DOI:
10.1039/C7RA09899E
(Paper)
RSC Adv., 2017,
7, 53236-53246
Anticancer potency of nitric oxide-releasing liposomes†
Received
5th September 2017
, Accepted 5th November 2017
First published on 20th November 2017
Abstract
In this study, fast and slow nitric oxide (NO)-releasing liposomes (half-lives of 2.5 and >72 h, respectively) were prepared by encapsulation of N-propyl-1,3-propanediamine/NO (PAPA/NO) and diethylenetriamine/NO (DETA/NO), respectively, via reverse phase evaporation. The anticancer activity of the otherwise equivalent fast and slow NO-releasing systems was evaluated against several distinct pancreatic, colorectal, and breast cancer cell lines. The anticancer assays (via cytotoxicity) over 72 h revealed that the slower NO-releasing liposomes consistently required lower NO payloads (LD50 < 3 μg mL−1) relative to the fast NO-release system (LD50 > 6 μg mL−1) to elicit cytotoxicity. The mechanism of intracellular NO build-up in cancer cells was studied using confocal fluorescence microscopy and flow cytometry, the results of which indicated that a more gradual NO accumulation was characteristic of the slow NO-release system. Protein expression via western blot analysis revealed that slower NO release resulted in more necrotic/apoptotic cells, while faster release reduced the number of mitotic cells to a greater extent. Overall, these studies demonstrate the potential of NO-releasing liposomes for anticancer therapy and highlight the significance of release kinetics (and NO payloads) required to induce cell death.
Introduction
Small molecule chemotherapeutics (e.g., doxorubicin and cisplatin) often exhibit off-target cytotoxicity due to poor localization.1–5 The use of a large macromolecular carrier (e.g., polymer, nanoparticle) to deliver the therapeutic to the targeted site represents one method for mitigating the adverse side effects of small molecules. The leaky vasculature of tumors allows macromolecules to extravasate at the malignant site, with the delivery of the therapeutic payload via cellular uptake or triggered release (e.g., pH and temperature).6 Liposomes are among the most widely-investigated delivery systems available for drug delivery. Traditionally, liposomes are composed of an aqueous inner core separated from the external solution by a phospholipid bilayer. This unique architecture allows for the confinement of compounds with a wide range of molecular weights, hydrophobicities, and charges until delivery at a location of interest. The ability of liposomes to adsorb and fuse with cell membranes enhances the intracellular uptake of the therapeutic payload, a process that is not typically observed for other macromolecular vehicles (e.g., nanoparticles). Liposomes have thus been developed for a number of therapeutic agents, such as gemcitabine and DNA.6,7 The exterior surface properties of the liposome (e.g., charge) may be tuned independently of the encapsulant, a critical feature for impacting targeting capabilities and/or reducing aggregation in the bloodstream. In this manner, liposomes have improved the anticancer delivery of many chemotherapeutics, including doxorubicin, arsenic trioxide, and daunorubicin.8–10
Nitric oxide (NO), an endogenous diatomic free radical, is an important mediator of inflammation,11–13 vasodilation,14,15 biocidal action,16–18 cardio- and neuroprotection,19,20 and cancer cell proliferation/killing.21–23 Off-target toxicity of NO is mitigated by scavenging and/or reaction to nitrite/nitrate.14 Nitric oxide donors have been developed as a strategy for delivering NO to biological systems as dissolved NO rather than a gas. Examples of currently approved NO donors include sodium nitroprusside, isosorbide mononitrate, glyceryl trinitrate, and pentaerythrityl tetranitrate.24 N-diazeniumdiolates are a class of NO donors that spontaneously release NO under physiological conditions. The rate of NO release depends on the molecular structure of the amine precursor, facilitating diverse and tunable NO-release kinetics.25,26 With respect to chemotherapy, the pH-dependent release of N-diazeniumdiolates is advantageous because tumor microenvironments are generally more acidic (pH ∼ 6) than healthy tissue (pH 7.4).27,28 The lower pH promotes accelerated NO release at the tumor. A vast literature has proven that small molecule N-diazeniumdiolates are capable of eliciting potent anticancer action.29–35 Clinical utility has not been achieved because of the excessive loss of NO prior to reaching the tumor site.
In this study, two N-diazeniumdiolate-encapsulated liposome systems were prepared with distinct NO-release kinetics (fast and slow). The anticancer activity of these liposomes was evaluated against pancreatic, colorectal, and breast cancer cell lines. Confocal fluorescence and flow cytometry were used to measure both cellular uptake of the liposomes and intracellular NO delivery. The effect of the NO release on protein expression, specifically cleaved PARP, cyclin B1, and cyclin D1, was also examined via western blot analysis to assess apoptosis and cell cycle arrest/ejection.
Experimental
Materials
Dipalmitoylphosphatidylcholine (DPPC) and 1,2-dipalmitoyl-sn-glycerol-3-phosphoethanolamine-N-(lissamine rhoadmine B sulfonyl) ammonium salt (Rh-PE) were purchased from Avanti Polar Lipids (Alabaster, AL). Cholesterol (Chol), paraformaldehyde, propidium iodide, fetal bovine serum (FBS), penicillin streptomycin, 1× Dulbecco's modified Eagle's medium (DMEM), McCoy's 5A medium, RPMI 1640 medium, Dulbecco's phosphate-buffered saline (DPBS) for cell culture, N-propyl-1,3-propanediamine (PAPA), and diethylenetriamine (DETA) were obtained from Sigma (St. Louis, MO). Prolong diamond antifade mountant, 4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate (DAF-FM), trypsin, Accutase, Annexin V Pacific Blue conjugate, 1% (v/v) NP40 lysis buffer, chloroform, phenazine methosulfate (PMS), anhydrous acetonitrile, anhydrous diethyl ether, dimethyl sulfoxide (DMSO), anhydrous ethanol (EtOH), sulfuric acid (H2SO4), protein stripping buffer, sodium hydroxide (NaOH), calcium chloride (CaCl2), PageRuler Plus prestained protein ladder, and secondary antibodies (both mouse and rabbit) for western blotting were purchased from Fisher Scientific (Fair Lawn, NJ). Sephadex G-25 was obtained from GE Healthcare (Pittsburgh, PA). 3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophen-yl)-2H-tetrazolium, inner salt (MTS) was purchased from Promega (Madison, WI). Primary antibodies for total and cleaved PARP (rabbit), cyclin B1 (rabbit), cyclin D1 (rabbit), and vinculin (mouse) used in western blotting were obtained from Cell Signaling (Danvers, MA). Western Lightning ECL Pro substrate for western blot detection was from PerkinElmer (Waltham, MA). Phosphatase and protease inhibitors were purchased from Roche (Basel, Switzerland). Criterion TGX Gel, tris/glycine transfer buffer with sodium dodecyl sulfate (SDS), tris-buffered saline with Tween 20 (TBST), and polyvinylidene fluoride (PVDF) transfer membrane were obtained from Bio-Rad (Hercules, CA). Nitric oxide (NO; 99.5%), nitrogen (N2; 99.998%), argon (Ar; 99.995%), and NO calibration (26.80 ppm, balance N2) gases were obtained from Airgas National Welders (Durham, NC). A Millipore Milli-Q UV Gradient A10 System (Bedford, MA) was used to purify distilled water to a resistivity of 18.2 MΩ cm and a total organic content ≤6 ppb. MIA PaCa-2, AsPc1, and Pa14c pancreatic cancer cells were a gift from Dr Channing Der of the Department of Pharmacology at the University of North Carolina (Chapel Hill, NC). MDA-MB-231, MCF-7, MDA-MB-468, HCT116, HT-29, and SW480 breast and colorectal cancer cells were a gift from Dr Matthew Lockett of the Department of Chemistry at the University of North Carolina (Chapel Hill, NC).
Synthesis of N-diazeniumdiolate NO donors
A previously reported method was used to synthesize small molecule N-diazeniumdiolate NO donors.27 Briefly, DETA and PAPA were dissolved in anhydrous acetonitrile at a concentration of 33.3 mg mL−1. The solution was then purged with Ar to 100 psi inside a stainless steel Parr bomb. Six consecutive purges with Ar (three quick purges of 10 s each, followed by three slow purges of 10 min each) were carried out to remove dissolved oxygen. The solution was subsequently pressurized to 145 psi with NO for 3 d, after which the solution was purged again with Ar (100 psi) at least six times to remove residual NO. The precipitated product was filtered over a Hirsch funnel, washed three times with diethyl ether, and dried under vacuum overnight. The final NO donor product was stored at −20 °C until use.
Liposome synthesis
The liposomes were prepared using a reverse phase evaporation method.36,37 A 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of lipid to Chol (49.5 μmol lipid:49.5 μmol Chol) was dissolved in a mixture of diethyl ether (5.0 mL) and chloroform (5.0 mL) in a round-bottom flask under N2 atmosphere. Fluorescent liposomes were prepared using the above protocol, but with 1 mol% lipid of Rh-PE. The N-diazeniumdiolate was dissolved in 10 mM NaOH to make a 14 mM stock NO donor solution. This solution was injected into the flask, and then sonicated for 4 min at 45 °C. The organic phase was removed by rotoevaporation to yield the aqueous liposome suspension. Liposomes were incubated at 45 °C for an additional 30 min, after which the unencapsulated material was removed using four Sephadex G-25 spin columns packed in 10 mL syringes. The liposomes collected from the column were stored at 4 °C.
:1 molar ratio of lipid to Chol (49.5 μmol lipid:49.5 μmol Chol) was dissolved in a mixture of diethyl ether (5.0 mL) and chloroform (5.0 mL) in a round-bottom flask under N2 atmosphere. Fluorescent liposomes were prepared using the above protocol, but with 1 mol% lipid of Rh-PE. The N-diazeniumdiolate was dissolved in 10 mM NaOH to make a 14 mM stock NO donor solution. This solution was injected into the flask, and then sonicated for 4 min at 45 °C. The organic phase was removed by rotoevaporation to yield the aqueous liposome suspension. Liposomes were incubated at 45 °C for an additional 30 min, after which the unencapsulated material was removed using four Sephadex G-25 spin columns packed in 10 mL syringes. The liposomes collected from the column were stored at 4 °C.
Characterization of liposomes
Dynamic light scattering (DLS; Malvern Zetasizer Nano; UK) was used to determine liposome size distribution in water. Transmission electron microscopy (TEM) was used to confirm liposome formation. Liposome samples for TEM analysis were prepared by diluting the stock solution with Milli-Q water (1:1 volumetric ratio) and casting the suspension onto Formvar-coated, square mesh copper TEM grids (Electron Microscopy Sciences, Hatfield, PA). The solvent was allowed to evaporate for 45 min prior to applying a negative-stain using a 2% (w/v) uranyl acetate solution. A drop of the stain was left on the grid for 30 s and then removed using filter paper. The grid was dried for 5 min prior to imaging using a JEOL 100CX II transmission electron microscope (100 kV accelerating voltage).
Nitric oxide release
Nitric oxide storage and NO-release kinetics from the liposomes were measured using a Sievers Chemiluminescence Nitric Oxide Analyzer (NOA; Boulder, CO).38,39 Studies to evaluate NO-release kinetics were performed in 10 mM PBS (pH 7.4) at 37 °C. The instrument was calibrated using air passed through a NO zero filter (0 ppm NO) and a 26.80 ppm NO standard (balance N2). Nitric oxide storage for encapsulation efficiency (i.e., the extent to which the NO donor is entrapped within the liposomal aqueous core) was performed in a 2:1 volumetric ratio of ethanol to 0.183 M sulfuric acid (30 mL total volume) at 37 °C. The encapsulation efficiency was calculated by comparing the liposome NO storage to the amount of NO in the free donor solution used during liposome preparation. All presented data is from n ≥ 3 separate liposome preparations. Nitric oxide release measurements were terminated when the NO concentration dropped below 10 ppb per 300 μL liposomes.
Cytotoxicity assays
Pa14c and MIA PaCa-2 cells were cultured in DMEM. HCT116 and HT-29 cells were cultured in McCoy's 5A medium. MDA-MB-231, MCF-7, MDA-MB-468, SW480, and AsPc1 cells were cultured in RPMI medium. All media were supplemented with 10 vol% FBS and 1 wt% penicillin/streptomycin. Cells were maintained at 37 °C in a humidified incubator with 5 vol% CO2. For cell viability evaluations, the MTS assay was used as previously described with cells plated in triplicate.40 Briefly, cells (2 × 103 cells per well) in DMEM were treated with various volumes of liposomes in a 96-well plate (100 μL total volume per well). Following a 72 h incubation period at 37 °C, the supernatant of each well was removed, rinsed with PBS (100 μL), and replaced with fresh DMEM (100 μL). The cells were further incubated with 20 μL of MTS/PMS reagent (20:1 v/v MTS to PMS) at 37 °C for ∼90 min. The absorbances of the resulting solutions at 490 nm were measured using a Thermoscientific Multiskan EX plate reader (Waltham, MA). The ratio of absorbance values between the samples and the controls was represented as the percent cell viability. Dose response curves and LD50 values were plotted and tabulated using GraphPad Prism 6 software (La Jolla, CA) and non-linear regression (three-parametric Hill function), respectively. All presented data are from n ≥ 3 separate experiments.
Confocal fluorescence microscopy
Cells were plated in 10 × 10 mm cloning cylinders (VWR, Atlanta, GA) secured to no. 1.5 glass cover slips (VWR, Atlanta, GA) using silicone grease. The slips were placed in a Petri dish prior to the addition of cell media. After 24 h, the medium within the cylinders was replaced with a 10 μM DAF-FM solution. An additional 30 min incubation period at 37 °C was then carried out before removing the DAF-FM solution. Fresh cell media was subsequently added and allowed to incubate for another 15 min at 37 °C to allow for saponification of the probe. Liposomes were added to the cells and incubated for 2 h, followed by rinsing twice with 100 μL DPBS. A 100 μL aliquot of 4 vol% paraformaldehyde solution (diluted with DPBS) was injected into the wells and incubated for 15 min at room temperature. The cover slips were then mounted on glass slides using a droplet of mounting media, sealed with nail polish (Electron Microscopy Sciences; Hatfield, PA), and imaged after 1 h using a Zeiss LSM 700 laser scanning confocal microscope. The excitation/emission wavelengths for DAF-FM and Rh-PE were 495/515 and 560/583 nm, respectively. Fiji software was used for image processing and densitometry calculations. Autoquant X3 software (Media Cybernetics; Warrendale, PA) generated orthogonal views of the z-stacked images. All images were collected under constant exposure times. The images were also processed equivalently and normalized to a single brightness level.
Western blot analysis
Cells were added to a 6-well plate (3 × 105 cells per well) and incubated for 24 h. Media was then removed and replaced with fresh media containing liposomes. At specified timepoints, the plates were placed on ice, washed once with cold DPBS, and incubated for 15 min with 1 vol% NP40 lysis buffer (50 μL) containing protease and phosphatase inhibitors. The wells were then scraped and the solution added to cold microcentrifuge tubes. Cellular debris was removed by centrifugation (4 °C, 5 min). Protein concentrations in samples were determined using the Bradford assay. Equal total protein amounts (∼30 μg) were added to each lane of the gel (4–20% gradient). After electrophoresis, the proteins were transferred onto PVDF membranes, blocked with 5% (w/v) milk, and stained with primary antibody overnight. After incubation with the secondary antibody for 1 h, the membrane was incubated with the Western Lightning ECL Pro substrate (10 min) and then imaged using a ChemiDoc chemiluminescence imaging system (Bio-Rad; Hercules, CA). Western blot images were processed using Image Lab software (Bio-Rad; Hercules, CA). Fiji software was used for densitometry calculations. Loading controls were used as a normalization factor for densitometric calculations.
Results and discussion
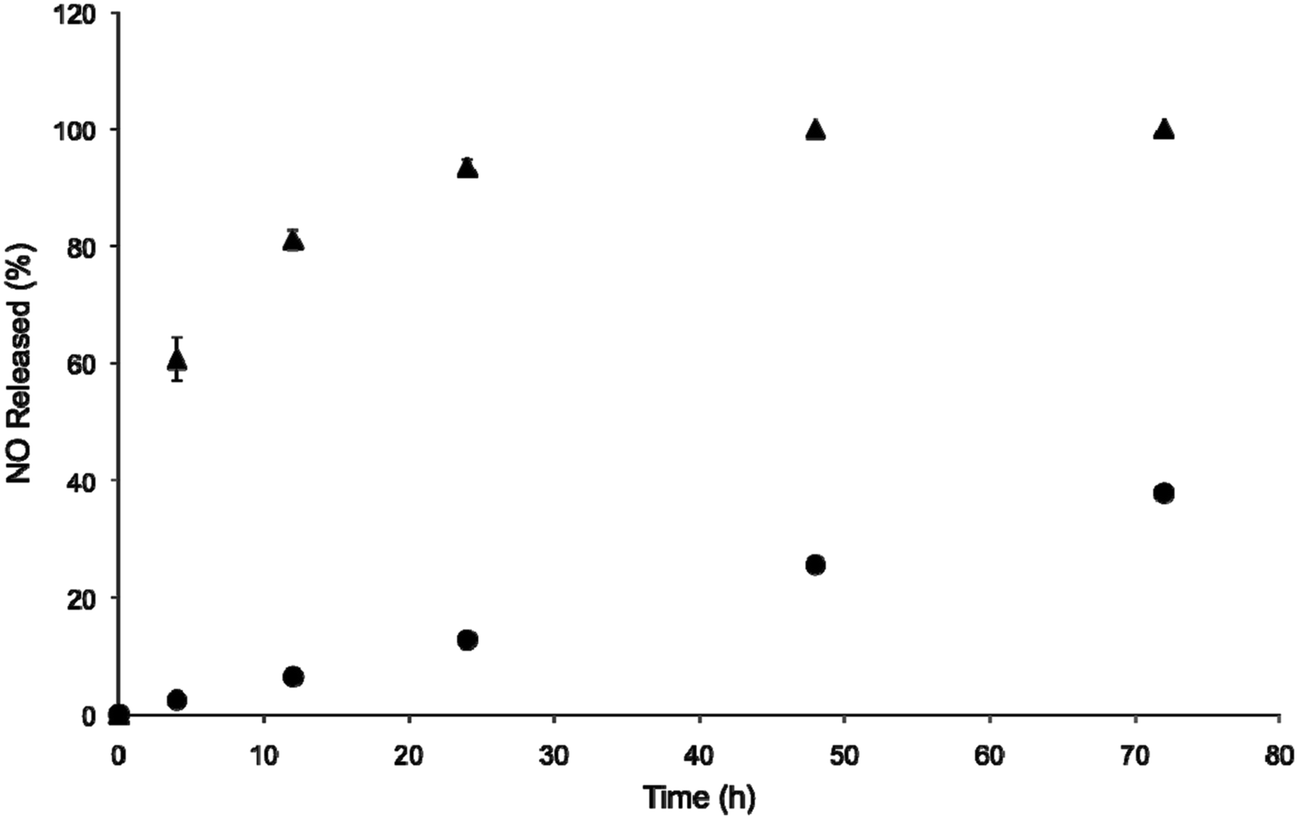
The N-diazeniumdiolates used in this study (PAPA/NO and DETA/NO; Fig. S1†) were selected because of their dissimilar NO-release half-lives in PBS at pH 7.4 (0.25 h and 20 h, respectively). On the basis of our previous work and that of others,28,41 we hypothesized that the resulting NO-releasing liposomes would have different NO-release kinetics. Liposome formation was confirmed by dynamic light scattering (DLS) measurements. As shown in Fig. 1, DETA/NO and PAPA/NO liposomes exhibited hydrodynamic sizes typical of liposomes synthesized via reverse-phase evaporation (Table 1).37 The slight difference in size between the systems should not appreciably affect their anticancer activity as liposomes with sizes of approximately 150 to 400 nm exhibit similar cellular uptake.42 Transmission electron microscopy (TEM) corroborated the DLS measurements and indicated negligible liposome–liposome fusion (Fig. S2†). Real-time NO release measurements demonstrated that the NO donor encapsulation efficiency was similar to efficiencies of other reverse-phase evaporated liposomes and consistent between the two liposome formulations (Table 1), likely the result of similar size of the NO donors.37 As expected, the liposomes released NO more slowly at physiological pH (7.4) than the corresponding small molecule NO donor alone (Fig. 1). The PAPA/NO liposomes released ∼50% of their total NO in 2.5 h, a ten-fold longer NO-release half-life than the free NO donor. As the rate of NO release impacts NO's toxicity,29–35 the use of two distinct N-diazeniumdiolates as encapsulants allows for the study of the anticancer therapeutic potential of the liposomes as a function of NO-release kinetics.
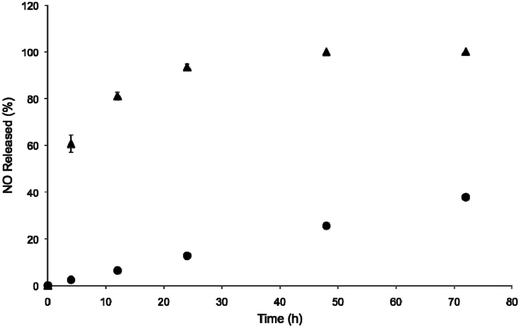 |
| | Fig. 1 Nitric oxide-release profiles from liposomal (●) DETA/NO and (▲) PAPA/NO in 10 mM PBS (pH 7.4, 37 °C) over the first 72 h. | |
Table 1 Properties of NO-releasing liposomes
| NO donor |
Hydrodynamic sizea (nm) |
Encapsulation efficiencyb (%) |
Total NOc (μg mL−1) |
| Z-Average size measured using DLS. Ratio of μmol of NO inside liposomes to μmol used for synthesis, multiplied by 100. Total amount of NO released in acid normalized to the injected liposome volume. |
| PAPA/NO |
377 ± 52 |
19.0 ± 3.5 |
125.7 ± 41.1 |
| DETA/NO |
246 ± 32 |
20.6 ± 3.2 |
133.2 ± 26.7 |
Cytotoxicity of the liposomes
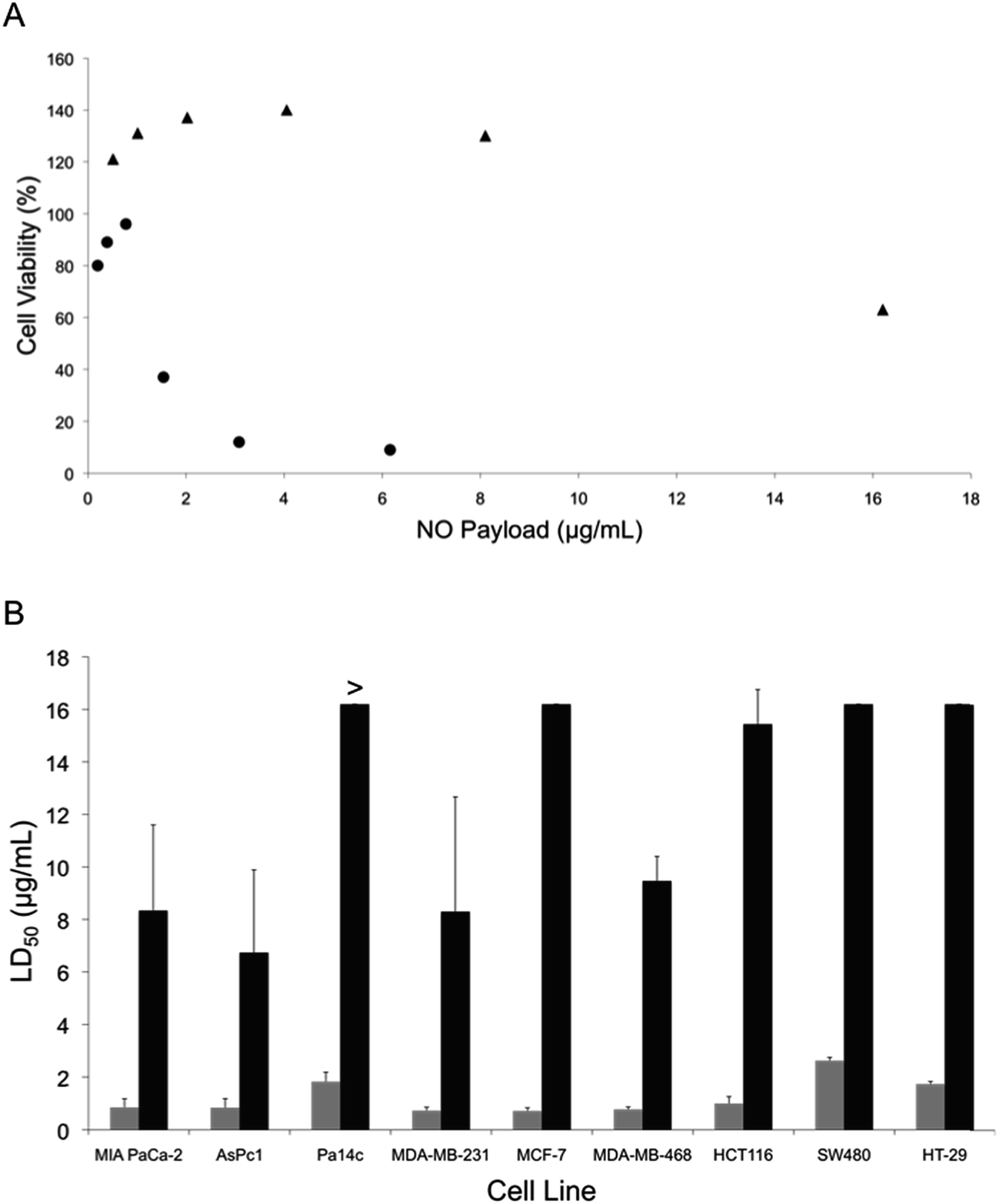
The potential anticancer activity of the NO-releasing liposomes was initially tested against Pa14c pancreatic cancer cells, an aggressive pancreatic cancer cell line. The PAPA/NO and DETA/NO liposome systems showed a pronounced toxicity difference, attributable to the NO release (Fig. 2A). At low NO payloads (∼0.9 μg mL−1), the viability was slightly enhanced for each liposomal system. Previous research has reported that low levels of NO induces EGF-dependent cell proliferation.14,43 At NO payloads >1.5 μg mL−1, the slower NO-release system (DETA/NO liposomes) was markedly more toxic towards the Pa14c cells. The less effective PAPA/NO liposomes required larger NO payloads to induce toxicity likely because of the faster release rate, resulting in the release of the majority of the NO payload before reaching the cell and/or cellular uptake. Significantly less toxicity (killing) was observed (Fig. S3†) when using a noncancerous epithelial cell line (HPNE), indicating that NO may elicit greater cytotoxicity towards cancer cells by further enhancing the existing oxidative and nitrosative stresses that such cells are already experiencing.44
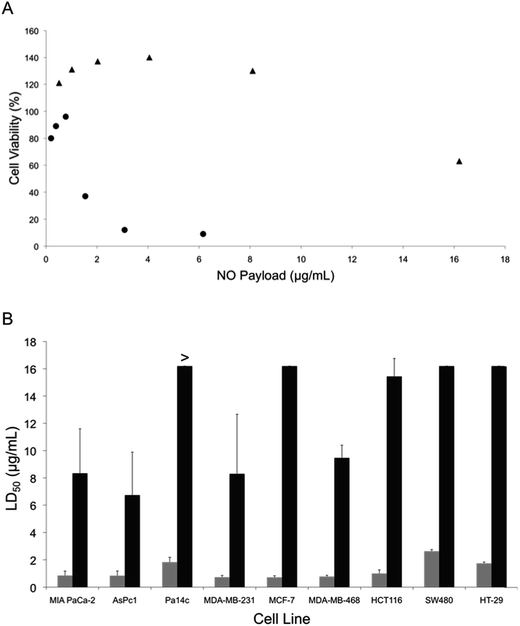 |
| | Fig. 2 (A) Cytotoxicity of liposomal (●) DETA/NO and (▲) PAPA/NO as a function of NO payload against human Pa14c pancreatic cancer cells after 72 h of exposure. (B) Calculated LD50 values for (grey bars) DETA/NO and (black bars) PAPA/NO liposomes against pancreatic, breast, and colorectal cancer cell lines. Of note, the LD50 of PAPA/NO liposomes against Pa14c cells was >16.2 μg mL−1. NO payloads were calculated based on the total amount of NO released from the liposomes over 72 h in PBS. | |
The cytotoxicity of the liposomes was next evaluated against a number of malignant lines from pancreatic, breast, and colorectal cancers to ascertain if the observed dependence on NO-release kinetics applied to other cell lines. The slow NO-releasing liposomes (DETA/NO) consistently required lower NO payloads to elicit cytotoxic effects, regardless of cancer type or cell line (Fig. 2B). In fact, the LD50 was <3 μg mL−1 NO for the DETA/NO liposomes against all cancer cell lines investigated, while the faster-releasing PAPA/NO liposomes required >6 μg mL−1 NO to elicit cytotoxic action. These results agree with prior work that demonstrated that free NO donors exhibiting slow NO-release kinetics required lower anticancer payloads relative to their fast-releasing counterparts.29,33,45
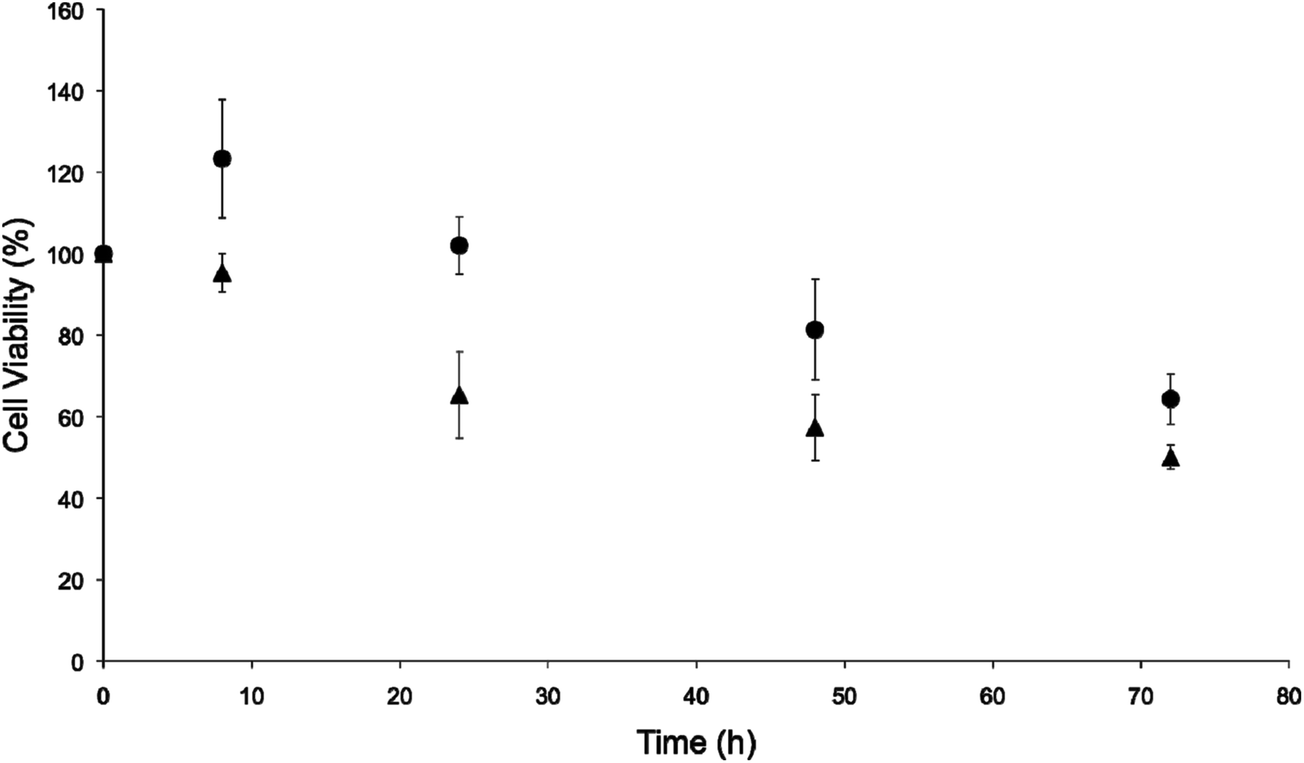
A human breast cancer cell line (MCF-7) was chosen as a representative model for further evaluation of the NO-release kinetics and associated cytotoxicity because of the stark differences in the LD50 values for the PAPA/NO and DETA/NO liposomes. Our immediate goal was to determine if the PAPA/NO liposomes induced cytotoxicity earlier in the assay (i.e., before 72 h). Cells were exposed to the 72 h LD50 concentrations of the DETA/NO and PAPA/NO liposomes (0.75 μg mL−1 and 16.2 μg mL−1 after 72 h exposure, respectively) for 8, 24, 48, and 72 h. As shown in Fig. 3, neither the fast nor slow NO-releasing liposome system exhibited cytotoxic effects at early timepoints (8 h). Rather, mild cell proliferation was noted for both. After 24 h, cell viability diminished greatly (up to 60%) for cells exposed to the PAPA/NO system (16.2 μg mL−1 NO), with no further change through 72 h. PAPA/NO liposomes deliver ∼90% of the NO payload by 24 h (Fig. 1), correlating with this observed cytotoxicity. The NO liberated from the DETA/NO liposomes (0.75 μg mL−1) displayed a more consistent cell viability profile with a steady drop over the 72 h period. Cytotoxicity for PAPA/NO liposomes at 0.75 μg mL−1 payloads was not induced at any time point (negligible toxicity relative to controls). Collectively, this data suggests that faster NO release (i.e., using shorter half-life NO donors) elicits cytotoxicity more rapidly than corresponding slower release, but necessitates larger NO payloads. Relative to the slower NO-releasing liposomes, the greater levels of NO from the fast release liposomes may work to increase the entropy within the cells through protein denaturation.
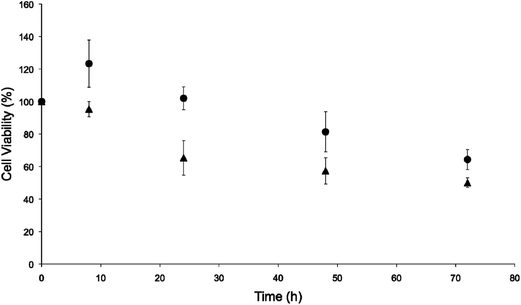 |
| | Fig. 3 Time-course cytotoxicity study of liposomal (●) DETA/NO and (▲) PAPA/NO at their respective LD50 values against human MCF-7 breast cancer cells. | |
Intracellular liposome uptake and NO delivery
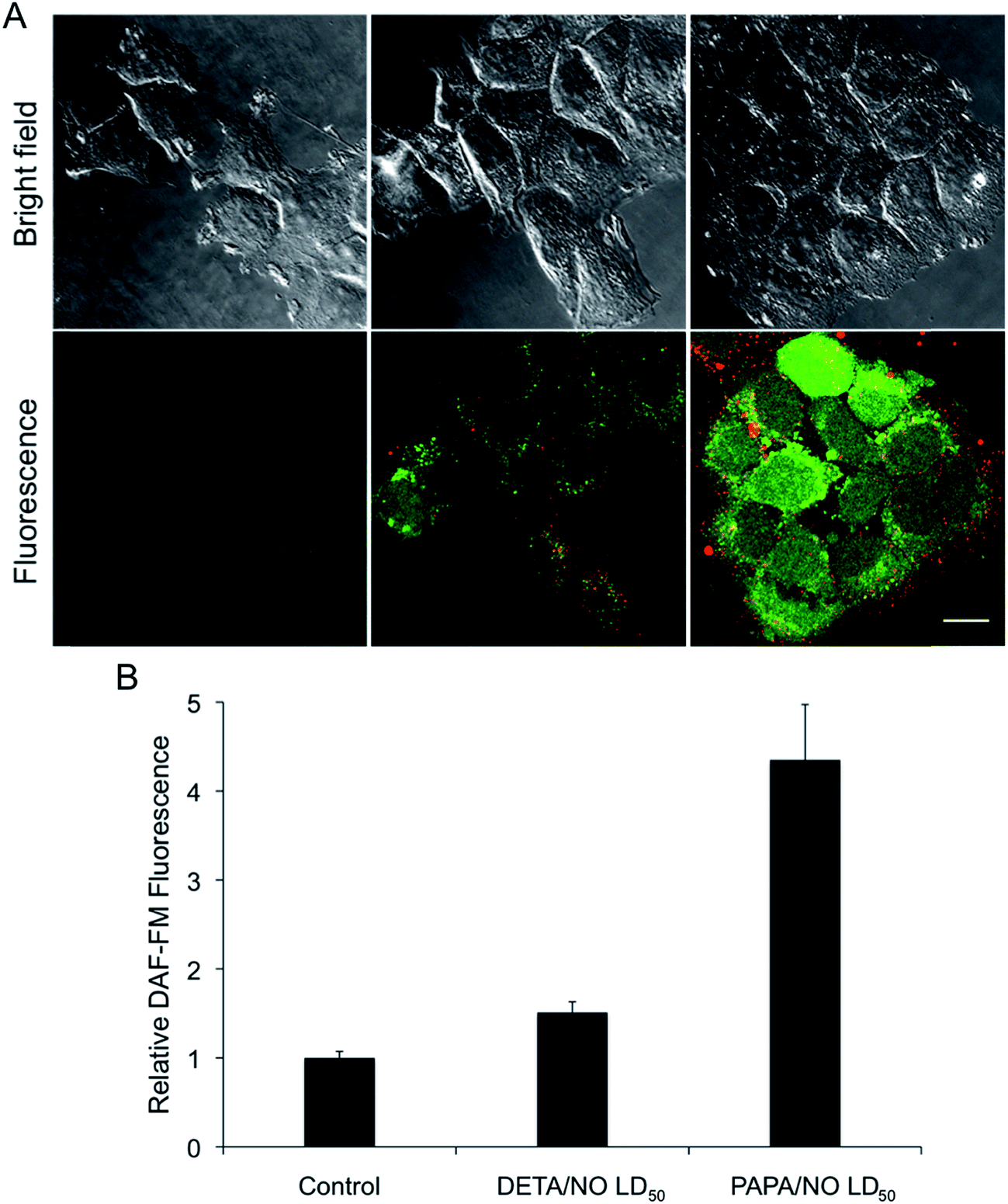
The observed relationship between liposome NO-release kinetics and anticancer action was hypothesized to be the result of intracellular NO accumulation. Confocal fluorescence microscopy was employed to measure intracellular NO build-up over time for the two systems using DAF-FM, a molecular probe that selectively reacts with NO to form a fluorescent benotriazole compound.46 Additionally, cellular uptake of the liposomes was visualized by incorporating a fluorescent phospholipid (Rh-PE) into the lipid bilayer.47 A 2 h exposure period was initially selected for this study as cells have been shown to initiate liposomal uptake within this timeframe,48,49 allowing for visualization of delivered NO. The bright field and fluorescence images of MCF-7 cells after exposure to DETA/NO and PAPA/NO liposomes (at their LD50 values) are provided in Fig. 4A. Of note, the amount of NO released during the confocal experiment is significantly lower than the corresponding LD50 values due to the shorter exposure time (2 vs. 72 h). Liposome uptake was clearly observed after 2 h, with z-stack images revealing intracellular localization of the NO-releasing liposomes (Fig. 5). The rapid uptake of the liposomes results from their ability to adsorb to and then fuse with the cell membrane, a phenomenon that does not readily occur with other delivery vehicles (e.g., nanoparticles).6 Relative to DETA/NO liposomes, cells exposed to PAPA/NO liposomes had substantially elevated levels of intracellular NO. Densitometry calculations were carried out to quantify intracellular NO levels for the two liposomes (Fig. 4B). Treatment with PAPA/NO liposomes resulted in a 4-times larger fluorescence signal relative to DETA/NO after 2 h, supporting the results observed in the time-course study where the fast release system elicited more rapid cytotoxicity (at 24 h). The lack of cytotoxicity observed for the PAPA/NO liposomes at the same NO payloads as the DETA/NO liposomes (0.75 μg mL−1) was supported by negligible intracellular NO accumulation at 2 h (data not shown). Collectively, the greater NO accumulation and NO exposure observed for the PAPA/NO liposomes leads to more rapid anticancer action.
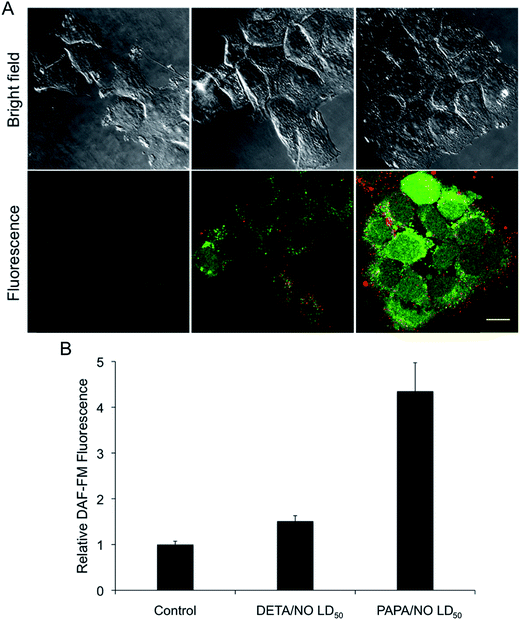 |
| | Fig. 4 (A) Confocal fluorescence images of MCF-7 cells incubated with DAF-FM (green) and treated with NO-releasing liposomes (red) for 2 h. Scale bar represents 15 μm. Column 1 is controls. Column 2 and 3 are cells exposed to the LD50 values of DETA/NO and PAPA/NO liposomes, respectively. By 2 h, DETA/NO and PAPA/NO liposomes released ∼1 and 30% of their NO payloads, respectively. (B) Densitometric analysis of intracellular DAF-FM levels relative to untreated controls. | |
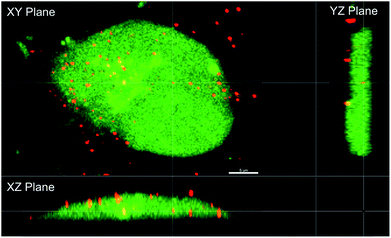 |
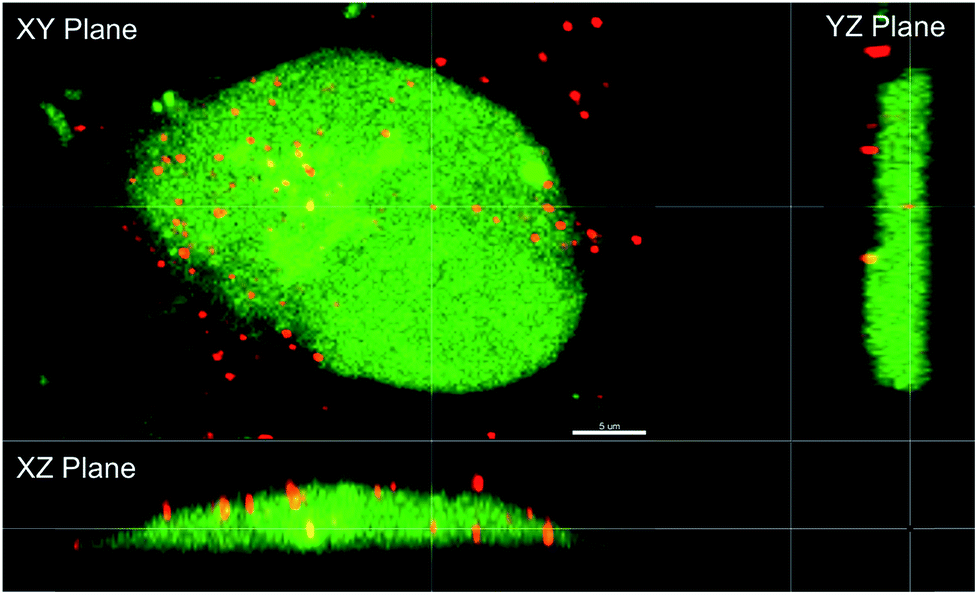
| | Fig. 5 Orthogonal view of MCF-7 cells after treatment with PAPA/NO liposomes. Scale bar represents 5 μm. | |
Kinetics of intracellular NO accumulation
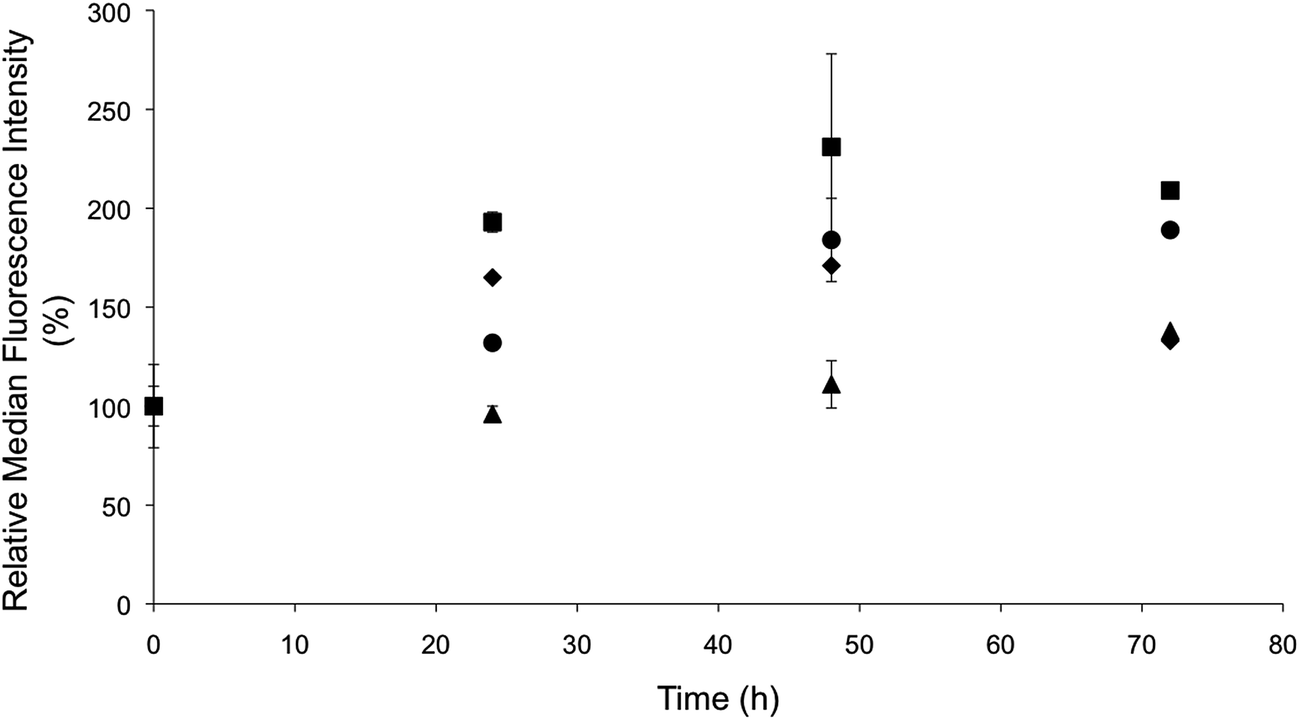
The rapid NO delivery from the PAPA/NO liposomes was hypothesized to be key in eliciting cytotoxicity at short time periods (i.e., 24 h). The more gradual cell killing observed using the DETA/NO liposomes would be expected to parallel the build-up of intracellular NO over time. Flow cytometry was utilized to quantify NO accumulation within the MCF-7 cells over a 72 h period using the same DAF-FM probe (Fig. 6). A large increase in intracellular fluorescence was observed by 24 h for cells treated with PAPA/NO liposomes (16.2 μg mL−1 NO). At 48 and 72 h, the fluorescence essentially remained at the same level, signaling that no more NO was delivered to the cell. In contrast, the fluorescence within cells treated with the slower NO-releasing DETA/NO liposomes (0.75 μg mL−1 NO) continued to increase steadily over the entire 72 h period. The therapeutic action of both slow and fast NO-releasing liposomes followed the same trend observed in the cytotoxicity time-course study, where cytotoxicity was elicited more rapidly with PAPA/NO liposomes and the DETA/NO liposomes required the full 72 h before eliciting toxicity. At equal NO payloads (0.75 μg mL−1), minimal intracellular NO accumulation was observed for the PAPA/NO liposomes due to premature NO loss prior to cellular uptake (Fig. S4†), which is in agreement with cytotoxicity findings.
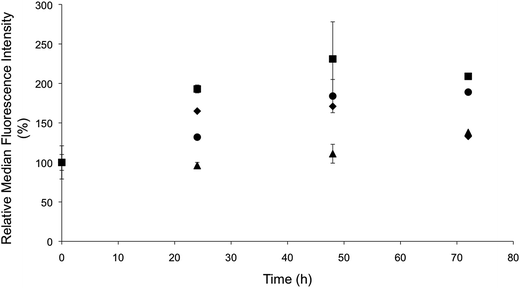 |
| | Fig. 6 Change in median fluorescence intensity over time indicating intracellular NO accumulation, as determined by flow cytometry, after treating MCF-7 cells with 0.75 μg mL−1 NO from (●) liposomal and (▲) free DETA/NO, and 16.2 μg mL−1 NO from (■) liposomal and (◆) free PAPA/NO. | |
A median fluorescence intensity comparison between free and liposomal NO donors was performed to highlight the benefits of using NO-releasing liposomes over the low molecular weight NO donors. Cells treated with NO-releasing liposomes exhibited greater intracellular NO accumulation (Fig. 6), as a result of enhanced NO donor stability (within the liposomes) and targeted cellular uptake. Similar behavior has been observed for other small molecule drugs (e.g., gemcitabine and doxorubicin) encapsulated within liposomes.8,50
Effect of NO-releasing liposomes on intracellular signaling
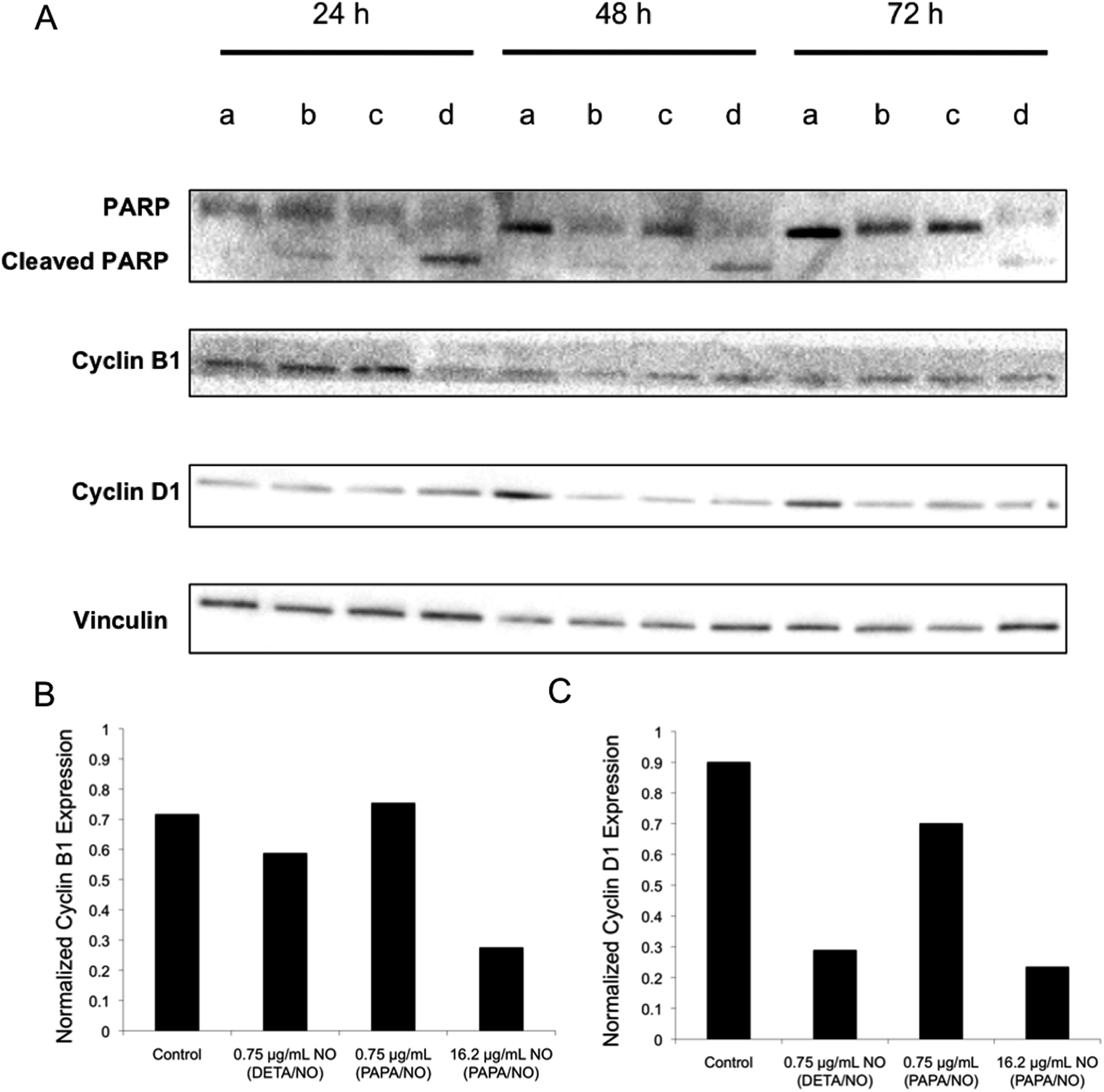
Western blot analysis was used to evaluate if any differences existed in protein expression levels. Poly(ADP-ribose) polymerase (PARP) is a critical mediator of DNA repair and upon cleavage by caspase-3 initiates cellular breakdown and apoptosis.51 A measurement of increased PARP levels after treatment would indicate that cells underwent PARP-mediated apoptosis. Cyclin analysis would facilitate understanding the cell cycle and whether cells were arrested or ejected.52,53 The expressions of these regulator proteins in MCF-7 cells were measured after NO exposure at 24, 48, and 72 h (Fig. 7). Cleaved PARP levels were the greatest for cells treated with 16.2 μg mL−1 NO from PAPA/NO liposomes, indicating apoptosis, especially at early timepoints (i.e., 24 and 48 h). This data correlates well with the rapid cytotoxicity observed from the fast NO-release system (Fig. 3). Densitometric calculations were performed on the blots to more accurately compare protein levels between exposure conditions. Even though cells treated with DETA/NO liposomes exhibited reduced cyclin B1 levels relative to controls by 24 h (Fig. 7), the levels were still greater than cells treated with PAPA/NO liposomes. At 72 h, the two systems, at their respective LD50 values, had similar expressions of cyclin D1 (Fig. 7), suggesting an equivalent capacity to either inactivate transcription factors that drive cell proliferation (i.e., prevent cell growth) or initiate cyclin D1 destruction. Of note, minimal changes in protein expression were observed for cells exposed to PAPA/NO liposomes at NO payloads (0.75 μg mL−1) equivalent to the DETA/NO liposomes, corroborating insufficient NO delivery and low toxicity. These results suggest that both types of NO-releasing liposomes trigger the same anticancer pathways, but to different degrees depending on the exposure time and NO concentration. Slower NO release elicits a more gradual increase in cleaved PARP levels (i.e., apoptosis) and arrested cells in the cell cycle, while faster NO release promotes rapid PARP cleavage and prevention of mitosis.
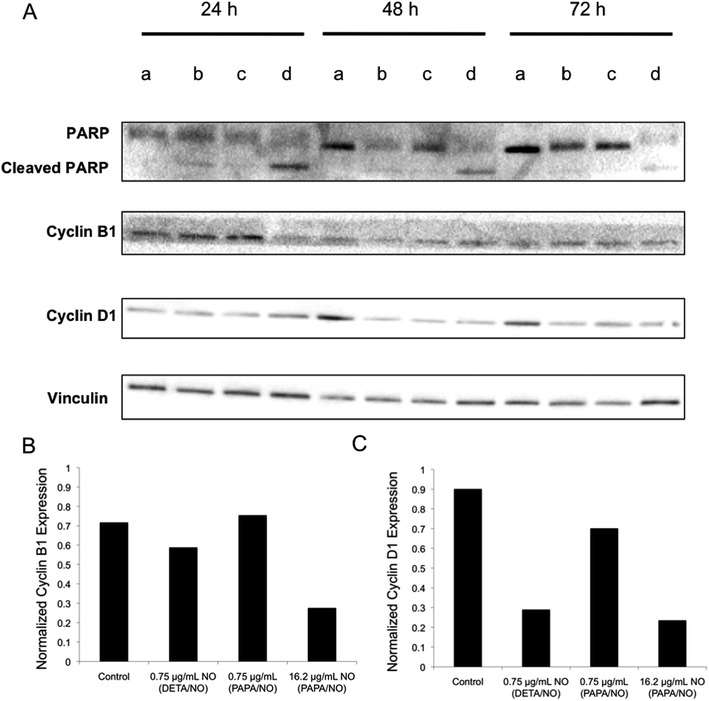 |
| | Fig. 7 (A) Western blot of MCF-7 cells after no treatment (lane a), 0.75 μg mL−1 NO from DETA/NO liposomes (lane b), 0.75 μg mL−1 NO from PAPA/NO liposomes (lane c), and 16.2 μg mL−1 NO from PAPA/NO liposomes (lane d). (B) Densitometric analysis of cyclin B1 levels after 24 h exposure. (C) Densitometric analysis of cyclin D1 levels after 72 h exposure. | |
Conclusions
The transition from low molecular weight NO donors to macromolecular NO-release systems for anticancer treatments may represent an important step in creating more effective chemotherapies. Two NO-releasing liposome systems with distinct NO-release kinetics were used to study cytotoxicity against pancreatic, colorectal, and breast cancer cell lines. Through the encapsulation of the low molecular weight NO donors within liposomes, greater intracellular NO accumulation was observed due to enhanced uptake. The preliminary cell studies herein suggest that NO-release kinetics play an important role in eliciting cell death, with a direct relationship to intracellular NO accumulation. Fast NO-releasing liposomes represent a less effective anticancer therapeutic as the NO is liberated too rapidly in advance of intracellular uptake. The ability of NO to further increase the oxidative/nitrosative stresses that cancer cells experience is a unique mechanism to enhance killing action on cancer cells over healthy cells. Future studies will evaluate the benefits of targeting ligands (e.g. folate) on directing the NO-releasing liposomes to tumors.
Conflicts of interest
The authors declare no competing financial interests.
Acknowledgements
Funding for this research was provided by the National Institutes of Health (DE025207). This work was performed in part at the Chapel Hill Analytical and Nanofabrication Laboratory (CHANL), a member of the North Carolina Research Triangle Nanotechnology Network (RTNN), which is supported by the National Science Foundation, Grant ECCS-1542015, as part of the National Nanotechnology Coordinated Infrastructure (NNCI). The UNC Flow Cytometry Core Facility is supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center. The Intellicyt iQue Screener PLUS is supported in part by the North Carolina Biotech Cancer Institutional Support Grant 2015-IDG-1001. We would like to thank Dr Pablo Ariel for his guidance in analyzing the confocal fluorescence microscopy images, Sébastien Coquery for help with the collection and processing of the flow cytometry data, and Samuel George and Nathan Whitman for their assistance in the Western blot experiments.
References
- A. Bender, J. Schieber, M. Glick, J. W. Davies, K. Azzaoui, J. Hamon, L. Urban, S. Whitebread and J. L. Jenkins, Analysis of pharmacology data and the prediction of adverse drug reactions and off-target effects from chemical structure, ChemMedChem, 2007, 2, 861–873 CrossRef CAS PubMed.
- B. K. Park, A. Boobis, S. Clarke, C. Goldring, D. Jones, J. G. Kenna, C. Lambert, H. G. Laverty, D. J. Naisbitt, S. Nelson, D. A. Nicoll-Griffith, R. S. Obach, P. Routledge, D. A. Smith, D. J. Tweedie, N. Vermeulen, D. P. Williams, I. D. Wilson and T. A. Baillie, Managing the challenge of chemically reactive metabolites in drug development, Nat. Rev. Drug Discovery, 2011, 10, 292–306 CrossRef CAS PubMed.
- C. Widakowich, G. De Castro, E. De Azambuja, P. Dinh and A. Awada, Review: Side effects of approved molecular targeted therapies in solid cancers, Oncologist, 2007, 12, 1443–1455 CrossRef CAS PubMed.
- L. Galluzzi, I. Vitale, J. Michels, C. Brenner, G. Szabadkai, A. Harei-Bellan, M. Castedo and G. Kroemer, Systems biology of cisplatin resistance: Past, present and future, Cell Death Dis., 2014, 5, 1–18 Search PubMed.
- A. De Angelis, K. Urbanek, D. Cappetta, E. Piegari, L. Ciuffreda, A. Rivellino, R. Russo, G. Esposito, F. Rossi and L. Berrino, Doxorubicin cardiotoxicity and target cells: A broader perspective, Cardio Oncol., 2016, 2, 1–8 CrossRef PubMed.
- V. P. Torchilin, Recent advances with liposomes as pharmaceutical carriers, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS PubMed.
- T. M. Allen and P. R. Cullis, Liposomal drug delivery systems: From concept to clinical applications, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed.
- Y. C. Barenholz, Doxil – The first FDA-approved nano-drug: Lessons learned, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- H. Chen, R. C. MacDonald, S. Li, N. L. Krett, S. T. Rosen and T. V. O'Halloran, Lipid encapsulation of arsenic trioxide attenuates cytotoxicity and allows for controlled anticancer drug release, J. Am. Chem. Soc., 2006, 128, 13348–13349 CrossRef CAS PubMed.
- P. P. Piccaluga, G. Visani, G. Martinelli, A. Isidori, M. Malagola, M. Rondoni, M. Baccarani and S. Tura, Liposomal daunorubicin (daunoXome) for treatment of relapsed meningeal acute myeloid leukemia, Leukemia, 2002, 16, 1880–1881 CrossRef CAS PubMed.
- E. L. Taylor, I. L. Megson, C. Haslett and A. G. Rossi, Nitric oxide: A key regulator of myeloid inflammatory cell apoptosis, Cell Death Differ., 2003, 10, 418–430 CrossRef CAS PubMed.
- D. A. Wink, H. B. Hines, R. S. Cheng, C. H. Switzer, W. Flores-Santana, M. P. Vitek, L. A. Ridnour and C. A. Colton, Nitric oxide and redox mechanisms in the immune response, J. Leukocyte Biol., 2011, 89, 873–891 CrossRef CAS PubMed.
- J. N. Sharma, A. Al-Omran and S. S. Parvathy, Role of nitric oxide in inflammatory diseases, Inflammopharmacology, 2007, 15, 252–259 CrossRef CAS PubMed.
- D. D. Thomas, L. A. Ridnour, J. S. Isenberg, W. Flores-Santana, C. H. Switzer, S. Donzelli, P. Hussain, C. Vecoli, N. Paolocci, S. Ambs, C. A. Colton, C. C. Harris, D. D. Roberts and D. A. Wink, The chemical biology of nitric oxide: Implications in cellular signaling, Free Radical Biol. Med., 2008, 45, 18–31 CrossRef CAS PubMed.
- S. Moncada, R. Palmer and E. Higgs, Nitric oxide: Physiology, pathophysiology, and pharmacology, Pharmacol. Rev., 1991, 43, 109–142 CAS.
- A. W. Carpenter and M. H. Schoenfisch, Nitric oxide release: Part II. Therapeutic applications, Chem. Soc. Rev., 2011, 41, 3742–3752 RSC.
- F. C. Fang, Perspective series: Host/pathogen interactions. Mechanisms of nitric oxide-related antimicrobial activity, J. Clin. Invest., 1997, 99, 2818–2825 CrossRef CAS PubMed.
- F. C. Fang, Antimicrobial reactive oxygen and nitrogen species: Concepts and controversies, Nat. Rev. Microbiol., 2004, 2, 820–832 CrossRef CAS PubMed.
- S. P. Jones and R. Bolli, The ubiquitous role of nitric oxide in cardioprotection, J. Mol. Cell. Cardiol., 2006, 40, 16–23 CrossRef CAS PubMed.
- V. Calabrese, C. Mancuso, M. Calvani, E. Rizzarelli, D. A. Butterfield and A. M. Stella, Nitric oxide in the central nervous system: Neuroprotection versus neurotoxicity, Nat. Rev. Neurosci., 2007, 8, 766–775 CrossRef CAS PubMed.
- D. A. Wink, Y. Vodovotz, J. Laval, F. Laval, M. W. Dewhirst and J. B. Mitchell, The multifaceted roles of nitric oxide in cancer, Carcinogenesis, 1998, 19, 711–721 CrossRef CAS PubMed.
- L. Wang and K. Xie, Nitric oxide and pancreatic cancer pathogenesis, prevention, and treatment, Curr. Pharm. Des., 2010, 16, 421–427 CrossRef CAS PubMed.
- R. Sullivan and C. H. Graham, Chemosensitization of cancer by nitric oxide, Curr. Pharm. Des., 2008, 14, 1113–1123 CrossRef CAS PubMed.
- M. R. Miller and I. L. Megson, Recent developments in nitric oxide donor drugs, Br. J. Pharmacol., 2007, 151, 305–321 CrossRef CAS PubMed.
- J. A. Hrabie, J. R. Klose, D. A. Wink and L. K. Keefer, New nitric oxide-releasing zwitterions derived from polyamines, J. Org. Chem., 1993, 58, 1472–1476 CrossRef CAS.
- L. K. Keefer, Fifty years of diazeniumdiolate research. From laboratory curiosity to broad-spectrum biomedical advances, ACS Chem. Biol., 2011, 6, 1147–1155 CrossRef CAS PubMed.
- U. Shamim, S. Hanif, A. Albanyan, F. Beck, B. Bao, Z. Wang, S. Banerjee, F. H. Sarkar, R. M. Mohammad, S. M. Hadi and A. S. Azmi, Resveratrol-induced apoptosis is enhanced in low pH environments associated with cancer, J. Cell. Physiol., 2012, 227, 1493–1500 CrossRef CAS PubMed.
- I. F. Tannock and D. Rotin, Acid pH in tumors and its potential for therapeutic exploitation, Cancer Res., 1989, 49, 4373–4384 CAS.
- C. M. Maragos, J. M. Wang, J. A. Hrabie, J. J. Oppenheim and L. K. Keefer, Nitric oxide/nucleophile complexes inhibit the in vitro proliferation of A375 melanoma cells via nitric oxide release, Cancer Res., 1993, 53, 564–568 CAS.
- D. J. Stuehr and C. F. Nathan, A macrophage product responsible for cytostasis and respiratory inhibition of tumor target cells, J. Exp. Med., 1989, 169, 1543–1555 CrossRef CAS PubMed.
- S. Tamir, R. S. Lewis, T. Walker, W. M. Deen, J. S. Wishnok and S. R. Tannenbaum, The influence of delivery rate on the chemistry and biological effects of nitric oxide, Chem. Res. Toxicol., 1993, 6, 895–899 CrossRef CAS PubMed.
- D. L. Mooradian, T. C. Hutsell and L. K. Keefer, Nitric oxide (NO) donor molecules: Effect of NO release rate on vascular smooth muscle cell proliferation in vitro, J. Cardiovasc. Pharmacol., 1995, 25, 674–678 CrossRef CAS PubMed.
- M. Kielbik, M. Klink, M. Brzezinska, I. Szulc and Z. Sulowska, Nitric oxide donors: Spermine/NO and diethylenetriamine induce ovarian cancer cell death and affect STAT3 and AKT signaling proteins, Nitric Oxide, 2013, 35, 93–109 CrossRef CAS PubMed.
- E. L. Taylor, I. L. Megson, C. Haslett and A. G. Rossi, Dissociation of DNA fragmentation from other hallmarks of apoptosis in nitric oxide-treated
neutrophils: Differences between individual nitric oxide donor drugs, Biochem. Biophys. Res. Commun., 2001, 289, 1229–1236 CrossRef CAS PubMed.
- U. K. Meßmer and B. Brüne, Nitric oxide (NO) in apoptotic versus necrotic RAW 264.7 macrophage cell death: The role of NO-donor exposure, NAD+ content, and p53 accumulation, Arch. Biochem. Biophys., 1996, 327, 1–10 CrossRef PubMed.
- D. J. Suchyta and M. H. Schoenfisch, Encapsulation of N-diazeniumdiolates within liposomes for enhanced nitric oxide donor stability and delivery, Mol. Pharm., 2015, 12, 3569–3574 CrossRef CAS PubMed.
- F. Szoka Jr and D. Papahadjopoulos, Procedure for preparation of liposomes with large internal aqueous space and high capture by reverse-phase evaporation, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 4194–4198 CrossRef CAS.
- R. J. Soto, L. Yang and M. H. Schoenfisch, Functionalized mesoporous silica via an aminosilane surfactant ion exchange reaction: Controlled scaffold design and nitric oxide release, ACS Appl. Mater. Interfaces, 2016, 8, 2220–2231 CAS.
- B. V. Worley, K. M. Schilly and M. H. Schoenfisch, Anti-biofilm efficacy of dual-action nitric oxide-releasing alkyl chain modified poly(amidoamine) dendrimers, Mol. Pharm., 2015, 12, 1573–1583 CrossRef CAS PubMed.
- S. Ganguly, S. Bandyopadhyay, A. Sarkar and M. Chatterjee, Development of a semi-automated colorimetric assay for screening of anti-leishmanial agents, J. Microbiol. Methods, 2006, 66, 79–86 CrossRef CAS PubMed.
- M. A. Elnaggar, R. Subbiah, D. K. Han and Y. K. Joung, Lipid-based carriers for controlled delivery of nitric oxide, Expert Opin. Drug Delivery, 2017, 14, 1341–1353 CrossRef CAS PubMed.
- S. Ong, L. C. Ming, K. S. Lee and K. H. Yuen, Influence of the encapsulation efficiency and size of liposomes on the oral bioavailability of griseofulvin-loaded liposomes, Pharmaceutics, 2016, 8, 25 CrossRef PubMed.
- A. Villalobo, Nitric oxide and cell proliferation, FEBS J., 2006, 273, 2329–2344 CrossRef CAS PubMed.
- H. Pelicano, D. Carney and P. Huang, ROS stress in cancer cells and therapeutic implications, Drug Resist. Updates, 2004, 7, 91–110 CrossRef PubMed.
- P. J. Shami, D. L. Sauls and J. B. Weinberg, Schedule and concentration-dependent induction of apoptosis in leukemia cells by nitric oxide, Leukemia, 1998, 12, 1461–1466 CrossRef CAS PubMed.
- J. Sheng, D. Wang and A. P. Braun, DAF-FM (4-amino-5-methylamino-2′,7′-difluorescein) diacetate detects impairment of agonist-stimulated nitric oxide synthesis by elevated glucose in human vascular endothelial cells: Reversal by vitamin C and L-sepiapterin, J. Pharmacol. Exp. Ther., 2005, 315, 931–940 CrossRef CAS PubMed.
- N. Skalko, R. Peschka, U. Altenschmidt, A. Lung and R. Schubert, pH-sensitive liposomes for receptor-mediated delivery to chicken hepatoma (LMH) cells, FEBS Lett., 1998, 434, 351–356 CrossRef CAS PubMed.
- E. Mastrobattista, G. Storm, L. van Bloois, R. Reszka, P. Bloemen, D. Crommelin and P. Henricks, Cellular uptake of liposomes targeted to intercellular adhesion molecule-1 (ICAM-1) on bronchial epithelial cells, Biochim. Biophys. Acta, 1999, 1419, 353–363 CrossRef CAS.
- G. Thurston, J. W. McLean, M. Rizen, P. Baluk, A. Haskell, T. J. Murphy, D. Hanahan and D. M. McDonald, Cationic liposomes target angiogenic endothelial cells in tumors and chronic inflammation in mice, J. Clin. Invest., 1998, 101, 1401–1413 CrossRef CAS PubMed.
- D. Cosco, A. Bulotta, M. Ventura, C. Celia, T. Calimeri, G. Perri, D. Paolino, N. Costa, P. Veri, P. Tagliaferri, P. Tassonem and M. Fresta, In vivo activity of gemcitabine-loaded PEGylated small unilamellar liposomes against pancreatic cancer, Cancer Chemother. Pharmacol., 2009, 64, 1009–1020 CrossRef CAS PubMed.
- M. Javle and N. J. Curtin, The role of PARP in DNA repair and its therapeutic exploitation, Br. J. Cancer, 2011, 105, 1114–1122 CrossRef CAS PubMed.
- A. Hwang, A. Maity, W. G. McKenna and R. J. Muschel, Cell cycle-dependent regulation of the cyclin B1 promoter, J. Biol. Chem., 1995, 270, 28419–28424 CrossRef CAS PubMed.
- D. W. Stacey, Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells, Curr. Opin. Cell Biol., 2003, 15, 158–163 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09899e |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*