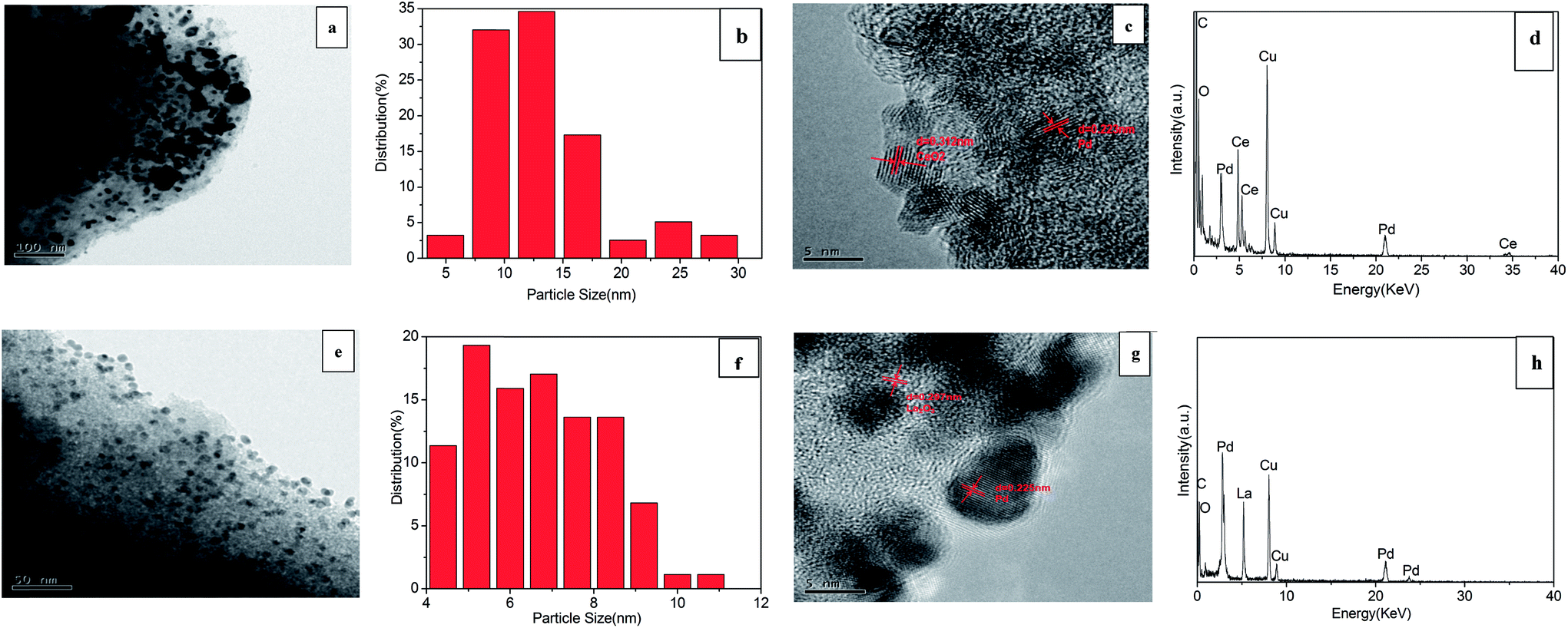 | ||
| Fig. 2 (a) TEM image of 3% Pd/@-10% CeO2/AC, (b) size distribution of Pd on 3% Pd/@-10% CeO2/AC, (c) HRTEM image of 3% Pd/@-10% CeO2/AC, (d) EDX spectrum of 3% Pd/@-10% CeO2/AC, (e) TEM image of 3% Pd/@-10% La2O3/AC, (f) size distribution of Pd on 3% Pd/@-10% La2O3/AC, (g) HRTEM image of 3% Pd/@-10% La2O3/AC, (h) EDX spectrum of 3% Pd/@-10% La2O3/AC. | ||
The relevant physicochemical parameters of the AC, 10% CeO2/AC, 10% La2O3/AC, 3% Pd/@-10% CeO2/AC and 3% Pd/@-10% La2O3/AC were summarized in Table 1. The AC had a high specific surface area of 1418.6 m2 g−1, however, the specific surface area decreased to 1309.2 m2 g−1 and 1349.7 m2 g−1 after doping CeO2 and La2O3, respectively. The surface area decreased gradually with further loading of Pd. Thus, the catalysts 3% Pd/@-10% CeO2/AC and 3% Pd/@-10% La2O3/AC exhibit the surface area of 1138.1 m2 g−1 and 1349.7 m2 g−1, respectively. This indicated that the loading of metal oxide and Pd within the pores, which is consistent with the reports.44
| Entry | Catalysts | Surface areaa (m2 g−1) | Pd loading (wt%) | Pd particle sizec (nm) | |
|---|---|---|---|---|---|
| Theoretical | Experimentalb | ||||
| a BET surface area.b Determined by ICP.c Observed from TEM images. | |||||
| 1 | AC | 1418.6 | — | — | — |
| 2 | 10% CeO2/AC | 1309.2 | — | — | — |
| 3 | 3% Pd/@-10% CeO2/AC | 1138.1 | 3 | 2.985 | 5–30 |
| 4 | 10% La2O3/AC | 1410.4 | — | — | — |
| 5 | 3% Pd/@-10% La2O3/AC | 1349.7 | 3 | 2.977 | 4–12 |
XPS was performed on the 3% Pd/@-10% CeO2/AC and 3% Pd/@-10% La2O3/AC catalysts and the results were shown in Fig. 3. From Fig. 3a, Pd, Ce, C, O and Pd, La, C, O were detected in two samples, respectively. Indicated the presence of CeO2 and La2O3 in Pd/@-CeO2/AC and Pd/@-La2O3/AC, respectively. And Pd species was loaded on the composite support by photochemical route. In Fig. 3b, the Pd 3d XPS spectra of the two catalysts show four asymmetric broad peaks. Peaks detected at around 335.9 eV and 341.1 eV can be readily assigned to the Pd 3d5/2 and Pd 3d3/2 electronic states of Pd(0),45 respectively. Compared with standard values (335 eV and 340.3 eV), the measured values are higher than their standard values, the electron move from Pd to CeO2, the dissociatively chemisorbed hydrogen on Pd may diffuse from Pd surface to CeO2 and reduce Ce4+ to Ce3+. That means the strong metal-support interaction between nano-Pd and nano-CeO2/AC. Peaks at 337.1 eV and 342.6 eV correspond to the Pd 3d5/2 and Pd 3d3/2 electronic states of Pd(2+) species, respectively. The presence of Pd(2+) species was probably due to the interaction of Pd and surface adsorbed oxygen, which is beneficial for the enhancement of adherence of Pd on the support.46 The Ce 3d XPS spectrum (Fig. 3c) consists of a spin orbit split doublet, the peaks located at 883.0 eV, 887.2 eV and 898.8 eV represent the Ce 3d5/2 and the peaks located at 901.6 eV, 907.2 eV and 917.1 eV represent the Ce 3d3/2 peaks. The peaks at 883.0 eV, 898.8 eV, 907.2 eV and 917.1 eV could be attributed to Ce4+ oxidation states while the peaks at 887.2 eV and 901.6 eV emanate from Ce3+ oxidation states of cerium.47,48 Hence, from the XPS spectrum it was confirmed that the sample contains mixed oxidation states (Ce4+ and Ce3+). The La 3d XPS spectrum was shown in Fig. 3d, the peaks located at 835.8 eV, 838.2 eV and 839.8 eV represent the La 3d5/2 and the peaks located at 852.6 eV, 854.6 eV and 856.5 eV represent the La 3d3/2 peaks. And the peaks at 835.8 eV and 838.2 eV could be attributed to La3+.49
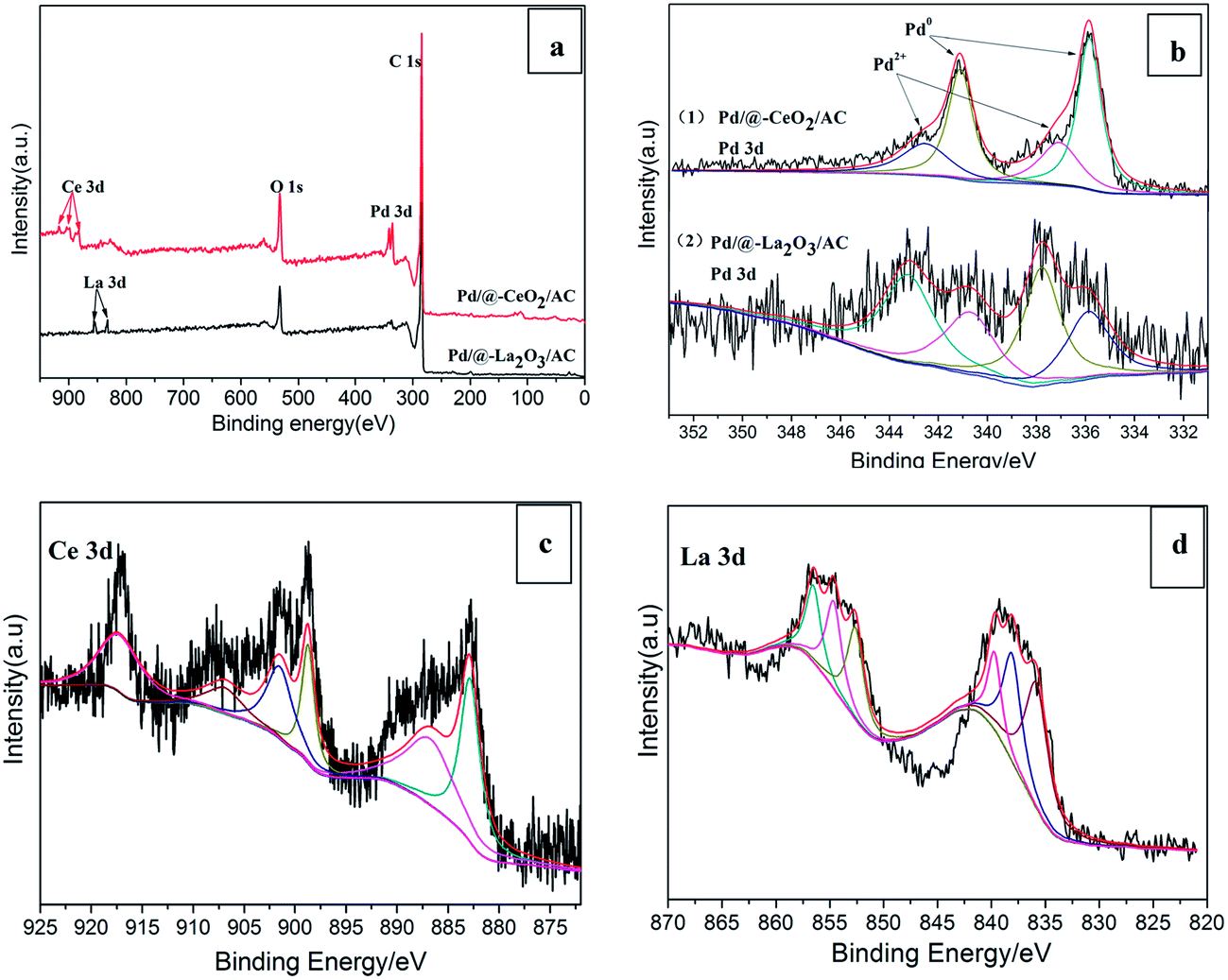 | ||
| Fig. 3 (a) XPS scans survey for 3% Pd/@-10% CeO2/AC and 3% Pd/@-10% La2O3/AC, (b) Pd 3d XPS spectra of 3% Pd/@-10% CeO2/AC and 3% Pd/@-10% La2O3/AC, (c) Ce 3d XPS spectra, (d) La 3d XPS spectra. | ||
3.2 Hydrogenation of phenol
The Pd complex nanostructured catalysts Pd/@-CeO2/AC and Pd/@-La2O3/AC were first applied to selective hydrogenation of phenol. For comparison, the CeO2/AC, La2O3/AC, Pd/AC, Pd/La2O3 and Pd/CeO2 were also used as reference catalysts, the results are shown in Table 2. As we can see, only metal oxide supported on AC samples CeO2/AC or La2O3/AC have no catalytic activity (Table 2, entry 5 and 6). Obviously, the Pd played a major role in reaction. Whereas these reference catalysts 3% Pd/CeO2 (Table 2, entry 3) and 3% Pd/La2O3 (Table 2, entry 4) almost no catalytic activity, with a phenol conversion of less than 10%, the selectivity of cyclohexanone were over than 99%. The 3% Pd/AC (Table 2, entry 1) catalyst have a better catalytic activity for selective hydrogenation of phenol, conversion of phenol could achieved to 76.6%. Wang Yong et al.49 have reported that hydrogenation of phenol to cyclohexanone can achieve 2% phenol conversion and 100% cyclohexanone selectivity at 65 °C and 6 h in water with commercial Pd@CeO2 catalyst. Chen Jinzhu et al.18 achieve 55% phenol conversion and 89% cyclohexanone selectivity with 10% Pd/AC catalyst at 80 °C and 9 h. Obviously, the activity of Pd/metal oxide and Pd/AC catalysts were far less than satisfactory. See the complex nanostructured catalysts 3% Pd/@-10% CeO2/AC and 3% Pd/@-10% La2O3/AC catalysts (Table 2, entry 9 and 15), they exhibited remarkable catalytic performance for selective hydrogenation of phenol, conversion of phenol were achieved up to 100%, selectivity of cyclohexanone were higher than 96%. According to the above results, the complex nanostructured catalysts showed a high activity because of the synergistic effect between metal nanoparticles and nano-semiconductors support, which enhanced effect of electronic promote the excellent charge transfer, promoted the synthesis of cyclohexanone and increase its selectivity. Then, the nature of the support plays an important role on activity and selectivity of Pd catalysts, because it has a large specific surface area and the add of Ce or La could increase the alkaline of catalyst,27,50–52 which provided a large enough place for the hydrogenation of phenol and avoid the co-planar adsorption of phenol, thus improved the selectivity of cyclohexanone. These results indicated that the Pd complex nanostructured catalysts have superiority catalytic activity.| Entry | Catalyst | Phenol conversion (%) | Selectivityb (%) | |
|---|---|---|---|---|
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
C–OH | |||
a Reaction conditions: phenol (0.1 g), n(Pd)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n(phenol) = 0.75%, CH2Cl2 (20 mL), 70 °C, 3 h, 0.7 MPa H2.b CO indicates cyclohexanone and C–OH indicates cyclohexanol.c Reaction conditions: phenol (40 mg), 5 mol% Pd relative to phenol, H2O (3 mL), 80 °C, 9 h, 0.1 MPa H2.18 :n(phenol) = 0.75%, CH2Cl2 (20 mL), 70 °C, 3 h, 0.7 MPa H2.b CO indicates cyclohexanone and C–OH indicates cyclohexanol.c Reaction conditions: phenol (40 mg), 5 mol% Pd relative to phenol, H2O (3 mL), 80 °C, 9 h, 0.1 MPa H2.18 |
||||
| 1 | 3% Pd/AC | 76.62 | 96.21 | 3.79 |
| 2c | 10% Pd/AC | 55 | 89 | 11 |
| 3 | 3% Pd/CeO2 | 4.50 | 99.20 | 0.8 |
| 4 | 3% Pd/La2O3 | 8.15 | 100 | 0 |
| 5 | 10% CeO2/AC | — | — | — |
| 6 | 10% La2O3/AC | — | — | — |
| 7 | 1% Pd/@-10% CeO2/AC | 32.6 | 96.30 | 3.70 |
| 8 | 2% Pd/@-10% CeO2/AC | 70.00 | 95.80 | 4.20 |
| 9 | 3% Pd/@-10% CeO2/AC | 97.22 | 96.32 | 3.68 |
| 10 | 3% Pd/@-5% CeO2/AC | 72.12 | 96.91 | 3.09 |
| 11 | 3% Pd/@-2% CeO2/AC | 66.73 | 97.14 | 2.86 |
| 12 | 3% Pd/@-1% CeO2/AC | 60.90 | 96.93 | 3.07 |
| 13 | 1% Pd/@-10% La2O3/AC | 10.98 | 100 | 0 |
| 14 | 2% Pd/@-10% La2O3/AC | 49.24 | 97.12 | 2.88 |
| 15 | 3% Pd/@-10% La2O3/AC | 100 | 96.82 | 3.18 |
| 16 | 3% Pd/@-5% La2O3/AC | 96.52 | 97.07 | 2.93 |
| 17 | 3% Pd/@-2% La2O3/AC | 88.08 | 97.16 | 2.84 |
| 18 | 3% Pd/@-1% La2O3/AC | 79.69 | 97.35 | 2.65 |
The loading of metal oxide has a great influence on the structure and catalytic performances of the complex nanostructured catalyst. Only Pd nanoparticles have been equably anchored on the nano-CeO2 or nano-La2O3 film layer, which was spread on the high specific surface AC, these complex nanostructured Pd catalysts exhibit synergistic catalysis and remarkable catalytic performances for the hydrogenation reaction of phenol. According to principles of the spontaneous monolayer distribution, metal oxide would disperse on the support at the state of the spontaneous monolayer distribution, when the content of metal oxide is less than a certain threshold and surface area of the support is large enough, so an appropriate amount of CeO2 or La2O3 can form a single or multi-layer nano-semiconductor layer on AC. From Table 2, when the loading of metal oxide was 10%, the conversion of phenol was achieved 100% and 97.22% by using 3% Pd/@-10% La2O3/AC and 3% Pd/@-10% CeO2/AC as catalyst, respectively. The conversion of phenol was 72.12% when the loading of CeO2 was 5% (Table 2, entry 10), while the conversion decreased to 60.9% when CeO2 content was 1% (Table 2, entry 12). The 3% Pd/@-xLa2O3/AC catalyst was observed same result, the lower loading of La2O3, the lower conversion (Table 2, entry 15–18). Compared the 3% Pd/@-1% La2O3/AC (Table 2, entry 18) with the 3% Pd/AC (Table 2, entry 1), it shown that add a few La2O3 could enhanced catalytic performance. The reason was that the nano-La2O3 improve the dispersity of the Pd particles, consistent with the result from the TEM images (Fig. 2e). But a few loading of La2O3 could not formed the rare-earth oxide layer, therefore Pd nanoparticles may be mostly supported on activated carbon directly rather than loaded on composite support with metal oxides. Moreover, Table 2 was also confirmed that a low loading of Pd was unfavorable for the formation of cyclohexanone. For Pd/@-CeO2/AC catalyst, the phenol conversion decreased quickly from 97.22% (Table 2, entry 9) to 32.6% (Table 2, entry 7) when the loading of Pd from 3% to 1%. For Pd/@-La2O3/AC catalyst, the phenol conversion decreased from 100% (Table 2, entry 15) to 10.98% (Table 2, entry 13) when the loading of Pd from 3% to 1%.
These results also shown that the catalytic performance difference between 3% Pd/@-10% La2O3/AC and 3% Pd/@-10% CeO2/AC, the possible reason could be attribute to the specific surface area. The specific surface area of 3% Pd/@-10% La2O3/AC (1349.667 m2 g−1) was more than 3% Pd/@-10% CeO2/AC (1138.102 m2 g−1), so it was more beneficial to material transfer. The other reason can be find in TEM, the Pd particles of 3% Pd/@-10% La2O3/AC was more uniformly dispersed and more smaller than 3% Pd/@-10% CeO2/AC. As we all know, the high dispersed noble metal catalyst often exhibit high catalytic performances.
In order to fully illustrate the superiority catalytic performance of the complex nanostructured catalysts, the catalytic performance of Pd/@-CeO2/AC and Pd/@-La2O3/AC were investigated under various reaction conditions (Table 3). When reaction temperature increased from 50 °C, 60 °C to 70 °C, the conversion of phenol increased quickly (Table 3, entry 1, 2, 5). Increasing reaction temperature could made the reactant molecular energy increased and the activated molecule number continually increased, and accelerated the reaction rate. At the same time, it was indicated that a high H2 pressure was more favorable for the formation of cyclohexanone, the selectivity only declined slightly (Table 3, entry 5, 6, 7). Considered that the higher H2 pressure, the more H2 participated in reaction, the cyclohexanone could not quickly out of active site, then lead to further hydrogenation for cyclohexanol. Also, the results showed that phenol conversion increased quickly as the reaction time was extended from 1 h to 3 h (Table 3, entry 3–5).
| Entry | Time (h) | Temperature (°C) | H2 pressure (MPa) | Pd/@-La2O3/AC | Pd/@-CeO2/AC | ||
|---|---|---|---|---|---|---|---|
| Conversion (%) | Selectivity (%) | Conversion (%) | Selectivity (%) | ||||
| a Reaction conditions: phenol (0.1 g), catalyst (0.353 g), n(Pd)/n(phenol) = 0.75%, CH2Cl2 (20 mL). | |||||||
| 1 | 3 | 50 | 0.7 | 52.26 | 96.79 | 42.62 | 95.35 |
| 2 | 3 | 60 | 0.7 | 95.16 | 97.11 | 46.48 | 95.75 |
| 3 | 1 | 70 | 0.7 | 55.34 | 98.11 | 43.25 | 96.51 |
| 4 | 2 | 70 | 0.7 | 92.59 | 97.61 | 71.70 | 95.60 |
| 5 | 3 | 70 | 0.7 | 100 | 96.82 | 98.28 | 96.33 |
| 6 | 3 | 70 | 0.6 | 85.6 | 96.77 | 58.10 | 96.67 |
| 7 | 3 | 70 | 0.5 | 79.14 | 97.3 | 45.32 | 96.70 |
Consider the environmental impact of the solvent, we also select H2O as a solvent, compared the performance of both Pd/@-CeO2/AC and Pd/@-La2O3/AC under the same conditions, the results were shown in Table 4. The phenol hydrogenation reaction used CH2Cl2 as a solvent was so much better than that of in H2O. The conversion of phenol were only 54.13% and 35.27% in water within 3 h by Pd/@-La2O3/AC and Pd/@-CeO2/AC catalyst, respectively. When the reaction time extends to 5 h, the Pd/@-La2O3/AC catalyst catalyzed phenol totally conversion, we can inferred that phenol hydrogenation reaction in water instead of organic solvent should not be affected by mass transfer resistance and the phenol conversion will achieve 100% in water when the reaction time was enough long. But whether Pd/@-La2O3/AC or Pd/@-CeO2/AC, the turnover frequency (TOF) in H2O was lower than it in CH2Cl2. The TOF of the Pd/@-La2O3/AC catalyst in CH2Cl2 could reach 44.35 h−1, and is higher than that of in H2O (24.01 h−1). For Pd@-CeO2/AC catalyst, the TOF in CH2Cl2 (36.98 h−1) is also higher than that of in H2O (15.64 h−1). Because the catalysts were dispersed in CH2Cl2 preferably, and H2 had a better solubility, so phenol was easier adsorbed on the surface of catalysts for hydrogenate to cyclohexanone.
| Time (h) | Solvent | Pd/@-La2O3/AC | TOF (h−1) | Pd/@-CeO2/AC | TOF (h−1) | ||
|---|---|---|---|---|---|---|---|
| Conversion (%) | Selectivity (%) | Conversion (%) | Selectivity (%) | ||||
| a Reaction conditions: phenol (0.1 g), catalyst (0.353 g), n(Pd)/n(phenol) = 0.75%, 70 °C, 0.7 MPa H2. Turnover frequency (TOF) defined as (mole of product/(moles of Pd × reaction time)). | |||||||
| 3 | CH2Cl2 | 100 | 96.82 | 44.35 | 98.28 | 96.33 | 36.98 |
| 3 | H2O | 54.13 | 96.29 | 24.01 | 35.27 | 96.70 | 15.64 |
| 5 | H2O | 100 | 94.88 | 26.61 | 54.36 | 97.60 | 14.47 |
Fig. 4 illustrates the TOF obtained in the hydrogenation of phenol with the different catalysts (include different metal oxide loading) in CH2Cl2. The TOF of the Pd/@-La2O3/AC catalysts was higher than Pd/@-CeO2/AC catalyst with different loading of rare-earth oxide as a whole. When the loading of rare-earth oxide achieved 10%, the TOF reached maximum, 3% Pd/@-10% La2O3/AC catalyst is 44.35 h−1 and 3% Pd/@-10% CeO2/AC is 36.98 h−1. For 3% Pd/La2O3 and 3% Pd/CeO2, only 3.59 h−1 and 1.99 h−1 TOF were obtained under the same conditions, respectively. Less than one-tenth of the complex nanostructured catalysts. The TOF of 3% Pd/AC was 33.98 h−1, less than the 3% Pd/@-10% La2O3/AC or 3% Pd/@-10% CeO2/AC. This result indicated that these novel nano-Pd complex catalysts were high efficient selective hydrogenation of phenol.
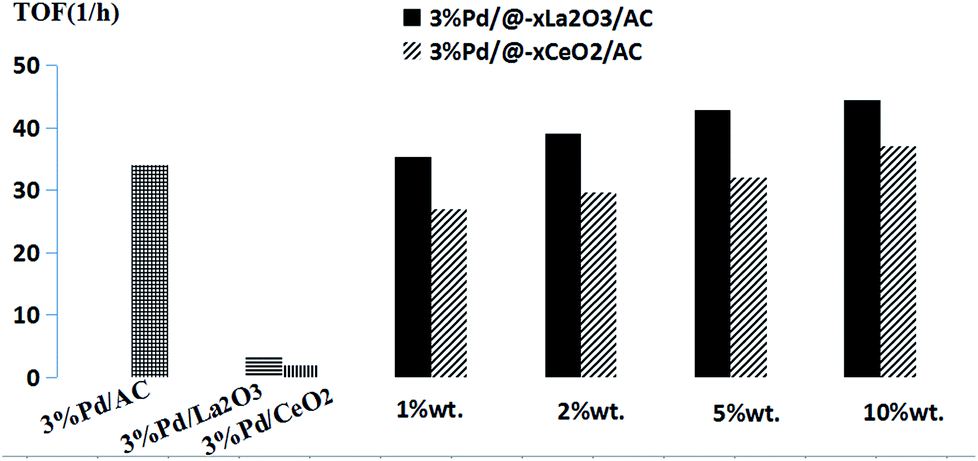 | ||
| Fig. 4 The TOF of hydrogenation of phenol with the different catalysts. The x represents the CeO2/AC and La2O3/AC loading by weight. The conditions are as follows: n(Pd)/n(phenol) = 0.75%, 20 mL CH2Cl2, 70 °C, 0.7 MPa H2, 3 h. Turnover frequency (TOF) defined as (mole of product/(moles of Pd × reaction time)). | ||
3.3 Catalytic activity
In Table 2, it was clear that the 3% Pd/@-10% La2O3/AC and 3% Pd/@-10% CeO2/AC catalysts exhibited high efficient catalytic activity for hydrogenation of phenol to cyclohexanone. To further understand the catalytic performance of the complex nanostructured catalysts, H2-TPR of the catalysts and AC, 10% CeO2/AC, 10% La2O3/AC, 3% Pd/@-10% La2O3/AC, 3% Pd/@-10% CeO2/AC were carried out. The results are shown in Fig. 5. Two peaks at 462.1 °C and 663.5 °C in the AC sample could be attributed to the oxygen-containing functional groups like –COOH, –OH,O, which is a feature of carbon, etc.51 The reduction peak was observed at 556.2 °C over CeO2/AC. For CeO2/AC, the low temperature peak at 400–600 °C was due to the reduction of surface oxygen species attached to surface Ce4+ ions in an octahedral coordination.22 H2-TPR of La2O3/AC show one reduction peak at 643.1 °C that may be ascribed to the reduction of La2O3.
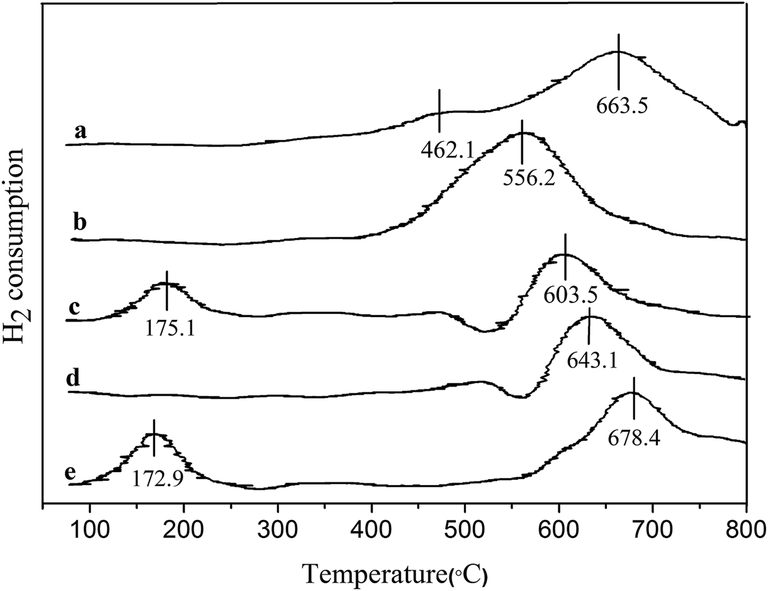 | ||
| Fig. 5 TPR profiles of catalysts. (a) AC, (b) CeO2/AC, (c) Pd/@-CeO2/AC, (d) La2O3/AC, (e) Pd/@-La2O3/AC. | ||
The TPR profiles of the CeO2/AC and La2O3/AC supported Pd catalysts changed significantly compared with those of CeO2/AC and La2O3/AC. For Pd/@-CeO2/AC, there were two other reduction peaks at 175.1 °C and 603.5 °C. The latter was ascribed to the reduction of the bulk oxygen of CeO2. The peak at 175.1 °C was assigned to the reduction of the species related to the PdO–CeO2 interaction, which suggests that PdO is well-dispersed on the support and CeO2 interacts with PdO.52 For Pd/@-La2O3/AC, there were two other reduction peaks at 172.9 °C and 678.4 °C. The latter was ascribed to the reduction of the La2O3. The former was considered the reduction of PdO which was affected by La2O3. Such results are also shown that the reduction temperature of PdO in Pd/@-CeO2/AC or Pd/@-La2O3/AC shifts to a higher value compared with the individual PdO (50 °C). It is further demonstrated that the complex nanostructured catalysts exhibit synergistic effect of both nano-Pd and nano-La2O3/AC or nano-CeO2/AC.
3.4 Compare with other catalyst
To unveil the underlying factors that provide the superior activity of the hydrogenation over these complex nanostructured catalysts (Pd/@-La2O3/AC and Pd/@-CeO2/AC), we compared the TOF for the hydrogenation of phenol over these complex nanostructured catalysts with other catalysts in Table 5. The result shown that the Pd/@-La2O3/AC catalyst catalytic the hydrogenation of phenol to cyclohexanone could be finished within 3 h, with a TOF reaching over 44.35 h−1. TOF of the Pd/@-La2O3/AC catalyst was about an order of magnitude higher than that of traditional catalysts and 2-ford higher than that of the best reported catalyst Pd/Al2O3-CWE (Table 5, entry 3). It is illustrated that the complex nanostructured catalyst was an efficient catalytic system and exhibited outstanding activity and selectivity for phenol hydrogenation.| Entry | Catalyst | n(Pd):n(phenol) (%) |
T (°C) | Time (h) | Conversion (%) | Selectivity (%) | TOF (h−1) | Note |
|---|---|---|---|---|---|---|---|---|
| a Turnover frequency defined as (mole of product/(moles of Pd × reaction time)). | ||||||||
| 1 | Pd/@-La2O3/AC | 0.75 | 70 | 3 | 100 | 96.82 | 44.35 | This work |
| 2 | Pd/@-CeO2/AC | 0.75 | 70 | 3 | 98.28 | 96.33 | 36.98 | This work |
| 3 | Pd/Al2O3-CWE | 2.5 | 100 | 2 | 100 | 98 | 20.05 | Ref. 53 |
| 4 | Pd/TiO2-AC | 1.6 | 50 | 4 | 47.3 | 96.4 | 15.27 | Ref. 54 |
| 5 | Pd/MIL-101 | 1.8 | 50 | 4 | 99.6 | 80.3 | 13.52 | Ref. 55 |
| 6 | Pd/SiO2 | 2 | 60 | 7.5 | 99 | 97 | 6.32 | Ref. 56 |
| 7 | Pd-HPW | 5 | 80 | 7 | >99 | >99 | 2.86 | Ref. 20 |
| 8 | Pd-PANI/CNT | 5 | 80 | 9 | >99.9 | >99 | 2.22 | Ref. 18 |
3.5 Possible reaction mechanism of phenol over complex nanostructured catalysts
In our study, the main product is cyclohexanone, a small amount of cyclohexanol (very slight). Generally, phenol was adsorbed by “non-planar” on alkaline support which was more advantage for the production of cyclohexanone. Whereas, it was more tend to produce cyclohexanol that phenol was adsorbed by “co-planar” on acidity support.28,57 As we all know, the adding of Ce or La could increase the alkaline of catalyst, then phenol was adsorbed by non-planar, inhibited the saturated hydrogenation of phenol to cyclohexanol. According to reaction results and the discussion above, possible reaction mechanism was proposed as the following. The process is shown in Scheme 3. The strong interaction between Pd and rare-earth oxide increases the ability to attract electron of hydroxyl group on phenol by non-planar, on which the H in the excited state could react on active benzene ring. After benzene ring hydrogenated to cyclohexanone, cyclohexanone was quickly dissociation of active site, otherwise it will further hydrogenation for cyclohexanol.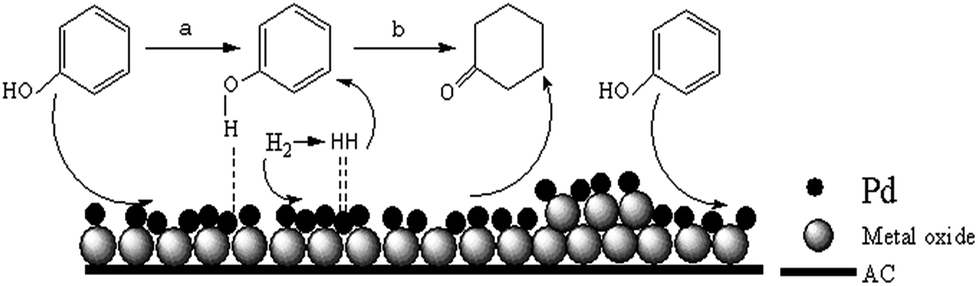 | ||
| Scheme 3 Mechanism of phenol hydrogenation with catalysis of Pd/@-CeO2/AC or Pd/@-La2O3/AC. | ||
After the reaction, the catalysts were recycled for phenol hydrogenation. The activity of Pd/@-La2O3/AC and Pd/@-CeO2/AC were decreased. The XRD patterns of the once-used Pd/@-La2O3/AC and once-used Pd/@-CeO2/AC were shown in Fig. 6. The Pd peaks of once-used catalysts were weakened, due to the destruction of the special structural and lost its portion activity component. Further study for the repeatability of the catalysts is still carried out.
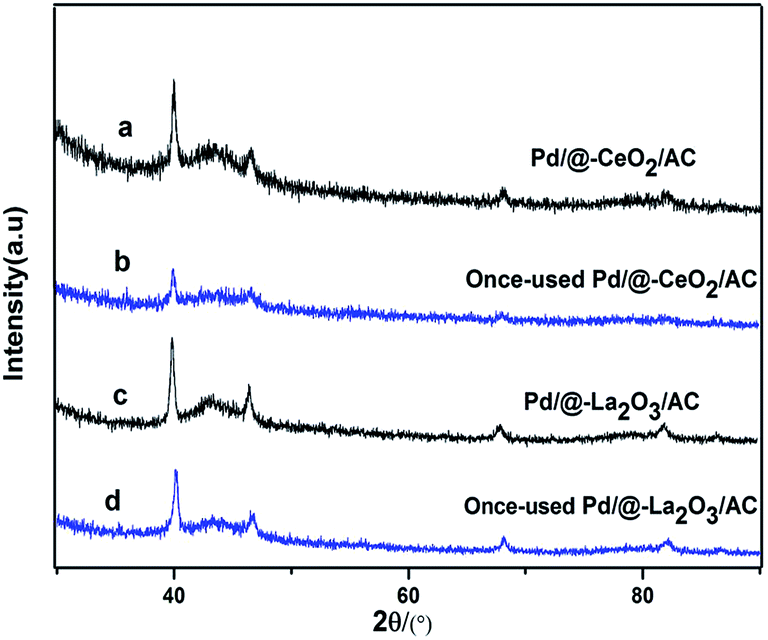 | ||
| Fig. 6 The XRD patterns of catalysts, (a) Pd/@-CeO2/AC, (b) once-used Pd/@-CeO2/AC, (c) Pd/@-La2O3/AC, (d) once-used Pd/@-La2O3/AC. | ||
4. Conclusions
Novel complex nanostructured catalysts Pd/@-La2O3/AC and Pd/@-CeO2/AC have been successfully fabricated via a photochemical route. The prepared catalysts Pd/@-La2O3/AC and Pd/@-CeO2/AC exhibited excellent performance of phenol hydrogenation under mild conditions. The results shown that 100% conversion of phenol and a selectivity higher than 96% were achieved within 3 h at 70 °C and 0.7 MPa H2 pressure, and the activity of Pd complex nanostructured catalyst was better than that of supported Pd on the metal oxide or AC directly. Furthermore, H2O was used as solvent, the prepared complex nanostructured catalysts also exhibited remarkable performance of phenol hydrogenation. It is indicated that the remarkable activity of these complex nanostructured catalysts may be ascribed to the synergistic effect of the interaction between active component nano-Pd and atom-layer rare-earth oxide, and enhance effect of electron of complex nanostructured catalysts, and formation of more contact interface due to the high dispersion of Pd particles on the large surface area support La2O3/AC and CeO2/AC. This work demonstrated that novel complex nanostructured catalysts Pd/@-La2O3/AC and Pd/@-CeO2/AC prepared via a photochemical route exhibited synergistic catalysis of nano-Pd and rare-earth oxide, and opened a new direction of complex nanostructured catalyst. The synergistic catalysis of complex nanostructured catalyst opened a promising direction to developing nanostructured catalyst for heterogeneous catalytic reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of China (21676227, 20976147), the Key Project of Hunan Provincial Natural Science Foundation of China (09JJ3021), the Key Laboratory Open Foundation of Higher Education Institutions of Hunan Province (12K047).References
- R. D. Patil and Y. Sasson, Appl. Catal., A, 2015, 499, 227–231 CrossRef CAS.
- X. Xuan, L. Haoran and W. Yong, ChemCatChem, 2015, 6, 3328–3332 Search PubMed.
- J. Hong, Q. Zhengyan, L. Ying, H. Jun, C. Rizhi and X. Weihong, Chem. Eng. J., 2016, 284, 724–732 CrossRef.
- H. Shuo, Z. Xiang, Q. Zhengyan, J. Hong, L. Yefei, H. Jun, X. Weihong and C. Rizhi, J. Ind. Eng. Chem., 2017, 53, 333–340 CrossRef.
- Z. Fengwei, C. Shuai, L. Huan, Z. Xianming and Y. Hengquan, RSC Adv., 2015, 5, 102811–102817 RSC.
- M. Juan and C. Avelino, Appl. Catal., A, 2011, 404, 103–112 CrossRef.
- N. Mahata, K. V. Raghavan, V. Vishwanathan, C. Parkb and M. A. Keane, Phys. Chem. Chem. Phys., 2001, 3, 2712–2719 RSC.
- G. S. Sheldon, D. Errun, P. Colin and A. K. Mark, J. Mol. Catal. A: Chem., 2004, 212, 291–300 CrossRef.
- W. Shingo and A. Venu, Top. Catal., 2010, 53, 1150–1152 CrossRef.
- M. Kohsuke, F. Ken, O. Shusuke and Y. Hiromi, Chem. Commun., 2012, 48, 8886–8888 RSC.
- Y. Hiroshi, N. Satomi, F. Shinichiro and A. Masahiko, J. Mol. Catal. A: Chem., 2013, 379, 80–85 CrossRef.
- M. S. Priscilla, C. R. N. Raimundo, E. P. B. Luiz, J. Gary, H. D. Burtron, M. G. Uschi, E. R. Daniel and B. N. Fabio, ACS Catal., 2015, 5, 7385–7398 CrossRef.
- G. Qingqing, Z. Yanhua, S. Junjun, C. XinSheng, G. Junjie, M. Rongrong, L. Bin and N. Ping, Chem. Eng. J., 2016, 299, 63–73 CrossRef.
- K. Sergey, M. Anton, Z. Anna and K. Eduard, Catal. Commun., 2016, 73, 63–68 CrossRef.
- L. Huizhen, J. Tao, H. Buxing, L. Shuguang and Z. Yinxi, Science, 2009, 326, 1250–1252 CrossRef PubMed.
- W. Yong, Y. Jia, L. Haoran, S. Dangsheng and A. Markus, J. Am. Chem. Soc., 2011, 133, 2362–2365 CrossRef PubMed.
- Z. Mengsi, S. Juanjuan and H. Zhaoyin, Chin. J. Catal., 2016, 37, 234–239 CrossRef.
- C. Jinzhu, Z. Wei, C. Limin, M. Longlong, G. Hui and W. Tiejun, ChemPlusChem, 2013, 78, 142–148 CrossRef.
- C. Aibing, L. Yonglei, C. Jinzhu, Z. Guoying, M. Longlong and Y. Yifeng, ChemPlusChem, 2013, 78, 1370–1378 CrossRef.
- C. Aibing, Z. Guoying, C. Jinzhu, C. Limin and Y. Yifeng, RSC Adv., 2013, 3, 4171–4175 RSC.
- X. Guangyue, G. Jianhua, Y. Zhang, Y. Fu, C. Jinzhu, M. Longlong and G. Qingxiang, ChemCatChem, 2015, 7, 2485–2492 CrossRef.
- Y. Ye, W. Zhimiao, A. Hualiang, X. Wei and W. Yanji, Chin. J. Catal., 2015, 36, 1142–1154 CrossRef.
- L. Lianjun, C. Yuan, S. Wenjing, Y. Zhijian, L. Bin, G. Fei and D. Lin, Catal. Today, 2011, 175, 48–54 CrossRef.
- W. Zili, L. Meijun and H. O. Steven, J. Catal., 2012, 285, 61–73 CrossRef.
- C. N. Nicholas, J. M. Sebastián, D. S. Aaron, H. O. Steven and I. S. Igor, ACS Catal., 2015, 5, 2051–2061 CrossRef.
- K. SangWoo and R. ShiWoo, J. Electrochem. Soc., 2002, 6, C345–C348 Search PubMed.
- S. Salvatore, M. Simona and C. Carmelo, Appl. Catal., A, 2002, 235, 21–31 CrossRef.
- S. Velu, M. P. Kapoor, S. Inagaki and K. Suzuki, Appl. Catal., A, 2003, 245, 317–331 CrossRef CAS.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- F. Qi, W. Deng, H. Saltsburg and M. Flytzani-Stephanopoulos, Appl. Catal., B, 2005, 56, 57–68 CrossRef.
- Y. Ming, L. F. Allard and F. S. Maria, J. Am. Chem. Soc., 2013, 135, 219–224 Search PubMed.
- Q. Botao, W. Aiqin, Y. Xiaofeng, L. F. Allard, J. Zheng, C. Yitao, L. Jingyue, L. Jun and Z. Tao, Nat. Chem., 2011, 3, 634–641 CrossRef PubMed.
- L. Jian, W. Aiqin, Q. Botao, L. Xiaoyan, Y. Xiaofeng, W. Xiaodong, L. Jinxia, L. Jun, L. Jingyue and Z. Tao, J. Am. Chem. Soc., 2013, 135, 15314–15317 CrossRef PubMed.
- Y. XiaoFeng, W. Aiqin, Q. Botao, L. Jun, L. Jingyue and Z. Tao, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef PubMed.
- Z. Jicheng, Y. Xiaofeng, W. Yaqiong and C. Weijun, Catal. Commun., 2014, 46, 228–233 CrossRef.
- L. Lin, Z. Jicheng, X. Zhibo and O. Wenbing, Acta Pet. Sin., 2013, 29, 975–983 Search PubMed.
- W. Yaqing, Z. Jicheng and Y. Xiaofeng, Chin. J. Process Eng., 2009, 6, 1186–1191 Search PubMed.
- G. Glaspell, L. Fuoco and M. S. El-Shall, J. Phys. Chem. B, 2005, 37, 17350–17355 CrossRef PubMed.
- S. F. Chen, J. P. Li and K. Qian, Nano Res., 2010, 3, 244–255 CrossRef CAS.
- P. Liu, Y. Zhao, R. Qin, S. Mo, G. Chen, L. Gu, D. M. Chevrier, P. Zhang, Q. Guo, D. Zang, B. Wu, G. Fu and N. Zheng, Science, 2016, 352(6287), 797 CrossRef CAS PubMed.
- H. Gu, Y. Yang, J. Tian and G. Shi, ACS Appl. Mater. Interfaces, 2013, 5, 6762–6768 CAS.
- G. K. Lau, T. S. Zhang and G. K. L. Goh, J. Nanosci. Nanotechnol., 2010, 10, 4733–4737 CrossRef CAS PubMed.
- X. L. Seoane, N. S. Figoli, P. C. L'Argentiere, J. A. Gonzalez and A. Arcoya, Catal. Lett., 1997, 47, 213–220 CrossRef CAS.
- S. Zheng and L. Gao, Mater. Chem. Phys., 2002, 78, 512–517 CrossRef CAS; S. A. Khan and A. Ahmad, Mater. Res. Bull., 2013, 48, 4134–4138 CrossRef.
- R. V. Gulyaev, E. M. Slavinskaya, S. A. Novopashin, D. V. Smovzh, A. V. Zaikovskii, D. Y. Osadchii, O. A. Bulavchenko, S. V. Korenev and A. I. Boronin, Appl. Catal., B, 2014, 147, 132–143 CrossRef CAS.
- D. Shuaishuai, Z. Chunhua, L. Yefei, J. Hong, X. Weihong and C. J. Rizhi, Ind. Eng. Chem., 2017, 46, 258–265 CrossRef.
- E. A. Anumol, P. Kundu, P. A. Deshpande, G. Madras and N. Ravishankar, ACS Nano, 2011, 5, 8049–8061 CrossRef CAS PubMed.
- H. Spod, M. Lucas and P. Claus, ChemCatChem, 2016, 8, 1–9 CrossRef.
- L. Yi, X. Xuan, Z. Pengfei, G. Yutong, L. Haoran and W. Yong, RSC Adv., 2013, 3, 10973–10982 RSC.
- L. Hui, L. Jianliang and L. Hexing, Mater. Lett., 2008, 62, 297–300 CrossRef.
- L. Hui, L. Jun, X. Songhai, Q. Minghua, D. Weilin, L. Yunfeng and L. Hexing, Adv. Funct. Mater., 2008, 18, 3235–3241 CrossRef.
- S. Chandra Shekar, J. Krishna Murthy, P. Kanta Rao and K. S. Rama Rao, J. Mol. Catal. A: Chem., 2003, 191, 45–59 CrossRef CAS.
- Z. Huaqing, Q. Zhangfeng, S. Wenjuan, S. Wenjie and W. Jianguo, J. Catal., 2004, 225, 267–277 CrossRef.
- Y. Pérez, M. Fajardo and A. Corma, Catal. Commun., 2011, 12, 1071–1074 CrossRef.
- Z. Damin, Y. Feiyang, X. Teng, G. Yejun and W. Yi Meng, Catal. Today, 2014, 234, 133–138 CrossRef.
- C. Ling, D. Qiguang, L. Hua and W. Xingyi, Catal. Commun., 2014, 57, 23–28 CrossRef.
- Y. Z. Chen, C. W. Liaw and L. I. Lee, Appl. Catal., A, 1999, 177, 1–8 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09917g |
| This journal is © The Royal Society of Chemistry 2017 |