
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzymatic hydrogelation of self-assembling peptide I4K2 and its antibacterial and drug sustained-release activities†
Jingkun Bai *ab,
Zhongying Gonga,
Jingxin Wangb and
Chengdong Wang*c
*ab,
Zhongying Gonga,
Jingxin Wangb and
Chengdong Wang*c
aShandong Provincial Key Laboratory of Biopharmaceuticals, School of Bioscience and Technology, Weifang Medical University, 7166 Baotong West Street, Weifang, 261042, P. R. China. E-mail: jkbaizg@163.com
bCentre for Bioengineering and Biotechnology, China University of Petroleum (East China), 66 Changjiang West Road, Qingdao 266580, P. R. China
cQingdao Industrial Energy Storage Research Institute, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, P. R. China. E-mail: wangcd@qibebt.ac.cn
First published on 16th October 2017
Abstract
Hydrogels provide great potential for biomedical applications. For clinical use, hydrogels could be used as scaffold materials for cell culture, regenerative medicine and drugs release with bactericidal properties. The amphiphilic peptide I4K2 is designed to inhibit bacterial growth through membrane permeation mechanisms. I4K2 is found to be able to self-assemble into nanofibers and form hydrogels in the presence of an enzyme (plasma amine oxidase, PAO). HPLC and MALDI-TOF-MS data show that PAO promoted the oxidation of the ε-amine of the lysine side chain. The cross-linking of I4K2 molecules catalyzed by PAO leads to a decrease in the amount of the positive charge of the system, which enhances the interaction between the self-assembled nanofibers and contributes to the formation of hydrogels. This self-supported hydrogel showed antibacterial activity against both G+ and G− bacteria and has low cytotoxicity, which enable it be consequently used as an antimicrobial agent or biological engineering scaffold material. The hydrogel also possesses good drug sustained-release activities. These advantages result in the great potential of this enzymatic I4K2 hydrogel for biomedical applications.
1. Introduction
Self-assembling peptide hydrogels have good biocompatibility and cell adhesion activity, which has led to their wide use in cell cultures and tissue engineering scaffolds in recent years.1–4 Contamination from contaminated media is a very common problem in cell cultures, some of which are contaminated by bacteria.5–7 The control of pollution rates are related to experimental costs, so obtaining a hydrogel with certain bacteriostatic effects for cell cultures can reduce the excessive use of antibiotics.Hydrogels such as conventional polymer hydrogels are usually formed by long, stable nanofibers, which are entangled by noncovalent hydrogen bonding and electrostatic interaction8–10 as well as covalent chemical cross-linking.11,12 Thus, the formation of long nanofibers is the key for the hydrogels formation.13–15 Some peptides, such as Nap-FFpY,16 Nap-GFFpY-OMe,17,18 Fmoc-FpY19 and Fmoc-pYGGDADA20 were reported to be able to form hydrogels in these systems, based on strong π–π stacking, peptide molecules self-assemble into nanofibers which were entangled in the response of phosphatase and subsequently formed hydrogels. These peptide materials has been successfully used for cell culture and drug release, yet their antibacterial properties have not been reported.
Antibacterial peptides can kill bacteria by rupturing their membranes,21–23 However, some of these reported antimicrobial peptides, such as G(IIKK)nI-NH2 (n = 2–4) are usually disordered in water and could not self-assemble to form nanofibers and supramolecular hydrogels in aqueous solutions,24–26 which hinders its application in tissue engineering.
During the past few decades, several unfavourable issues, such as the addition of chemical substances for chemical cross-linking of chemical residues, have been avoided by using enzymatic reactions of covalently cross-linked hydrogels.27,28 A number of previous studies have demonstrated that enzymes were employed to convert designed precursors into self-assembling peptides, forming supramolecular hydrogels under physiological conditions.29,30 Many enzymatic reactions occur in the human body. The reactions involved typically included enzyme-catalyzed bond cleavage31,32 and amide bond formation.33–35 Lysyl oxidase (LO) is an important enzyme for the formation and repair of the extracellular matrix, which is present in the blood of mammals and oxidizes primary amines on the side chains of lysine residues to form aldehydes.36 LO can stabilize collagen and elastin and promote the formation and repair of the respiratory tract, cardiovascular tissue and other connective tissue.37 Plasma amine oxidase (PAO),38 a similar and commercially available enzyme which also functions by oxidation of primary amines.36 PAO's molecular weight is 170![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, composed of two identical polypeptide chains,39 which have two pyridoxal phosphates and two atoms of Cu2+ per molecule.40,41
000, composed of two identical polypeptide chains,39 which have two pyridoxal phosphates and two atoms of Cu2+ per molecule.40,41
Our previous studies have shown that Ac–I4K2–NH2 (hereinafter called I4K2) self-assembles into nanofibers.42 I4K2 has two charged lysine residues as the substrate amino acid of PAO, and its self-assembled nanofibers could not form hydrogels in aqueous solution. We here investigate the self-assembly and hydrogelation of I4K2 in the absence or presence of PAO, by the combined use of transmission electron microscope (TEM), atomic force microscope (AFM), circular dichroism (CD) spectroscopy and rheology. Matrix assisted laser desorption/ionization time-of-flight mass spectroscopy (MALDI-TOF MS) and reversed-phase high performance liquid chromatography (RP-HPLC) measurements are also adopted to investigate the possible mechanism involved in the enzyme-mediated self-assembly and hydrogelation of I4K2. Peptide hydrogels composed of three-dimensional (3D) networks of nanofibers could be used for not only cell culture but also drug delivery.43–45 We tuned the pore structure of the hydrogel by controlling the crosslink density, and then we examined the sustained release properties of the hydrogels entrapped with different drug molecules. For practical applications, we finally assessed the cytotoxicity of the resulting I4K2 hydrogels by using the MTT and hemolysis assays.
2. Materials and methods
2.1. Materials
Fmoc-protected amino acids, coupling reagents, and Rink amide MBHA resin were obtained from GL Biochem Ltd. (Shanghai, China), and solvents were purchased from Bo Maijie Technology (Beijing, China). Calcein-AM and propidium iodide (PI) were obtained from KeyGen Biotechnology Co. (Nanjing, China), and SYTO 9 was bought from Life Technologies (Carlsbad, USA). Other chemical reagents were purchased from Sigma. Ultrapure water (resistivity of 18.2 MΩ cm), which was used in all experiments, was produced by a Milli-Q Biocel system (Millipore).Gram-negative (G−) strains Escherichia coli DH5a (E. coli DH5a) and Pseudomonas aeruginosa (P. aeruginosa) and Gram-positive (G+) strains Bacillus subtilis 168 (B. subtilis 168) and Staphylococcus aureus (S. aureus) were supplied by the China Center of Industrial Culture Collection. E. coli DH5a and P. aeruginosa were incubated in LB medium (tryptone 10 g L−1, yeast extract 5 g L−1, and NaCl 10 g L−1, pH of 7.0), while B. subtilis 168 and S. aureus were cultured in beef medium (glucose 60 g L−1, beef extract 10 g L−1, peptone 10 g L−1, yeast extract 10 g L−1, and NaCl 5 g L−1, pH 7.0).46 Human cervical carcinoma HeLa cells and NIH Swiss mouse embryo fibroblast NIH 3T3 cells were purchased from the Shanghai Institute for Biological Science of the Chinese Academy of Sciences and cultured in DMEM (Dulbecco's modified Eagle's medium, pH 7.4) with 10% (v/v) foetal bovine serum (FBS) at 37 °C under 5% CO2.46
2.2. Peptide synthesis and hydrogel preparation
The synthesis of peptide I4K2 (Fig. S1†) was performed on a CEM Liberty microwave peptide synthesizer and based on Fmoc solid-phase chemistry. Piperidine, dichloromethane and N,N′-dimethyl formamide (DMF) were redistilled before use. All the peptides were synthesized via reactions coupling from the C-terminus to the N-terminus. The Fmoc group was deprotected by 20% piperidine in DMF. Rink amide MBHA resin was used to make their C-terminus amidated. Carboxyl group of Fmoc-amino acid was activated by active reagent with O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluorophosphate (HBTU) and 1-hydroxybenzotriazole (HOBt) in DMF. The amino acid was sequenced into peptide. The N-terminus was capped with acetic anhydride before the product was cleaved from Rink amide MBHA resin. The peptide was cleaved from Rink amide MBHA resin by cleavage reagent with 95% TFA, 2.5% TIS, 2.5% H2O. The cleavage mixture was filtered and the filtrate was evaporated to remove cleavage reagent. The residue was precipitated in cold ether and precipitate was collected by centrifugation. Finally, the white solid was obtained by lyophilisation.47 The purity of final product was confirmed with MALDI-TOF MS and RP-HPLC analysis (Fig. S2†).I4K2 was directly dissolved in Hepes (25 mM, pH 7.4) to obtain an 8 mM (6.15 mg mL−1) stock solution, which was incubated for 1 day at room temperature prior to use. FBS and plasma amine oxidases (PAOs) were added to a stock solution of I4K2, which was then incubated at 37 °C for 15 days. The changes in peptide concentration and pH after these treatments were found to be negligible.
2.3. Self-assembly of I4K2 in solution and hydrogels
Atomic force microscopy (AFM) height images were acquired on a Veeco Nanoscope IVa MultiMode AFM instrument (Digital Instruments, Santa Barbara, CA), in tapping mode using TESP silicon probes. The samples were prepared on a freshly cleaved mica surface.Transmission electron microscopy (TEM) measurements were performed at an accelerating voltage of 200 kV on a JEOL JEM-2100 UHR instrument. The samples were negatively stained with uranyl acetate (2%, w/v) on 300-mesh copper grids.
The rheological properties of I4K2 hydrogels were recorded at 25 °C on a Haake MARS III modular rheometer (Thermo Fisher). The cone-plate geometry was used with a 2.0° cone angle. Strain sweeps (from 0.01% to 100% strain, 1 Hz) were performed to determine the linear viscoelastic region,47 and dynamic frequency sweeps were performed from 0.01 to 10 Hz and at 1% strain.
The turbidity measurements were performed on a UV-Vis spectrophotometer (Perkin-Elmer Lambda 25) at 25 °C. The turbidity of I4K2 solution at different time was monitered by absorbance at 350 nm using a quartz cell with 1 cm optical length.
CD measurements were performed at room temperature on a MOS-450/AF-CD spectrometer (Biologic, France). The CD spectra were scanned from 260 to 190 nm with 0.5 nm steps in a 0.1 mm quartz cell. The resulting CD spectra were the average of six independent measurements of each sample.
2.4. MALDI-TOF MS and RP-HPLC
Mass spectra were recorded on a Microflex MALDI-TOF mass spectrometer (Bruker Daltonics) in the positive-ion mode with a mass range from m/z 250 to 3500. 8 mM I4K2 solution and the enzymatic hydrogel were diluted 50 times using 25 mM Hepes (pH 7.4). 1 μL of I4K2 solution or hydrogel was mixed with 1 μL 4-hydroxy-α-cyanocinnamic acid (HCCA) solution. Then, 1 μL of the mixture was dropped on a polished steel sample target and air-dried, immediately followed by MS measurements.35RP-HPLC chromatograms of I4K2 before and after adding PAO were recorded by C18 reverse phase column on a Waters 2696 Alliance HPLC system. RP-HPLC analysis was performed from 95% water (with 0.1% (v/v) TFA)/5% acetonitrile (with 0.1% (v/v) TFA) to 5% water (with 0.1% (v/v) TFA)/95% acetonitrile (with 0.1% (v/v) TFA) from 1 to 50 min. Note that 8 mM I4K2 solution and enzymatic I4K2 hydrogel were diluted 8 times with 25 mM Hepes (pH 7.4), then they were filtered with 0.22 μm filter membrane for an analysis.35
2.5. Bacterial viability assays
I4K2 hydrogels used in the assessment of bacterial viability were prepared in a 96-well tissue-culture polystyrene (TCPS) plate and an 8-well borosilicate glass plate. Aliquots (100 μL) of bacterial suspensions (1 × 106 CFU mL−1) were added onto the hydrogel surface. After these suspensions were incubated for 24 h at 37 °C, 100 μL of solution containing 15 μM SYTO 9 and 30 μM PI was introduced into each well of the 8-well plate, and staining occurred for 30 min at ambient temperature. The bacteria in solution above the hydrogels were collected and observed using an inverted fluorescence microscope (Leica DMI3000B) at 100× magnification. The bacteria in the wells without peptide were used as the control.To quantitatively assess the proliferation of bacteria, we transferred the bacterial suspension above the hydrogels to individual wells of a new 96-well TCPS plate. The absorbance was read at 600 nm on a microplate reader (M2e, Molecular Device). The bacterial suspension without hydrogels was used as a control, and the wells without bacteria served as a blank for the absorbance measurements.
2.6. MTT assay and hemolytic activity
The cytotoxicity of these peptides in vitro was measured by an MTT assay using NIH 3T3 cells. Briefly, NIH 3T3 cells were pre-cultured in cell culture bottles. After 48 h incubation, cells (∼1 × 105 cells per mL, 100 μL) were collected and transferred onto the top of the I4K2 hydrogels. After incubated for further 24 h or 72 h, 100 μL of MTT solution (in PBS, 5 mg mL−1) was added to each well and incubated for 4 h. Subsequently, the supernatants of NIH 3T3 cells were removed. 150 μL of dimethyl sulfoxide (DMSO) was added into each well and the supernatants of the wells were transfer to a new 96 well plate and the absorbance at 490 nm was recorded using a microplate reader (M2e, Molecular Devices). The wells without peptides were used as controls, and the wells without cells were used as blanks for the spectrophotometer. Experiments were independently performed at least three times.46The anti-tumor properties of peptide hydrogel coated with drugs in vitro was measured by an MTT assay using Hela cells. 106.9 mg of FITC-G(IIKK)3I-NH2 was added to an I4K2 solution, this sample was incubated in a 37 °C oven for 24 h, and 10 U mL−1 PAO was then added. After 2 weeks, MTT assay results with Hela cells on I4K2 gels coated with FITC-G(IIKK)3I-NH2, I4K2 gels and TCPS plates were recorded using a microplate reader (M2e, Molecular Devices) after different incubation times.
The hemolysis of I4K2 solution was assessed using red blood cells (RBCs). The study was performed according to National Institutes of Health guidelines and the study protocol was approved by Weifang Medical University Institutional Animal Care and Use Committee. We have obtained written informed consent from all human participants of this study. RBCs were prepared from fesh human blood (self donor) via centrifugation at 1000g for 5 minutes, washed three times with PBS, and then 100 μL of RBCs suspended in PBS at 8% (v/v) were mixed with 100 μL of 2 times the concentration of I4K2 solutions in a 96-well plate and incubated for 1 h at 37 °C. Then, the 96-well plate was centrifugated at 1000g for 10 min, and 100 μL of the supernatants of the wells were transferred into a new 96-well plate. Finally, the absorbance at 540 nm (A540 nm) using the M2e microplate autoreader was recorded as the hemoglobin release. Hemoglobin release in PBS and in 0.1% (v/v) Triton X-100 was employed as the negative (0% release) and positive control (100% release), respectively.26
2.7. In vitro drug release test
First, 29 mg of adriamycin DOX (580 Da) or 106.9 mg of FITC-G(IIKK)3I-NH2 (2137 Da) was added to an I4K2 solution; this sample was incubated in a 37 °C oven for 24 h, and 10 U mL−1 PAO was then added. After 2 weeks, 1 mL of PBS buffer was soaked into the hydrogel. At every predetermined time interval, 100 μL of PBS was removed, and 100 μL of fresh PBS was then added. The concentrations of DOX and FITC-G(IIKK)3I-NH2 were quantified via a Horiba Jobin Yvon Fluoromax-4 fluorescence spectrophotometer at 590 and 520 nm,48,49 respectively. The release rate was defined as the ratio of the total amount of drug released to the total drug load at a given time.50 The experiments were repeated three times for each sample.3. Results and discussion
3.1. Fabrication and properties of I4K2 hydrogels
LO, which is present in FBS, can oxidize the primary amine on the lysine side chain.36 In our previous study, I4K2 self-assembled to form nanofibers that contain lysine residues,42 so I4K2 should be used as the substrate of the LO. We selected the amphiphilic peptide I4K2 as the target molecule for the following studies. The effects of FBS on the formation of I4K2 hydrogels were first investigated.When 8 mM I4K2 is dissolved in Hepes buffer, a clear solution is formed. When the volume fraction of FBS was as least 2%, the I4K2 solution transformed into a hydrogel (Fig. 1A and S3†) at 15 days of incubation at 37 °C after the addition of FBS. Since LO is not commercially produced, and PAO is commercially available and has the same oxidation effect as LO,51–53 the experiments were carried out using commercially available PAO. When the concentration of PAO is up to 10 U mL−1, I4K2 could self-assemble to form a self-supporting hydrogel (Fig. 1B).
 | ||
| Fig. 1 Optical images of 8 mM I4K2 aqueous solutions in the presence of (A) FBS (0%, 1%, 2% and 4%) and (B) 10 μM Cu2+ and PAO (0, 5, 10 and 20 U mL−1) after incubation for 15 days at 37 °C. (C) Frequency sweeps of I4K2 samples after the addition of 10 μM Cu2+ and 10 U mL−1 PAO. All rheological measurements were performed at 25 °C. | ||
The rheological data reliably reflect the observed changes in I4K2 self-assembly into a hydrogel, which indirectly verifies the self-assembly behaviour of peptide molecules.54 After I4K2 solution converted into a hydrogel after the addition of 10 μM Cu2+ and 10 U mL−1 PAO, the viscoelasticity of the peptide hydrogel was investigated using a thermo rheometer. The value of the storage modulus (G′) was approximately 700–900 Pa, and the energy consumption modulus (G′′) was approximately 70–100 Pa, so the storage modulus (G′) exceeds the energy consumption modulus (G′′) by an order of magnitude in the linear viscoelastic regime (Fig. 1C), indicating that I4K2 forms a typical elastomeric hydrogel at a low peptide concentration.55 Consistent with the occurrence of the formation of I4K2 hydrogel, the turbidity (OD350 nm) of the I4K2 suspension was gradually enhanced after PAO addition (Fig. S4†).
The morphology of self-assembly was characterized by negative-strain TEM. The amphiphilic peptide I4K2 self-assembled to form nanofibers in solution (Fig. 2A). After the addition of PAO, the self-assembled morphology did not significantly change. The difference was that multi-bundle aggregation and the entanglement of nanofibers began to occur (Fig. 2B). As shown in Fig. 2C, the diameter of I4K2 that self-assembled in solution mostly ranged from 9–12 nm, while the nanofiber width was mostly 10–13 nm after the addition of PAO and 15 days of incubation. The heights of nanofibers increased significantly after enzyme addition (Fig. S5†), indicating that the addition of PAO contributes to the aggregation of the self-assembled peptides. The entanglement among nanofibers was deduced to cause the formation of I4K2 gel state.56
 | ||
| Fig. 2 TEM images of I4K2 (A) before and (B) after the addition of PAO. (C) Diameter summary of I4K2 self-assembled nanostructures before and after the addition of PAO. (D) CD spectra of 8 mM I4K2 before and after addition of PAO. All measurements were performed after 15 days of incubation at 37 °C. | ||
The CD spectra were performed to characterize the molecular conformation in self-assembly. The negative peak in CD spectra around 217 nm showed that I4K2 predominantly adopted a β-sheet secondary structure in self-assemble fibres and that it did not significantly change when PAO was added (Fig. 2D). The molecular arrangement in nanofibers did not change before and after the addition of PAO.
3.2. Induction of the formation of I4K2 hydrogels by PAO
To study the role of PAO in the formation of hydrogels by I4K2, the product of I4K2 and PAO was analysed by RP-HPLC. According to the RP-HPLC analysis (Fig. 3 and S6†), the intensity of the peptide peak (at approximately 22.3 min) significantly decreased after the addition of PAO and incubation for 15 days, and the peak area decreased 78.3%. Approximately 80% of I4K2 molecules reacted in the presence of PAO, and a small product peak appeared at 24.8 min. However, the product peak did not increase significantly, some of the product might be removed by filtration during the filtration process. The product was more hydrophobic than I4K2, and it was most likely the result of I4K2 oxidation. The addition of PAO reduced the amount of positive charge carried by the I4K2 peptide, reduced the electrostatic repulsion between molecules, and promoted the formation and aggregation of hydrogels.57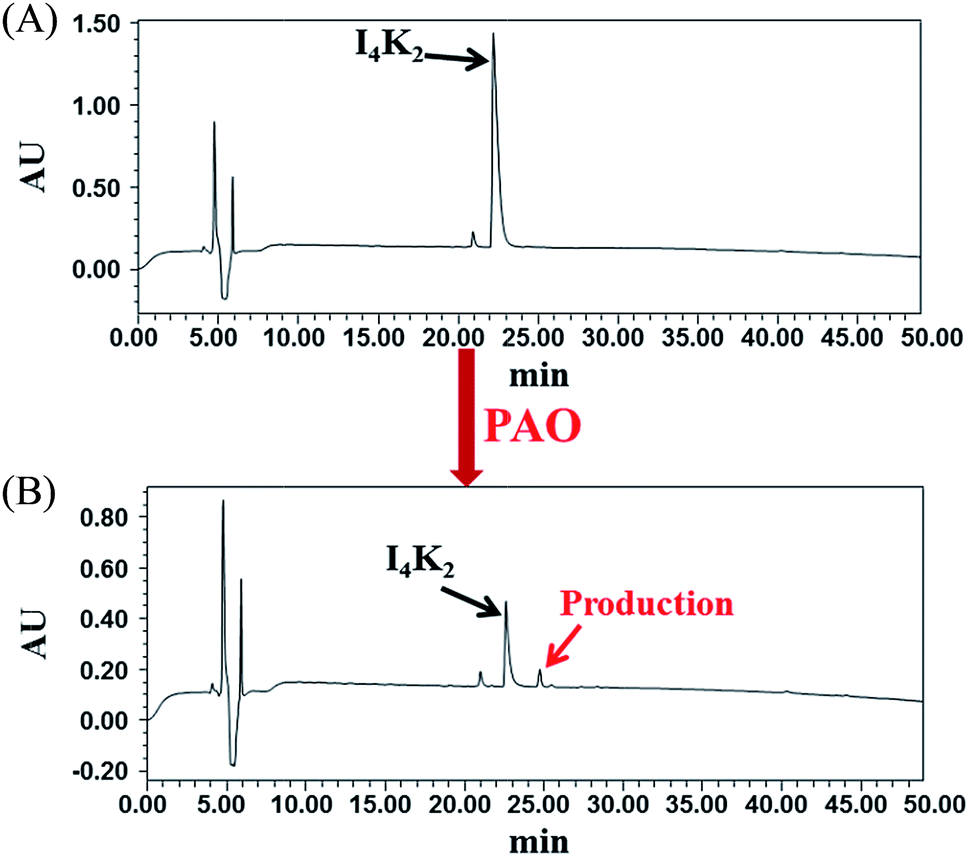 | ||
| Fig. 3 RP-HPLC of I4K2 samples (A) before and after (B) the addition of PAO and 15 days of incubation at 37 °C. | ||
To further study the mechanism of hydrogel formation, we used MALDI-TOF mass spectrometry to analyse the peptide molecules after the addition of PAO.35 The theoretical molecular weight of I4K2 is 768.5, and a molecular weight of 791.7 is attributable to its Na+ cationization (Fig. 4A). MALDI-TOF MS spectra showed the formation of intermolecular cross-linked products after the addition of PAO to the samples. Lysines on an I4K2 molecule and another I4K2 molecule coupled to produce cross-linked products whose theoretical molecular weight is 1520.0 (Fig. 4C). The mass spectrum indicated a molecular weight of 1521.4, which is attributable to its H+ cationization (Fig. 4B).
 | ||
| Fig. 4 (A) MALDI-TOF-MS spectra of I4K2 before and after addition of PAO. Molecular structure of (B) I4K2 and (C) inter-molecular product of I4K2 catalyzed by PAO. | ||
The ε-amino group in the lysine side chain of the I4K2 molecule can oxidize to an aldehyde group under the action of PAO according to the work of Rucker et al.58 Two aldehydes or an aldehyde and an amino group can undergo a series of condensation reactions. This mechanism produced Schiff bases that then proceeded to form the intermolecular product lysinonorleucine (Fig. 4C),58 which ultimately triggered the conversion of I4K2 from sol to gel.
3.3. In vitro cell cytotoxicity
To further explore whether I4K2 hydrogels can be used as a biological scaffold in tissue engineering, we assessed the cytotoxicity of hydrogels by an MTT assay. The MTT method can be used to determine the amount of exogenous MTT that is converted into formazan by the reduction activity of living cells, and the increase in formazan concentration can be used as an indicator of cell survival.46,59 NIH 3T3 cells were cultured on top of the hydrogels for 24 or 72 h, and MTT determination was then performed. The amount of formazan derived from cells that were cultured on the surface of I4K2 hydrogels was slightly smaller than that from cells directly cultured on the surface of TCPS. In addition, NIH 3T3 cells were cultured on I4K2 hydrogels, so a small amount of the formazan resulting from the MTT assay permeated into the hydrogels and could not be sufficiently dissolved into DMSO. The measured OD value was therefore slightly lower than the real value (Fig. 5A). MTT assays showed that I4K2 hydrogels had a low cytotoxicity. | ||
| Fig. 5 (A) MTT assay results with NIH 3T3 cells on I4K2 gels and TCPS plates after different incubation times. (B) Human red blood cell hemolysis of I4K2 solution. | ||
In clinical applications, I4K2 hydrogel would be used for wound healing and hence, so we assessed the hemolytic activity of I4K2 solution. As shown in Fig. 5B, the hemolysis of I4K2 incubated with human red blood cells even at higher concentrations of 32 mM was below 3%, showed little hemolysis.
3.4. Antibacterial activity of I4K2 hydrogels
Based on the differences in cell wall structure between G+ and G− bacteria, the growth of G+ (S. aureus and B. subtilis 168) and G− bacteria (E. coli DH5a and P. aeruginosa) on I4K2 hydrogels was examined. 1 × 106 CFU mL−1 bacteria were incubated on the surface of the hydrogel or on a TCPS plate for 24 h, and the optical density at 600 nm was read on a microplate reader.60 The mortality% (= (1 - OD600 nm gel/OD600 nm TCPS) * 100) of these four strains of bacteria was 56.7, 67.5, 57.9 and 60.9% respectively. All bacterial mortality rates were above 55% (Fig. 6A). I4K2 hydrogels formed with the addition of PAO have antibacterial activity against G+ and G− bacteria. | ||
| Fig. 6 (A) OD of TCPS control surface or I4K2 hydrogel surface challenged with G+ bacteria and G− bacteria for 24 h. (B) Fluorescence microscopy images of E. coli DH5a in solution above the I4K2 hydrogel surface (right) and the TCPS plates (left) after incubation for 24 h. The bacteria were subjected to live (green)/dead (red) staining with SYTO 9 and PI prior to imaging. | ||
To better determine the antimicrobial effects of I4K2 hydrogel, the viability of E. coli DH5a cells was observed with live/dead bacterium staining. The fluorescent dyes SYTO 9 and PI were selected to stain bacteria. SYTO 9 can pass through intact cell membranes and specifically binds DNA, staining all cells with green fluorescence. PI cannot pass through the intact cell membranes of living cells but stains dead cells with red (635 nm) fluorescence. When SYTO 9 and PI were mixed in appropriate proportions, bacteria with intact cell membranes were stained green and bacteria with the incomplete cell membranes were stained red, enabling us to determine the live/dead state of bacteria.61,62
E. coli DH5a cells on the surface of the hydrogel or on the surface of the TCPS plate were stained for 24 h and observed by using a fluorescence inverted microscope (Leica DMI3000B). Most of the bacteria incubated on the I4K2 gel exhibited the red PI fluorescence, although a small number of bacteria showed the green fluorescence of SYTO 9, indicating that most E. coli DH5a cells on the I4K2 gel died. Meanwhile, most of E. coli DH5a cells that were incubated on the control TCPS plates survived (Fig. 6B).
The membranes of E. coli and other bacteria are mainly composed of negatively charged phospholipids.63 The ε-amino group of the lysine side chains in the I4K2 peptide carries a positive charge, so it could bind the surface of the bacterial membrane by electrostatic interaction. It subsequently inserted into the phospholipid bilayer of the bacterial membrane, increasing its permeability and eventually leading to bacterial death.25,64–66
3.5. In vitro drug release from the I4K2 hydrogels
The porous structures of hydrogels can be effectively regulated by controlling the cross-linking density of the matrix in the gel. Their porosity allows drugs to be loaded into the gel matrix, and the rate of drug release depends on the diffusion coefficient of the drug molecule through the gel network.12 The above results confirmed that I4K2 hydrogels have good mechanical strength and low cytotoxicity and that they could serve as a potential drug delivery system for sustained release. I4K2 hydrogels were coated with FITC-G(IIKK)3I-NH2 or DOX. The release curve of DOX, with a lower molecular weight, reflected two stages: rapid release and slow release. The DOX release reached 50% of the total in the first 50 h. The amount of DOX that was released gradually increased with time and finally reached 80% of the total amount. The half-release time of DOX in I4K2 hydrogels was approximately 46 h, while the half-release time of FITC-G(IIKK)3I-NH2 was approximately 168 h (Fig. 7A). FITC-G(IIKK)3I-NH2 was released more slowly, which may be caused by the interaction between the small pore size and the hydrogen bonding of peptide molecules. Gel-controlled release of DOX greatly improved the utilization rate of the drug and resulted in a sustained antibacterial material.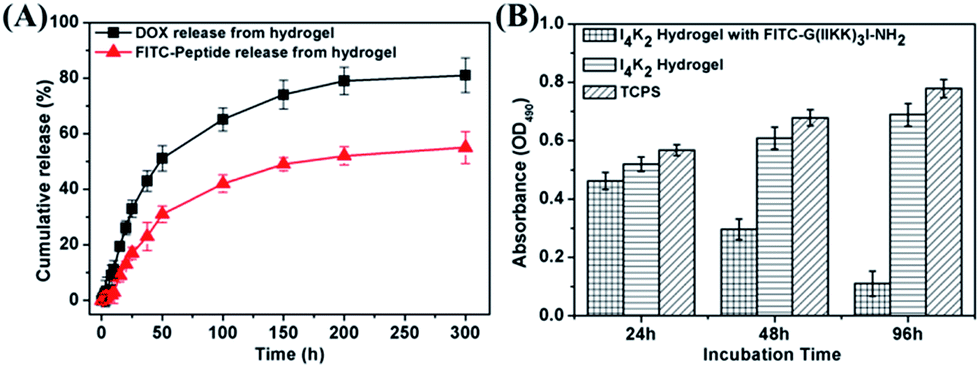 | ||
| Fig. 7 (A) The percentage of fluorescent molecules released from I4K2 hydrogels into buffer over time. (B) MTT assay results with Hela cells on I4K2 gels coated with FITC-G(IIKK)3I-NH2, I4K2 gels and TCPS plates after different incubation times. | ||
Approximately 52% of the total DOX was released from the I4K2 hydrogel in 50 h. In contrast, the amount of FITC-G(IIKK)3I-NH2 released from the I4K2 hydrogel was more moderate. This release sustained at a steady rate and lasted up to 300 hours, but the concentration of the helical peptide at this release rate resulted in good antibacterial and antitumour activity.25,26 The anti-tumor properties of I4K2 hydrogel encapsulated helical peptide was further investigated. Survival rate of Hela cells on I4K2 gels coated with FITC-G(IIKK)3I-NH2 was measured by an MTT assay. After 24 h, 48 h, 96 h of incubation, the survival rates of Hela cells were 89.0%, 48.7%, 16.1%, respectively. The release rate was about 42% after 96 h of incubation in the hydrogel supernatant, and more than 80% of the Hela cells grown on the surface of the hydrogel died.
The process exhibited a rapid release of 52 h and a sustained release of 12 d, which could be effectively applied to the early stage of sterilization and the later stage of anti-infection. This result demonstrated the potential of this hydrogel as a long-acting sustained-release drug material. The interaction of the gel with the helical peptide greatly prolonged the release of the drug, and it is promising as a sustained antibacterial and antitumour material.
4. Conclusions
In this paper, we designed the amphiphilic peptide I4K2 that forms a self-supporting hydrogel upon addition of PAO, and this process is accompanied by the formation of the entangled nanofibers. PAO catalyses the oxidation of primary amines of lysine side chains in I4K2, reducing the electrostatic repulsion between peptides and inducing the formation of hydrogels. I4K2 hydrogels effectively inhibit bacterial growth by electrostatic adsorption and rupturing membranes, and they have low cytotoxicity to normal mammalian cells. The hydrogel also has a drug sustained-release ability, so it can be used for the long-term healing of wounds to prevent wound infections after surgery. These advantages give I4K2 hydrogels a great potential for cell culture and biomedical applications.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge the supervision and help by Prof. Hai Xu and Prof. Cuixia Chen, and the useful discussion with Dr Peng Zhou in China University of Petroleum (East China), and the financial support of Doctoral Fund of Weifang Medical University.Notes and references
- J. Kisiday, M. Jin, B. Kurz, H. Hung, C. Semino, S. Zhang and A. Grodzinsky, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9996–10001 CrossRef CAS PubMed.
- M. W. Tibbitt and K. S. Anseth, Biotechnol. Bioeng., 2009, 103, 655–663 CrossRef CAS PubMed.
- A. Dasgupta, J. H. Mondal and D. Das, RSC Adv., 2013, 3, 9117–9149 RSC.
- W. Wang, G. Li, W. Zhang, J. Gao, J. Zhang, C. Li, D. Ding and D. Kong, RSC Adv., 2014, 4, 30168–30171 RSC.
- F. J. van Kuppeveld, K. E. Johansson, J. M. Galama, J. Kissing, G. Bölske, V. D. L. Jt and W. J. Melchers, Appl. Environ. Microbiol., 1994, 60, 149–152 CAS.
- C. Leifert, J. Y. Ritchie and W. M. Waites, World J. Microbiol. Biotechnol., 1991, 7, 452–469 CrossRef CAS PubMed.
- C. Leifert and A. C. Cassells, In Vitro Cell. Dev. Biol.: Plant, 2001, 37, 133–138 CrossRef.
- B. S. Kim, S. W. Park and P. T. Hammond, ACS Nano, 2008, 2, 386–392 CrossRef CAS PubMed.
- B. J. B. Folmer, R. P. Sijbesma, R. M. Versteegen, J. A. J. van der Rijt and E. W. Meijer, Adv. Mater., 2000, 12, 874–878 CrossRef CAS.
- B. Xing, C. W. Yu, K. H. Chow, P. L. Ho, D. Fu and B. Xu, J. Am. Chem. Soc., 2002, 124, 14846–14847 CrossRef CAS PubMed.
- Y. Okumura and K. Ito, Adv. Mater., 2001, 13, 485–487 CrossRef CAS.
- T. R. Hoare and D. S. Kohane, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- N. T. Phuoc, J. F. Quinn, M. R. Whittaker and T. P. Davis, Polym. Chem., 2016, 7, 4295–4312 RSC.
- N. T. Phuoc, J. F. Quinn, A. Anastasaki, M. Rolland, V. Mai, D. Haddleton, M. R. Whittaker and T. P. Davis, Polym. Chem., 2017, 8, 1353–1363 RSC.
- M. Ma, Y. Kuang, Y. Gao, Y. Zhang, P. Gao and B. Xu, J. Am. Chem. Soc., 2010, 132, 2719–2728 CrossRef CAS PubMed.
- Q. G. Wang, Z. M. Yang, Y. Gao, W. W. Ge, L. Wang and B. Xu, Soft Matter, 2008, 4, 550–553 RSC.
- H. M. Wang, C. H. Ren, Z. J. Song, L. Wang, X. M. Chen and Z. M. Yang, Nanotechnology, 2010, 21, 225606 CrossRef PubMed.
- H. Wang, Z. Luo, Y. Wang, T. He, C. Yang, C. Ren, L. Ma, C. Gong, X. Li and Z. Yang, Adv. Funct. Mater., 2016, 26, 1822–1829 CrossRef CAS.
- J. W. Sadownik, J. Leckie and R. V. Ulijn, Chem. Commun., 2011, 47, 728–730 RSC.
- J. Shi, X. Du, D. Yuan, R. Haburcak, D. Wu, N. Zhou and B. Xu, Chem. Commun., 2015, 51, 4899–4901 RSC.
- K. Bazaka, M. V. Jacob, W. Chrzanowski and K. Ostrikov, RSC Adv., 2015, 5, 48739–48759 RSC.
- K. A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238 CrossRef CAS PubMed.
- R. E. Hancock and A. Rozek, FEMS Microbiol. Lett., 2002, 206, 143–149 CrossRef CAS PubMed.
- C. Chen, Y. Chen, C. Yang, P. Zeng, H. Xu, F. Pan and J. R. Lu, ACS Appl. Mater. Interfaces, 2015, 7, 17346–17355 CAS.
- C. Chen, J. Hu, P. Zeng, Y. Chen, H. Xu and J. R. Lu, ACS Appl. Mater. Interfaces, 2014, 6, 16529–16536 CAS.
- C. Chen, J. Hu, P. Zeng, F. Pan, M. Yaseen, H. Xu and J. R. Lu, Biomaterials, 2014, 35, 1552–1561 CrossRef CAS PubMed.
- S. Sakai and K. Kawakami, Acta Biomater., 2007, 3, 495–501 CrossRef CAS PubMed.
- A. A. Aimetti, A. J. Machen and K. S. Anseth, Biomaterials, 2009, 30, 6048–6054 CrossRef CAS PubMed.
- J. Li, Y. Gao, Y. Kuang, J. Shi, X. Du, J. Zhou, H. Wang, Z. Yang and B. Xu, J. Am. Chem. Soc., 2013, 135, 9907–9914 CrossRef CAS PubMed.
- Y. Zhang, Y. Kuang, Y. Gao and B. Xu, Langmuir, 2011, 27, 529–537 CrossRef CAS PubMed.
- A. R. Hirst, S. Roy, M. Arora, A. K. Das, N. Hodson, P. Murray, S. Marshall, N. Javid, J. Sefcik and J. Boekhoven, Nat. Chem., 2010, 2, 1089–1094 CrossRef CAS PubMed.
- Z. Yang, P.-L. Ho, G. Liang, K. H. Chow, Q. Wang, Y. Cao, Z. Guo and B. Xu, J. Am. Chem. Soc., 2007, 129, 266–267 CrossRef CAS PubMed.
- C. Chen, Y. Zhang, R. Fei, C. Cao, M. Wang, J. Wang, J. Bai, H. Cox, T. Waigh and J. R. Lu, ACS Appl. Mater. Interfaces, 2016, 8, 17833–17841 CAS.
- M. Hughes, P. W. J. M. Frederix, J. Raeburn, L. S. Birchall, J. Sadownik, F. C. Coomer, I. H. Lin, E. J. Cussen, N. T. Hunt, T. Tuttle, S. J. Webb, D. J. Adams and R. V. Ulijn, Soft Matter, 2012, 8, 5595–5602 RSC.
- J. Bai, C. Chen, J. Wang, Y. Zhang, H. Cox, J. Zhang, Y. Wang, J. Penny, T. Waigh and J. R. Lu, ACS Appl. Mater. Interfaces, 2016, 8, 15093–15102 CAS.
- E. L. Bakota, L. Aulisa, K. M. Galler and J. D. Hartgerink, Biomacromolecules, 2011, 12, 82–87 CrossRef CAS PubMed.
- H. M. Kagan and W. Li, J. Cell. Biochem., 2003, 88, 660–672 CrossRef CAS PubMed.
- B. G. Malmstrom, L. E. Andreasson and B. Reinhammar, Enzymes, 1975, 12, 507–579 CAS.
- F. M. Achee, C. H. Chervenka, R. A. Smith and K. T. Yasunobu, Biochemistry, 1968, 7, 4329–4335 CrossRef CAS PubMed.
- H. Yamada, K. Yasunobu, T. Yamano and H. S. Mason, Nature, 1963, 198, 1092–1093 CrossRef CAS PubMed.
- H. Yamada and K. T. Yasunobu, J. Biol. Chem., 1963, 238, 2669–2675 CAS.
- L. Deng, P. Zhou, Y. Zhao, Y. Wang and H. Xu, J. Phys. Chem. B, 2014, 118, 12501–12510 CrossRef CAS PubMed.
- A. Altunbas, S. J. Lee, S. A. Rajasekaran, J. P. Schneider and D. J. Pochan, Biomaterials, 2011, 32, 5906–5914 CrossRef CAS PubMed.
- D. Das and S. Pal, RSC Adv., 2015, 5, 25014–25050 RSC.
- R. Liang, Z. Luo, G. Pu, W. Wu, S. Shi, J. Yu, Z. Zhang, H. Chen and X. Li, RSC Adv., 2016, 6, 76093–76098 RSC.
- C. Chen, J. Hu, S. Zhang, P. Zhou, X. Zhao, H. Xu, X. Zhao, M. Yaseen and J. R. Lu, Biomaterials, 2012, 33, 592–603 CrossRef CAS PubMed.
- C. Chen, Y. Gu, L. Deng, S. Han, X. Sun, Y. Chen, J. R. Lu and H. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 14360–14368 CAS.
- J. H. Park, L. Gu, G. V. Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Nat. Mater., 2009, 8, 331–336 CrossRef CAS PubMed.
- M. Saito, S. J. Korsmeyer and P. H. Schlesinger, Nat. Cell Biol., 2000, 2, 553 CrossRef CAS PubMed.
- J. Zhao, B. Guo and P. X. Ma, RSC Adv., 2014, 4, 17736–17742 RSC.
- G. A. Lyles, Int. J. Biochem. Cell Biol., 1996, 28, 259–274 CrossRef CAS PubMed.
- C. R. Kothapalli and A. Ramamurthi, J. Tissue Eng. Regener. Med., 2009, 3, 655–661 CrossRef CAS PubMed.
- Y.-K. I. Lau, A. M. Gobin and J. L. West, Ann. Biomed. Eng., 2006, 34, 1239–1246 CrossRef PubMed.
- R. Picou, J. P. Moses, A. D. Wellman, I. Kheterpal and S. D. Gilman, Analyst, 2010, 135, 1631–1635 RSC.
- A. P. Nowak, V. Breedveld, L. Pakstis, B. Ozbas, D. J. Pine, D. Pochan and T. J. Deming, Nature, 2002, 417, 424–428 CrossRef CAS PubMed.
- N. P. Truong, J. F. Quinn, A. Anastasaki, D. M. Haddleton, M. R. Whittaker and T. P. Davis, Chem. Commun., 2016, 52, 4497–4500 RSC.
- C. L. Bell and N. A. Peppas, J. Controlled Release, 1996, 39, 201–207 CrossRef CAS.
- R. B. Rucker, T. Kosonen, M. S. Clegg, A. E. Mitchell, B. R. Rucker, J. Y. Uriu-Hare and C. L. Keen, Am. J. Clin. Nutr., 1998, 67, 996S–1002S CAS.
- K. Abe and N. Matsuki, Neurosci. Res., 2000, 38, 325–329 CrossRef CAS PubMed.
- C. H. Park, E. V. Valore, A. J. Waring and T. Ganz, J. Biol. Chem., 2001, 276, 7806–7810 CrossRef CAS PubMed.
- W. K. Jung, H. C. Koo, K. W. Kim, S. Shin, S. H. Kim and Y. H. Park, Appl. Environ. Microbiol., 2008, 74, 2171–2178 CrossRef CAS PubMed.
- M. Berney, F. Hammes, F. Bosshard, H. U. Weilenmann and T. Egli, Appl. Environ. Microbiol., 2007, 73, 3283–3290 CrossRef CAS PubMed.
- Y. Shai, Biochim. Biophys. Acta, Biomembr., 1999, 1462, 55–70 CrossRef CAS.
- C. Chen, F. Pan, S. Zhang, J. Hu, M. Cao, J. Wang, H. Xu, X. Zhao and J. R. Lu, Biomacromolecules, 2010, 11, 402–411 CrossRef CAS PubMed.
- A. S. Veiga, C. Sinthuvanich, D. Gaspar, H. G. Franquelim, M. A. Castanho and J. P. Schneider, Biomaterials, 2012, 33, 8907–8916 CrossRef CAS PubMed.
- D. A. Salick, J. K. Kretsinger, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2007, 129, 14793–14799 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09743c |
| This journal is © The Royal Society of Chemistry 2017 |