
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot enzyme-catalyzed synthesis of dual-functional polyester macromers towards surface-active hydrophobic films†
Samer Nameer‡
 a,
Stefan Semlitsch‡b,
Mats Martinelleb and
Mats Johansson*a
a,
Stefan Semlitsch‡b,
Mats Martinelleb and
Mats Johansson*a
aKTH Royal Institute of Technology, Department of Fibre and Polymer Technology, Division of Coating Technology, SE-100 44 Stockholm, Sweden. E-mail: matskg@kth.se
bKTH Royal Institute of Technology, School of Biotechnology, Division of Industrial Biotechnology, SE-106 91 Stockholm, Sweden
First published on 27th October 2017
Abstract
Selective enzyme catalysis is a valuable tool for the processing of monomers into value-added materials. In the present study natural resources were used to retrieve an ω-hydroxy fatty acid monomer containing an epoxide functionality. A procedure was developed for the synthesis of dual-functional oligomers by utilizing lipase catalysis in a one-pot synthesis route. The chemoselectivity of the enzyme allowed addition of thiol monomers to the retrieved epoxy monomers, without harming the epoxides, achieving a thiol–epoxy functional polyester resin. The synthesis reached full conversion (>99%) after 8 h. It was possible to selectively crosslink the resin through UV-initiated cationic polymerization of the epoxides into thiol–functional thermosets. The curing performance was followed in situ by real-time FTIR. The thiol groups on the surface of the film were accessible for post-modification.
Introduction
The development of the bio-refinery concept has started to introduce totally new building blocks for future advanced materials.1,2 This also implies that new chemistries and technologies need to be further developed in order to utilize these sources optimally. Nature provides monomers containing wide variety of functional groups ranging from, hydroxyls, carboxylic acids, amines, epoxides, thiols etc.2 Combining these would allow totally new multifunctional structures to be made. These multifunctional structures have a great potential in applications where the surface properties needs to be tailored such as wetting,3 adhesion,4 post-functionalization,5,6 adhesion promoters (primers),7 microfluidics8 and biosensors9 just to mention a few.One interesting renewable source of building blocks is epoxidized fatty acids either from vegetable oils10 or other sources such as suberin and cutin.11 The physical properties of thermosets based on these building blocks can be changed by connecting the epoxidized fatty acids to a more rigid core than in the naturally occurring triglycerides12,13 or by reducing the dangling-ends inside the network.14
One specific fatty acid structure in the context of multifunctional building blocks is 9,10-epoxy-18-hydroxyoctadecanoic acid (EFA) extracted from outer birch bark. EFA stands for about 10 wt% of the total dried outer birch bark.15,16 The molecule contains three functional groups hence; making it prone to many types of reactions.
Thermoset resins based on epoxide monomers have been extensively studied in the past. The epoxide group is considered versatile due to the fact that it is easily reacted with various functional groups.4 Epoxides can react in both chain-wise and stepwise polymerization techniques i.e. via cationic polymerization17–20 or amine coupling reactions21,22 respectively. The advantage of cationic polymerization is that it is considered as oxygen insensitive and has rapid curing rates. In addition the polymerization of epoxides proceeds for a long period of time due to the effect of post-polymerization i.e. propagating species proceeds after e.g. UV irradiation.17,19
Another type of chemistry that has recently received significant interest as a tool to enable the use of bio-derived monomers is the so-called thiol-ene reaction.23 This has been combined with fatty acids to make polyols24 and enabled the use of terpenes.25 Conventional thiol-ene chemistry is used for different applications such as surface modification,6 biomedical applications,26 microfluidics,8 and optics and electronics.23 The thiol-ene reaction proceeds via a free-radical step-growth process and is considered fast and insensitive to oxygen.23,27
Introducing thiol end-groups in resins enable post-functionalization reactions. However, such an introduction could be a tedious process when using conventional polymer synthesis because the thiol group requires protection and deprotection chemistry.28–30 In order to overcome this issue one can use biocatalysis e.g. enzymes.
Enzymes are considered environmentally friendly, selective, effective and operate under mild conditions.31 One of the most extensively used enzymes in polymer chemistry is Novozyme 435 (immobilized Candida antarctica lipase B (CALB)). CALB is considered versatile and robust due to its ability to catalyze acyl transfer reactions using a large number of different substrates and tolerate a variation of experimental conditions.32–34 It has previously been shown that end-capped thiol–functional resins can be synthesized by CALB.35–37 Moreover, CALB is more chemo-selective towards alcohols than thiols.38 Furthermore, CALB has been utilized to catalyze reactions with EFA effectively while preserving the mid-chain epoxy group39 and to add additional functionalities while forming the oligoesters.40–42 These additional functionalities play an important role in the final properties of the materials depending on the crosslinking method used. Biocatalysis broadens the spectrum of catalysts in polymer chemistry. Moreover, it is possible to obtain new bio-based building blocks by biocatalysis otherwise hard to obtain by conventional polymer chemistry.
In this paper we describe an enzymatic route for the synthesis of a thiol–epoxy-functional resin starting from renewable monomers. These resins are then cationically polymerized to lead to films with a thiol-functionality available for post-functionalization.
Experimental
Materials
The fatty acids were retrieved from natural resources; 9,10-epoxy-18-hydroxyoctadecanoic acid (EFA, 1) from birch bark provided by Holmen AB (c/o Holmen Energi, SE-89180) and epoxidized methyl oleate (EMO) from epoxidized linseed oil provided by Ackross Chemicals. 11-Mercaptoundecanol (98%) (2), dimethyl adipate (≥99%) (3), Novozyme 435 (immobilized CALB), 5,5′-dithiobis-(2-nitrobenzoic acid) (≥98%, Bioreagent) (DTNB), phosphate buffer pH 7.0 (1 M, 20 °C), and molecular sieves 4 Å (8–12 mesh) were purchased from Sigma Aldrich. The photoinitiators UVAcure 1600 and benzophenone (≥99%) were supplied by CYTEC and Fluka respectively. 1,6-Hexanediol mono vinyl ether (HMVE) was supplied by BASF. All materials were used as received, unless otherwise noted.Analytical methods
Procedures
EMO was retrieved by methanolysis at 60 °C from epoxidized linseed oil. The obtained methyl esters were first extracted in n-heptane/deionized water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) 4 times and then purified by column chromatography in n-heptane/EtOAc gradient solution. 1H NMR (400 MHz, CDCl3, δ): 3.66 (3H, s, –O–CH3), 2.89 (2H, bs, –CH–O–CH–epoxide), 2.30 (2H, t, –CH2–CO–, J = 7.5 Hz), 1.62 (2H, p, –CH2–CH2–CH2–CO–, J = 7.1 Hz), 1.54–1.20 (26H, m, –CH2–aliphatic), 0.90 (3H, t, CH3–, J = 6.7 Hz).
:1) 4 times and then purified by column chromatography in n-heptane/EtOAc gradient solution. 1H NMR (400 MHz, CDCl3, δ): 3.66 (3H, s, –O–CH3), 2.89 (2H, bs, –CH–O–CH–epoxide), 2.30 (2H, t, –CH2–CO–, J = 7.5 Hz), 1.62 (2H, p, –CH2–CH2–CH2–CO–, J = 7.1 Hz), 1.54–1.20 (26H, m, –CH2–aliphatic), 0.90 (3H, t, CH3–, J = 6.7 Hz).
The other methyl esters after methanolysis were methyl stearate, epoxidized methyl linoleate and epoxidized methyl linolenate. These fatty acids were not used in this study.
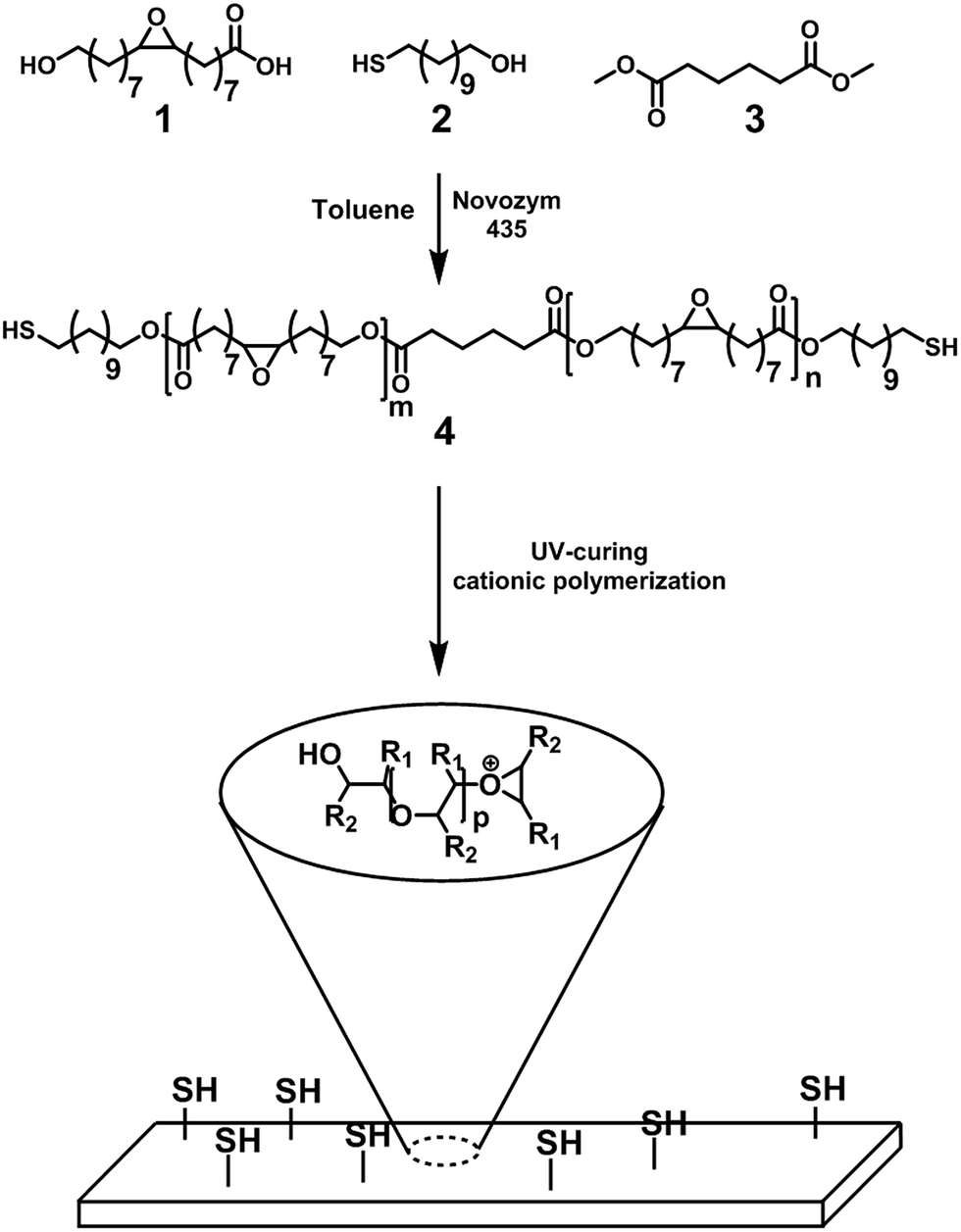 | ||
| Scheme 1 Schematic overview of the synthesis of the epoxy–thiol dual-functional oligomer (4) based on EFA (1). | ||
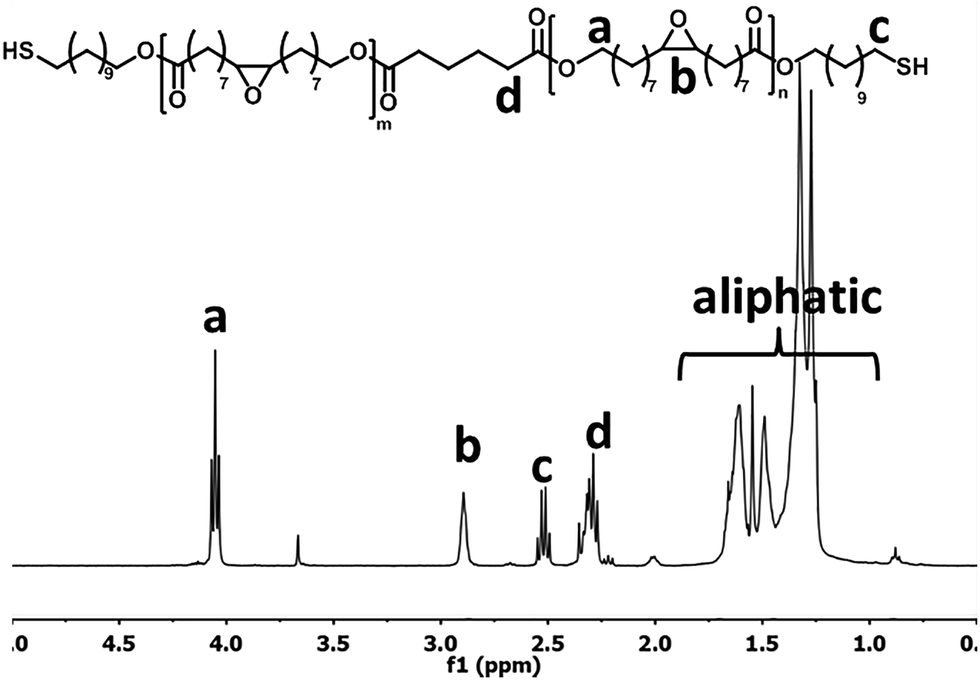 | ||
| Fig. 1 1H NMR of the synthesised EFA based oligomer (4). | ||
Results and discussion
Monomers retrieved from natural resources
Natural resources were used in order to isolate the fatty acid EFA. Birch bark was subjected to alkali hydrolysis at 100 °C. The alkali hydrolysis breaks the ester bonds in the suberin structure and a mixture of different free fatty acids was obtained. The solution was then acidified to pH 5.7 to protonate the carboxylic group of EFA. Subsequently a precipitate occurred. However, it is not recommended to acidify below pH 5.5 since the epoxide could ring-open and form 9,10,18-trihydroxyoctadecanoic acid.44 In a last step EFA was recrystallized from toluene. According to literature outer birch bark suberin structure contains 99 mg EFA per g dry outer birch bark.45 Comparing with the yield from literature our yield was 60–70%. The EFA monomer was confirmed by 1H NMR and FTIR (ESI Fig. 1†).Synthesis of the multifunctional oligomer based on EFA
The purified EFA was used for an enzyme-catalyzed polymerization utilizing CALB as the catalyst and 11-mercaptoundecanol acid as end-capper. The one-pot enzymatic catalysis occurred for 8 h and reached conversions of >99% for the OH-groups in EFA and the 11-mercaptoundecanol. NMR analysis confirmed the structure of the product (Fig. 1). Furthermore, NMR showed that the DP was 2.5 and the Mw was 1189.85 g mol−1. The functional groups were unaffected during the synthesis (peaks b and c).The MALDI-TOF-MS spectrum shows the main component with end-cappers on both sides, while showing a minor amount of the oligomer with end-capping only on one side (Fig. 2). This is also confirmed by 1H NMR as there is a CH3-peak at about 3.6 ppm (Fig. 1). The mass difference between the major peaks of the MALDI-TOF-MS spectrum is 296 Da which corresponds to an EFA-repeating-unit. A reaction in bulk at 85 °C resulted in an insoluble network. At this temperature the combination of a carboxylic acid, a thiol and epoxide groups led to reactions creating a network.
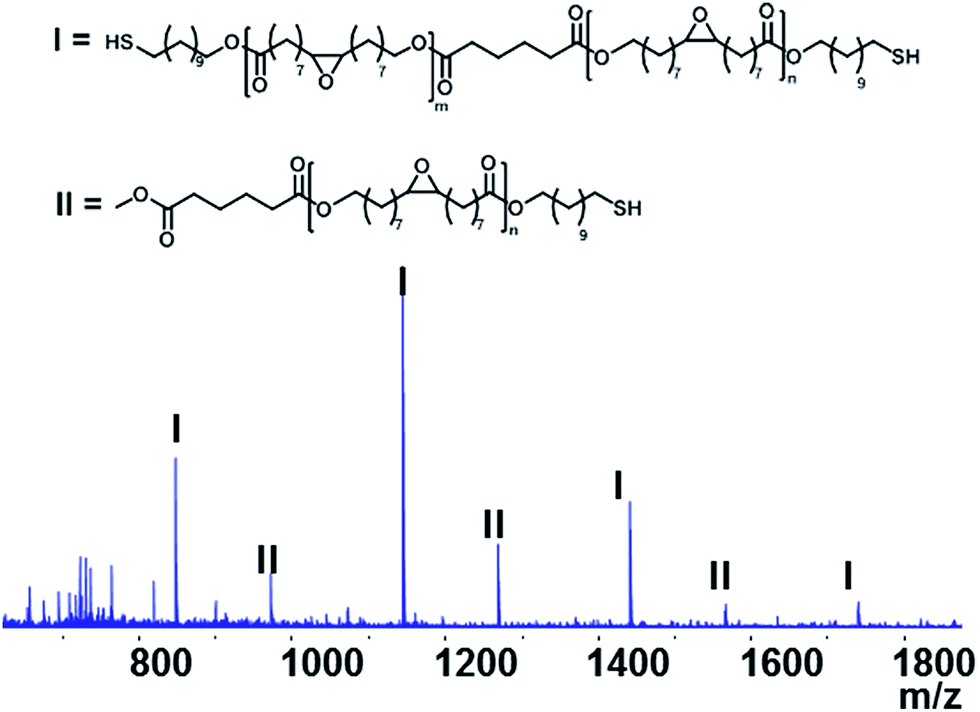 | ||
| Fig. 2 MALDI-TOF-MS of the synthesized EFA based oligomer (4). | ||
Network formation and polymerization performance of the oligomers
The synthesized oligomer contained two functional groups, epoxides and thiols as confirmed by 1H NMR and MALDI (Fig. 1 and 2). In order to preserve the thiol groups, cationic polymerization initiated by UVAcure 1600 was chosen to selectively propagate the epoxides as indicated by model experiment A. The polymerization performance was followed by RT-FTIR (Fig. 3). The epoxide functional group at 827 cm−1 disappeared over time while the peak at 1073 cm−1 increased representing the formation of ether bonds. Furthermore, the result indicated that it was a moderately fast reaction. The epoxide was fully converted after approximately 10 min of UV-light exposure. A transparent film was obtained after the reaction. The thiol group has low absorbance in FTIR mode hence, FT Raman analysis was made in order to confirm the thiol groups. Fig. 4 shows FT Raman spectra of before and after UV-curing. The figure shows that the thiol groups at 2570 cm−1 were still intact after UV-curing. Furthermore, the FT Raman analysis of the oligomer further confirms the 1H NMR results, both analysis methods shows that the thiol groups were still intact after enzyme catalysis. However, FT Raman analysis provided information about the bulk properties of the film. In order to distinguish if there were any thiol groups on the surface of the film, Ellman's reagent was used. During such a test Ellman's reagent (DTNB) reacts with aliphatic thiol compounds and a highly yellow colored anion (p-nitrophenol anion) is produced.46 During this study DTNB was dissolved in an aqueous phosphate buffer solution (pH 7) and was used to confirm any presence of thiol groups. Crosslinked films were put in a vial with 1 mL DTNB solution. After approximately 1 min the color of the solution changed to yellow. Since the bulk of the film is hydrophobic due to ether bonds formed from the epoxide reaction, it was hypothesized that the reaction of the DTNB with thiol producing the highly yellow colored p-nitrophenol occurred on the surface of the film.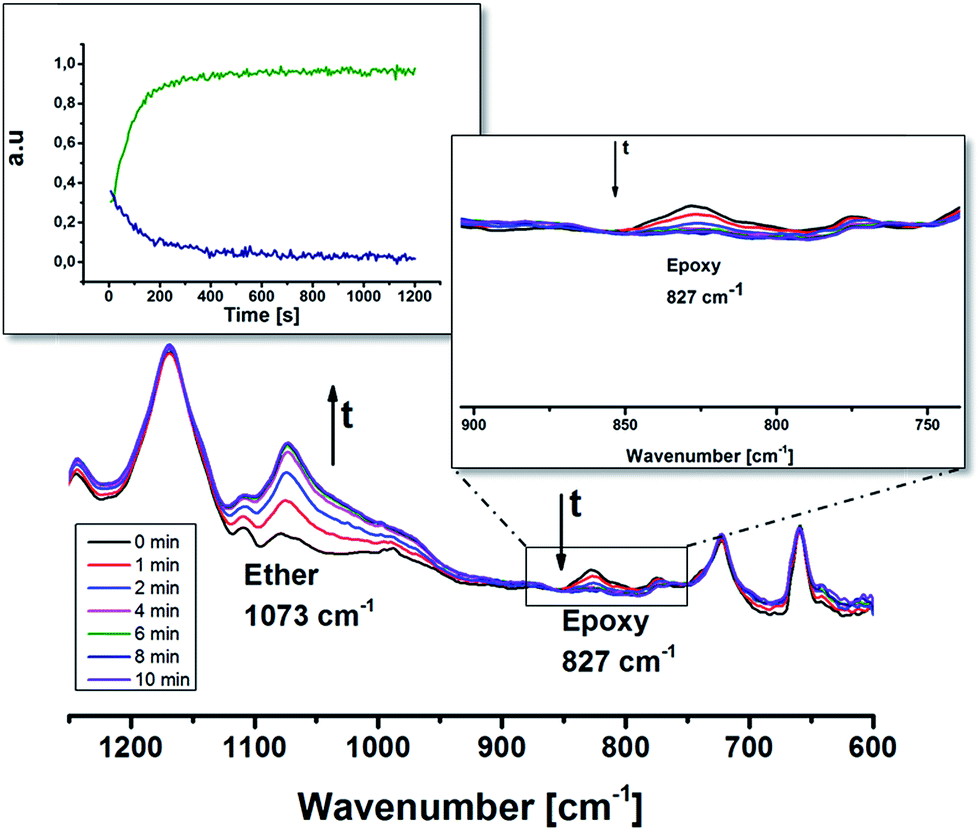 | ||
| Fig. 3 UV curing of EFA-DMA-SH (4) followed by RT-FTIR. | ||
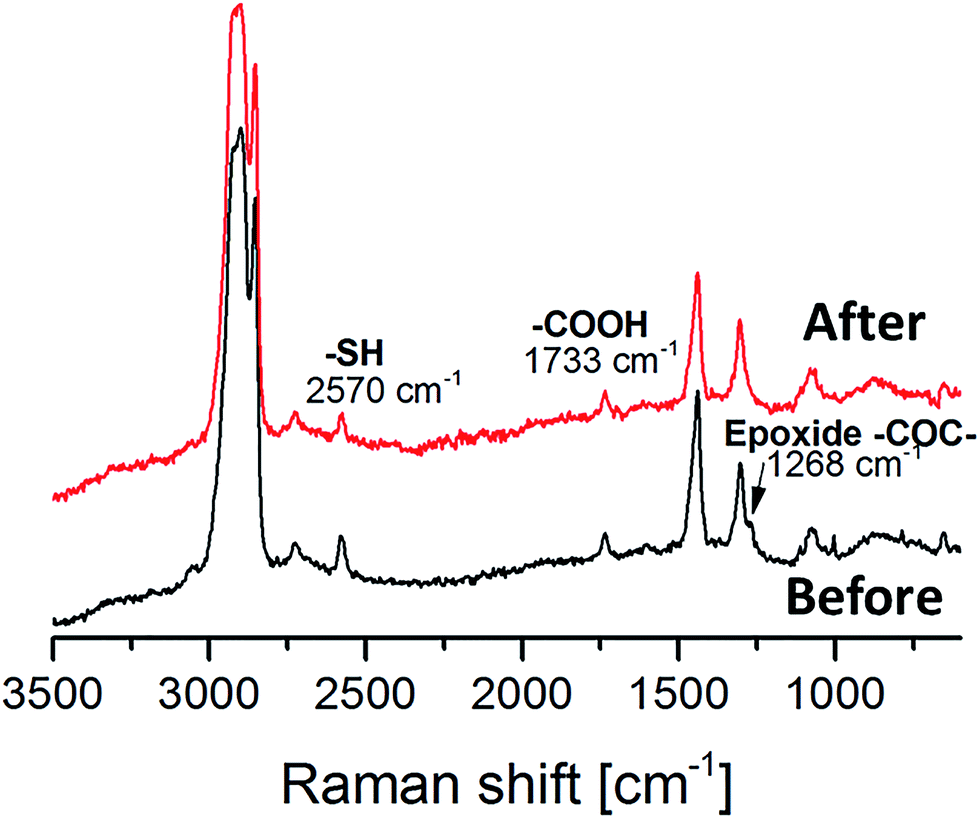 | ||
| Fig. 4 FT Raman before and after UV curing of compound 4. | ||
Modification of surface properties
As previously mentioned, the FT Raman analysis and Ellman's reagent test indicated that the thiol groups were unharmed during UV-light exposure. Model experiment B proved that with HMVE no homopolymerization occurs when initiated by benzophenone. Furthermore, when thiol groups were presented to the reaction mixture HMVE reacted with the thiol (ESI Fig. 4†). To further investigate if the thiol groups are available on the surface of the films a thiol-ene reaction with HMVE was proposed. Fig. 5 shows the contact angle of a water droplet on a surface of crosslinked film before and after thiol-ene reaction. The measurement revealed that the surface properties of the film changed after the reaction. Moreover, the surface became more hydrophilic since the thiol groups were exchanged with hydroxyl groups on the surface. This further confirms the hypothesis that the thiol groups were accessible on the surface of the films. | ||
| Fig. 5 Contact angle before (to the left) and after (to the right) modifying the surface. | ||
Conclusions
The present study demonstrates a chemo-enzymatic route to make multifunctional thermoset resins with both epoxy and thiol functionalities. The resins can be readily polymerized to form thermoset films with retained thiol functionalities active for subsequent post surface modifications. A thiol–epoxy functional oligomer was synthesized by utilizing a chemo-enzymatic route. The epoxy monomer was bio-based and extracted from outer birch bark (Betula pendula). By utilizing the enzyme CALB it was possible to selectively add the thiol group to the bio-based epoxide monomer under mild reaction conditions without harming the epoxide. Furthermore, it was possible to crosslink the synthesized thiol–epoxy oligomer by selectively reacting the epoxides. The remaining thiols where reacted to confirm their activity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Swedish research council, FORMAS (Grant Number 211-2013-70), Lantmännen Research Foundation and the European Union's Seventh Framework Programme for research, technological development and demonstration under grant agreement no. 289253.Notes and references
- J. H. Clark, J. Chem. Technol. Biotechnol., 2007, 82, 603–609 CrossRef CAS.
- B. Kamm and M. Kamm, Adv. Biochem. Eng./Biotechnol., 2007, 105, 175–204 CrossRef CAS PubMed.
- Y. Arima and H. Iwata, Biomaterials, 2007, 28, 3074–3082 CrossRef CAS PubMed.
- B. Ellis, Chemistry and technology of epoxy resins, Springer, 1993 Search PubMed.
- M. A. Gauthier, M. I. Gibson and H. A. Klok, Angew. Chem., Int. Ed. Engl., 2009, 48, 48–58 CrossRef CAS PubMed.
- S. Mongkhontreerat, K. Oberg, L. Erixon, P. Lowenhielm, A. Hult and M. Malkoch, J. Mater. Chem. A, 2013, 1, 13732–13737 CAS.
- V. Granskog, O. C. J. Andren, Y. K. Cai, M. Gonzalez-Granillo, L. Fellander-Tsai, H. von Holst, L. A. Haldosen and M. Malkoch, Adv. Funct. Mater., 2015, 25, 6596–6605 CrossRef CAS.
- Z. T. Cygan, J. T. Cabral, K. L. Beers and E. J. Amis, Langmuir, 2005, 21, 3629–3634 CrossRef CAS PubMed.
- N. Vasylieva, B. Barnych, A. Meillerd, C. Maucler, L. Pollegioni, J. S. Lin, D. Barbier and S. Marinesco, Biosens. Bioelectron., 2011, 26, 3993–4000 CrossRef CAS PubMed.
- M. A. R. Meier, J. O. Metzger and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1788–1802 RSC.
- J. Graca, Front. Chem., 2015, 3, 62 Search PubMed.
- X. Pan, P. Sengupta and D. C. Webster, Biomacromolecules, 2011, 12, 2416–2428 CrossRef CAS PubMed.
- K. Huang, P. Zhang, J. W. Zhang, S. H. Li, M. Li, J. L. Xia and Y. H. Zhou, Green Chem., 2013, 15, 2466–2475 RSC.
- S. Torron, S. Semlitsch, M. Martinelle and M. Johansson, Biomacromolecules, 2016, 17, 4003–4010 CrossRef CAS PubMed.
- A. Gandini, C. Pascoal and A. J. D. Silvestre, Prog. Polym. Sci., 2006, 31, 878–892 CrossRef CAS.
- R. Ekman, Holzforschung, 1983, 37, 205–211 CrossRef CAS.
- J. V. Crivello, Adv. Polym. Sci., 1984, 62, 1–48 CrossRef CAS.
- J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 4241–4254 CrossRef CAS.
- C. Decker and K. Moussa, J. Polym. Sci., Part C: Polym. Lett., 1990, 28, 3429–3443 CrossRef CAS.
- C. Decker, T. T. N. Viet and H. P. Thi, Polym. Int., 2001, 50, 986–997 CrossRef CAS.
- J. M. Barton, in Epoxy Resins and Composites I, Springer Berlin Heidelberg, Berlin, Heidelberg, 1985, pp. 111–154, DOI:10.1007/3-540-15546-5_5.
- B. A. Rozenberg, in Epoxy Resins and Composites II, ed. K. Dušek, Springer Berlin Heidelberg, Berlin, Heidelberg, 1986, pp. 113–165, DOI:10.1007/BFb0017916.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- L. M. de Espinosa and M. A. R. Meier, Eur. Polym. J., 2011, 47, 837–852 CrossRef.
- M. Claudino, M. Jonsson and M. Johansson, RSC Adv., 2013, 3, 11021–11034 RSC.
- G. J. Chen, S. Amajjahe and M. H. Stenzel, Chem. Commun., 2009, 10, 1198–1200 RSC.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- M. Liras, I. Quijada-Garrido, M. Palacios-Cuesta, S. Munoz-Durieux and O. Garcia, Polym. Chem., 2014, 5, 433–442 RSC.
- G. Carrot, J. Hilborn, J. L. Hedrick and M. Trollsas, Macromolecules, 1999, 32, 5171–5173 CrossRef CAS.
- A. Southan, E. Hoch, V. Schonhaar, K. Borchers, C. Schuh, M. Muller, M. Bach and G. E. M. Tovar, Polym. Chem., 2014, 5, 5350–5359 RSC.
- S. Shoda, H. Uyama, J. Kadokawa, S. Kimura and S. Kobayashi, Chem. Rev., 2016, 116, 2307–2413 CrossRef CAS PubMed.
- S. Kobayashi, Proc. Jpn. Acad., Ser. B, 2010, 86, 338–365 CrossRef CAS PubMed.
- R. A. Gross, M. Ganesh and W. H. Lu, Trends Biotechnol., 2010, 28, 435–443 CrossRef CAS PubMed.
- B. Yeniad, H. Naik and A. Heise, Biofunctionalization of Polymers and Their Applications, 2011, vol. 125, pp. 69–95 Search PubMed.
- C. Hedfors, E. Ostmark, E. Malmstrom, K. Hult and M. Martinelle, Macromolecules, 2005, 38, 647–649 CrossRef CAS.
- M. Finnveden, S. Nameer, M. Johansson and M. Martinelle, Macromol. Chem. Phys., 2016, 217, 1335–1341 CrossRef CAS.
- M. Takwa, N. Simpson, E. Malmstrom, K. Hult and M. Martinelle, Macromol. Rapid Commun., 2006, 27, 1932–1936 CrossRef CAS.
- C. Hedfors, K. Hult and M. Martinelle, J. Mol. Catal. B: Enzym., 2010, 66, 120–123 CrossRef CAS.
- A. Olsson, M. Lindstrom and T. Iversen, Biomacromolecules, 2007, 8, 757–760 CrossRef CAS PubMed.
- S. Torron, S. Semlitsch, M. Martinelle and M. Johansson, Macromol. Chem. Phys., 2014, 215, 2198–2206 CrossRef CAS.
- S. Torron and M. Johansson, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2258–2266 CrossRef CAS.
- S. Semlitsch, S. Torron, M. Johansson and M. Martinelle, Green Chem., 2016, 4003–4010 Search PubMed.
- S. Nameer and M. Johansson, J. Coat. Technol. Res., 2017, 14, 757–765 CrossRef CAS.
- P. A. Krasutsky, Nat. Prod. Rep., 2006, 23, 919–942 RSC.
- P. C. R. O. Pinto, A. R. Sousa, A. J. D. Silvestre, C. P. Neto, A. Gandini, C. Eckerman and B. Holmbom, Ind. Crops Prod., 2009, 29, 126–132 CrossRef CAS.
- G. L. Ellman, Arch. Biochem. Biophys., 1958, 74, 443–450 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: ESI One-pot enzyme-catalyzed synthesis of dual-functional polyester macromers towards surface active hydrophobic films. See DOI: 10.1039/c7ra09828f |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2017 |