
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-component Diels–Alder based polyurethanes: a unique way to self-heal†
B. Willocq a,
F. Khelifaa,
J. Brancartb,
G. Van Asscheb,
Ph. Duboisac and
J.-M. Raquez*a
a,
F. Khelifaa,
J. Brancartb,
G. Van Asscheb,
Ph. Duboisac and
J.-M. Raquez*a
aLaboratory of Polymeric and Composite Materials (LPCM), Center of Innovation and Research in Materials and Polymers (CIRMAP), University of Mons, Place du Parc 23, B-7000 Mons, Belgium. E-mail: jeanmarie.raquez@umons.ac.be
bPhysical Chemistry and Polymer Science (FYSC), Vrije Universiteit Brussel, Pleinlaan 2, Brussels, Belgium
cDepartment of Materials Research and Technology, National Composite Center-Luxembourg, Luxembourg Institute of Science and Technology, 41 rue du Brill, Belvaux, Luxembourg
First published on 12th October 2017
Abstract
In this work, we introduce single-component self-healing polyurethanes based on reversible maleimide/furan Diels–Alder reactions with good physical integrity on healing. Diols bearing thermo-reversible furfuryl and maleimide moieties were synthesized by a four-step coupling strategy starting from commercially available 2,2-bis(hydroxymethyl)propionic acid (bis-MPA) and thermo-responsive mono-alcohol (with furfuryl or maleimide functions). These diols were reacted in presence of 1,6-hexamethylene diisocyanate and polypropylene glycol (PPG), in order to obtain amorphous polyurethanes with good chain mobility for the self-healing processes (Tg < RT). The cross-linking density, related to the healing process, could be readily tuned with the amount of thermo-responsive moieties as well as the PPG chain length. Swelling test and FTIR spectroscopy confirmed the formation of reversible networks. Qualitative micro-scratch test on thin films indicated tunable healing efficiencies based on the polyurethane composition as a function of the content, the chemical nature of the thermo-responsive moieties (reversible vs. irreversible), the PPG nature (hydroxyl or amino terminated) and chain length. This new structural concept is of great interest for thermally mendable, self-healing and adhesive polyurethanes.
Introduction
Polyurethanes are versatile polymers and they can be found in many applications such as coatings, adhesives, automotive and building construction.1–3 During their life-time, these polymers can suffer from mechanical or photo-thermal stimulations leading to micro-cracks, greatly affecting the performance and, over time, the reliability of the material. Recently, many efforts have been made to develop self-healing polymers, with autonomous healing capabilities to repair (micro-)cracks and regain the original material properties.4 Among the different strategies used to confer healing abilities to polyurethanes, the use of intrinsic reversible chemistry in the polymeric backbone is the most preferred one since the healing (reversibility) can be governed by ionic or supramolecular interactions5,6 or activated by stimuli such as light,7,8 pH9 or temperature changes.10–15Thermo-reversible [4 + 2] Diels–Alder reactions between maleimide and furan functions are widely implemented in intrinsic self-healing polymers due to their high efficiency without catalyst.16 Upon thermal activation, the two moieties form, at low temperature (TDA < 50 °C), a cyclo-adduct, which reversibly regenerates the initial furan and maleimide groups at higher temperatures (TrDA > 120 °C).
In general, the design of self-healing architectures is based on low molecular weight cross-linkers or complementary oligomers, enabling sufficient chain mobility for healing cracks. In recent work on furan-maleimide based reversible covalent networks for applications in soft robotics,17 it was shown that millimetre-sized cuts could be healed at 80 °C, below the degelation temperature, ensuring the pneumatic actuators retained their shape during the healing process. However, at retro-scission temperatures (T > 120 °C), the material viscosity dramatically decreases, affecting their overall structural integrity (and thus limiting their applicability).
In an alternative approach, Leibler et al. reported the concept of vitrimers, which are permanently cross-linked systems at all temperature (except for degradation temperature) but displaying network malleability thank to exchange reactions within the network.18–20 Therefore, vitrimers present unique properties such as recyclability or re-shaping, and, in contrast to conventional thermally mendable networks, prevent the dramatic drop of viscosity with temperature increase.21
Recently, Kötteritzsch et al. were the first to report on the original synthesis of a one-component self-healing terpolymer containing both furan and maleimide moieties in the same chain as well as an acrylic ter-monomer and it was shown that the chain mobility, ensuring the healing, could be tuned by the nature of the acrylic monomer (methyl, butyl or lauryl methacrylate).22,23 Such approach is of great interest when self-healing polyurethanes are considered. Due to the presence of additional urethane hydrogen bonds (N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), the material integrity may be preserved even at high temperatures, similarly to the vitrimers' behavior, while the Diels–Alder based healing processes are taking place, recovering the damaged material properties. In the literature, the use of the Diels–Alder chemistry in polyurethane has been reported for few examples11–15,24,25 but, so far, no example of a one-component polyurethane has been reported.
C), the material integrity may be preserved even at high temperatures, similarly to the vitrimers' behavior, while the Diels–Alder based healing processes are taking place, recovering the damaged material properties. In the literature, the use of the Diels–Alder chemistry in polyurethane has been reported for few examples11–15,24,25 but, so far, no example of a one-component polyurethane has been reported.
In the present work, we synthesized one-component self-healing polyurethanes based on thermo-reversible furan/maleimide Diels–Alder reactions. For this, furan and maleimide-based diol monomers were synthesized in four steps and incorporated in the polyurethane in the presence of 1,6-hexamethylene diisocyanate (HMDI) and polypropylene glycol soft segments (PPG). Maleimide functions remained protected to avoid premature cross-linking during the synthesis and then, they were deprotected to obtain the thermo-reversible networks. The cross-linking density was readily tuned by the content of thermo-reversible moieties or the length of PPG and it was possible to establish a correlation between cross-linking and healing properties. As a non-reversible reference network, an maleimide/anthracene couple was selected in this work.
Experimental section
Materials and instrumentation
All chemicals were purchased from Aldrich, Fluka, Alfa Aesar, VWR as well as Acros Organics and were used without further purification. 1H-NMR spectra were recorded on a Bruker AMX-500 (500 MHz) at 25 °C in DMSO-d6 or CDCl3. Size-exclusion chromatography analyses were carried out on an Agilent 1200 apparatus in THF (2% NEt3). Sample solutions (1 mg ml−1) were injected with a 1 ml min−1 flow rate at 35 °C, in a pre-column PL gel 10 μm (50 × 7.5 mm) and in two gradient columns PL gel 10 μm mixed-B (300 × 7.5 mm). Thermo-gravimetric analyses (TGA) were performed using TGA Q500 TA Instruments under nitrogen and with a heating rate of 20 °C min−1 from room temperature to 800 °C. Differential scanning calorimetry (DSC) measurements were carried out using DSC Q2000 TA Instruments with modulated heat/cool/heat program from −80 °C to 200 °C at 10 °C min−1. FTIR spectra were recorded on a ATR-mode Bruker Tensor 17 spectrometer from 4000 to 600 cm−1. Self-healing experiments were performed on 300 μm-thick films using an optical microscope Leica DMRXP. Cross-linked films were scratched with a fresh razor blade and heated with a controlled hot stage Mettler FP82HT (20 min at 140 °C). Swelling tests were performed on cross-linked films in DMF during 24 h at 25 °C. Dynamic mechanical analyses (DMA) were performed on DMA Q800 TA Instruments using a cyclic program and a frequency of 1 Hz (cycle conditions: ramp 2 °C min−1 from −80 to 140 °C, followed by an isotherm at 50 °C for 1 h). Tensile tests were performed using a Zwick universal tensile testing machine (speed = 10 mm min−1 and preload = 1 N)Synthesis
The synthesis of maleimide, furfuryl and anthracene diol monomers (respectively named MAL(OH)2, FUR(OH)2 and ANT(OH)2 hereafter) were inspired by the four-step synthesis strategy proposed by Tonga et al.26 All syntheses details are reported in ESI 1.† 4-(2-Hydroxy-propyl)-10-oxa-4-aza-tricyclo-dec-8-ene-3,5-dione (MAL(OH)) was synthesized based on the procedure described by Gramlich et al.27 The synthesis of the one-component polyurethanes (ESI 2†) was adapted from the work of Billiet et al.28General procedure for the synthesis of one-component polyurethane
In a conditioned flask were added MAL(OH)2, FUR(OH)2 (or ANT(OH)2), polypropylene glycol (PPG) (1 eq.) and 20 ml of anhydrous N,N-dimethylformamide (DMF). The mixture was stirred till complete dissolution of the reagents and a certain amount of hexamethylene diisocyanate (1 eq.) was added under nitrogen flow. The solution was stirred during 1 min then, 100 μl of dibutyltin dilaurate (DBTDL) were added. The reaction was carried out at 70 °C for 6 h. Then the solution was placed in a Petri-dish and the solvent was removed under vacuum (48 h at 60 °C) to obtain the final polymer (ESI 2†).Films preparation
Films for DMA, optical microscopy and adhesion test characterizations were obtained by solvent casting (Fig. 1). Typically, 1 g of protected polyurethane was dissolved in 10 ml of DMF, then the polymer solution was poured in a 7 cm diameter Petri dish and placed in a ventilated oven for 12 h at 60 °C. The films were thermally deprotected under vacuum at 130 °C for 2 h, then placed in an oven at 50 °C for 12 h to enable the film cross-linking.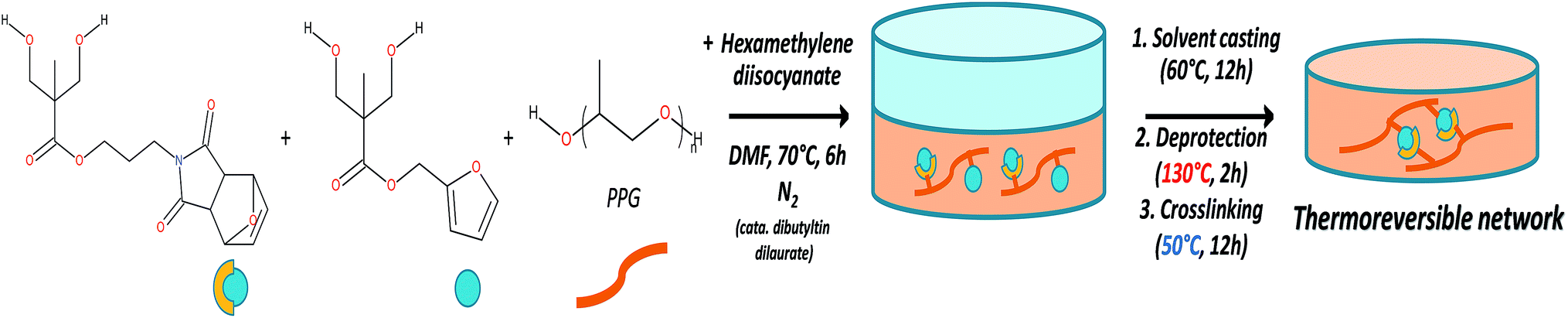 | ||
| Fig. 1 Schematic representation of the synthesis of the one-component self-healing polyurethanes. | ||
Scratch recovery quantification
A Rockwell 100 μm-diameter diamond tip apparatus was used to scratch the samples and for the evaluation of associated recoveries. Superficial surface scratches (Ps) were performed at 25 °C by applying a load of 2 N and post-scans were subsequently carried out to estimate recoveries. Surface profiles prior and after heating treatment (respectively dundamaged and dhealed) and surface profile after damage (ddamaged) were used to calculate the scratch healing efficiency η, eqn (1):| η = (dhealed − ddamaged)/(dundamaged − ddamaged) | (1) |
For each scratch a hundred of depth recoveries were calculated at different points of the scratch and an average was taken.
Results and discussion
To achieve the elaboration of the single component self-healing polyurethanes, new Diels–Alder based thermo-responsive diol monomers were synthesized. These monomers contain furfuryl, furan-protected maleimide or anthracene pendant groups. In this strategy, detailed in the ESI 1 (see Fig. S1†), the first step consists in the protection of the two hydroxyl functions of commercially available 2,2-bis(hydroxymethyl)propionic acid (bis-MPA) with acetonide group. The obtained molecule is then converted in an acetonide-protected anhydride derivative of bis-MPA in presence of dicyclohexylcarbodiimide (DCC). Esterification reaction between the anhydride and a specific alcohol (MAL(OH); furfuryl alcohol or 9-methanolanthracene) enables the addition of thermo-responsive groups in the molecules. The last step is the deprotection of the acetonide group to reform a diol bearing a thermo-responsive function. The obtained monomers MAL(OH)2, FUR(OH)2 and ANT(OH)2 were characterized by 1H-NMR spectroscopy (ESI 1†).The general strategy for the synthesis of maleimide/furan reversible one-component self-healing polyurethanes is depicted on Fig. 1. The “one-pot” polymerization step proceeded through the polyaddition reaction between furan-protected maleimide and furfuryl diols (namely MAL(OH)2 and FUR(OH)2), a polypropylene glycol soft segment (PPG) and a diisocyanate coupling agent (hexamethylene diisocyanate). Advantageously, the presence of protective groups on maleimide derivatives prevents undesirable cross-linking reactions during the polymerization process, enabling the polymer characterization by solvent-based techniques and improving the processability for film preparation (i.e. solvent casting). 1H-NMR characterization of the obtained protected polyurethanes and quantification of thermoresponsive moieties (maleimide and furan) are presented on ESI 2.† The experimental ratio furan/maleimide determined by 1H-NMR indicates that the stoichiometry between the two entities is respected. The furan-protecting group was subsequently thermally removed during the film preparation procedure (130 °C, 2 h) and films were cross-linked by forming the cyclo-adducts using a moderated temperature (50 °C, 12 h). The thermal deprotection of furan protective group was attested by TGA measurements (ESI 3†) on which a clear weight loss, corresponding to the furan removal, is observed around 137 °C on the protected polyurethane. This removal was further confirmed using FTIR spectroscopy by the appearance of maleimide characteristic bands centered at 695 and 830 cm−1 (ESI 4†). It is worth noting that the preparation of irreversible polyurethane networks was performed using the same aforementioned strategy, replacing FUR(OH)2 by ANT(OH)2 in the polymerization step. As maleimide/anthracene couple undergoes thermal reversion at 210 °C, these networks can be considered as “irreversible” in the temperature range investigated for maleimide/furan couple (i.e. 50–120 °C) and be used as a non-reversible reference network.29
The proposed strategy allows for a high modularity of the material properties, playing on parameters such as the PPG chain length, PPG end-groups (–NH2 or –OH), the content and the ratio of thermo-responsive moieties. In this work, particular attention was paid on the three compositions detailed in Table 1. For all compositions the nMAL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :nFUR and nNCO:nOH ratio was kept constant (1:1) and, in order to tune the cross-linking density and the resulting material properties, the variable parameters were either the nDA-diol:nPPG ratio (PU1 and PU2) or the PPG chain length (PU2 and PU3). The cross-link density could not be determined precisely as the solvent–polymer interaction parameter (χ) is unknown for this complex system.30 However, it was shown that the swelling degree could be decreased either by increasing the content of thermo-responsive groups (from PU1 to PU2) or by reducing the PPG soft segment chain length (from PU2 to PU3). As a result, the glass transition temperature (Tg) of the networks increased with increasing cross-linking due to restricted motions of the polymer chains.
:nFUR and nNCO:nOH ratio was kept constant (1:1) and, in order to tune the cross-linking density and the resulting material properties, the variable parameters were either the nDA-diol:nPPG ratio (PU1 and PU2) or the PPG chain length (PU2 and PU3). The cross-link density could not be determined precisely as the solvent–polymer interaction parameter (χ) is unknown for this complex system.30 However, it was shown that the swelling degree could be decreased either by increasing the content of thermo-responsive groups (from PU1 to PU2) or by reducing the PPG soft segment chain length (from PU2 to PU3). As a result, the glass transition temperature (Tg) of the networks increased with increasing cross-linking due to restricted motions of the polymer chains.
| Polymer | nDA-diol:nPPG |
Mw PPG (g mol−1) | Mn (g mol−1)a | PDIa | Degree of swelling (%)c | Tg (°C)d |
|---|---|---|---|---|---|---|
| a GPC in THF (eq. PS).b Not soluble in THF.c Swelling test in DMF (24 h), detailed in ESI 5.d DSC from −80 to 150 °C, 10 °C min−1 heating/cooling rate. | ||||||
| PU1 | 75:25 |
2000 | 9500 | 2.9 | 248 | −58.3 |
| PU2 | 90:10 |
2000 | 8700 | 3.1 | 145 | −56.6 |
| PU3 | 90:10 |
1000 | NDb | NDb | 114 | −53.3 |
The thermal reversibility, crucial point to get a healing network, was investigated (Fig. 2). The results are presented only for the intermediate network PU2, as PU1 and PU3 presented similar behavior. As seen in Fig. 2a, concentrated solutions of PU2 (0.5 g ml−1) exhibit reversible gel formation upon heating/cooling cycles. When heated (130 °C, 2 h), thermal dissociation of Diels–Alder adducts results in the network de-reticulation and solubilization in DMF. On the other hand, once the solution is kept at room temperature overnight, reversible gel formation through Diels–Alder reticulation is attested. This reversibility, repeated twice, is of great interest for the polymer recyclability and reprocessability. ATR-FTIR spectroscopy was used to follow up chemical changes during the heating/cooling cycles on PU2 network (Fig. 2b). The maleimide characteristic bands are centered at 695 cm−1 and 830 cm−1 (i.e. C–H deformation of maleimide). After a heating at 130 °C for 3 h, the maleimide peak intensities increase indicating the occurrence of the retro-Diels–Alder process. Subsequently to the cooling step at 50 °C for 12 h, the decrease of the maleimide band intensities attesting for the network reformation through Diels–Alder reactions is clearly seen. As described on Fig. 2a the process could be, at least, extended to a second heating/cooling cycle. The evolution of the storage modulus (G′) of PU2 with respect to temperature was also monitored by DMA measurements (Fig. 2c). The analysis was performed on two heating/cooling cycles (detailed on Section 2.1) and during the first heating ramp, three transitions were observed. The first transition [1] at −55 °C is related to the PPG soft segment. The second transition [2] at 60 °C represents the glassy transition of the Diels–Alder polyurethane network. Finally, the third transition [3] observed at 130 °C is attributed to the network decrosslinking by retro-Diels–Alder process. Once the material is cooled down, an important increase of G′ value indicates that recrosslinking is taking place as demonstrated by FTIR data. Note that cooling under slow cooling rate present two transitions on the DMA curves which are attributed by the recrosslinking of the Diels–Alder adduct and of the PUR network respectively (see ESI 6†). During the isotherm at 50 °C [4], a slight increase in G′ value is observed, suggesting that cross-linking is going on during the analysis. Similar behavior observed for the second heating/cooling cycle confirms the good thermal reversibility of PU2. Moreover, the sample kept a good integrity upon thermal activation as the value of G′ after analysis was similar to the initial one [5]. It is important to note that similar DMA data could be obtained for PU1 and PU3 (ESI 7†).
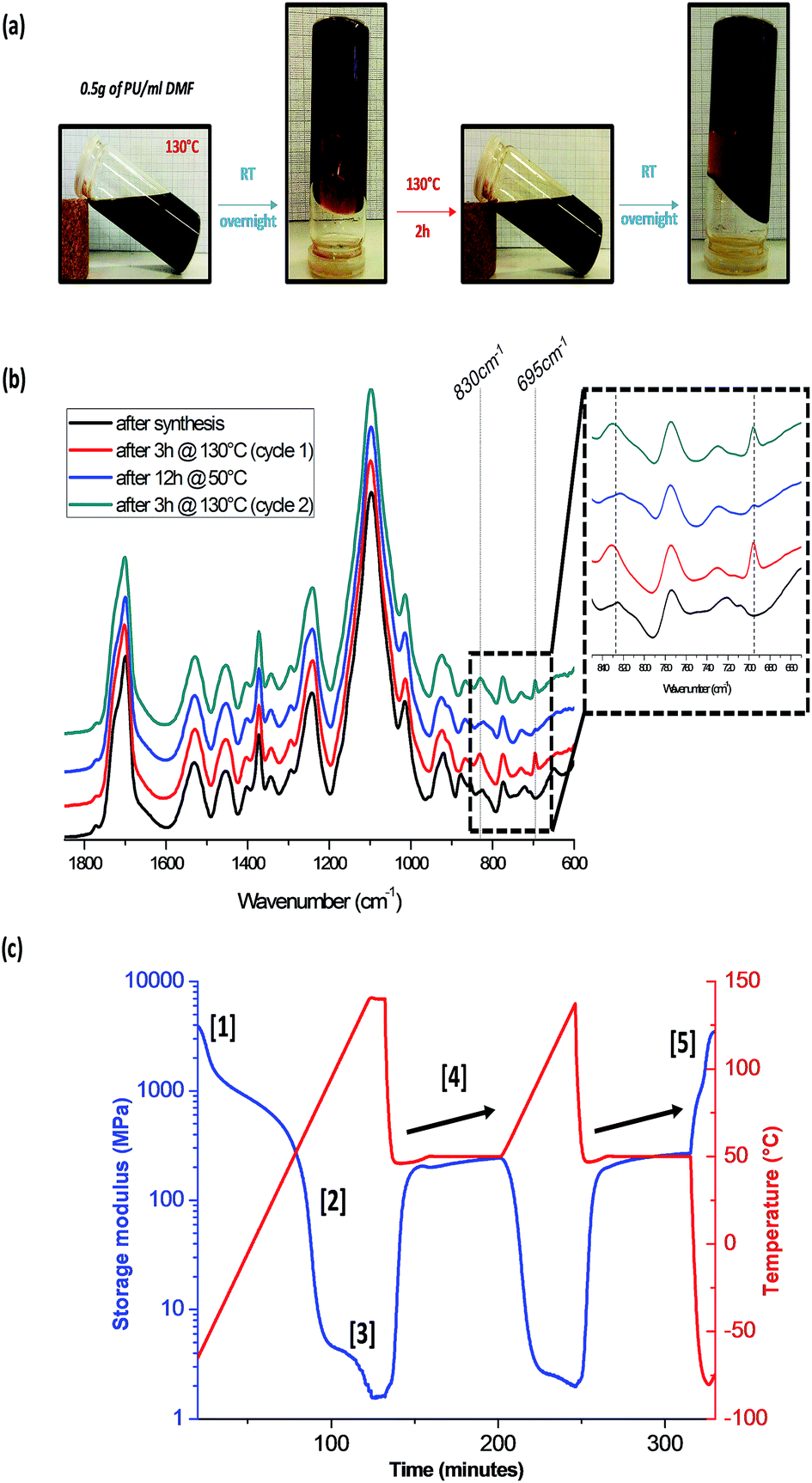 | ||
| Fig. 2 Thermal-reversibility of PU2. Reversible gel formation in DMF from a polymer solution (a), FTIR spectrum (b) and DMA curves of PU2 (c) after two heating/cooling cycles. | ||
For PU1, the modulus (G′) after two heating/cooling cycles was lower than the initial one due to the lower amount of Diels–Alder moieties inducing a lack of accessibility of these moieties limiting in turn their recombination and the recovery of material integrity. On the other hand, PU3 exhibited excellent recovery of G′ after the two heating/cooling cycles. Also, the increase in G′ during the 50 °C isotherm is more pronounced for PU3 since the concentration of Diels–Alder moieties is the highest. In summary, G′ at 25 °C during the first cycle for PU1, PU2 and PU3 (30, 80 and 110 MPa respectively) increases since the network becomes more crosslinked when passing from PU1 to PU2 to PU3 (see Table 1).
Then, in order to evaluate the healing performances of the films, scratch recovery tests were performed after heating treatments and monitored by optical microscopy (Fig. 3). For this, 300 μm-thick films (see Section 2.4) were scratched superficially using a fresh razor blade and heated during 20 min at 140 °C using an in situ heating stage coupled with the optical microscope. Among the three different PU compositions, only PU2 exhibited excellent scratch recovery capabilities while PU1 and PU3 remained unhealed after thermal-treatment. This different healing response is attributed to the content in thermo-responsive moieties in the obtained networks as well as, the elasticity of the material. The three polyurethane compositions behave as doubly cross-linked networks: physically (hydrogen bonds) and covalently (Diels–Alder) cross-linked. Upon heating at 130 °C, the proportion of free and hydrogen-bonded urethanes (respectively assigned as the FTIR bands at 1710 and 1680 cm−1, Fig. 2 for PU2)31 participating to the physical network does not dramatically change, indicating that the physical network is preserved for the three compositions. The content of thermoresponsive moieties (quantified on ESI 2†) is a key parameter for the material to self-heal. For PU1 the thermal response is most likely dominated by the elastic response of the polyurethane and not by the dissociation of Diels–Alder moieties present in low amount in the material (Fig. 3 a and b). In contrast, PU3 has poor healing capabilities, which is most probably due to the restricted motions of polymers chains in the highly crosslinked network (Fig. 3 e and f). PU2 seems to represent a good comprise between those two extreme compositions and, for this particular composition, full scratch recovery was observed after the heating treatment. To quantify further the scratch healing of PU2, diamond-tip profilometry was performed and the healing was defined as the ratio (dhealed − ddamaged)/(dundamaged − ddamaged), where dundamaged, dhealed and ddamaged represent respectively the surface profile before being scratched, after scratch healing and the scratch surface profile (see Section 2.5). Hence, the recovery capability of PU2 for a 50 μm-depth scratch was evaluated at (102.2 ± 0.6)%.
 | ||
| Fig. 3 Healing performances of the thermo-reversible one-component polyurethanes. Scratch recovery test prior and after 20 min at 140 °C for PU1 (a, b), PU2 (c, d) and PU3 (e, f). Quantification of the scratch recovery for PU2 by diamond-tip profilometry (g). | ||
In addition to the excellent scratch healing capability of PU2, interesting for coating applications, the concept was extended to free-standing films. Recovery of mechanical properties prior and after healing was investigated by tensile test. Samples were cut into two pieces, reconnected and thermally treated (30 min at 130 °C and 24 h at 50 °C). The breaking strain and tensile stress recoveries were respectively of 40 and 68% after healing process (see ESI 8†). These efficiencies of mechanical recovery may be related to the lack of complete accessibilities and incomplete reconnection of furan and maleimide moieties at the repaired interface but it is important to note that the ability to recover up to 17 MPa tensile strength is greater compared to the majority of reported polymers.32 In addition of mechanical testing, adhesion tests on both irreversible and reversible PU2 cross-linked films were performed (Fig. 4). In practice, two rectangular-shaped films were placed under a slight pressure (1 cm2 contact between the two films) and submitted to a thermal treatment (130 °C for 30 min and 50 °C for 12 h). For irreversible films (i.e. maleimide/anthracene), no decrosslinking and network reshuffling are expected in the aforementioned thermal conditions as maleimide/anthracene reversibility occurs at 210 °C (Fig. 4a). The absence of chemical decrosslinking, confirmed by FTIR spectroscopy (ESI 9†), restricts the polymer chain motion and, as a consequence, the two network films did not present adhesion after heating treatment. It is worth noting that this absence of mobility also greatly reduced the scratch recovery of the irreversible network (ESI 10†). At contrast, for the reversible PU2 (i.e. maleimide/furan), when heated-up above retro-Diels–Alder temperature (T > 130 °C for 30 min) and subsequently cooled down to 50 °C for 12 h, a network gets formed through the chemical association of interfacial Diels–Alder moieties reinforcing mechanically the newly formed film. After the heating process, the two pieces of films presented a good adhesion and the bilayer could be submitted to a load of 250 g (up to 2500 times of the bilayer weight and which represents a shear stress of approximately 2.5 × 104 Pa [(250 g × 10 N/1000 g)/1 cm2]). This adhesion test demonstrates the potential applicability of the single-component self-healing and thermally adhesive polyurethane for free-standing healable materials.
 | ||
| Fig. 4 Adhesion recovery of PU2. Schematic representation of adhesion test for irreversible (a) and reversible (b) networks and digital images of films submitted to a 250 g load. | ||
Conclusions
In conclusion, the concept of single-component self-healing polymers introduced in a previous study23 was extended to polyurethanes by synthesizing new thermo-responsive diols (with furfuryl, maleimide and anthracene functions). It was shown that the composition and degree of crosslinking have to be finely tuned to achieve scratch recovery properties upon heating treatment. The obtained polyurethanes were thermally reprocessable and presented excellent healing capabilities (PU2). Furthermore, the possibility to get free-standing healable films was shown. The strategy developed in the present work is of great interest for the fine tuning of healing properties of polyurethanes. As an example, hydroxyl-terminated polypropylene glycol (PPG(OH)2 2000 g mol−1) was replaced by amino-terminated polypropylene glycol (PPG(NH2)2 2000 g mol−1) in PU2, introducing additional hydrogen bonds through formation of urea functions, while keeping excellent scratch healing properties (ESI 11†). Finally, the synthesis of new thermo-responsive diols opens a way to the preparation of other single component self-healing polymers based on polyester or polycarbonate matrices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research in Mons is supported by the Science Policy Office of the Belgian Federal Government (PAI 7/5), the European Commission/Walloon Region (FEDER – BIORGEL RF project – Valicell), and by the Belgian National Fund for Scientific Research (FNRS). B. W. and J. M. R. are FRS - FNRS research fellows. The authors warmly acknowledge Dr Ranjita K. Bose and Dr Santiago J. Garcia from Novel Aerospace Materials Group (TU Delft, the Netherland) for providing scratch recovery test data.Notes and references
- G. Oertel and L. Abele, Polyurethane Handbook: Chemistry, Raw Materials, Processing, Application, Properties, Hanser, 1994 Search PubMed.
- Z. S. Petrović and J. Ferguson, Prog. Polym. Sci., 1991, 16, 695–836 CrossRef.
- C. Hepburn, Polyurethane Elastomers, Springer, Netherlands, 1992 Search PubMed.
- D. Y. Wu, S. Meure and D. Solomon, Prog. Polym. Sci., 2008, 33, 479–522 CrossRef CAS.
- Y. J. Kim, P. H. Huh and B. K. Kim, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 468–474 CrossRef CAS.
- S. Chen, F. Mo, Y. Yang, F. J. Stadler, S. Chen, H. Yang and Z. Ge, J. Mater. Chem. A, 2015, 3, 2924–2933 CAS.
- J. Ling, M. Z. Rong and M. Q. Zhang, Polymer, 2012, 53, 2691–2698 CrossRef CAS.
- B. Ghosh and M. W. Urban, Science, 2009, 323, 1458–1460 CrossRef CAS PubMed.
- V. Yesilyurt, M. J. Webber, E. A. Appel, C. Godwin, R. Langer and D. G. Anderson, Adv. Mater., 2016, 28, 86–91 CrossRef CAS PubMed.
- C. e. Yuan, M. Z. Rong and M. Q. Zhang, Polymer, 2014, 55, 1782–1791 CrossRef CAS.
- Y. Heo and H. A. Sodano, Adv. Funct. Mater., 2014, 24, 5261–5268 CrossRef CAS.
- P. Du, X. Liu, Z. Zheng, X. Wang, T. Joncheray and Y. Zhang, RSC Adv., 2013, 3, 15475–15482 RSC.
- Y. Zhong, X. Wang, Z. Zheng and P. Du, J. Appl. Polym. Sci., 2015, 132, 41944–41953 Search PubMed.
- P. Du, M. Wu, X. Liu, Z. Zheng, X. Wang, T. Joncheray and Y. Zhang, J. Appl. Polym. Sci., 2014, 131, 40234–40241 Search PubMed.
- J. Li, G. Zhang, L. Deng, S. Zhao, Y. Gao, K. Jiang, R. Sun and C. Wong, J. Mater. Chem. A, 2014, 2, 20642–20649 CAS.
- A. Gandini, Prog. Polym. Sci., 2013, 38, 1–29 CrossRef CAS.
- S. Terryn, J. Brancart, D. Lefeber, G. Van Assche and B. Vanderborght, Science Robotics, 2017, 2, 4268–4280 CrossRef.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- R. K. Bose, J. Kötteritzsch, S. J. Garcia, M. D. Hager, U. S. Schubert and S. van der Zwaag, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1669–1675 CrossRef CAS.
- J. Kötteritzsch, S. Stumpf, S. Hoeppener, J. Vitz, M. D. Hager and U. S. Schubert, Macromol. Chem. Phys., 2013, 214, 1636–1649 CrossRef.
- S. Yu, R. Zhang, Q. Wu, T. Chen and P. Sun, Adv. Mater., 2013, 25, 4912–4917 CrossRef CAS PubMed.
- G. Rivero, L.-T. T. Nguyen, X. K. D. Hillewaere and F. E. Du Prez, Macromolecules, 2014, 47, 2010–2018 CrossRef CAS.
- M. Tonga, N. Cengiz, M. M. Kose, T. Dede and A. Sanyal, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 410–416 CrossRef CAS.
- W. M. Gramlich, M. L. Robertson and M. A. Hillmyer, Macromolecules, 2010, 43, 2313–2321 CrossRef CAS.
- S. Billiet, K. De Bruycker, F. Driessen, H. Goossens, V. Van Speybroeck, J. M. Winne and F. E. Du Prez, Nat. Chem., 2014, 6, 815–821 CrossRef CAS PubMed.
- B. Gacal, H. Durmaz, M. A. Tasdelen, G. Hizal, U. Tunca, Y. Yagci and A. L. Demirel, Macromolecules, 2006, 39, 5330–5336 CrossRef CAS.
- B. Willocq, R. K. Bose, F. Khelifa, S. J. Garcia, P. Dubois and J. M. Raquez, J. Mater. Chem. A, 2016, 4, 4089–4097 CAS.
- M. M. Coleman, D. J. Skrovanek, J. Hu and P. C. Painter, Macromolecules, 1988, 21, 59–65 CrossRef CAS.
- Y. Yang, X. Ding and M. W. Urban, Prog. Polym. Sci., 2015, 49–50, 34–59 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09898g |
| This journal is © The Royal Society of Chemistry 2017 |