
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Blue-white screening as a new readout for deoxyribozyme activity in bacterial cells†
S. Sadeghi,
N. Ahmadi,
A. Esmaeili * and
F. Javadi-Zarnaghi*
* and
F. Javadi-Zarnaghi*
Cell, Molecular Biology and Biochemistry Division, Department of Biology, Faculty of Sciences, University of Isfahan, Isfahan, Iran. E-mail: aesmaeili@sci.ui.ac.ir; fa.javadi@sci.ui.ac.ir
First published on 1st December 2017
Abstract
The 10–23 deoxyribozyme is considered as sequence-specific “molecular scissors” for RNA molecules. Extensive investigations have been reported for this deoxyribozyme in vitro and in eukaryotic host cells. However, few investigations are reported in the literature on the activity of this deoxyribozyme inside bacterial cells. The available reports focus on the cleavage of target mRNAs that encode for proteins which are responsible for the viability of bacterial colonies. Hence, the growth of bacterial cells was blocked and the main readouts in such studies were colony counts or optical density at 600 nm. In the current study, blue-white screening was utilized as a novel readout for analysis of the activity of the 10–23 deoxyribozymes in viable bacterial cells. Two deoxyribozymes were designed to target the α-peptide fragment from β-galactosidase (lacZ) mRNA at two different positions, i.e. 5′ untranslated region and translated region. Control experiments were performed utilizing DNA oligos that lacked the catalytic core. The 3′–3 inverted thymidine modified deoxyribozymes were compared with unmodified ones to analyze the effect of such modification in prokaryotic cells. The activity of the designed deoxyribozymes caused a significant retardation in the formation of the blue-color in colonies with deoxyribozymes. Miller assay confirmed the blue-white screening results. This report showed a proof of concept for application of blue-white screening as a readout system for the activity of the 10–23 deoxyribozyme that is a model RNA-cleaving deoxyribozyme. The result of this report can promote future investigations on the activity of deoxyribozymes in prokaryotic cells.
Introduction
Protein enzymes were known as the sole biological catalysts until the 1980s. During the 1980s two natural RNA catalysts (ribozymes) were discovered for which a Nobel prize was shared between two scientists, Sidney Altman and Thomas Cech in 1989.1 A few years later, reports on oligodeoxyribonucleotide-mediated catalysis demonstrated catalytic ability of DNA molecules.2 Since then, a growing number of deoxyribozymes (DNAzymes, DNA catalysts, Dzs) have been selected in vitro.3 Early evolved deoxyribozymes were RNA-cleaving ones.2b,4 The catalytic cores of these deoxyribozymes, like many others, were flanked by two binding arms that complementarily bind specific RNA substrates.RNA-cleaving deoxyribozymes can be considered as sequence-specific “molecular scissors” for RNA molecules. The 10–23 deoxyribozyme which consists of a 15 nucleotide long catalytic core is a well-known example of an RNA-cleaving deoxyribozyme. It was shown that this deoxyribozyme can cleave biologically relevant RNA molecules in vitro in a site specific manner.5 The activity of the 10–23 deoxyribozyme was also monitored in vivo.6 The enzyme was shown to be capable to modulate gene expression at post-transcriptional level.7
The in vivo targets of 10–23 deoxyribozyme were mostly viral RNA8 or mRNA of eukaryotic cells.9 Many genes from HIV-1,10 hepatitis B and C,11 influenza A and B,12 human papilloma virus,13 severe acute respiratory syndrome coronavirus (SARS)14 and Epstein-Barr virus have been the targets for 10–23 deoxyribozyme RNA cleavage.15 Handful of genes responsible for cardiovascular, inflammatory or central nervous system diseases were down-regulated with 10–23 deoxyribozyme.6 Many cancer related genes such as multidrug resistance gene (MDR) have been targeted with this enzyme.16 Currently, the 10–23 deoxyribozyme is a candidate therapeutic for several human diseases such as allergic bronchial asthma with TH2 molecular signature.17 The GATA-3 targeting 10–23 deoxyribozyme is now under clinical trial investigations by sterna biologicals and phase IIa for early-phase and late phase asthmatic responses after allergen provocation has been completed.18
Despite advances in application of 10–23 deoxyribozyme in eukaryotic and mammalian cells, limited numbers of reports are available on investigation of the 10–23 deoxyribozyme activity inside the bacterial cells.6 A major challenge in application of deoxyribozymes in bacterial cells is dilution of the transfected oligonucleotide inside the fast dividing cells. Thus, all present reports used the 10–23 deoxyribozyme in an antibiotic-like manner. Such reports focus on targeting mRNAs of genes that were essential for viability and proliferation of the bacterial cells. The mRNAs for genes that are responsible for antibiotic resistance such as, mecR1 (ref. 23b) and bla1 (ref. 23a) related to the β-lactamase pathway are the most targets for application of 10–23 deoxyribozyme in bacterial cells. Besides, Tan et al. reported cleavage of ftsZ mRNA by 10–23 deoxyribozyme.24 FtsZ gene is responsible for binary division of E. coli cells and its downregulation prevents bacterial colony formation. Therefore, in all the reported cases for prokaryotic application of 10–23 deoxyribozyme, inhibition of cell growth was the main result of the activity of the deoxyribozyme. Counting colonies on agar plate or analyzing OD600 were the main readout systems for such studies.
Due to the aforementioned technical issues, the current applications of deoxyribozymes in prokaryotic cells are limited to 10–23 and cleavage of vital mRNAs. Although inhibition of bacterial growth has advantages and permits application for controlling bacterial populations, indeed it does not grant investigation of the activity of the deoxyribozyme inside of the prokaryotic cells. Cells where the deoxyribozyme are active die due to the blockade of the target genes and thus no means are available to understand the mechanism of action of the deoxyribozyme in viable bacterial cells. Fundamentally, it would be helpful to have a new readout system for investigation of the activity of various deoxyribozymes inside vital prokaryotic cells. In addition, with deeper understanding on the kinetics of deoxyribozymes inside prokaryotic cells, the scope is capable of broadening e.g. to utilize the prokaryotic-active deoxyribozymes as biological elements of biosensors.
The lactose (lac) operon consists of three genes for production of β-galactosidase (Lac Z), permease (Lac Y) and transacetylase (Lac A). The lac operon is widely used as a reporter of gene insertion in cloning at molecular biology labs by simple and visual blue-white screening. The enzyme β-galactosidase is a tetramer with identical four polypeptides and is responsible for formation of a blue color upon presence of a chromogenic substrate like 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal). Deletion of a few amino acids from amino-terminus of monomers of the enzyme, causes the formation of a dimer which may also further breaks down to monomers. Administration of the “missing” amino acids in an oligopeptide, also called α-peptide results in re-structuring of the enzyme in active tetramer form. Molecular biologists hijacked this mechanism to analyze cloning efficiency.26 To this endeavor, the host must be an E. coli strain with missing amino acids from amino-terminus of β-galactosidase. Upon transformation, the α-peptide gets provided into the cells. For a standard blue-white screening, the gene of interest is inserted in a multiple cloning site which is located inside the coding sequence of the α-peptide. Thus, upon gene insertion, the α-peptide is disrupted and α-complementation may not occur. In contrast, vectors with complete and active α-peptide will perform α-complementation. As a result, the colonies with vector that contains the gene of interest do not bear active tetramer β-galactosidase and cannot produce a blue color from X-gal, while colonies with vectors that lack the gene of interest will form a blue color. Accordingly, the color of the colonies will be a marker for of the activity of the β-galactosidase and consequently presence of the gene of interest.
This report provides a novel application for the well-known method of blue-white screening. The presented method doesn't utilize the screening as a readout of gene insertion but rather it pursues the catalytic activity of the 10–23 deoxyribozyme. Here, the fully active α-peptides were transformed into bacterial hosts. The 10–23 deoxyribozymes targeted the mRNA of the α-peptides. The post-transcriptional cleavage of the mRNA of the α-peptide was expected to diminish α-complementation and formation of the blue color. Hence, the proposed method is able to report the activity of the β-galactosidase in viable cells. Proof of principle results were shown for the activity of the 10–23 deoxyribozyme for site specific cleavage at 5′-UTR and translated regions.
In most of the few available reports on prokaryotic cells, the oligodeoxyribonucleotide sequences of 10–23 deoxyribozyme were transfected to the bacterial cell as naked single stranded DNA. As an example, in a case report, oligonucleotides bearing the sequence of 10–23 deoxyribozyme as monomers and dimers designed against β-lactamase gene and was investigated in TEM-1 producing E. coli.19
Naked unmodified oligonucleotides are prone to degradation by nucleases. Various types of DNA modifications are available to block nuclease degradation of oligonucleotides in vivo. Addition of a 3′ inverted nucleotide at the 3′ end of the oligo,20 phosphorothioation of the oligo backbone,21 modification at 2′ position of the ribose sugar such as 2′-O-methyl modification22 and application of locked nucleic acid (LNA)13 on deoxyribozymes' binding arms are common examples. Among such a wide spectrum of DNA modifications, only phosphorothioation of the 10–23 deoxyribozyme was investigated in prokaryotic cells. Phosphorothioated DNA oligos were applied to cleave β-lactamase in oxacillin and methicillin resistant S. aureus, WHO-2 strains.23 In another strategy, vectors have been designed to transform the bacterial cells with deoxyribozymes 10–23. pssXGa vector24 and pBlue-script-II KS (+) phagemid25 were designed for E. coli. In such strategies, the deoxyribozymes are induced to be synthesized inside bacteria and thus there is less concern for nuclease degradation. In this report, modified and unmodified DNA oligos with 3′–3′ inverted thymidine were compared for blockade of the activity of the β-galactosidase. In addition, the effect of the deoxyribozymes was dissected from antisense effect, by comparing the results of full deoxyribozymes and the ones without catalytic core.
Results
Design of deoxyribozymes
The promoter of the lacZ-α peptide fragment was located in the reverse orientation from nucleotides 210–239 of the pGEM-T vector. Thus, the transcript template starts from position 209 in the reverse complement form down to position 2832, encompassing multiple cloning site and SP6 and m13 primer binding sites. The complete pGEM-T vector sequence with assigned fragments is depicted in ESI Appendix SI.† The mRNA transcript of the α-peptide fragment with pGEM-T vector was 378 nt long. The sequence of the transcript is provided in ESI Appendix SII.† The transcript coded for a fragment of lacZ α-peptide from amino acid 67–115. The sequence of the α-peptide fragment from pGEM-T is presented in ESI Appendix SIII.† The start codon on the transcribed mRNA was at position 44–47.The predicted secondary structure of the transcript was taken as a guide to choose the best cleavage sites. To support effective approach of the deoxyribozymes's binding arms to the target mRNA, positions at which no extensive stems were present have been selected as target sites. Extended binding arms would thermodynamically inhibit binding of the deoxyribozymes to the target. To address the effect of the position of the cleavage site, two target sites were chosen; one in the 5′-untranslated region (5′-UTR), upstream of the first AUG and the second in the translated region. The 10–23 deoxyribozyme is known to be specific for cleavage at purine–pyrimidine junctions. The A39–C40 junction (inside 5′ UTR) and A106–C107 junction (inside translated region) were selected for cleavage by the deoxyribozymes. The deoxyribozymes were applied in two formats; with 3′-OH and with 3′–3′ inverted thymidine. The latter was used to investigate protection potentials of such modification in prokaryotic host cells. The effect of three control DNAs was analyzed as well; (1) to dissect the catalytic activity of the designed deoxyribozymes from antisense effect, DNA oligos without catalytic cores (WOCC) were analyzed in parallel. WOCC had three thymidines instead of the catalytic core. (2) Additionally, a DNA oligonucleotide that do not target lacZ mRNA was co-transformed with pGEM-T vector as non-target DNA control (NTC). (3) Besides, in a negative control experiment, pGEM-T was transformed solely to E. coli competent cells. Fig. 1 and Table 2 depict the schematic structure of the target mRNA and sequences of the designed deoxyribozymes respectively.
 | ||
| Fig. 1 (A) Schematic presentation of the secondary structure of the α-peptide mRNA from pGEM-T vector. The secondary structure was predicted by mfold. Two target sites for cleavage with 10–23 deoxyribozyme were A39, prior to the start codon (5′ UTR) and A106 in the translated region. The sequences with which designed deoxyribozymes bind are colored light green and purple. The start codon is colored orange. (B) The non-modified 10–23 deoxyribozyme (NMS) designed for A39. The cleavage site is shown by an arrow. (C) The modified 10–23 (MS) deoxyribozyme has the same sequence as NMS, except with an inverted thymidine at 3′ end. (D) The control DNA without catalytic core (WOCC) had three thymidines instead of catalytic core and was modified with 3′–3′ inverted thymidine as MS. (E) The structure of the modification used in this report. | ||
Magnesium chloride optimization
Kinetic studies on 10–23 deoxyribozyme, similar to many others, confirmed strong dependence of the enzyme on bivalent metal ions e.g. Mg2+.4 The activity of this deoxyribozyme is increased upon increment in Mg2+ concentration in a hyperbolic manner.4 However, cellular concentration of Mg2+ is limited to sub-millimolar concentrations. Thereupon, the activity of deoxyribozymes gets hampered. Specific pumps, channels etc. guarantee a balanced concentration of Mg2+ inside cells, and the Mg2+ concentration of the culture media do not directly represent its cellular concentration. However, to assure that adequate amount of Mg2+ is available, the bacterial growth at various concentrations of Mg2+ was analyzed. The effect of Mg2+ on the cell viability of E. coli colonies were investigated on agar plates. The highest concentration of Mg2+ with no inhibition of cell growth was considered to be optimal. To this end, we investigated at which concentration of Mg2+ the cell viabilities start to drop. Our result showed that Mg2+ concentration above 40 mM reduces colony formation and thus, we decided to perform experiments with 35 mM Mg2+ present in the culture media (Fig. 2). | ||
| Fig. 2 Optimization of the Mg2+ concentration for formation of E. coli colonies. | ||
Blue-white screening
Upon transformation and subsequent incubation on ampicillin containing agar medium, white colonies get formed in a time span of ca. 12 h. After appearance of white colonies, a solution containing both IPTG and X-gal (IPTG–X-gal) was sprayed over the agar plates. The solution was sprayed to ensure even distribution of the solution. The color of the colonies started to change from white to blue. The complete color change took about 12 h and the colonies had shown a range of blue color intensities in the meantime. ESI Fig. SI and SII† show exemplary agar plates prior and after IPTG–X-gal spray. The percentages of the blue color of samples with active deoxyribozymes were clearly less than control samples at early time points (Fig. SIII†). The photographs and analyzed data for the case of the 10–23 deoxyribozyme for untranslated region, A39, are shown in Fig. 3. The best effect was observed 1.5 h after the spray. The difference faded at later time points in a way that at 12 h after spray, almost no difference of the blue color was detectable in colonies. Bacterial colonies with different ranges of blue-color were grouped and plotted in Fig. 3B. Such presentation shows that the number of colonies with higher cyan percentage are reduced in MS and NMS in comparison to negative control.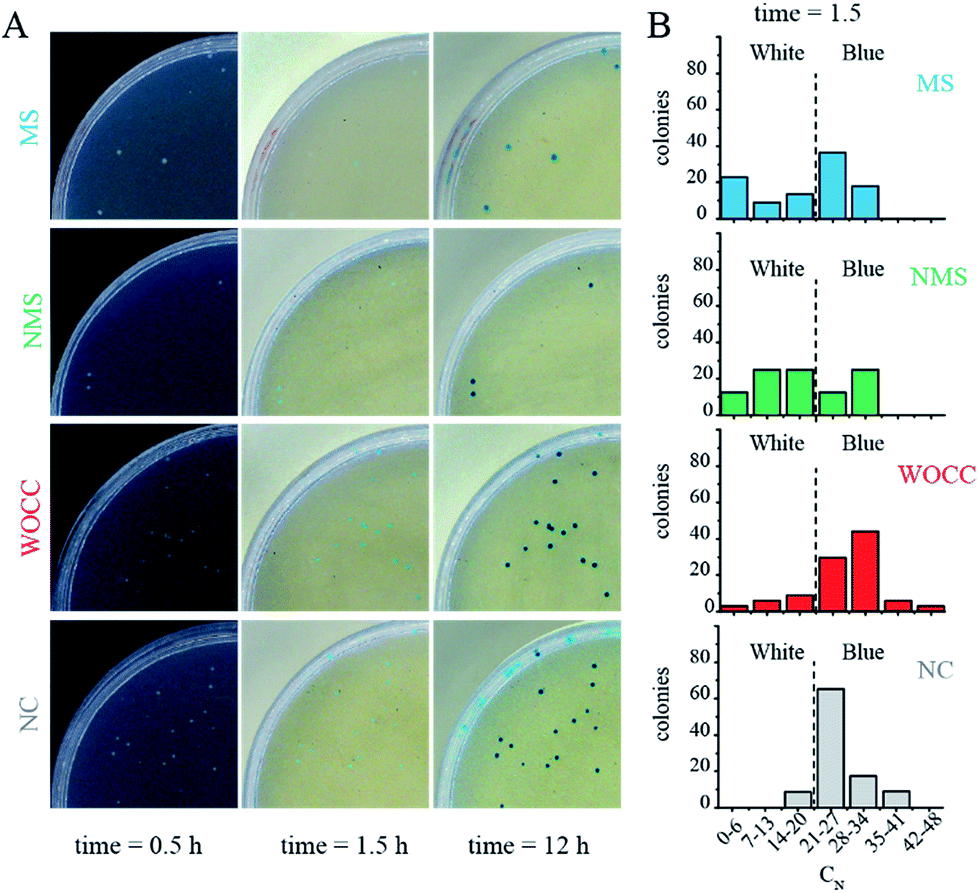 | ||
| Fig. 3 (A) Photos of the agar plates 0.5, 1.5 and 12 h after IPTG–X-gal spray. The background at time point 0.5 is black for better illustration of the white colonies. The blue color percentage of some colonies are marked in an exemplary photo of an agar plate in Fig. SI.† (B) Histogram of populations with various blue percentages at 1.5 h after spraying IPTG–X-gal solution for samples with deoxyribozymes against the untranslated region. The dashed lines indicate the threshold for transition to blue color colony. NC: negative control, WOCC: without catalytic core, NMS: non-modified sequence, MS: modified sequence with 3′–3′ inverted thymidine. Full plate images are shown in ESI Fig. SIV.† | ||
In the case of the assays for untranslated region, the in negative control (NC), which contained only pGEM-T blue color started to appear rapidly and the number of colonies with high cyan percentage shifted to about 60% of the population within 1.5 h. Such rapid color change was in favor of successful transcription of the mRNA of the α-peptide, fruitful translation of the mRNA and active complementation of the β-galactosidase. Formation of the blue color in samples with WOCC had no significant change in comparison to the negative control and ca. 70% of the colony population had a cyan color between 14–27% after 1.5 h. The color change in the other two samples, i.e. NMS and MS were stalled at early time points and the effect was more pronounced for MS. In a way that ca. 20% of the population of the colonies had minimal cyan color (0–6%) after 1.5 h (Fig. 3A, second column). All the bacterial colonies could transform to complete blue color at longer time points, i.e. 12 h after spray (Fig. 3A, last column).
Fig. 4 depicts the average of the percentage of the cyan color in the colonies of agar plates at three-time points. For both cases of assays, with 10–23 deoxyribozymes for untranslated region, A30 and translated region, A106, the most difference between the experimental setups was observed at 1.5 h after spraying IPTG–X-gal. At this time point significant reduction of the blue color was observed with modified sequences. Presence of the 3′-inverted thymidine at the 3′ end of the deoxyribozymes was turned out to improve the catalytic performance of both deoxyribozymes that inside bacterial cells. The non-modified sequence could only reduce the cyan color in the translated region. In both cases, the WOCC DNA had no meaningful effect in comparison to the control experiments (Fig. 4).
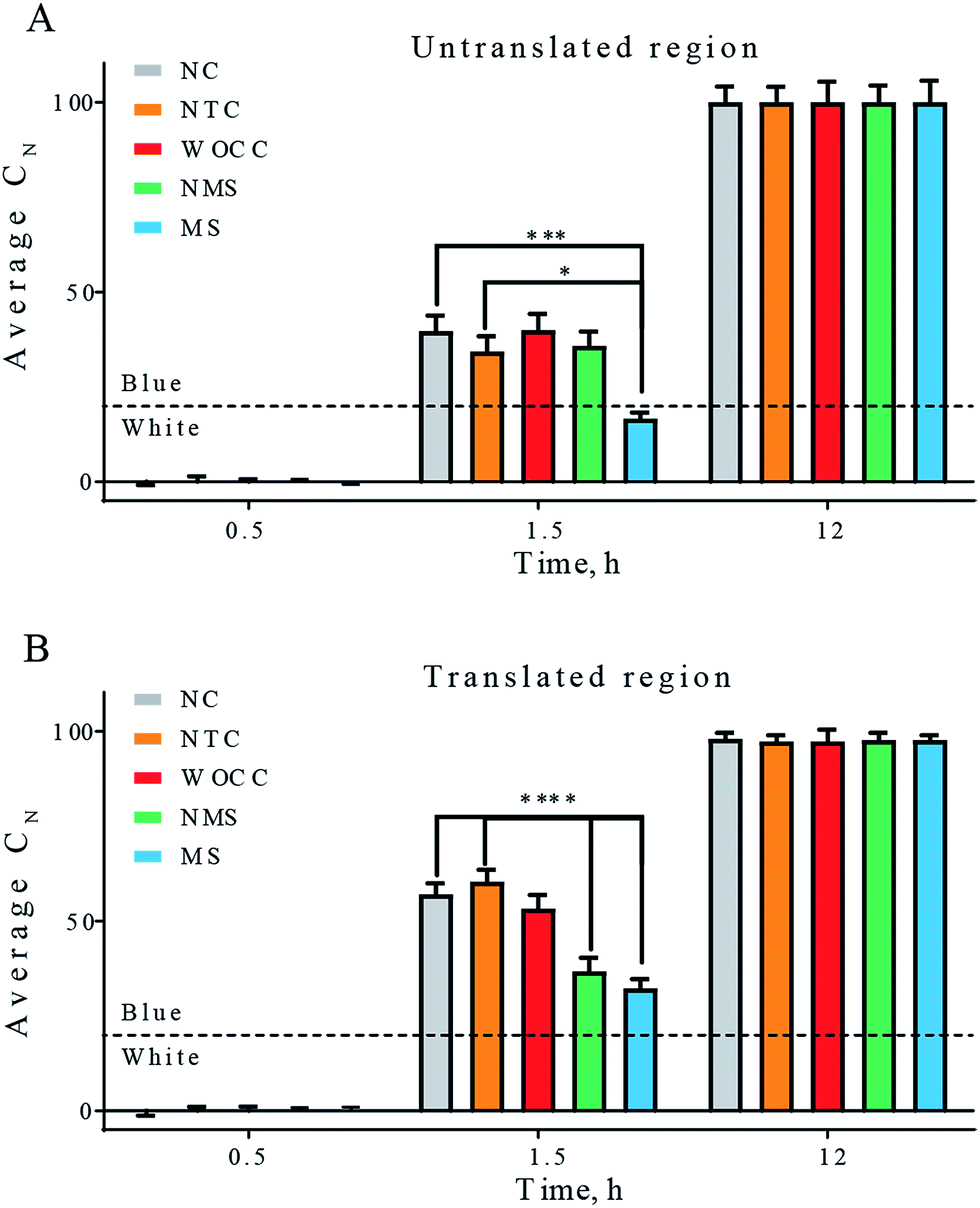 | ||
| Fig. 4 Average percentage of cyan color in total number of colonies. The first column is negative control (NC) in which pGEM-T vector was transformed in the absence of additional oligonucleotide. The next four columns are the result of co-transformation of pGEM-T vector and according oligonucleotides. Non-target DNA control (NTC), WOCC, NMS and MS sequences are as in Table 2. Panel (A) untranslated region as target, panels (B) translated region as target. The indicated times are the time points after solution spray. The dashed line depicts the threshold for transition to blue color. All values reported are mean ± SEM. [* for p < 0.05, ** for p < 0.01, *** for p < 0.005 and **** for p < 0.001]. | ||
Miller assay
The enzyme β-galactosidase is able to hydrolyze a wide range of artificial β-D-galactopyranosides including the chromogenic substrate ortho-nitrophenyl-β-D-galactopyranoside (ONPG). The hydrolysis of the glycosidic bond results in formation of ortho-nitrophenol which is yellow. A method that measures the specific activities of the β-galactosidase has been presented by Miller in 1972. The assay reports the enzymatic activity as miller units.The β-galactosidase activity of the transformed bacterial colonies were analyzed by Miller assay in this study. The yellow color was started to appear ca. 17 h after transformation. Since then, the Miller assays were performed for negative control, non-target control DNA, WOCC, NMS and MS. As in blue-white screening, the significant differences among the experimental setups were observed transiently. The most difference was at the 19 h time point. Consistent with blue-white screening results, at this time point, modified sequences of deoxyribozymes had significant reduction effect in comparison to both negative control and non-target control in both target sites. Non-modified sequence of 10–23 was effective for reduction of the blue color in translated region, but no meaningful reduction was observed with non-modified sequence in the untranslated region, consistent with blue-white screening results. WOCC could not show significant difference in either of the cases (Fig. 5).
 | ||
| Fig. 5 Miller assay for activity of 10–23 deoxyribozyme and controls for untranslated (A) and translated (B) region. NC: negative control, NTC: non-target DNA control WOCC: without catalytic core, NMS: non-modified sequence, MS: modified sequence with 3′–3′ inverted thymidine. [**** for p < 0.001]. | ||
Discussion
The RNA-cleaving 10–23 deoxyribozyme has been widely used for gene silencing in eukaryotic cells.27 This deoxyribozyme was extensively applied for cleavage of mRNAs that are responsible for several diseases such as allergic reactions, inflammation and cancer.28 A few 10–23 deoxyribozymes have been gone through clinical trials and currently passed phase I and II. In addition, many viral mRNAs have been targeted for cleavage with 10–23 deoxyribozymes inside eukaryotic cells.5 Despite such advances in application of 10–23 deoxyribozymes in eukaryotic cells, there are only few reports on the activity of this deoxyribozyme inside prokaryotic cells (Table 1). In addition, the available reports only target bacterial growth and survival. The β-lactamase pathway was the main target for 10–23 deoxyribozymes mediated cleavage in these reports.| Target pathway | Target gene | Host | Delivery method | Transfected molecule | Read outs | Ref. |
|---|---|---|---|---|---|---|
| a Carrying 10–23 deoxyribozyme sequence.b Plus helper phage VCSM13 to acquire single strand recombinant vector.c Standard heat-shock transformation of chemically competent cells. | ||||||
| Binary fission | ftsZ | E. coli | Not defined | pssXGa vectora | Cell growth, Western blot | 24 |
| β-Lactamase | mecR1 | S. aureus | Electroporation | Phosphorothioated 10–23 | Cell growth, reveres transcription-PCR | 23b |
| β-Lactamase | blaR1 | S. aureus | Electroporation | Phosphorothioated 10–23 | Cell growth, real time-PCR | 23a |
| β-Lactamase | TEM spectrum β-lactamase | E. coli | Electroporation | Unmodified 10–23 | Cell growth | 19 |
| β-Lactamase | β-Lactamase | E. coli | Electroporation | pBlue-script-II KS (+) phagemida,b | Cell growth | 25 |
| lacZ | β-Galactosidase | E. coli | Heat shockc | 3′-inverted T 10–23 | Blue-white screening | This report |
To the best of our knowledge, the current report is the first one for cleavage of an mRNA of a reporter gene with 10–23 deoxyribozymes in a viable prokaryotic cell. The target mRNA in this study was not intrinsically present in the host genome and was transformed simultaneously with the deoxyribozyme's oligonucleotide or its controls. An equivalent study for eukaryotic cells has been reported by Ackermann et al. for human embryonic kidney, HEK293 cell lines.29 The target gene was provided with a eukaryotic expression vector. The transformation of the expression vector and deoxyribozyme's oligonucleotide was carried out simultaneously. The ratio of vector to oligonucleotide was 8500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Although the strength of the promoters and the cell division rates of the two studies are not directly comparable, this ratio was taken as a rule of thumb for this study.
:1. Although the strength of the promoters and the cell division rates of the two studies are not directly comparable, this ratio was taken as a rule of thumb for this study.
In our study, we applied 0.02 pmol vector and 600 pmol deoxyribozyme for co-transformation into 50 μl of competent cells with total number of ca. 375000000 cells. The ratio of vector to oligonucleotide was ca. 32000:1, and the ratio of deoxyribozyme to bacterial cells was 1000000:1. Despite excessive application of deoxyribozymes in this study, the transformed RNA-cleaving 10–23 deoxyribozyme was in a big challenge inside rapidly growing E. coli colonies. In other words, besides nuclease-mediated degradation of the transformed deoxyribozymes, 10–23 oligonucleotides were prone to dilute and vanish in bacterial descendants. Hence, we expected a transient knock-down effect which was confirmed by our results.
The blockade of formation of blue-color from X-gal was transient. In other words, the colonies with active enzyme were as blueish as control colonies at long time span (above 4 h from induction). The reason could be due to two parallel phenomena; (1) dilution of the deoxyribozymes upon bacterial division, (2) nuclease degradation. In this study, the deoxyribozymes had no antibiotic-like effect; therefore, the colonies were growing during the assays. This means, while the number of transformed DNA oligonucleotides was constant, the number of cells was increasing. Thus, it would be logical to deduce that within a colony there would be some bacterial cells that get a low concentration of the deoxyribozyme. At such low concentrations, the target mRNA (α-peptide) was not efficiently cleaved and cells scape from blockade of α-complementation. If only a few cells in a colony form the active β-galactosidase the presence of blue color will override the color of the colony i.e. the colony becomes blue, in spite of probable presence of bacterial cells that lack active β-galactosidase.
A thorough study must have been performed to find the best time point with maximal difference with control samples. With 30 min intervals, we comprehensively investigated for the best time to visualize the effect. The optimal time for blue-white screening was between 1–2 h after induction by spraying IPTG–X-gal solution. This time was indeed 11–12 hours after transformation, due to the fact that it took about 10 h for formation of detectable colonies. The best time point for observing the difference in the Miller assay was 19 h, since it took about 17 h for initial observation of the yellow color from Miller assay.
Regarding the nuclease dependent degradation of oligonucleotides, various modifications have been applied for 10–23 deoxyribozyme to increase the half-life of the DNA within eukaryotic cells. In contrast, the only modification in prokaryotic system was phosphorothioation. Here, we investigated the effect of the 3′–3′ inverted thymidine. Our result demonstrated that MS (the 10–23 deoxyribozyme with modification) could knock-down β-galactosidase more efficiently in comparison to NMS (non-modified sequence). Formation of the blue color from X-gal and formation of yellow color from ONPG was meaningfully reduced in MS, although the effect was transient. NMS was effective only in translated region, while the MS could stall blue color formation in both target sites. Thus, we conclude that 3′–3′ inverted thymidine is an efficient modification for application of deoxyribozymes in prokaryotic cells.
The DNA oligonucleotides at which the catalytic cores were replaced by three thymidines could not cause a decrease in the readout of any of the assays, expectedly. These oligonucleotides were modified with 3′–3′ inverted thymidine and thus the absence of the effect was not the result of nuclease degradation. Hence, the blue- or yellow-color reductions were deduced to be the direct effect of 10–23 deoxyribozyme catalysis and cleavage of the mRNA of the α-peptide. In the absence of α-peptide, the four monomers of β-galactosidase could not form the active tetramer and the enzyme was blocked. Our result showed that similar reduction extent for translated and untranslated regions when modified deoxyribozymes were used. However, with non-modified sequences, the 10–23 deoxyribozyme could not efficiently cleave and block translation of α-peptide in untranslated region.
Experimental
Materials and methods
The secondary structure of the mRNA transcript of the α-peptide was predicted with mfold from the DINAmelt web server.30 The default parameters were used, and the secondary structure with the lowest free energy was exploited to identify the regions within the mRNA that are accessible to the deoxyribozyme. The deoxyribozymes with 9 nucleotides long binding arms at each site have been designed against target sequences at A39–C40 and A106–C107. The left binding arms fully hybridize to the target DNA sequence except at the cleavage site's adenosines. The catalytic core of the 10–23 deoxyribozyme with the sequence of GGCTAGCTACAACGA were substituted the base pair at this adenosine. The adjacent cytosines at the cleavage sites and its 3′ nucleotides were hybridized to the right binding arms. The designed binding arms were blasted to assure absence of non-target binding of the deoxyribozymes. The designed deoxyribozyme sequences were either modified (here mentioned as MS = modified sequence) by adding a reversed 3′-thymidine or not modified (NMS).
An oligonucleotide sequence from designed deoxyribozyme without catalytic core (WOCC) was considered as a control. The catalytic domain of 10–23 deoxyribozyme was substituted with three consecutive thymidines in WOCC oligonucleotides. Additionally, non-enzymatic, and non-target control DNA (NTC) which was a random oligonucleotide sequences was used to control the effect of transformation. Table 2 depicts the oligonucleotide sequences that have been used in this study.
| Target | Oligonucleotide | Sequence |
|---|---|---|
| a NMS: non-modified 10–23 deoxyribozyme sequence, MS: modified 10–23 deoxyribozyme sequence with 3′–3′ inverted thymidine, WOCC: without catalytic core. The sequence of the catalytic core or its substitutes are bold and underlined. | ||
| Untranslated region A39–C40 | NMS |  |
| MS |  |
|
| WOCC |  |
|
| Translated region A106–C107 | NMS |  |
| MS |  |
|
| WOCC |  |
|
| Non-target control | NTC | TTTAAGCTTGCTTTGGAACAGAACCAG |
| Miller unit = 1000 × [(A420 − 1.75 × A550)/(A600 × volume × reaction time)] | (1) |
Conclusions
In summary, this report showed a proof of concept for application of blue-white screening as a readout system for activity of the model 10–23 deoxyribozyme. The result of this report can promote future investigations on activity of RNA-cleaving deoxyribozymes in vivo. The applied condition provided a transient inhibition effect on β-galactosidase activity. Stable knock-down effect is expected by employing methods that provide in situ production of single stranded deoxyribozymes in prokaryotic cells e.g. pBlue-scriptIIKS (+)25 or MMLV-RT system.7Conflicts of interest
None of the authors declare conflict of interest.Acknowledgements
The authors would very much like to thank University of Isfahan for its support of this study.Notes and references
- T. R. Cech, The Ribosome Is a Ribozyme, Science, 2000, 289(5481), 878–879 CrossRef CAS PubMed.
- (a) P. Chartrand, S. C. Harvey, G. Ferbeyre, N. Usman and R. Cedergren, An oligodeoxyribonucleotide that supports catalytic activity in the hammerhead ribozyme domain, Nucleic Acids Res., 1995, 23(20), 4092–4096 CrossRef CAS PubMed; (b) R. R. Breaker and G. F. Joyce, A DNA enzyme that cleaves RNA, Chem. Biol., 1994, 1(4), 223–229 CrossRef CAS PubMed.
- Y. Lee, P. C. Klauser, B. M. Brandsen, C. Zhou, X. Li and S. K. Silverman, DNA-Catalyzed DNA Cleavage by a Radical Pathway with Well-Defined Products, J. Am. Chem. Soc., 2017, 139(1), 255–261 CrossRef CAS PubMed.
- S. W. Santoro and G. F. Joyce, A general purpose RNA-cleaving DNA enzyme, Proc. Natl. Acad. Sci. U. S. A., 1997, 94(9), 4262–4266 CrossRef CAS.
- N. Singh, A. Ranjan, S. Sur, R. Chandra and V. Tandon, Inhibition of HIV-1 Integrase gene expression by 10-23 DNAzyme, J. Biosci., 2012, 37(3), 493–502 CrossRef CAS PubMed.
- A. A. Fokina, D. A. Stetsenko and J. C. Francois, DNA enzymes as potential therapeutics: towards clinical application of 10-23 DNAzymes, Expert Opin. Biol. Ther., 2015, 15(5), 689–711 CrossRef CAS PubMed.
- J. Li, N. Wang, Q. Luo and L. Wan, The 10-23 DNA enzyme generated by a novel expression vector mediate inhibition of taco expression in macrophage, Oligonucleotides, 2010, 20(2), 61–68 CrossRef CAS PubMed.
- L. Robaldo, A. Berzal-Herranz, J. M. Montserrat and A. M. Iribarren, Activity of core-modified 10-23 DNAzymes against HCV, ChemMedChem, 2014, 9(9), 2172–2177 CrossRef CAS PubMed.
- K. Yehl, J. P. Joshi, B. L. Greene, R. B. Dyer, R. Nahta and K. Salaita, Catalytic Deoxyribozyme-Modified Nanoparticles for RNAi-Independent Gene Regulation, ACS Nano, 2012, 6(10), 9150–9157 CrossRef CAS PubMed.
- H. Unwalla and A. C. Banerjea, Novel mono- and di-DNA-enzymes targeted to cleave TAT or TAT-REV RNA inhibit HIV-1 gene expression, Antiviral Res., 2001, 51(2), 127–139 CrossRef CAS PubMed.
- S. Kim, S.-R. Ryoo, H.-K. Na, Y.-K. Kim, B.-S. Choi, Y. Lee, D.-E. Kim and D.-H. Min, Deoxyribozyme-loaded nano-graphene oxide for simultaneous sensing and silencing of the hepatitis C virus gene in liver cells, Chem. Commun., 2013, 49(74), 8241–8243 RSC.
- B. Kumar, P. Kumar, R. Rajput, L. Saxena, M. K. Daga and M. Khanna, Sequence-specific cleavage of BM2 gene transcript of influenza B virus by 10-23 catalytic motif containing DNA enzymes significantly inhibits viral RNA translation and replication, Nucleic Acid Ther., 2013, 23(5), 355–362 CrossRef CAS PubMed.
- P. Reyes-Gutierrez and L. M. Alvarez-Salas, Cleavage of HPV-16 E6/E7 mRNA mediated by modified 10-23 deoxyribozymes, Oligonucleotides, 2009, 19(3), 233–242 CrossRef CAS PubMed.
- S. Wu, J. Xu, J. Liu, X. Yan, X. Zhu, G. Xiao, L. Sun and P. Tien, An efficient RNA-cleaving DNA enzyme can specifically target the 5'-untranslated region of severe acute respiratory syndrome associated coronavirus (SARS-CoV), J. Gene Med., 2007, 9(12), 1080–1086 CrossRef CAS PubMed.
- Y. Cao, L. Yang, W. Jiang, X. Wang, W. Liao, G. Tan, Y. Liao, Y. Qiu, D. Feng and F. Tang, Therapeutic evaluation of Epstein-Barr virus-encoded latent membrane protein-1 targeted DNAzyme for treating of nasopharyngeal carcinomas, Mol. Ther., 2014, 22(2), 371–377 CrossRef CAS PubMed.
- A.-Y. Xing, D.-B. Shi, W. Liu, X. Chen, Y.-L. Sun, X. Wang, J.-P. Zhang and P. Gao, Restoration of chemosensitivity in cancer cells with MDR phenotype by deoxyribozyme, compared with ribozyme, Exp. Mol. Pathol., 2013, 94(3), 481–485 CrossRef CAS PubMed.
- U. Homburg, H. Renz, W. Timmer, J. M. Hohlfeld, F. Seitz, K. Lüer, A. Mayer, A. Wacker, O. Schmidt, J. Kuhlmann, A. Turowska, J. Roller, K. Kutz, G. Schlüter, N. Krug and H. Garn, Safety and tolerability of a novel inhaled GATA3 mRNA targeting DNAzyme in patients with TH2-driven asthma, J. Allergy Clin. Immunol., 2015, 136(3), 797–800 CrossRef CAS PubMed.
- N. Krug, J. M. Hohlfeld, A.-M. Kirsten, O. Kornmann, K. M. Beeh, D. Kappeler, S. Korn, S. Ignatenko, W. Timmer, C. Rogon, J. Zeitvogel, N. Zhang, J. Bille, U. Homburg, A. Turowska, C. Bachert, T. Werfel, R. Buhl, J. Renz, H. Garn and H. Renz, Allergen-Induced Asthmatic Responses Modified by a GATA3-Specific DNAzyme, N. Engl. J. Med., 2015, 372(21), 1987–1995 CrossRef PubMed.
- F. Chen, Z. Li, R. Wang, B. Liu, Z. Zeng, H. Zhang and J. Zhang, Inhibition of ampicillin-resistant bacteria by novel mono-DNAzymes and di-DNAzyme targeted to beta-lactamase mRNA, Oligonucleotides, 2004, 14(2), 80–89 CrossRef CAS PubMed.
- A. Abdelgany, M. K. Uddin, M. Wood, K. Taira and D. Beeson, Design of efficient DNAzymes against muscle AChR alpha-subunit cRNA in vitro and in HEK 293 cells, J. RNAi Gene Silencing, 2005, 1(2), 88–96 CAS.
- M. Chakravarthy, M. T. Aung-Htut, B. T. Le and R. N. Veedu, Novel Chemically-modified DNAzyme targeting Integrin alpha-4 RNA transcript as a potential molecule to reduce inflammation in multiple sclerosis, Sci. Rep., 2017, 7, 1613 CrossRef PubMed.
- A. A. Fokina, M. I. Meschaninova, T. Durfort, A. G. Venyaminova and J. C. Francois, Targeting insulin-like growth factor I with 10-23 DNAzymes: 2'-O-methyl modifications in the catalytic core enhance mRNA cleavage, Biochemistry, 2012, 51(11), 2181–2191 CrossRef CAS PubMed.
- (a) Z. Hou, J. R. Meng, J. R. Zhao, B. Q. Hu, J. Liu, X. J. Yan, M. Jia and X. X. Luo, Inhibition of beta-lactamase-mediated oxacillin resistance in Staphylococcus aureus by a deoxyribozyme, Acta Pharmacol. Sin., 2007, 28(11), 1775–1782 CrossRef CAS PubMed; (b) Z. Hou, J. R. Meng, C. Niu, H. F. Wang, J. Liu, B. Q. Hu, M. Jia and X. X. Luo, Restoration of antibiotic susceptibility in methicillin-resistant Staphylococcus aureus by targeting mecR1 with a phosphorothioate deoxyribozyme, Clin. Exp. Pharmacol. Physiol., 2007, 34(11), 1160–1164 CAS.
- X. X. Tan, K. Rose, W. Margolin and Y. Chen, DNA enzyme generated by a novel single-stranded DNA expression vector inhibits expression of the essential bacterial cell division gene ftsZ, Biochemistry, 2004, 43(4), 1111–1117 CrossRef CAS PubMed.
- Y. Sheng, Z. Zeng, W. Peng, D. Jiang, S. Li, Y. Sun and J. Zhang, Design and switch of catalytic activity with the DNAzyme-RNAzyme combination, FEBS Lett., 2007, 581(9), 1763–1768 CrossRef CAS PubMed.
- D. H. Juers, B. W. Matthews and R. E. Huber, LacZ β-galactosidase: Structure and function of an enzyme of historical and molecular biological importance, Protein Sci., 2012, 21(12), 1792–1807 CrossRef CAS PubMed.
- C. R. Dass, P. F. Choong and L. M. Khachigian, DNAzyme technology and cancer therapy: cleave and let die, Mol. Cancer Ther., 2008, 7(2), 243–251 CrossRef CAS PubMed.
- M. Zhang, G. P. Drummen and S. Luo, Anti-insulin-like growth factor-iiP3 Dnazymes inhibit cell proliferation and induce caspase-dependent apoptosis in human hepatocarcinoma cell lines, Drug Des., Dev. Ther., 2013, 7, 1089 Search PubMed.
- J. M. Ackermann, S. Kanugula and A. E. Pegg, DNAzyme-mediated silencing of ornithine decarboxylase, Biochemistry, 2005, 44(6), 2143–2152 CrossRef CAS PubMed.
- N. R. Markham and M. Zuker, DINAMelt web server for nucleic acid melting prediction, Nucleic Acids Res., 2005, 33(Web Server issue), W577–W581 CrossRef CAS PubMed.
- T. Tschirhart, X. Y. Zhou, H. Ueda, C.-Y. Tsao, E. Kim, G. F. Payne and W. E. Bentley, Electrochemical Measurement of the β-Galactosidase Reporter from Live Cells: A Comparison to the Miller Assay, ACS Synth. Biol., 2016, 5(1), 28–35 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09679h |
| This journal is © The Royal Society of Chemistry 2017 |