
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthetic routes for a variety of halogenated (chiral) acetic acids from diethyl malonate†
Manuel R. Mazenauera,
Stole Manova,
Vanessa M. Galatia,
Philipp Kappelera and
Jürgen Stohner *ab
*ab
aInstitute of Chemistry and Biotechnology, Zurich University of Applied Sciences, Einsiedlerstrasse 31, CH-8820 Wädenswil, Switzerland. E-mail: sthj@thaw.ch
bGuest Scientist, ETH zürich, Laboratory for Physical Chemistry, CH-8093 zürich, Switzerland. E-mail: just@phys.chem.ethz.ch
First published on 7th December 2017
Abstract
Chiral halomethane is the smallest stable molecule with a single asymmetric C-atom and halogenated acetic acids often serve as precursors. We focus on a synthetic route to synthesise chiral halogenated acetic acids with F, Cl, Br, and H/D isotopic substitution at the α-C-atom starting from diethyl malonate. This reactant is easily available, cheap and allows the obtainment of target acids in a few reaction steps with great versatility. Among all of the possible fully halogenated acetic acids (more than one hundred, which are, in principle, accessible by this route), there are only a small number of chiral halogenated acetic acids, which have been synthesized following the devised synthetic route.
Introduction
Small chiral molecules are excellent prototype candidates for the investigation of molecular parity violation,1,2 the determination of absolute configurations by new direct methods based on Coulomb explosion imaging,3–6 and the study of frequency dependent chiroptical properties.7,8 Due to the small number of atoms, they are also well suited to high-quality ab initio calculations9–12 that are relevant for a detailed understanding of the spectroscopic investigations of chiral molecules.13–15The synthesis of chiral acetic acid with all three hydrogen isotopes at the chiral C-atom and known handedness was truly pioneering work for the investigation of enzyme reaction mechanisms back in 1969.16–18 Here we focus on chiral halogenated acetic acids,19 CXYZCOOH, which are of general interest in the fields mentioned above. They serve as precursors to halomethanes, specifically CHBrClF,20–23 CHBrFI,24 and isotopically chiral CHDXY.25–28 CHBrClF (7) itself has been subject to very early attempts to measure parity violation effects in molecules.29 Since the prediction and experimental verification of the weak interaction in particle physics in around 1956/57,30,31 it became immediately clear that this interaction also has consequences in chemistry: for an isolated enantiomer, racemization following R = S leads to a tiny heat of reaction, ΔrH°, in the order of 10−15 kJ mol−1 (ref. 32 and 33) which can be related to the parity violating energy difference, ΔpvE, between both enantiomers according to ΔpvE ≈ ΔrH°, thus leading to slightly different absorption spectra of isolated R and S molecules. Attempts to measure these effects have failed until today,32–34 but molecular parity violation is still an active field of research.
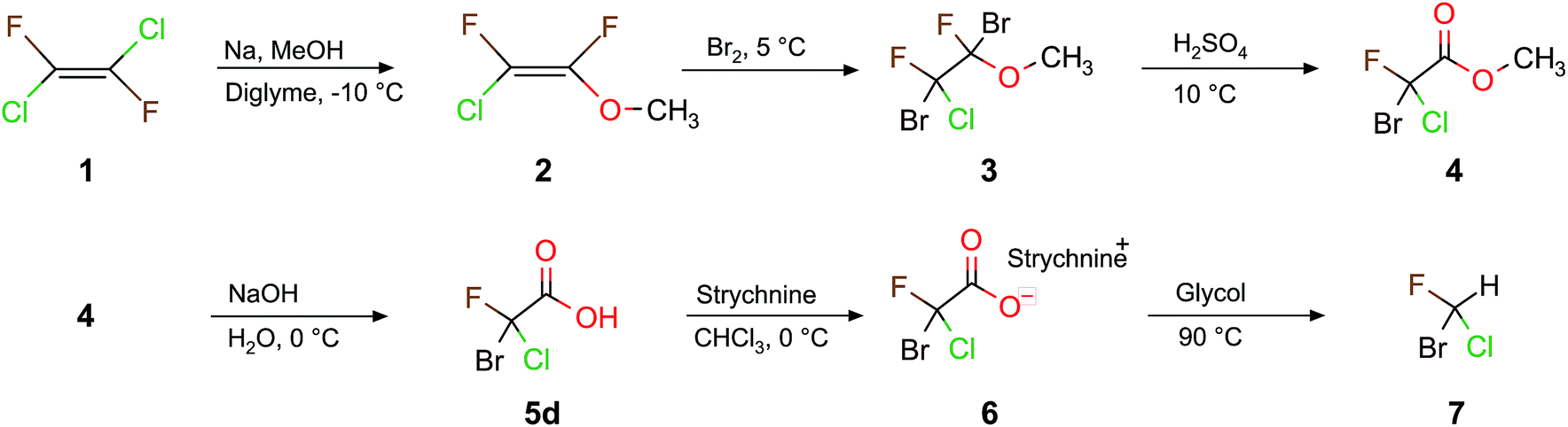
The synthesis of these halomethanes usually starts from perhalogenated ethene (see Scheme 1).21,23 This reactant is not very well suited for the synthesis of a large variety of halogenated acetic acids since it has a low boiling point and is subject to producing undesirable byproducts. Furthermore, due to the CFC(chlorofluorocarbon)-ban in 1989, its commercial availability is very limited. We therefore examined a synthetic pathway starting from an easily available diethyl malonate.35–37 We explored the reaction scheme (Scheme 2) for the synthesis of a whole series of halogenated chiral (and also achiral) acetic acids with high yields and purity.38 Some of these lead directly to the corresponding chiral halomethane upon decarboxylation.39,40
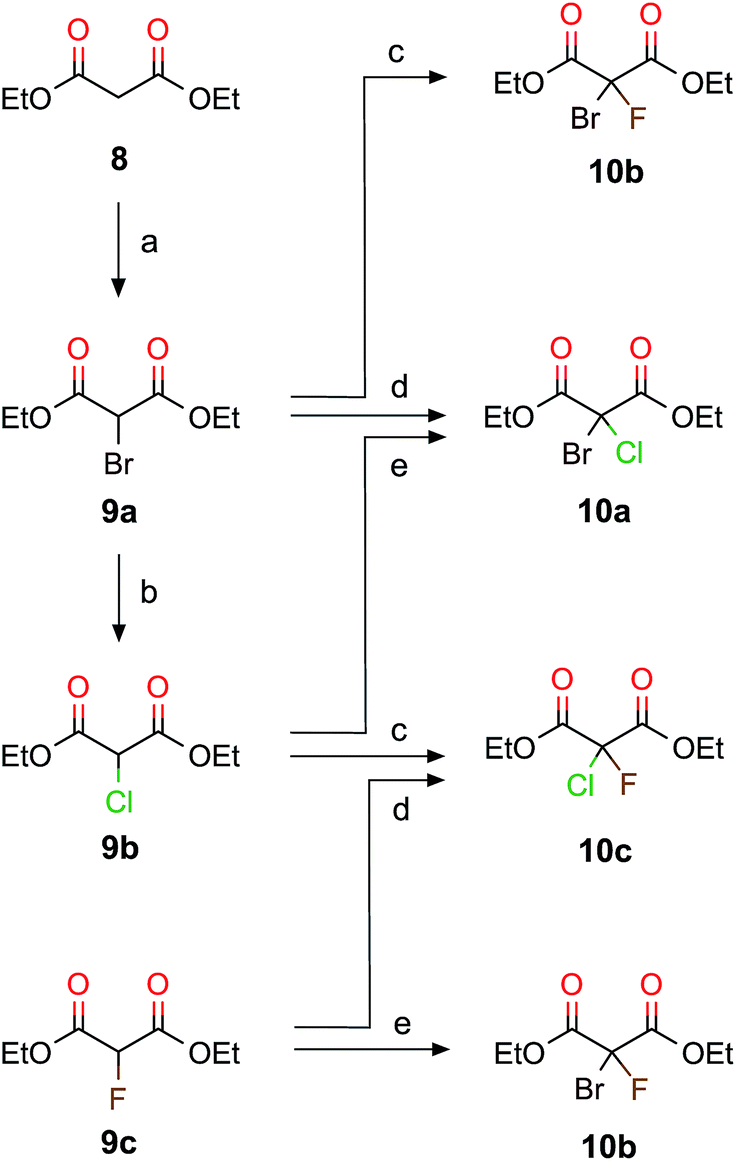 | ||
Scheme 2 The halogenated diethyl malonates 10a–c were obtained by selective stepwise halogenation. (a) (1) NaH, rt, 2.5 h, (2) CuBr2, DMSO, 40 °C, 1.5 h; (b) N-chlorosuccinimide, DMSO, rt, 24 h; (c) (1) NaH, rt, 3 h, (2) N-fluoro-2,4,6-trimethylpyridinium triflate, THF, 0 °C, 10 min; (d) NaOCl, AcOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetone (2:5), 0 °C, 1–3 h and (e) NaOBr, AcOH:acetone (2:5), 0 °C, 12 h. :acetone (2:5), 0 °C, 1–3 h and (e) NaOBr, AcOH:acetone (2:5), 0 °C, 12 h. | ||
Results and discussion
First we focus on CHBrClF (7) and its corresponding acetic acid (5d). A complete multistep synthesis for 7 was described by Doyle and Vogl22,39 and optimized by Beil et al.21 a few years later. Both research groups21,22,39 used the same experimental procedure as shown in Scheme 1 but instead of using 1,2-dichloro-1,2-difluoroethylene (1) they started with chlorotrifluoroethylene with different corresponding reaction conditions for some steps. Our modified synthetic route (Scheme 1) has some advantages compared to the past approaches that use chlorotrifluoroethene as the starting material, namely 1 has a higher boiling point of about 19 °C (−27 °C for chlorotrifluoroethene). Furthermore, the symmetric ethene 1 leads to fewer byproducts than when using chlorotrifluoroethene. The overall yields, however, do not differ much.In this work we present pathways to obtain chiral haloacetic acids starting from diethyl malonate 8. In comparison to past procedures starting from perhalogenated ethene20–22 for the synthesis of chiral halogenated acetic acids as just described in Scheme 1, the procedure advocated here has several advantages: (1) the reactant 8 is a common chemical reagent, cheap and easily accessible; (2) due to its CH acidity at the α-carbon atom it is a perfect starting material for halogenation reactions and (3) only a small number of reaction steps are needed. This paves the way to synthesize different dihalogenated diethyl malonates and, through decarboxylation, the corresponding halogenated acetic acids. Scheme 2 shows the halogenation reactions explored to synthesize various combinations of brominated, chlorinated and fluorinated diethyl malonates together with the main reaction details. The yields from our reaction in Scheme 2 are summarized and compared to previous work in Table 1.
| Species | Yield (in %) and reference | Yield (in %) in this work |
|---|---|---|
| 9a | 83 (ref. 43) (mw.); 62 (ref. 45) (l.i.); 96 (ref. 46) (l.i.) | 99 |
| 9b | 81 (ref. 43) (mw.); 97 (ref. 47) (from ethyl 3,3-diethoxyacrylate) | 99 |
| 10a | 99 (ref. 48) (l.i.); 90 (ref. 44) (from diazo compound) | 98 (from 9a); 100 (from 9b) |
| 10b | — | 86 (from 9a); 85 (from 9c) |
| 10c | 78 (ref. 49) | 73 (from 9b); 86 (from 9c) |
| 5a | n.s.37 (no deuteration, from 10a) | 76 (deuterated ∼ 89%, from 10a) |
| 5b | 32 (ref. 50 and 51) (deuterated ∼ 95%, total 3 steps, from halogenated ethene) | 51 (deuterated ∼ 97%, from 10b) |
| 5c | 41 (ref. 51–53) (deuterated ∼ 87%, total 3 steps, from halogenated ethene) | 49 (deuterated ∼ 94%, from 10c) |
| 5d | 19 (ref. 22); 36 (ref. 21) (total 4 steps, from halogenated ethene) | 26 (from 10c) |
| 7 | 50 to 70 (ref. 39) (from 5d) | 46 (from 5d) |
The halogenation of 8 with bromine leads to diethyl bromomalonate (9a) (experimental conditions are detailed in Scheme 2); the same reaction has been studied in the past by irradiating with light in the presence of Br2.36 Following Scheme 2, diethyl chloromalonate (9b) was obtained from 9a by replacing bromine with chlorine (99% yield), whereas diethyl fluoromalonate (9c) was inaccessible with usual mild fluorination agents. Fluorination is often performed with HF and triethylamine,41,42 which we did not try to reproduce in our laboratory since 9c is commercially available. 9a and 9b have also been obtained from 8 in the presence of MgX2 (X = Br, Cl, and I) under microwave irradiation.43 We tested different methods to obtain 9b directly from 8, but these attempts resulted in poor yields and mainly the formation of diethyl dichloromalonate. However, the two step procedure from 8 to 9b via 9a is an efficient alternative.
When the acidic hydrogen in 9a is substituted with chlorine, we obtain the diethyl bromochloromalonate (10a, 97% yield). When optimizing the procedure described by Read and McMath,35 Zimmer et al.37 started from 8 and added bromine to synthesise 9a and added SO2Cl2 to synthesize 10a. Recently, 10a was obtained from the diazo compound of diethyl malonate with a yield of about 90% (ref. 44) in the presence of a Cu-catalyst. Another way to synthesize 10a is by the bromination of 9b (100% yield). To obtain diethyl bromofluoromalonate (10b), we fluorinated 9a (86% yield) or brominated 9c (82% yield). Two routes are possible to synthesize diethyl chlorofluoromalonate (10c), either by fluorinating 9b (73% yield) or chlorinating 9c (86% yield). Thus, we successfully explored at least two different routes to synthesize diethyl dihalogenmalonates which enabled us to choose the pathway with the higher yield (see Table 1) and the most suitable reaction conditions.
Scheme 3 summarizes the synthetic pathways from a dihalogenated diethylmalonate to the corresponding dihalo acetic acid upon introducing D or H. Decarboxylation to the deuterated acetic acids 5a–c was successfully performed for deutero bromochloroacetic acid 5a with a yield of 67%. Deutero bromofluoroacetic acid (5b, 51% yield) and deutero chlorofluoroacetic acid (5c, 49% yield) were also obtained.
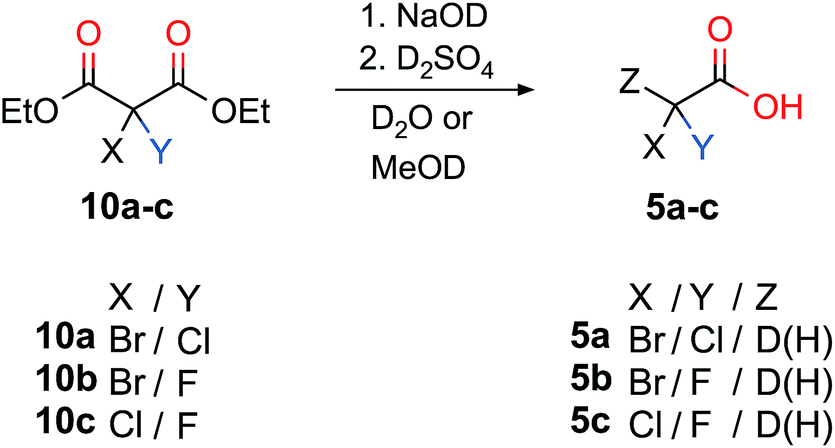 | ||
| Scheme 3 The chiral acetic acids (5a–d) were obtained via the unilateral decarboxylation of the halogenated diethylmalonates (10a–c). For 5b and 5c the decarboxylation was carried out without NaOD to obtain Z = H. | ||
Scheme 4 depicts the complete synthetic pathway from 8 to chiral halomethane 7. The decarboxylation of 10c to the bromochlorofluoroacetic acid 5d (Scheme 4) requires two more reaction steps compared to the decarboxylation when D or H are inserted (see Scheme 2). Firstly, 10c was saponified specifically to the monoethylester potassium salt. The Hunsdiecker decarboxylation54 was carried out with the corresponding silver salt (11c), which was obtained in a second step using a silver loaded ion exchange resin. Secondly, the resulting silver salt was then decarboxylated to obtain bromochlorofluoroacetate as product. The overall yield of these two steps was approximately 26% and the purity of the crude product was better than 80%. The decarboxylation was carried out with up to 20 mmol (≈6 g) of silver salt. Thirdly, a simple saponification of bromochlorofluoroacetate (4) with NaOH results in 5d with a yield of 99%. Table 1 summarizes our results with respect to recent synthetic attempts to obtain monohalogenated (9), and dihalogenated (10) malonates, as well as the corresponding chiral halogenated acetic acids.
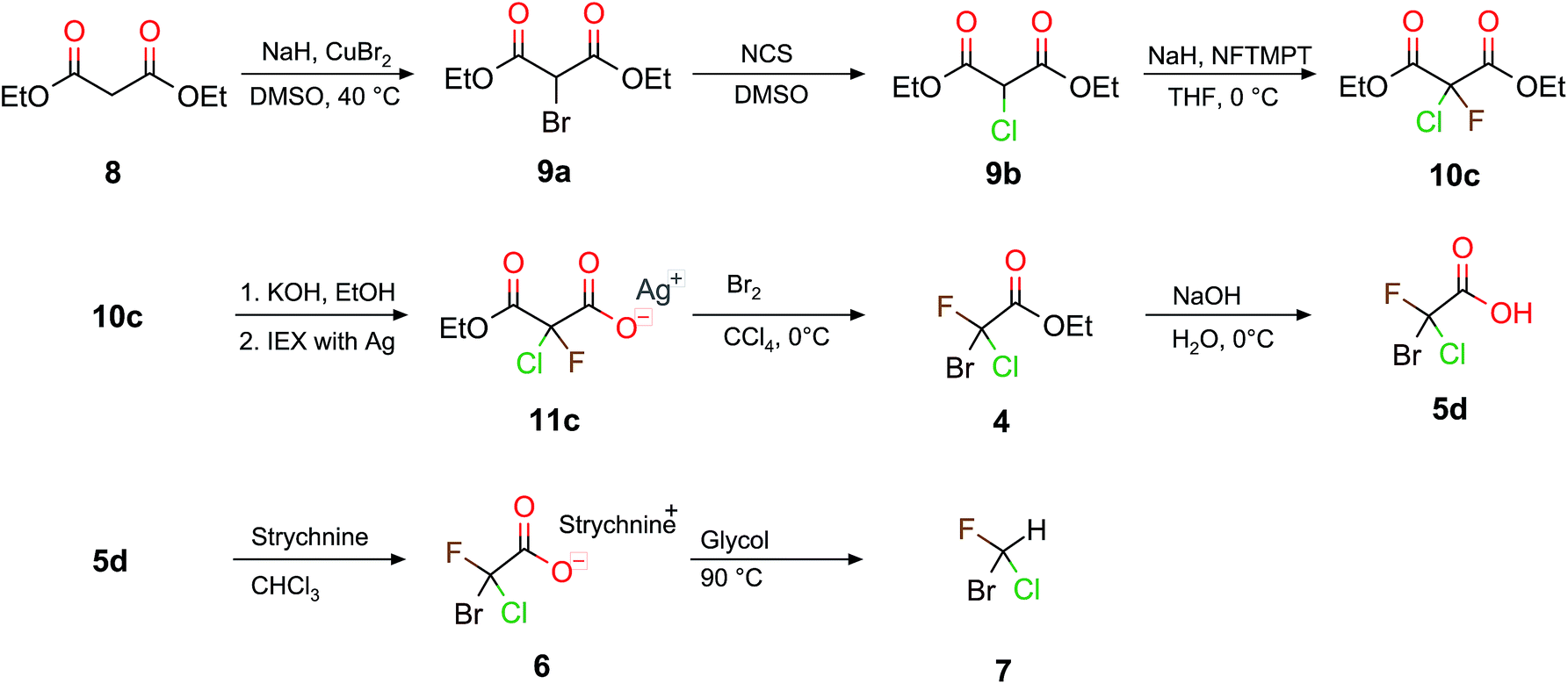 | ||
| Scheme 4 The synthesis pathway of this work to obtain 7 from 8 over a 9 step route. The steps from 8 to 10c are a branch of Scheme 2. | ||
Based on the chiral halogenated acetic acids, the corresponding halomethanes were, in principle, accessible. The required decarboxylation procedure was described in the literature for the example of the bromochlorofluoroacetic acid 5d to the corresponding halomethane CHBrClF (7).39 We have repeated this reaction here with a yield of 48% and a purity of 99% compared to a 50 to 70% yield reported by Doyle and Vogl.39 Although the overall yield of this 9 step synthesis is only approximately 12% (which is strongly affected by the 26% yield of the Hunsdiecker decarboxylation from 11c to 5d), it turned out that the 11c to 5d step formed the reactant 10c in a byreaction, therefore, 10c could be reused again.
Experimental procedures
General conditions
A flow or an atmosphere of dry nitrogen was used with a Schlenk-apparatus for all water- and/or air-sensitive reactions. For the same purpose, the glassware was dried with a heat gun. Quantitative and qualitative characterization was performed using GC-MS and NMR (500 MHz). For the gas chromatography with mass spectrometry an InertCap 5MS/NP capillary column was used and the signals were recorded as the total ion current of the ions between m/z 50 and 500. The temperature method started as isothermal at 40 °C for 1 minute and was increased to 150 °C by 15 °C per minute. At a rate of 40 °C per minute, the oven was heated to 300 °C and was kept at this temperature for 1 minute. This resulted in a method length of 11 minutes and 45 seconds. 1H, 13C and 19F NMR were recorded in parts per million and the data were reported as chemical shifts (δ) in parts per million (ppm), multiplicity, coupling constants (J) in Hz and integrals. The 13C NMR spectra were 1H decoupled.Commercial reagents were used with the given purity of the supplier. The purification of the intermediates was done using column chromatography if necessary. The given yields are the 1H NMR purity corrected values. For comparison we list the 1H NMR and GC-MS purities.
Diethyl bromomalonate 9a
The reaction apparatus was heated under vacuum and flushed with nitrogen before usage. Sodium hydride (12.9 g, 60%, 321.8 mmol) was suspended in dimethylsulfoxide (500 ml, anhydrous). Diethylmalonate 8 (50.2 g, 99%, 310.3 mmol) was slowly dropped into the suspension. The resulting mixture was added dropwise to a vigorously stirred solution of CuBr2 (150.3 g, 99%, 666.0 mmol) in dimethylsulfoxide (275 ml, anhydrous). After the addition of ice water (200 ml) and H2SO4 (200 ml, 5%), the mixture was extracted with dichloromethane (3 × 200 ml). The combined organic layer was washed with water (3 × 200 ml), dried over Na2SO4 and then completely evaporated. The total yield of the colorless liquid was 81.0 g (99%) with a purity of 91.7% according to 1H NMR (90.9% GC-MS).1H NMR (500 MHz, CDCl3): δ 4.84 (s, 1H), 4.31 (q, J = 7.1 Hz, 4H), 1.33 (t, J = 7.1 Hz, 6H). 13C NMR (126 MHz, CDCl3): δ 164.58 (s, 2C), 63.23 (t, 2C), 42.41 (d, 1C), 13.88 (q, 2C). GC-MS tR 8.60 min.
Diethyl chloromalonate 9b
The reaction apparatus was heated under vacuum and flushed with nitrogen before usage. Diethyl bromomalonate 9a (6.9 g, 98%, 28.2 mmol) was dissolved in dimethylsulfoxide (25 ml, anhydrous). N-Chlorosuccinimide (6.7 g, 99%, 49.0 mmol) was added and the mixture was stirred at room temperature overnight. After the addition of saturated ammonium chloride solution (20 ml) and water (50 ml), the mixture was extracted with ethyl acetate (3 × 20 ml). The combined organic layers were washed with saturated NaCl solution (3 × 20 ml). The solvent was completely evaporated after drying with Na2SO4. The crude product was purified by column chromatography (cyclohexane:ethyl acetate, 5:1). The yield was 6.2 g (99.4%) with a purity of 88.0% according to 1H NMR (83.4% GC-MS).
1H NMR (500 MHz, CDCl3): δ 4.86 (s, 1H), 4.32 (dq, J = 7.1 Hz; 1.1 Hz, 4H), 1.34 (t, J = 7.1 Hz, 6H). 13C NMR (126 MHz, CDCl3): δ 164.50 (s, 2C), 63.18 (t, 2C), 55.47 (d, 1C), 13.91 (q, 2C). GC-MS tR 7.89 min.
Diethyl bromochloromalonate 10a via chlorination
Diethyl bromomalonate 9a (5.1 g, 89%, 19.0 mmol) was dissolved in acetone (40.0 ml) and acetic acid (16.0 ml). Sodium hypochlorite solution (19.0 ml, 10%, 25.5 mmol) was added dropwise during 30 min at 0 °C. Saturated NaHCO3 solution (100 ml) was added to the reaction mixture. When the CO2 evolution stopped (after approximately 15 min) the organic product layer was separated and the aqueous layer was extracted with dichloromethane (3 × 30 ml). The combined organic layers were washed with water (3 × 30 ml), dried over Na2SO4 and were completely evaporated. The yield was 5.5 g (98.0%) with a purity of 92.6% according to 1H NMR (91.8% GC-MS).1H NMR (500 MHz, CDCl3): δ 4.39 (q, J = 7.1 Hz, 4H), 1.36 (t, J = 7.1 Hz, 6H). 13C NMR (126 MHz, CDCl3): δ 163.05 (s, 2C), 65.00 (s, 1C), 64.68 (t, 2C), 13.74 (q, 2C). GC-MS tR 9.40 min.
Diethyl bromochloromalonate 10a via bromination
For the preparation of the sodium hypobromite solution, sodium hydroxide (3.0 g, 85%, 63.8 mmol) was dissolved in water (50 ml) and cooled to 0 °C. Bromine (3.3 g, 99%, 20.3 mmol) was added to the ice cold mixture and stirred for 30 min. In a separate flask diethyl chloromalonate 9b (3.4 g, 99%, 17.3 mmol) was dissolved in acetone (10 ml) and acetic acid (5 ml) and cooled to 0 °C. The prepared sodium hypobromite solution was added dropwise to 9b. The reaction mixture was stirred at RT overnight. The reaction mixture was added to saturated NaHCO3 solution (20 ml) and extracted with dichloromethane (3 × 25 ml). The combined organic layers were washed with water (2 × 20 ml) and with saturated NaHCO3 solution (2 × 20 ml), dried over Na2SO4 and were then completely evaporated. The total yield of the light brown liquid was 4.8 g (99.9%) with a purity of 98.5% according to 1H NMR (99.1% GC-MS).1H NMR (500 MHz, CDCl3): δ 4.38 (q, J = 7.1 Hz, 4H), 1.35 (t, J = 7.1 Hz, 6H). 13C NMR (126 MHz, CDCl3): δ 163.02 (s, 2C), 64.99 (s, 1C), 64.67 (t, 2C), 13.73 (q, 2C). GC-MS tR 9.40 min.
Diethyl bromofluoromalonate 10b via fluorination
Sodium hydride (80.0 mg, 60%, 2.0 mmol) was dissolved in tetrahydrofurane (5.0 ml, anhydrous). To the mixture diethyl bromomalonate 9a (0.50 g, 99%, 2.0 mmol) was carefully added. After 3 h hydrogen evolution stopped and a clear solution was obtained. This solution was cooled to 0 °C. Then N-fluoro-2,4,6-trimethylpyridinium triflate (0.605 g, 99%, 2.1 mmol) was added. The reaction mixture was stirred at 0 °C for 30 min. To the reaction mixture water (10 ml) and hydrochloric acid (10 ml, 1 M) were added and it was then extracted with dichloromethane (3 × 20 ml). The combined organic layers were washed with saturated NaHCO3 solution (2 × 10 ml) and water, sequentially. They were subsequently dried with Na2SO4 and evaporated completely. The yield of the light brown liquid was 0.57 g (85.5%) with a purity of 81.0% according to 1H NMR (93.4% GC-MS).1H NMR (500 MHz, CDCl3): δ 4.40 (q, J = 7.1 Hz, 4H), 1.37 (t, J = 7.1 Hz, 6H). 13C NMR (126 MHz, CDCl3): δ 162.33 (d, JCF = 26.46 Hz, 2C), 89.95 (d, JCF = 270.9 Hz, 1C), 64.29 (t, 2C), 13.78 (q, 2C). 19F NMR (470 MHz, CDCl3): δ −122.38 (s, 1F). GC-MS tR 7.45 min.
Diethyl bromofluoromalonate 10b via bromination
The reaction procedure was similar to that of the bromination of 9b to 10a. Diethyl fluoromalonate 9c (1.0 g, 97%, 5.4 mmol) was the reactant. 2 equivalents of NaOBr were used. The total yield of the light brown liquid was 1.25 g (84.9%) with a purity of 94.3% according to 1H NMR (97.8% GC-MS).1H NMR (500 MHz, CDCl3): δ 4.38 (q, J = 7.1 Hz, 4H), 1.34 (t, J = 7.1 Hz, 6H). 13C NMR (126 MHz, CDCl3): δ 162.28 (d, JCF = 26.46 Hz, 2C), 89.93 (d, JCF = 270.9 Hz, 1C), 64.27 (t, 2C), 13.74 (q, 2C). 19F NMR (470 MHz, CDCl3): δ −122.39 (s, 1F). GC-MS tR 7.45 min.
Diethyl chlorofluoromalonate 10c via fluorination
The reaction procedure was similar to that of the fluorination of 9a to 10b. Diethyl chloromalonate 9b (0.52 g, 97%, 2.6 mmol) was the reactant. The yield of the light brown liquid was 0.42 g (72.7%) with a purity of 97.5% according to 1H NMR (97.4% GC-MS).1H NMR (500 MHz, CDCl3): δ 4.38 (q, J = 7.2 Hz, 4H), 1.35 (t, J = 7.2 Hz, 6H). 13C NMR (126 MHz, CDCl3): δ 161.83 (s, 2C), 97.89 (d, JCF = 261.4 Hz, 1C), 64.24 (t, 2C), 13.76 (q, 2C). 19F NMR (470 MHz, CDCl3): δ −161.83 (s, 1F). GC-MS tR 6.68 min.
Diethyl chlorofluoromalonate 10c via chlorination
The reaction procedure was similar to that of the chlorination of 9a to 10a. Diethyl fluoromalonate 9c (103.8 g, 97%, 565 mmol) was the reactant. The yield of the light brown liquid was 106.2 g (85.8%) with a purity of 97.1% according to 1H NMR (93.9% GC-MS).1H NMR (500 MHz, CDCl3): δ 4.38 (q, J = 7.2 Hz, 4H), 1.35 (t, J = 7.2 Hz, 6H). 13C NMR (126 MHz, CDCl3): δ 161.83 (s, 2C), 97.89 (d, JCF = 261.4 Hz, 1C), 64.24 (t, 2C), 13.76 (q, 2C). 19F NMR (470 MHz, CDCl3): δ −161.83 (s, 1F). GC-MS tR 6.68 min.
Deutero bromochloroacetic acid 5a
NaOD (0.75 g, 99%, 99% d-atom, 18.6 mmol) was dissolved in MeOD (10 ml) and added dropwise at room temperature to a solution of diethyl bromochloromalonate 10a (1.5 g, 99%, 5.4 mmol) in MeOD (5 ml). The reaction mixture was stirred at room temperature for 30 min and a white suspension was formed. D2SO4 solution (5 g, 30%, 99% d-atom, 15.0 mmol) was added to the reaction mixture and the solid dissolved. H2SO4-solution (20 ml, 10%) was added and the reaction mixture was extracted with diethyl ether (3 × 20 ml). The combined organic layers were dried over Na2SO4 and evaporated to dryness. The yield of the light brown liquid was 0.75 g (75.7%) with a purity of 95.1% according to 1H NMR (95.5% GC-MS). The degree of deuteration was ∼89% and it was determined using the abundance ratio of m/z 172 and 173.1H NMR (500 MHz, CDCl3): δ 8.80 (s, 1H). 13C NMR (126 MHz, CDCl3): δ 169.27 (s, 1C), 48.09 (t, JCD = 27.7 Hz, 1C). GC-MS tR 9.35 min.
Deutero bromofluoroacetic acid 5b
Diethyl bromofluoromalonate (1.25 g, 94%, 4.8 mmol) was added dropwise at room temperature to a solution of D2SO4 (5.0 g, 99%, 99% d-atom, 50.0 mmol) and D2O (5.0 g). The reaction mixture was stirred at 120 °C for 5 hours. A solution of H2SO4 (20 ml, 5%) was added to the mixture and then it was extracted with diethyl ether (3 × 20 ml). The combined organic layers were dried over Na2SO4 and evaporated to dryness. The yield of the light brown liquid was 0.44 g (50.8%) with a purity of 84.0% according to 1H NMR (90.3% GC-MS). The degree of deuteration was ∼97% and it was determined using the abundance ratio of m/z 156 and 157.1H NMR (500 MHz, CDCl3): δ 8.08 (s, 1H). 13C NMR (126 MHz, CDCl3): δ 166.50 (d, JCF = 25.5 Hz, 1C), 81.25 (dt, JCF = 262.1 Hz, JCD = 29.0 Hz, 1C). 19F NMR (470 MHz, CDCl3): δ −151.96 (t, JCDF = 9.4, 1F). GC-MS tR 8.11 min.
Deutero chlorofluoroacetic acid 5c
Diethyl chlorofluoromalonate (1.5 g, 98%, 6.9 mmol) was added dropwise at room temperature to a solution of D2SO4 (7.0 g, 99%, 99% d-atom, 70.0 mmol) and D2O (7.0 g). The reaction mixture was stirred at 120 °C overnight. A solution of H2SO4 (20 ml, 5%) was added to the mixture and then it was extracted with diethyl ether (3 × 20 ml). The combined organic layers were dried over Na2SO4 and evaporated to dryness. The yield of the light brown liquid was 0.41 g (48.5%) with a purity of 92.0% according to 1H NMR (94.2% GC-MS). The degree of deuteration was ∼94% and it was determined using the abundance ratio of m/z 61 and 62.1H NMR (500 MHz, CDCl3): δ 10.81 (s, 1H). 13C NMR (126 MHz, CDCl3): δ 168.99 (d, JCF = 27.7 Hz, 1C), 90.15 (dt, JCF = 253.3 Hz, JCD = 29.0 Hz, 1C). 19F NMR (470 MHz, CDCl3): δ −147.77 (t, JCDF = 4.7, 1F). GC-MS tR 7.11 min.
Potassium monoethyl chlorofluoromalonate
Diethyl chlorofluoromalonate 10c (87.2 g, 93.9%, 385.3 mmol) was dissolved in absolute ethanol (250 ml). Potassium hydroxide (24.5 g, 86%, 374.9 mmol) was dissolved in absolute ethanol (250 ml) and added dropwise at room temperature to the vigorously stirred solution of 3c within 4 h. The resulting suspension was filtered and the residue was washed with absolute ethanol (100 ml). The filtrate was completely evaporated. The solid product was washed with dichloromethane (150 ml) to remove the starting material. The pure product was obtained by a second filtration and drying. The yield of the white crystalline solid was 52.3 g (61%) with a purity of approximately 100%.1H NMR (500 MHz, D2O): δ 4.31 (q, J = 7.1 Hz, 2H), 1.25 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, D2O): δ 166.71 (d, JCF = 24.1 Hz, 1C), 165.62 (d, JCF = 28.4 Hz, 1C), 99.85 (d, JCF = 260.0 Hz, 1C), 65.01 (t, 1C), 13.11 (q, 1C). 19F NMR (470 MHz, D2O): δ −114.85 (s, 1F).
Silver monoethyl chlorofluoromalonate 11c
A highly acidic ion exchange resin (100 ml, Amberlite IR 120, Na+ form) was filled into a glass column. A solution of silver nitrate (400 ml, 0.4 M) was used to load the resin with silver ions. Before usage, the resin was washed until the eluting water was free of silver ions. An aqueous solution of potassium monoethyl chlorofluoromalonate (4.0 g, 0.018 mol, 0.03 gl−1) was passed through the resin. The complete evaporation of the aqueous fraction gave 5.2 g (99%) of the silver monoethyl chlorofluoromalonate 11c as a grey solid with a purity of 100% (XRF and 19F NMR).1H NMR (500 MHz, D2O): δ 4.44 (q, J = 7.1 Hz, 2H), 1.40 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, D2O): δ 165.70 (d, JCF = 28.5 Hz, 1C), 161.81 (d, JCF = 31.3 Hz, 1C), 97.29 (d, JCF = 262.3 Hz, 1C), 65.00 (t, 1C), 13.72 (q, 1C). 19F NMR (470 MHz, D2O): δ −121.17 (s, 1F).
Ethyl bromochlorofluoroacetate 4
Bromine (3.8 g, 99%, 23.7 mmol) was dissolved in CCl4 (40 ml). At 0 °C, silver monoethyl chlorofluoromalonate 11c (6.0 g, 98%, 20.2 mmol) was added portion-wise (about 1.5 g per hour) while the reaction mixture was prevented from humidifying. After 1.5 h a continuous evolution of CO2 started. When all of the reactant was added the mixture it was allowed to warm to room temperature and was then held at that temperature overnight. The precipitate was filtered and washed with CCl4 (20 ml). Bromine was removed with NaHSO3 solution (8%, 3 × 10 ml) and washed with saturated NaHCO3 (3 × 10 ml). After drying with Na2SO4 the solvent was completely evaporated. The yield of the light brown liquid was 1.39 g (26.1%) with a purity of 82.6% according to 1H NMR (82.0% GC-MS).1H NMR (500 MHz, CDCl3): δ 4.45 (q, J = 7.1 Hz, 2H), 1.43 (t, J = 7.1 Hz, 4H). 13C NMR (126 MHz, CDCl3): δ 161.90 (d, JCF = 27.7 Hz, 1C), 96.12 (s, 1C), 64.83 (t, 1C), 13.75 (q, 1C). 19F NMR (470 MHz, CDCl3): δ −64.23 (s, 1F). GC-MS tR 4.38 min.
Bromochlorofluoroacetic acid 5d
NaOH (3.5 g, 85%, 74.4 mmol) was dissolved in water (30 ml). At 0 °C methyl bromochlorofluoroacetate 4 (12.9 g, 59%, 34.7 mmol) was added. After 3 h the acetate was quantitatively converted. The basic solution was acidified with concentrated H2SO4 (3 ml) and extracted with diethylether (3 × 20 ml). After drying with Na2SO4 the organic layer was completely evaporated. The yield of the colorless liquid was 9.0 g (99%) with a purity of 73.0% according to 1H NMR (73.0% GC-MS).1H NMR (500 MHz, CDCl3): δ 11.36 (s, 1H). 13C NMR (126 MHz, CDCl3): δ 166.36 (d, JCF = 28.0 Hz, 1C), 94.47 (d, JCF = 315.0 Hz, 1C). 19F NMR (470 MHz, CDCl3): δ −66.02 (s, 1F). GC-MS tR 6.69 min.
Bromochlorofluoromethane 7
Bromochlorofluoroacetic acid 5d (9.0 g, 74%, 34.7 mmol) was dissolved in CHCl3 (30 ml). Strychnin (11.6 g, 98%, 34.0 mmol) was dissolved in CHCl3 (80 ml) and added to the acid at 0 °C over 30 minutes. After 1 h the CHCl3 was evaporated and the strychnin salt was obtained quantitatively as a brownish solid. A portion of the strychnine bromochlorofluoroacetate (5.2 g, 99%, 8.76 mmol) was suspended in ethylene glycol (10 ml). The apparatus, which consisted of an Anschuetz head, Liebig condenser, collecting flask and cooling trap, was heated and flushed with nitrogen. The collecting flask and cooling trap were cooled with a mixture of acetone and dry ice to −78 °C. The suspension was heated to 80 °C and slowly increased to 100 °C as the gas formation also increased. After 45 min the gas formation stopped and the suspension was stirred at 140 °C for 1 h. The yield of the colorless liquid was 0.6 g (46.2%) with a purity of 95.2% according to 1H NMR (99.0% GC-MS).1H NMR (500 MHz, CDCl3): δ 7.64 (d, J = 52.0 Hz, 1H). 13C NMR (126 MHz, CDCl3): δ 89.72 (dd, JCF = 304.6 Hz, 1C). 19F NMR (470 MHz, CDCl3): δ −81.66 (d, J = 52.0 Hz, 1F). GC-MS tR 1.81 min.
Conclusion
In conclusion, we have presented an efficient and versatile pathway to obtain a large variety of chiral haloacetic acids from a common reagent which is easily commercially available. Further decarboxylation provides, in principle, access to the corresponding halomethanes. Currently, this has been explored only for the trihalogenated chiral acetic acids with success. Furthermore, the ability of obtaining the different haloacetic acids from a single reagent is another advantage. Our reagent 8 is also more easy to use compared to a differently halogenated ethene. The drawbacks of halogenated ethenes are clearly the low boiling points and their decreasing availability due to the CFC (chlorofluorocarbon) ban. CHBrClF 7 has been obtained starting from 8 by exploring the full reaction pathway (Scheme 4) described here. Chiral acetic acid has been obtained as a racemic mixture, however, it can be derivatized for enantio-separation (e.g. by preparative gas-chromatography55 or by diastereomeric resolution19) along the reaction pathways shown in Schemes 3 and 4. This will then also remove minor impurities which may be present in the target compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is part of the master thesis of Manuel R. Mazenauer38 and the bachelor thesis of Philipp Kappeler.56 We thank Dr Hans Hollenstein, Manuela Meister, Fabian Deuber and Benjamin Spenger for their help and discussion. Our work is supported financially by the ZHAW through grants ASF-2016 and ASF-2017.References
- M. Quack, J. Stohner and M. Willeke, Annu. Rev. Phys. Chem., 2008, 59, 741 CrossRef CAS PubMed.
- M. Quack and J. Stohner, Phys. Rev. Lett., 2000, 84, 3807 CrossRef CAS PubMed.
- M. Pitzer, M. Kunitski, A. S. Johnson, T. Jahnke, H. Sann, F. Sturm, L. P. H. Schmidt, H. Schmidt-Böcking, R. Dörner, J. Stohner, J. Kiedrowski, M. Reggelin, S. Marquardt, A. Schießer, R. Berger and M. S. Schöffler, Science, 2013, 341, 1096 CrossRef CAS PubMed.
- M. Pitzer, G. Kastirke, M. Kunitski, T. Jahnke, T. Bauer, C. Goihl, F. Trinter, C. Schober, K. Henrichs, J. Becht, S. Zeller, H. Gassert, M. Waitz, A. Kuhlins, H. Sann, F. Sturm, F. Wiegandt, R. Wallauer, L. P. H. Schmidt, A. S. Johnson, M. Mazenauer, B. Spenger, S. Marquardt, S. Marquardt, H. Schmidt-Böcking, J. Stohner, R. Dörner, M. Schöffler and R. Berger, ChemPhysChem, 2016, 17, 2465 CrossRef CAS PubMed.
- P. Herwig, K. Zawatsky, M. Grieser, O. Heber, B. Jordon-Thaden, C. Krantz, O. Novotný, R. Repnow, V. Schurig, D. Schwalm, Z. Vager, A. Wolf, O. Trapp and H. Kreckel, Science, 2013, 342, 1084 CrossRef CAS PubMed.
- K. Zawatsky, P. Herwig, M. Grieser, O. Heber, B. Jordon-Thaden, C. Krantz, O. Novotný, R. Repnow, V. Schurig, D. Schwalm, Z. Vager, A. Wolf, H. Kreckel and O. Trapp, Chem.–Eur. J., 2014, 20, 5555 CrossRef PubMed.
- P. Lahiri, K. B. Wiberg and P. H. Vaccaro, J. Phys. Chem. A, 2015, 119, 8311 CrossRef CAS PubMed.
- C. Lux, M. Wollenhaupt, C. Sarpe and T. Baumert, ChemPhysChem, 2015, 16, 115 CrossRef CAS PubMed.
- A. A. Fokin, P. R. Schreiner, R. Berger, G. H. Robinson, P. Wei and C. F. Campana, J. Am. Chem. Soc., 2006, 128, 5332 CrossRef CAS PubMed.
- R. Berger and J. Stuber, Mol. Phys., 2007, 105, 41 CrossRef CAS.
- R. Berger, J. Chem. Phys., 2008, 129, 154105 CrossRef PubMed.
- S. Nahrwold and R. Berger, J. Chem. Phys., 2009, 130, 214101 CrossRef PubMed.
- F. Merkt and M. Quack, Handbook of High-Resolution Spectroscopy: Volume 1 Fundamentals, 2011, vol. 1, p. 1 Search PubMed.
- F. Hobi, R. Berger and J. Stohner, Mol. Phys., 2013, 111, 2345 CrossRef CAS.
- R. Berger, M. Quack and J. Stohner, Angew. Chem., Int. Ed., 2001, 40, 1667 CrossRef CAS PubMed.
- J. Lüthy, J. Rétey and D. Arigoni, Nature, 1969, 221, 1213 CrossRef.
- J. W. Cornforth, J. W. Redmond, H. Eggerer, W. Bückel and C. Gutschow, Nature, 1969, 221, 1212 CrossRef CAS PubMed.
- C. A. Townsend, T. Scholl and D. Arigoni, J. Chem. Soc., Chem. Commun., 1975, 921 RSC.
- S. Manov, V. Galati, M. Meister, M. Mazenauer, B. Spenger and J. Stohner, SASP 2016, Proceedings of the XXth Symposium on Atomic, Cluster and Surface Physics, ed. J. Stohner and C. Yeretzian, Innsbruck University Press, Innsbruck, Austria, 2016, p. 199 Search PubMed.
- R. Meier and F. Böhler, Chem. Ber., 1957, 90, 2342 CrossRef CAS.
- A. Beil, D. Luckhaus and M. Quack, Ber. Bunsenges. Phys. Chem., 1996, 100, 1853 CrossRef CAS.
- T. R. Doyle and O. Vogl, Monatsh. Chem., 1990, 121, 31 CrossRef CAS.
- R. N. Haszeldine, J. Am. Chem. Soc., 1952, 4259 RSC.
- D. B. Li, S.-C. Ng and I. Novak, Tetrahedron, 2002, 58, 5923 CrossRef CAS.
- A. N. Takata, K. V. Narasimham, A. G. Meister, J. M. Dowling, F. F. Cleveland, S. Sundaram, E. A. Piotrowski, R. B. Bernstein and S. I. Miller, J. Mol. Spectrosc., 1965, 15, 319 CrossRef CAS.
- R. A. Niedrich and D. M. Grant, J. Chem. Phys., 1965, 42, 3733 CrossRef CAS.
- S. Sugie, H. Takeo and C. Matsumura, Bull. Chem. Soc. Jpn., 1978, 51, 3065 CrossRef.
- C. Puzzarini, G. Cazzoli, A. Baldacci, A. Baldan, C. Michauk and J. Gauss, J. Chem. Phys., 2007, 127, 164302 CrossRef PubMed.
- V. Letokhov, Phys. Lett. A, 1975, 53, 275 CrossRef.
- T. D. Lee and C. N. Yang, Phys. Rev., 1956, 104, 254 CrossRef CAS.
- C. S. Wu, E. Ambler, R. W. Hayward, D. D. Hoppes and R. P. Hudson, Phys. Rev., 1957, 105, 1413 CrossRef CAS.
- M. Quack and J. Stohner, Chimia, 2005, 59, 530 CrossRef CAS.
- M. Quack and J. Stohner, Chirality, 2001, 13, 745 CrossRef CAS PubMed.
- S. K. Tokunaga, C. Stoeffler, F. Auguste, A. Shelkovnikov, C. Daussy, A. Amy-Klein, C. Chardonnet and B. Darquié, Mol. Phys., 2013, 111, 2363 CrossRef CAS.
- J. Read and A. M. McMath, J. Chem. Soc., 1926, 129, 2183 RSC.
- C. S. Palmer and P. W. McWherter, Org. Synth., 1927, 7, 34 CrossRef CAS.
- H. Zimmer, A. Amer and M. Rahi, Anal. Lett., 1990, 23, 735 CrossRef CAS.
- M. Mazenauer, Master thesis, ZHAW, Wädenswil, 2015.
- T. R. Doyle and O. Vogl, J. Am. Chem. Soc., 1989, 111, 8510 CrossRef CAS.
- H. Boussac, J. Crassous, J.-P. Dutasta, L. Grosvalet and A. Thozet, Tetrahedron: Asymmetry, 2002, 13, 975 CrossRef CAS.
- A. Günther, H. Weintritt and S. Böhm, Internat. Patentamt, WO2005019154A1, 2005, p. 1.
- T. Kitamura, K. Muta and J. Oyamada, Synthesis, 2015, 47, 3241 CrossRef CAS.
- J. C. Lee, J. Y. Park, S. Y. Yoon, Y. H. Bae and S. J. Lee, Tetrahedron Lett., 2004, 45, 191 CrossRef CAS.
- N. Shariff, S. Mathi, C. Rameshkumar and L. Emmanuvel, Tetrahedron Lett., 2015, 56, 934 CrossRef CAS.
- S. Wolfe, S. Ro, C.-K. Kim and Z. Shi, Can. J. Chem., 2001, 79, 1238 CrossRef CAS.
- L. Delon, P. Laurent and H. Blancou, J. Fluorine Chem., 2005, 126, 1487 CrossRef CAS.
- R. Winter and G. Gard, J. Fluorine Chem., 2000, 102, 79 CrossRef CAS.
- M. L. Meketa, Y. R. Mahajan and S. M. Weinreb, Tetrahedron Lett., 2005, 46, 4749 CrossRef CAS.
- R. D. Chambers and J. Hutchinson, J. Fluorine Chem., 1998, 92, 45 CrossRef CAS.
- H.-J. Tsai and C.-W. Hsieh, J. Chin. Chem. Soc., 2007, 54, 749 CrossRef CAS.
- M. F. Wempe, W. B. Anderson, H.-F. Tzeng, P. G. Board and M. W. Anders, Biochem. Biophys. Res. Commun., 1999, 261, 779 CrossRef CAS PubMed.
- B. Englund, Org. Synth., Coll., 1963, 4, 184 Search PubMed.
- B. Englund, Org. Synth., Coll., 1963, 4, 423 Search PubMed.
- H. Hunsdiecker and C. Hunsdiecker, Ber. Dtsch. Chem. Ges., 1942, 75, 291 CrossRef.
- B. Spenger and J. Stohner, Europ. Patentamt, EP 3069777A1, 2016, p. 1.
- P. Kappeler, Bachelor thesis, ZHAW, Wädenswil, 2017.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section including synthesis, characterization, NMR analysis and additional physico-chemical data. See DOI: 10.1039/c7ra09727a |
| This journal is © The Royal Society of Chemistry 2017 |