
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanism of asphaltene aggregation induced by supercritical CO2: insights from molecular dynamics simulation†
Bing Liu *,
Jiawei Li,
Chao Qi,
Xiaoqi Li,
Tingyi Mai and
Jun Zhang*
*,
Jiawei Li,
Chao Qi,
Xiaoqi Li,
Tingyi Mai and
Jun Zhang*
College of Science, China University of Petroleum, 266580 Qingdao, People's Republic of China. E-mail: liubing19720115@gmail.com; zhangjun.upc@gmail.com
First published on 1st November 2017
Abstract
The mechanisms of asphaltene aggregation leading to damage and wellbore plugging are still unknown, which is not conducive to finding effective solutions to the relevant problems of asphaltene precipitation in supercritical CO2 (scCO2) enhanced oil recovery processes. Therefore, molecular dynamics simulations were performed on an oil–scCO2 system to investigate the behavior and mechanism of asphaltene aggregation. The results show that the selective extraction of crude oil in scCO2 leads to the formation of asphaltene nanoaggregates. The vdW interactions among asphaltenes in scCO2 are stronger than those under vacuum, which aggravates aggregation. Free energy calculations indicate that the remaining binding energy between asphaltenes in scCO2 is the driving force of asphaltene aggregation. Interestingly, we observed a two-step process of separation and aggregation for asphaltene nanoaggregates, which demonstrates that the efficiency of inhibiting asphaltene aggregation can be improved by adding an inhibitor in the first step of aggregation in scCO2 flooding.
1. Introduction
CO2 flooding is one of the successful methods for enhanced oil recovery (EOR) applied in oil fields.1,2 The main mechanisms contributing to oil recovery by CO2 are the decrease in viscosity and interfacial tension and the increase in mobility of crude oil in oil reservoirs.3,4 Moreover, supercritical CO2 (scCO2) has been revealed to be useful for oil extraction due to its properties of low viscosity and high diffusivity.5 Rudyk et al. achieved a total oil recovery ranging from 44–77% when scCO2 was injected into dead oil with a density of 0.8573 kg L−1.6 However, one of the major drawbacks of CO2 injection in oil fields is associated with the well-documented asphaltene deposition. This may cause pore throat plugging, permeability reduction, and wettability alteration in reservoirs, further resulting in a reduction in the recovery factor.7 In addition, deposited asphaltenes can also lead to severe plugging of the wellbore, tubing, and pumps, which must be remediated by constant washing and scrapping.8 Consequently, this will generate a considerable increase in maintenance costs in the petroleum industry. Therefore, it is important to investigate asphaltene aggregation in the presence of scCO2 to understand the asphaltene aggregation mechanisms and find solutions to the relevant problems.Asphaltenes, the heaviest and most complicated components in crude oil, consist of polyaromatic species with alkyl side chains and some traces of heteroatoms. Asphaltene molecules tend to aggregate and form larger agglomerates, which is correlated with their molecular structure.9 According to the recognized Yenn–Mullins model,10 the aggregation of asphaltenes is a complex hierarchical process. Moreover, asphaltene aggregation is related to the pressure, temperature, and composition of the crude oil. With respect to CO2-EOR, there have been increasing efforts to study the aggregation behavior of asphaltenes induced by CO2. Verdier et al.11 investigated the phase behavior of asphaltenes after CO2 injection into two dead crude oil samples using a high-pressure cell. They observed that the asphaltenes were more stable at high pressure and low temperature. Gonzalez et al.12 investigated the asphaltene phase behavior in a live reservoir fluid and a dead oil, and they found that the perturbed chain-statistical associating fluid theory equation of state model can explicitly account for the effects of CO2 on asphaltene aggregation behavior. Zanganeh et al.13,14 studied the effects of temperature and pressure on asphaltene deposition during CO2 injection processes via high-pressure cell and image processing techniques. They observed that the amount of deposited asphaltene decreases with a decrease in pressure and temperature has a prominent impact on the size of asphaltene particles. Behbahani and Ghotbi15 investigated asphaltene aggregation and adsorption using a sandstone core sample during miscible CO2 injection. They reported that the asphaltenes are adsorbed as a multilayer, with formation of large clusters.
In recent years, numerous efforts have been made to understand asphaltene aggregation at the molecular level using molecular simulations. Using molecular mechanics methods, Murgich et al.16 confirmed that asphaltene aggregation occurs because of stacking of aromatic rings. Rogel17 studied the interactions of asphaltene aggregates under vacuum and in different solvents. He reported that the stabilization energy is more favorable for molecules with a lower hydrogen-to-carbon ratio, higher aromaticity, and higher aromatic condensation degree. From molecular dynamics simulations, Headen et al.18 reported that asphaltene nanoaggregation occurs in toluene and heptane. They observed that asphaltenes have a stronger tendency to aggregate in scCO2 than in heptane and toluene.19 Goual et al.20 studied asphaltene aggregation under vacuum, and in toluene and heptane by using an umbrella sampling method to calculate the free energies of asphaltene dimerization. They revealed the important effect of the size of the polynuclear aromatic core, the length of the aliphatic side chains, and heteroatoms on the asphaltene interaction. Furthermore, using molecular dynamics simulations, Headen and Goual19,21 investigated the prevention of asphaltene deposition in scCO2 by inhibitors. They put forward suggestions for inhibitor selection to limit asphaltene aggregation in scCO2 injection processes. In fact, crude oil is a complex mixture that consists of different hydrocarbons, such as saturated hydrocarbons, aromatics, resins, and asphaltenes (SARA). When the injected scCO2 comes into contact with the crude oil, it leads to the modification of the equilibrium conditions and components of the crude oil. This modification is mainly caused by the intermolecular interactions in oil–scCO2 systems, favoring asphaltene precipitation. Hence, understanding the intermolecular interactions in the system is fundamental to unveiling the mechanism of asphaltene precipitation, which in the CO2 flooding process is still subject to debate.
In this work, molecular dynamics simulations were performed on an oil–scCO2 system to address primarily the mechanism of asphaltene aggregation upon scCO2 injection in crude oil with SARA. Firstly, the structure of the crude oil was examined in the absence of scCO2. Then 2-D density maps and the radius of gyration were calculated to examine structural variations of the oil components in the presence of scCO2. Finally, the non-bond interaction energy and the potential of mean force (PMF) profile of the molecules were calculated to reveal the mechanism of asphaltene aggregation induced by scCO2.
2. Simulation method
All of the molecular dynamics simulations were performed in the GROMACS 4.5.5 software package.22 VMD software23 was used to visualize the results. The CHARMM-27 force field (FF)24 was used for the oil molecules, which is one of the popular FFs and can reproduce the results in good agreement with experiments for oil molecules.25–27 CHARMM FF parameters for oil molecules were generated in Swissparam software.28 The classical three-site EPM2 model29 was employed to describe carbon dioxide molecules. The long-range electrostatic interactions were treated with the particle mesh Ewald (PME) method.30 The non-bonded potential truncation was performed with a cut-off radius of 1.0 nm for the Lennard-Jones potential.31 The temperature was controlled by the Nose–Hoover thermostat algorithm with a coupling constant of 0.2 ps.32,33 The pressure was controlled by the Parrinello–Rahman algorithm. All simulations were carried out at a temperature of 323.15 K and a pressure of 15 MPa, which are reasonable reservoir conditions and can keep CO2 in the supercritical state. The initial atomic velocities of the systems were set by the commonly used Maxwell–Boltzmann distribution. The time step was set to 1 fs for all the simulations. Periodic boundary conditions were employed in all three directions.2.1 Model molecules
According to differences of polarity and molecular weight, the crude oil is separated into SARA, which is a standard analysis method for crude oil composition.34,35 Although asphaltenes do not have a specific chemical formula, simplification is still feasible to study the main characteristics of asphaltenes. There is a consensus that a single, well-defined compound can model the asphaltene aggregation process successfully.36 In order to build a more realistic model of crude oil, a widely used continental-type model of asphaltenes (C54H65NO2S, MW: 792)37 from Venezuelan crude oil was chosen. The chemical structure and the molecular weight of the model are similar to those of asphaltenes in crude oil.38 The model of the resins (C48H76S, MW: 685)16 was also taken from the spectroscopic data of Venezuelan crude oil. Resins contain less aromatic nuclei and heteroatoms than asphaltenes. Benzoapyrene (C20H12, MW: 252) and dodecane (C12H26, MW: 170) represented the aromatic and saturate fractions of the crude oil, respectively. The molecular numbers of SARA were established on the basis of the mass ratio of 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30:35:20, referring to a real heavy crude oil sample.39,40 From the colloidal instability index (CII), asphaltene stability is mathematically expressed as the sum of asphaltenes and saturates divided by the sum of aromatics and resins.41 In this work, CII is equal to 0.54, meaning that asphaltenes are quite stable in the crude oil.42 The molecular configurations are shown in Fig. 1.
:30:35:20, referring to a real heavy crude oil sample.39,40 From the colloidal instability index (CII), asphaltene stability is mathematically expressed as the sum of asphaltenes and saturates divided by the sum of aromatics and resins.41 In this work, CII is equal to 0.54, meaning that asphaltenes are quite stable in the crude oil.42 The molecular configurations are shown in Fig. 1.
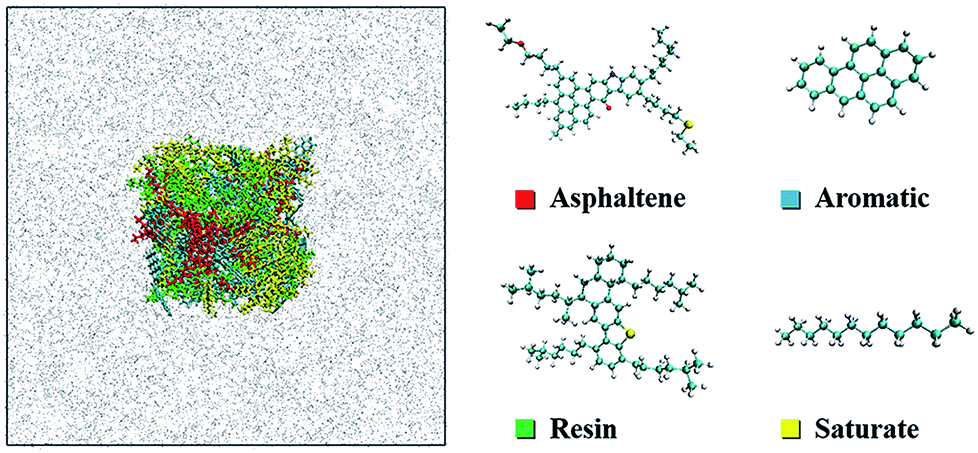 | ||
| Fig. 1 Sectional view of the simulation setup for the oil–scCO2 system (left) and the molecular structure of SARA components (right). Color scheme: cyan, carbon; white, hydrogen; red, oxygen; yellow, sulfur; blue, nitrogen; grey, CO2. | ||
2.2 Crude oil system
In order to obtain a proper molecular structure of crude oil, 8 asphaltenes, 18 resins, 60 aromatics and 50 saturates were put into a box randomly using the smart software package Packmol.43 The steepest descent method was applied for energy minimization. Then a 5 ns MD simulation was performed in the NVT ensemble. After that, a 5 ns MD simulation in the NPT ensemble was performed to represent the equilibrium process of the system and obtain a reasonable volume. Finally, an additional 5 ns MD simulation in the NVT ensemble was performed to collect data for the structural analysis of the crude oil.2.3 Crude oil–scCO2 system
Firstly, a bulk scCO2 box was built, which was kept in equilibrium by MD simulation in the NPT ensemble. Then, a volume the same size as the oil box was dug out in the center of the scCO2 box. After that, the oil box was placed in the hole. The size of the box is 12 × 12 × 12 nm3 containing 16970 scCO2 molecules. A 5 ns MD simulation in the NVT ensemble was performed, in which oil molecules were fixed. Next, the oil molecules were unfixed and a 20 ns MD simulation was performed on the crude oil–scCO2 system in the NVT ensemble.
2.4 Umbrella sampling
PMF is a valid parameter investigating the effective interaction between two molecules. The umbrella sampling method44 was used to compute the PMF along a given reaction coordinate. Through a series of umbrella sampling simulations, the binding energy between two molecules can be obtained from the PMF. To compute the PMF profile of the target molecules (asphaltene–asphaltene/asphaltene–resin/asphaltene–aromatic) versus the center-of-mass (COM) distance, three new simulation boxes including two target molecules were built and then energy minimization, MD simulation in the NPT ensemble, and MD simulation in the NVT ensemble were performed in sequence. The COM of one target molecule was pulled along the Z-axis while the other one was restrained. A series of frames were extracted from the trajectory. Each configuration corresponds to the desired COM spacing of less than 0.3 Å. Each of the separate simulations were run along the reaction coordinate ξ, biased by umbrella potentials wi(ξ). The umbrella potential is expressed as eqn (1):
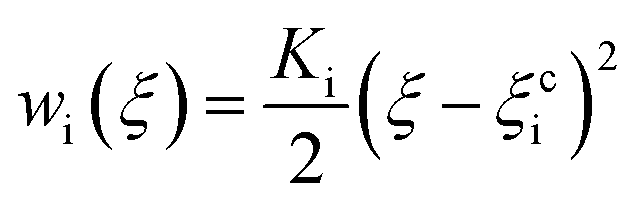 | (1) |
For each starting configuration of the sampling window, the data were collected for 5 ns in every sampling window. Three groups of target molecules were placed in scCO2 to perform the sampling simulation procedures again. The PMF profiles were obtained using the Weighted Histogram Analysis Method (WHAM).45,46
3. Results and discussion
3.1 Structure of the crude oil in the absence of scCO2
In order to show the effect of scCO2 on asphaltene aggregation, firstly, the structure of the crude oil was studied. When the crude oil system was in equilibrium, the calculated density of the oil was 0.95 g cm−3, coincident with that of actual heavy crude oil under reservoir conditions.47,48 The configuration of the crude oil is shown in Fig. 2. It shows that the resin molecules tend to aggregate with the asphaltene molecules, rather than coating the asphaltene nanoaggregates, leading to mixed asphaltene–resin aggregates. This result is in line with results from molecular thermodynamics49 and experiments.50 Aromatic molecules are distributed closer to the central area of the crude oil compared with the saturates. The saturates tend to be dispersed in the outer region of the heavy components. It should be noted that no parallel stacking configuration of asphaltenes occurs in the crude oil. This may result from the steric effect of the resin and aromatic molecules. Next, we will investigate the structure of the crude oil in the presence of scCO2.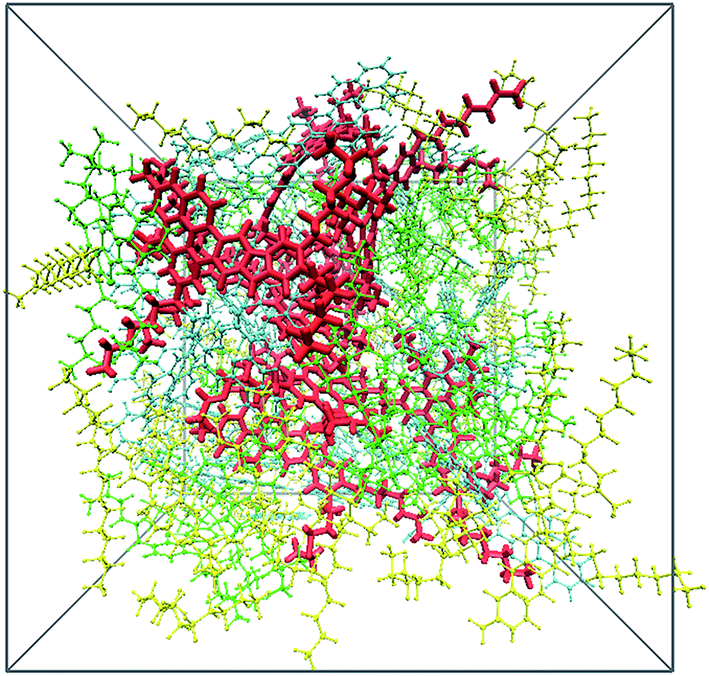 | ||
| Fig. 2 Perspective view of the crude oil system. Color scheme: red, asphaltenes; green, resins; blue, aromatics; yellow, saturates. | ||
3.2 Structure of the crude oil in the presence of scCO2
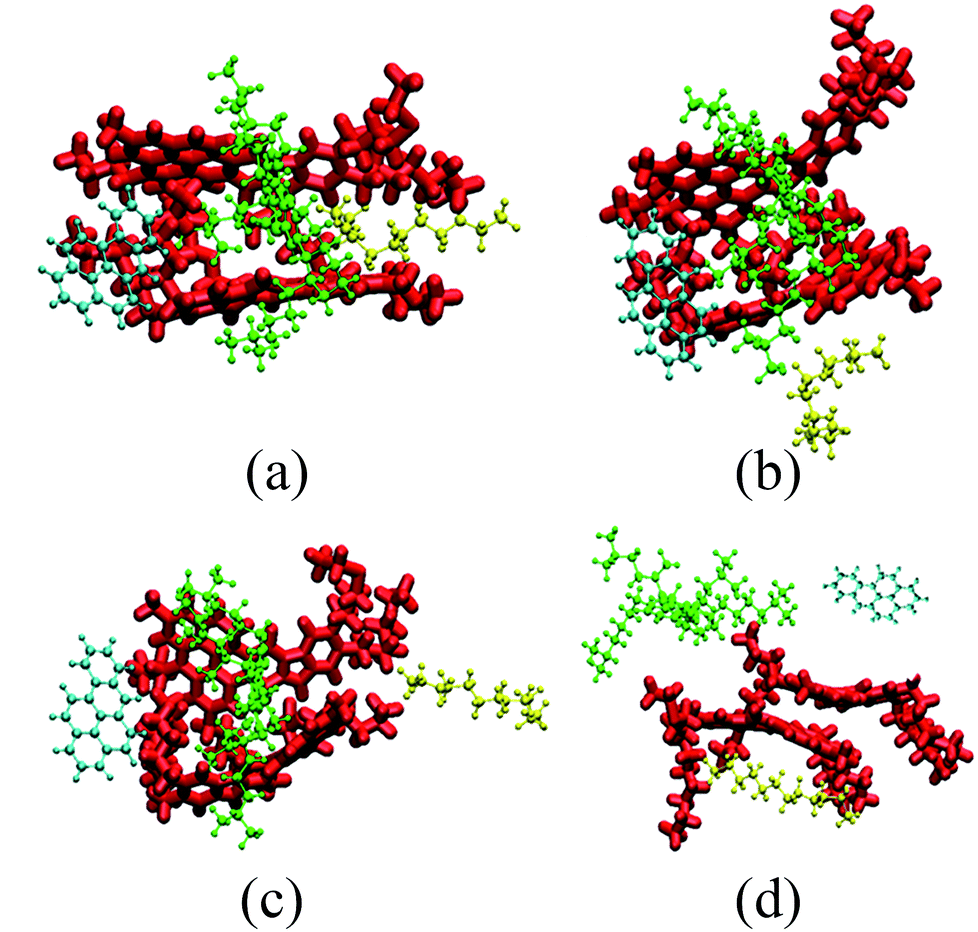 | ||
| Fig. 3 Representative snapshots of five typical molecules. Snapshots are taken at (a) 0 ns, (b) 1.2 ns, (c) 3.5 ns and (d) 10 ns. The color scheme used in the snapshots is the same as that used in Fig. 2. For clarity, scCO2 is not shown. | ||
Fig. 3 also shows the significant selective extraction of scCO2 in relation to EOR in the CO2 flooding process.52 The order of this extraction is saturate > aromatic > resin, due to the differences in molecular weight and polarity. This can also be described by the diffusibility of SARA in scCO2; the diffusion coefficients of SARA in scCO2 are equal to 9.4462, 3.5748, 2.1838, and 1.4199 (10−9 m2 s−1) respectively. The order of the diffusion coefficients of SARA is saturate > aromatic > resin, indicating that scCO2 has the strongest ability to dissolve saturates, i.e. the highest extracting power.
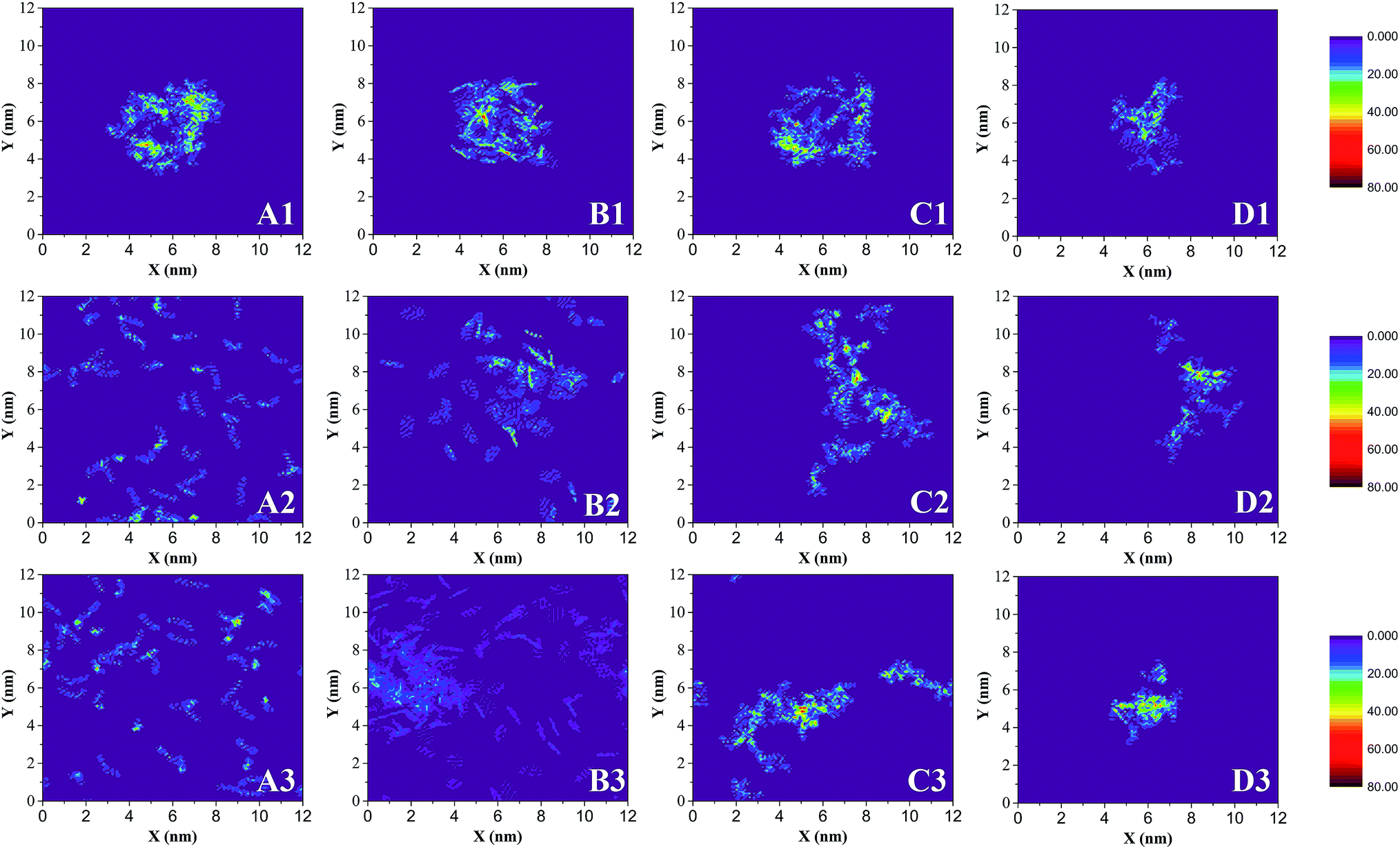 | ||
| Fig. 4 Planar density distribution of SARA. Z is the direction perpendicular to the surface, and the densities are expressed in number of atoms/molecules nm−3. (A) Saturates, (B) aromatics, (C) resins, and (D) asphaltenes. (1), (2) and (3) are taken at 0 ns, 3 ns and 20 ns, respectively. | ||
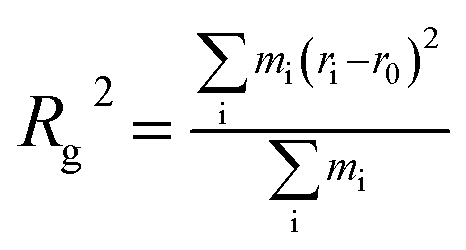 | (2) |
Fig. 5 shows the Rg of asphaltene aggregates as a function of time. In less than 7 ns, Rg increases quickly, indicating the rapid separation of asphaltene molecules due to the selective extraction of oil components by scCO2. At about 7 ns, an obvious Rg peak occurs, showing that asphaltenes are dispersed in scCO2 to the maximum degree. From 7 ns to 10 ns, Rg decreases steeply, implying the rapid association of the asphaltene molecules. At greater than 10 ns, Rg is essentially unchanged, indicating the stabilization of the asphaltene aggregates. The results are in agreement with the description in Fig. 4(D1–D3). This can be visualized by representative configurations of the contact surface between asphaltenes and scCO2, as shown in Fig. 5. With the extraction of saturates, aromatics, and resins into scCO2, more and more scCO2 occupies the accessible space between asphaltenes. This leads to increasing separation between asphaltenes and a subsequent increase in the contact area between asphaltenes and scCO2, as described by the configuration at 7 ns in Fig. 5. This result indicates that the selective extraction of oil components by scCO2 causes the asphaltenes to be dispersed first, rather than immediately aggregating to form clusters. After most of the components except for the asphaltenes in the crude oil are extracted into scCO2, the asphaltenes begin to associate and form dense nanoaggregates. This can be described by the configuration at 20 ns in Fig. 5. In this work, the diameter of the nanoaggregates is about 2 nm, with an aggregation number of 8, corresponding to the definition of a nanoaggregate in the Yen–Mullins model.10 We repeated the simulations with different asphaltene molecules and observed the same phenomenon.
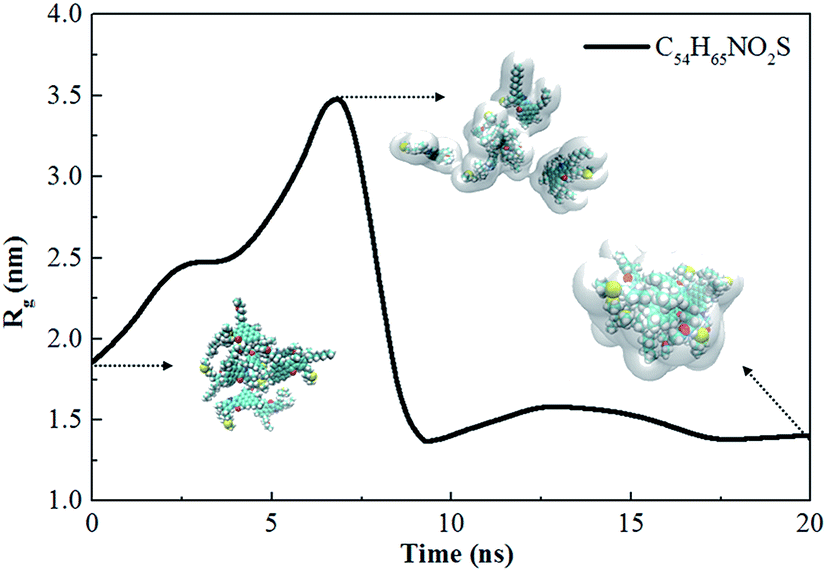 | ||
| Fig. 5 The time evolution of the gyration radius during a 20 ns simulation. The inset snapshots of asphaltene are taken at 0 ns, 7 ns and 20 ns. The color scheme used in the snapshots is the same as that used in Fig. 1, and the translucent silver surface is the contact surface between asphaltene and CO2. | ||
From the above analysis of the density distributions and Rg, a two-step process of asphaltene aggregation occurs when scCO2 is injected into crude oil. Asphaltenes experience a process of dispersion as the scCO2 selectively extracts the oil components. After the end of the extraction, the asphaltenes begin to associate with each other and form compact nanoaggregates. This finding demonstrates that the efficiency of inhibiting or mitigating asphaltene aggregation in scCO2 flooding can be significantly improved by controlling the process of selective extraction of oil components by scCO2. Moreover, the time of adding an inhibitor or dispersant into the oil–scCO2 system also influences the inhibition effect.
3.3 The mechanism of asphaltene aggregation
| Interaction energies for asphaltene–asphaltene | van der Waals energy (kcal mol−1) | Coulomb energy (kcal mol−1) | Total (kcal mol−1) |
|---|---|---|---|
| Without scCO2 | −154.54 ± 1.65 | 29.91 ± 0.52 | −124.63 ± 2.17 |
| With scCO2 | −173.66 ± 6.17 | 25.57 + 2.15 | −148.09 ± 8.32 |
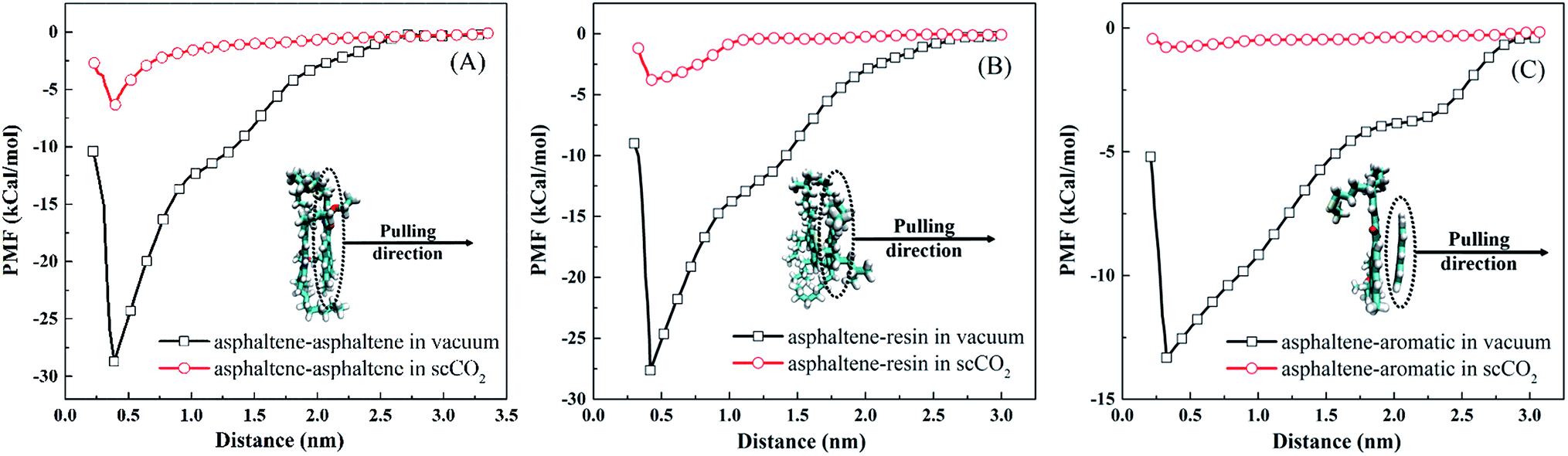 | ||
| Fig. 6 Potential of mean force profiles for (A) asphaltene–asphaltene, (B) asphaltene–resin, and (C) asphaltene–aromatic, under vacuum and in scCO2. The inset snapshots are the minimum energy state structures, and the color scheme used in the snapshots is the same as that used in Fig. 1. | ||
| Binding energy (kcal mol−1) | Asphaltene–asphaltene | Asphaltene–resin | Asphaltene–aromatic |
|---|---|---|---|
| Under vacuum | −28.82 ± 0.65 | −27.61 ± 0.62 | −13.94 ± 0.47 |
| In scCO2 | −6.90 ± 0.42 | −3.82 ± 0.38 | −0.51 ± 0.09 |
In Fig. 6(A), the asphaltene dimer with the lowest energy shows a diagonal structure. In this structure, the same heteroatoms are in opposite corners at the maximum distance away from each other. This is in agreement with the results obtained by recent quantum mechanical calculations.57 A shoulder occurs at about 0.9 nm for the PMF curve under vacuum, corresponding to the offset structure of the asphaltene dimer.20 The PMF reaches a steady value in scCO2 as the separation between them is about 0.6 nm. This indicates that asphaltene aggregates can be stably present in scCO2 as the separation of two asphaltene molecules is less than 0.6 nm. In Table 2, the asphaltene–asphaltene binding energy is −28.82 kcal mol−1 under vacuum. This value is slightly lower than that obtained by quantum mechanical calculations58 and higher than that from atomistic calculations.59 The difference may come from the difference in the asphaltene model used in these studies. π–π interaction is believed to be the driving force to form the face-to-face stacked structure, which was confirmed by recent studies.60,61 The binding energy of asphaltene–asphaltene in scCO2 drops to −6.9 kcal mol−1, which is much lower than that under vacuum. This indicates that asphaltenes are less prone to aggregation in scCO2 than under vacuum. The remaining strength of association between asphaltene molecules is the reason why the asphaltene aggregates are stable in scCO2, as shown in Fig. 4(D3) and Fig. 5.
In Fig. 6(B), the PMF profile of asphaltene–resin is similar to that of asphaltene–asphaltene under vacuum. The binding energy between asphaltenes and resins is −27.61 kcal mol−1, which is very close to that between two asphaltenes, as shown in Table 2. This indicates that resins tend to associate with asphaltenes and form mixed asphaltene–resin aggregates in crude oil. In scCO2, the binding energy between asphaltenes and resins drops to −3.82 kcal mol−1. This indicates that it is hard for the mixed asphaltene–resin aggregates to survive in scCO2. However, this nonzero binding energy implies that the resin molecules can be dragged together by asphaltenes. This can be observed in Fig. 3 and 4(C3).
In Table 2, the binding energy of asphaltene–aromatic under vacuum is −13.94 kcal mol−1, significantly lower than that of either asphaltene–asphaltene or asphaltene–resin. Thus, compared with the resins, the aromatics are more distant from the asphaltenes in crude oil. The binding energy between asphaltenes and aromatics is −0.51 kcal mol−1 in scCO2. This indicates that scCO2 can significantly reduce the effective interaction between asphaltenes and aromatics. Consequently, aromatics can be more easily extracted from the crude oil by scCO2 than resins.
It can be concluded that the order of the binding energy is asphaltene–asphaltene > asphaltene–resin > asphaltene–aromatic, both under vacuum and in scCO2. The ratio of the binding energies for asphaltene–asphaltene, asphaltene–resin and asphaltene–aromatic changes from 1:0.96:0.48 under vacuum to 1:0.55:0.07 in scCO2. The results also indicate that scCO2 can easily destroy the associations of asphaltene–resin and asphaltene–aromatic, which promotes asphaltene aggregation. From the above analysis, we can conclude that asphaltene aggregation in scCO2 results from the synergistic effect of selective extraction of oil components and the remaining strength of association between asphaltenes.
4. Conclusion
In this study, MD simulations were performed to investigate the dynamic process and microscopic mechanism of asphaltene aggregation in scCO2. The results show that resins tend to associate with asphaltenes in the absence of scCO2, and asphaltenes cannot form compact aggregates because of the steric effect of the resin and aromatic molecules. In the presence of scCO2, the order of selective extraction is saturate > aromatic > resin. This leads to asphaltenes accumulating and forming compact nanoaggregates in scCO2. In the process of asphaltene aggregation, asphaltenes firstly separate from each other, due to the extraction behavior of scCO2. After the separation stage, the asphaltenes associate with each other to form nanoaggregates that stably exist in scCO2.The aggregation mechanism was investigated from the point of view of energy, and it was observed that vdW interaction plays a predominant role in asphaltene aggregation, both in the absence and presence of scCO2. Furthermore, the coulomb interaction energy decreases and the vdW interaction energy increases due to the presence of scCO2, which aggravates asphaltene aggregation. The free energy calculations show that the binding energy of asphaltene–asphaltene is close to that of asphaltene–resin under vacuum, which implies that resins tend to mix with asphaltenes in crude oil. In scCO2, the binding energies of asphaltene–resin and asphaltene–aromatic were significantly decreased. Meanwhile the remaining −6.9 kcal mol−1 binding energy of asphaltene–asphaltene in scCO2 is responsible for the existence of asphaltene aggregates.
The dynamic process and microscopic mechanism are helpful in providing some useful information for guiding scCO2-enhanced oil recovery, especially for predicting and avoiding asphaltene aggregation. However, research on asphaltene inhibition is still inadequate, and our future work will focus on effective approaches to asphaltene inhibition.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the PetroChina Innovation Foundation (2017D-5007-0206), the National Basic Research Program of China (2014CB239204), and the Fundamental Research Funds for the Central Universities (15CX08003A).References
- F. Gozalpour, S. R. Ren, B. Tohidi and F. Gozalpour, Oil Gas Sci. Technol., 2005, 60, 537–546 CAS
.
- E. Tzimas, A. Georgakaki, C. G. Cortes and S. D. Peteves, Report EUR 21895 EN, European Commission, 2005 Search PubMed
- R. Farajzadeh, A. Andrianov, H. Bruining and P. L. J. Zitha, Ind. Eng. Chem. Res., 2009, 48, 4542–4552 CrossRef CAS
- M. L. Godec, V. A. Kuuskraa and P. Dipietro, Energy Fuels, 2013, 27, 4183–4189 CrossRef CAS
- R. G. Santos, W. Loh, A. C. Bannwart and O. V. Trevisan, Braz. J. Chem. Eng., 2014, 31, 571–590 CrossRef
- S. Rudyk, S. Hussain and P. Spirov, J. Supercrit. Fluids, 2013, 78, 63–69 CrossRef CAS
- R. Nguele, M. R. Ghulami, K. Sasaki, H. Said-Al Salim, A. Widiatmojo, Y. Sugai and M. Nakano, Energy Fuels, 2016, 30, 1266–1278 CAS
- B. Ju, T. Fan and Z. Jiang, J. Pet. Sci. Eng., 2013, 109, 144–154 CrossRef CAS
- I. David Borton, D. S. Pinkston, M. R. Hurt, X. Tan, K. Azyat, A. Scherer, R. Tykwinski, M. Gray, K. Qian and H. I. Kenttämaa, Energy Fuels, 2015, 24, 5548–5559 CrossRef
- O. C. Mullins, H. Sabbah, J. Eyssautier, A. E. Pomerantz, L. Barré, A. B. Andrews, Y. Ruizmorales, F. Mostowfi, R. Mcfarlane and L. Goual, Energy Fuels, 2012, 26, 3986–4003 CrossRef CAS
- S. Verdier, H. Carrier, S. I. Andersen and J.-L. Daridon, Energy Fuels, 2006, 20, 1584–1590 CrossRef CAS
- D. L. Gonzalez, F. M. Vargas, G. J. Hirasaki and W. G. Chapman, Energy Fuels, 2008, 22, 757–762 CrossRef CAS
- P. Zanganeh, S. Ayatollahi, A. Alamdari, A. Zolghadr, H. Dashti and S. Kord, Energy Fuels, 2012, 26, 1412–1419 CrossRef CAS
- P. Zanganeh, H. Dashti and S. Ayatollahi, Fuel, 2015, 160, 132–139 CrossRef CAS
- T. J. Behbahani, C. Ghotbi, V. Taghikhani and A. Shahrabadi, Fuel, 2014, 133, 63–72 CrossRef
- J. Murgich, J. Rodríguez and Y. Aray, Energy Fuels, 1996, 10, 68–76 CrossRef CAS
- E. Rogel, Energy Fuels, 2000, 14, 566–574 CrossRef CAS
- T. F. Headen, E. S. Boek and N. T. Skipper, Energy Fuels, 2009, 23, 1220–1229 CrossRef CAS
- T. F. Headen and E. S. Boek, Energy Fuels, 2010, 25, 503–508 CrossRef
- M. Sedghi, L. Goual, W. Welch and J. Kubelka, J. Phys. Chem. B, 2013, 117, 5765 CrossRef CAS PubMed
- E. Lowry, M. Sedghi and L. Goual, Energy Fuels, 2016, 30, 7187–7195 CrossRef CAS
- B. Hess, C. Kutzner, d. S. D. Van and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed
- P. Bjelkmar, P. Larsson, M. A. Cuendet, B. Hess and E. Lindahl, J. Chem. Theory Comput., 2010, 6, 459–466 CrossRef CAS PubMed
- W. Xie, H. Liu, Y. Sun, H. Fu and Y. Liang, Appl. Surf. Sci., 2017, 392, 747–759 CrossRef CAS
- Y. Yan, C. Li, Z. Dong, T. Fang, B. Sun and J. Zhang, Fuel, 2017, 190, 253–259 CrossRef CAS
- Y. Yu, I. A. Fursule, L. C. Mills, D. L. Englert, B. J. Berron and C. M. Payne, J. Mol. Graphics Modell., 2017, 32–42 CrossRef CAS PubMed
- V. Zoete, M. A. Cuendet, A. Grosdidier and O. Michielin, J. Comput. Chem., 2011, 32, 2359 CrossRef CAS PubMed
- J. G. Harris and K. H. Yung, J. Phys. Chem., 1995, 99, 12021–12024 CrossRef CAS
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS
- L. Verlet, Phys. Rev., 1967, 159, 98 CrossRef CAS
- D. J. Evans and B. L. Holian, J. Chem. Phys., 1985, 83, 4069–4074 CrossRef CAS
- G. J. Martyna, M. L. Klein and M. Tuckerman, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef
- K. A. Bissada, J. Tan, E. Szymczyk, M. Darnell, M. Mei and J. Zhou, Org. Geochem., 2016, 95, 21–28 CrossRef CAS
- N. Aske, H. Kallevik and J. Sjöblom, Energy Fuels, 2001, 15, 1304–1312 CrossRef CAS
- B. Breure, D. Subramanian, J. Leys, C. J. Peters and M. A. Anisimov, Energy Fuels, 2012, 27, 172–176 CrossRef
- E. Rogel, Colloids Surf., A, 1995, 104, 85–93 CrossRef CAS
- O. C. Mullins, B. Martínez-Haya and A. G. Marshall, Energy Fuels, 2008, 22, 1765–1773 CrossRef CAS
- P. Liu, Q. Shi, K. H. Chung, Y. Zhang, N. Pan, S. Zhao and C. Xu, Energy Fuels, 2010, 24, 5089–5096 CrossRef CAS
- S. Ashoori, M. Sharifi, M. Masoumi and M. M. Salehi, Egypt. J. Pet., 2017, 26(1), 209–213 CrossRef
- I. A. Wiehe and R. J. Kennedy, Energy Fuels, 2000, 14, 56–59 CrossRef CAS
- A. P. W. S. Asomaning, Heat Transfer Eng., 2000, 21, 10–16 CrossRef
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed
- J. Kästner, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 932–942 CrossRef
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS
- J. S. Hub, B. L. De Groot and D. Van Der Spoel, J. Chem. Theory Comput., 2010, 6, 3713–3720 CrossRef CAS
- S. E. Quiñones-Cisneros, S. I. Andersen and J. Creek, Energy Fuels, 2005, 19, 1314–1318 CrossRef
- B. Quail, G. Hill and K. N. Jha, Ind. Eng. Chem. Res., 1988, 27, 519–523 CrossRef CAS
- E. Rogel, Energy Fuels, 2008, 22, 3922–3929 CrossRef CAS
- M. Sedghi and L. Goual, Energy Fuels, 2010, 24, 2275–2280 CrossRef CAS
- A. T. Grogan, V. W. Pinczewski and G. J. Ruskauff, SPE Reservoir Eng., 1988, 3, 93–102 CrossRef CAS
- T. Fang, J. Shi, X. Sun, Y. Shen, Y. Yan, J. Zhang and B. Liu, J. Supercrit. Fluids, 2015, 113, 10–15 CrossRef
- R. Nguele, M. R. Ghulami, K. Sasaki, S. A. Salim, A. Widiatmojo, Y. Sugai and M. Nakano, Energy Fuels, 2016, 30(2), 1266–1278 CAS
- A. Alizadeh, H. Nakhli, R. Kharrat, M. H. Ghazanfari and M. Aghajani, Energy Sources, Part A, 2014, 36, 1523–1530 CrossRef CAS
- M. K. Shimamura and T. Deguchi, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2002, 65, 126 CrossRef PubMed
- P. Luo, X. Wang and Y. Gu, Fluid Phase Equilib., 2010, 291, 103–110 CrossRef CAS
- M. Mousavi, T. Abdollahi, F. Pahlavan and E. H. Fini, Fuel, 2016, 183, 262–271 CrossRef CAS
- A. Ortega-Rodriguez, C. Lira-Galeana, Y. Ruiz-Morales and S. Cruz, Pet. Sci. Technol., 2001, 19, 245–256 CrossRef CAS
- F. Alvarez-Ramirez, E. Ramirez-Jaramillo and Y. Ruiz-Morales, Energy Fuels, 2006, 20, 195–204 CrossRef CAS
- H. Wang, H. Xu, W. Jia, J. Liu and S. Ren, Energy Fuels, 2017, 31, 2488–2495 CrossRef CAS
- J. Liu, Y. Zhao and S. Ren, Energy Fuels, 2015, 29, 1233–1242 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09736k |
| This journal is © The Royal Society of Chemistry 2017 |