
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of sustainable lignin-derived mesoporous carbon for supercapacitors using a nano-sized MgO template coupled with Pluronic F127†
Yaoguang Song ,
Junli Liu*,
Kang Sun and
Wei Xu
,
Junli Liu*,
Kang Sun and
Wei Xu
Institute of Chemical Industry of Forest Products, CAF, National Engineering Lab. for Biomass Chemical Utilization, Key and Open Lab. on Forest Chemical Engineering, SFA, Key Lab. of Biomass Energy and Material, Jiangsu Province, Nanjing 210042, China. E-mail: liujunli@msn.com; Tel: +86-25-854828410
First published on 16th October 2017
Abstract
Herein, we describe the synthesis of meso-structured carbon from residual pre-cross-linked lignin from a wheat straw alkaline pulping process by employing nano-sized MgO and Pluronic F127 as templates. MgO nano-particles function as substrates and the main template by a space occupying effect, while F127 works as a dispersant preventing the agglomeration of MgO as well as a soft templating agent. To the best of our knowledge, no work concerning the synthesis of lignin-derived mesoporous carbon via a MgO template route has been reported. The mass ratio of lignin/MgO plays a major role in enriching the porosity of the carbon. Vacuum drying and freeze-drying technologies were applied during solvent evaporation to ensure the Mg template can be dispersed sufficiently. The Brunauer–Emmett–Teller specific surface area and total pore volume can reach 712 m2 g−1 and 0.90 cm3 g−1, with a mesopore content of over 83%. The electrochemical performance of the obtained carbons as supercapacitor electrode materials was investigated. Although the vacuum environment enhanced the porosity and pore uniformity, the corresponding carbon didn't exhibit better electrochemical behavior than the carbon derived without a vacuum environment.
1. Introduction
The irreversible depletion of fossil fuels and ever-increasing concentration of key greenhouse gases in the atmosphere have impelled the conversion of lignocellulosic biomass feedstock and the reutilization of waste biomass resources for energy applications to meet human's daily energy consumption demands.1,2 Besides cellulose and hemicellulose, lignin is one of the three recalcitrant components of lignocellulosic biomass and the most abundant heterogeneous aromatic structural biopolymer in the natural world. In addition, lignin, as an almost unlimited residual byproduct of the pulp and paper industry, with a total global yield of about 50 billion tons per year, and in particular over 5 million tons in China,3 constitutes 30% of non-fossil organic carbon and is inefficient as a solid fuel to supply energy by burning.4,5 Deriving more value from lignin therefore has successfully caught worldwide attention.6–10Lignin has a high carbon content over 50% (ref. 11) and becomes one of the most promising ideal precursors for carbonaceous materials. Microporous carbon materials (e.g. conventional activated carbons) are ideally suited to liquid and gas-phase adsorption but have industrial limited applications including biomedical devices, catalysis, and electrochemical partially due to the requirement for mesopores.2 Particularly, mesoporous carbons as electrode materials for supercapacitors show higher capacitance retention and excellent rate performance at high current density.12–15 Preparing mesoporous carbons from sustainable lignin therefore has interested researchers worldwide, especially through nano-casting techniques, which commonly employ sacrificial silica16–19 or amphiphilic surfactants20–22 to tailor the pore texture precisely.
Unlike linear polysaccharides, lignin is a complicated amorphous polymer, composed of phenylpropanolic units linked by ether and C–C bonds.6 Regardless of some negative aspects of hard template route such as multiple synthesis steps and the use of hazardous HF or NaOH,23 it is still challenging to ensure highly branched lignin to be impregnated into narrow pores of mesoporous silica. While the soft template route based on micelle formation possesses inherent limitations on choosing ideal template surfactants and carbon precursors,23,24 which are mostly phenolic monomers (e.g. phenol, resorcinol and phloroglucinol) or linear polysaccharides with lower molecular weight and specific molecular structure (e.g. glucose and furfural). Heterogeneous molecular architecture and highly branched structure can cause imperfect self-assembly of surfactant micelles in the randomly cross-linked matrix and result in disordered pore structure and small pore volume.22 Saha et al.20,22 recently employed a commercially available amphiphilic surfactant Pluronic F127 as template to fabricate mesoporous carbon from lignin in combination with formaldehyde (HCHO) as cross-linking agent. They compared it with carbons derived from a typical phenolic precursor (i.e. phloroglucinol) and natural hardwood pre-cross-linked lignin. The carbon with phloroglucinol precursor exhibited a narrower pore widths and a larger Brunauer–Emmett–Teller specific surface area (BET SSA) than those of lignin-derived mesoporous carbons. While, the carbon with pre-cross-linked lignin precursor showed a much smaller BET SSA than that of carbon with lower-molecular-weight fraction of lignin in combination with HCHO. The polymerization degree of lignin has a strong effect on the porosity and structure of carbon. Therefore, synthesizing lignin-derived carbons via porous silica template route or surfactant template route is lack of the versatility due to the highly branched structure and the diversity of lignin from different sources.
Some reports synthesized mesoporous carbons via hard template route employing oxides nanoparticles or salts as template, such as MgO,25–30 Fe2O3,15,31,32 and CaCO3,33,34 which is more convenient than surfactant template route or traditional porous silicon-containing templates. Particularly, the MgO template route is unconstrained by the polymerization degree of precursor, and throughout the whole carbonization, the MgO cannot react with the carbonaceous materials formed from the pyrolysis of carbon precursors and functions just as substrate.26,28 What's more, MgO template can be easily dissolved out by diluted acid after carbonization to isolate carbon products, and availably recovered and recycled for the production of porous carbons.26 To the best of our knowledge, no report about synthesizing lignin-derived mesoporous carbons by employing MgO as template has been found.
Herein, we describe the synthesis of mesoporous carbon from a kind of cheap and plentiful waste biomass, residual pre-cross-linked alkali lignin in wheat straw alkaline pulping process by using MgO as template additionally coupled with Pluronic F127 template (EO106PO70EO106, with an average molecular weight of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600). The space occupying effect of MgO template is critical to enlarge the porosity of carbons. Both the soft template Pluronic F127 and MgO precursor eventually show a negligible carbon yield after carbonization.24,26 Possible ideal synthesis mechanism schematic of this dual templates route is depicted roughly in Fig. 1. In this prevent work, Mg(CH3COO)2·4H2O was selected as the precursor of MgO particles, obtained by the pyrolysis of Mg acetate below 350 °C. The functions of nano MgO and Pluronic F127, precursors mixing mass ratios, pore textural properties and electrochemical performance of as-made carbons as electrode materials on supercapacitor were discussed in detail. Our objective was to provide a new thought to synthesize lignin-derived carbon materials and to enhance the sustainability by reutilizing waste biomass.
600). The space occupying effect of MgO template is critical to enlarge the porosity of carbons. Both the soft template Pluronic F127 and MgO precursor eventually show a negligible carbon yield after carbonization.24,26 Possible ideal synthesis mechanism schematic of this dual templates route is depicted roughly in Fig. 1. In this prevent work, Mg(CH3COO)2·4H2O was selected as the precursor of MgO particles, obtained by the pyrolysis of Mg acetate below 350 °C. The functions of nano MgO and Pluronic F127, precursors mixing mass ratios, pore textural properties and electrochemical performance of as-made carbons as electrode materials on supercapacitor were discussed in detail. Our objective was to provide a new thought to synthesize lignin-derived carbon materials and to enhance the sustainability by reutilizing waste biomass.
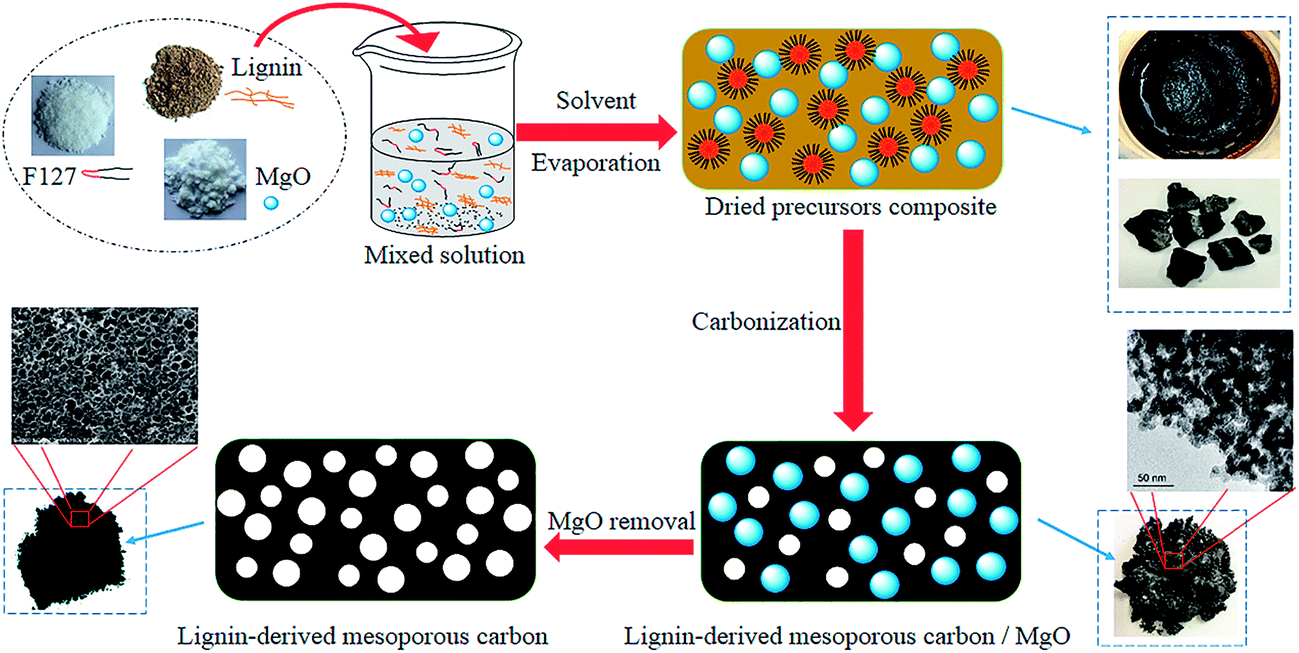 | ||
| Fig. 1 Schematic of lignin-derived mesoporous carbon via a dual-template strategy. | ||
2. Material and methods
2.1 Pretreatment of pre-cross-linked alkali lignin
80 g of initial alkali lignin powder (pH = 10.1, 3 wt% in water, Xinyi Feihuang Chemical Co., Ltd, China) was dissolved in 800 mL of deionized water and then filtered when pH was adjusted to ∼7.0 with hydrochloric acid (HCl, Nanjing Chemical Reagent Co., Ltd, China). The filtrate was subsequently stirred adequately in dilute slurry form after pH was adjusted to ∼2.0. Then lignin purified roughly was eventually obtained after being filtered, washed and dried at 105 °C.Detailed properties of the lignin purified are listed in Table 1. Note that the purity including acid-soluble and acid-insoluble lignin was measured according to American National Renewable Energy Laboratory's (NREL) Laboratory Analytical Procedures.
2.2 Synthesis of lignin-derived mesoporous carbons
For a typical run, the alkali lignin powder pretreated above and Pluronic F127 were dissolved in 200 mL of THF with additional 1.34 mL of 6 M HCl. Mg(CH3COO)2·4H2O was simultaneously dissolved in 300 mL of H2O, and slowly poured into the former solution. Afterward, the mixed solution was stirred at room temperature for 24 h, and placed at 45 °C for 1 day and maintained at 70 °C until the monolithic mass was exsiccated thoroughly. Then the dried mixture mass was collected in a quartz boat and carbonized in a tube furnace under flowing nitrogen (100 mL min−1). The heating profile was ramped from ambient temperature to 70 °C at 10 °C min−1, 70 °C to 400 °C at 1 °C min−1, 400 °C to 1000 °C at 2 °C min−1, and maintained at 1000 °C for 15 min, followed by cooling down to ambient temperature. Finally, the sample was washed by using 1 M HCl solution and H2O to dissolve the substrate MgO out, and dried at 105 °C for 24 h. The obtained carbons are labelled as LMCx–y–z, where the subscript x, y and z represent the mass of lignin, MgO and Pluronic F127, respectively.For comparison, two samples were also synthesized via the surfactant template route and the independent MgO template route, respectively. More concretely, the sample only using Pluronic F127 as template was fabricated by remaining all aforementioned experimental conditions identical except no Mg acetate aqueous solution added and labelled as LMCF127. While, the sample via the independent MgO template route was made by the same experimental conditions except no Pluronic F127 added and labelled as LMCMgO. Furthermore, a sample without any templating agent was also prepared by carbonizing alkali lignin powder only and labelled as LC.
2.3 Characterizations
The pore texture properties of all as-made carbons were investigated with an ASAP 2020 analyzer by N2 adsorption–desorption isotherms at 77 K. BET SSA and differential pore size distribution were calculated respectively by using the BET equation and the density functional theory (DFT). Morphology of carbons was performed by field emission scanning electron microscopy (FESEM, Hitachi S-4800) at an accelerating voltage of 3.0 kV and by transmission electron microscopy (TEM, JEM 2100) at an accelerating voltage of 200 kV. X-ray diffraction (XRD) patterns (10–80°) and Raman spectra were obtained by a D8 Focus X-ray diffractometer equipped with Cu Kα radiation (λ = 0.154 nm) and a Thermor DXR532 Raman spectrometer with a 532 nm excitation source, respectively. X-ray photoelectron spectroscopic (XPS) data was performed by a Kratos AXIS UltraDLD K-Alpha X-ray photoelectron spectrometer in the ultrahigh vacuum of 7 × 10−8 Pa equipped with hemisphere analyzer.2.4 Electrochemical test
The performance for supercapacitor of the obtained carbons was tested on a CHI 660D electrochemical analyzer at ambient temperature. The electrode was prepared by mixing 80 wt% carbon samples, 10 wt% acetylene black and 10 wt% PTFE, where the former two worked as an electrical conductor and the latter as a binder. In the three-electrode configuration, the counter electrode, reference electrode and electrolyte are a platinum wire, Ag/AgCl electrode, and 1 M H2SO4, respectively. The gravimetric capacitances (Cg, F g−1) was calculated by the formula of Cg = (I × Δt)/(m × V), where I (A) is discharge current, Δt (s) is the discharge time, V (V) is the potential window during discharge and m (g) is the mass of active material on electrode.3. Results and discussion
3.1 Characterization of carbons
We successfully synthesized mesoporous carbons from pre-cross-linked lignin particles by simultaneously employing MgO nanoparticles and Pluronic F127 as structure-directing agents. Detailed pore texture properties of as-made LMCs are listed in Table 2. Obviously, samples via single templating method (LMCF127 and LMCMgO) have higher porosities than LC, the sample without any templating agent. Moreover, the BET SSA and total pore volume of LMC10–1–16, the sample synthesized with 1 g of MgO and 16 g of F127, reached 356 m2 g−1 and 0.44 cm3 g−1, which are both much larger than those of LMCF127 and LMCMgO. For comparison with LMCF127, sample LMCF127–HCHO was simultaneously synthesized in the same way from lignin additionally in combination with HCHO as cross-linking agent, the BET SSA and total pore volume of which decreased to only 26 m2 g−1 and 0.029 cm3 g−1. Similarly, LMC10–1–16-HCHO with HCHO added was also prepared in the same way as LMC10–1–16 and exhibited a BET SSA of 234 m2 g−1 and total pore volume of 0.23 cm3 g−1. That the decreased porosities after adding HCHO can be attributed to the highly branched lignin structure utilized in this present work and the continuous over-cross-linked lignin matrix after the cross-linking reaction. However, as shown in Fig. S1,† the solvent evaporation process during synthesizing LMCMgO induced Mg acetate partially to recrystallize and agglomerate together, even in an extremely small lignin/Mg(CH3COO)2·4H2O mixing ratio, directly resulting in the poor porosity of LMCMgO, whose BET SSA and pore volume are still larger than LMCF127 though. Fig. S2† shows the TEM micrograph of sample LMC10–1–16 before template extraction. Some distorted aperiodic pores formed from the F127 and the carbonization of lignin itself can be clearly seen. Besides, nano-sized MgO particles with size of about 11 nm are distributed disorderly and randomly. Therefore, we infer that the Pluronic F127 is critical to disperse Mg acetate sufficiently as well as functioning as the soft template, and that the amount of MgO nanoparticles may play a major role to enrich the porosity of carbons.| Sample | Lignin (g) | MgO (g) | F127 (g) | BET SSA (m2 g−1) | Vmeso (cm3 g−1) | Vtotal (cm3 g−1) | Mesopores (%) |
|---|---|---|---|---|---|---|---|
| LC | 10 | 0 | 0 | 19 | 0.009 | 0.023 | 39.13 |
| LMCF127 | 10 | 0 | 16 | 59 | 0.070 | 0.087 | 80.46 |
| LMCF127–HCHO | 10 | 0 | 16 | 26 | 0.013 | 0.029 | 44.83 |
| LMCMgO | 10 | 1 | 0 | 85 | 0.071 | 0.103 | 68.93 |
| LMC10–1–16 | 10 | 1 | 16 | 356 | 0.35 | 0.44 | 79.55 |
| LMC10–1–16-HCHO | 10 | 1 | 16 | 234 | 0.14 | 0.23 | 60.87 |
| LMC10–2–16 | 10 | 2 | 16 | 419 | 0.54 | 0.67 | 80.60 |
| LMC10–4–16 | 10 | 4 | 16 | 563 | 0.65 | 0.77 | 84.42 |
| LMC10–6–16 | 10 | 6 | 16 | 610 | 0.67 | 0.81 | 82.72 |
| LMC10–8–8 | 10 | 8 | 8 | 555 | 0.56 | 0.67 | 83.58 |
| LMC10–8–16 | 10 | 8 | 16 | 642 | 0.72 | 0.86 | 84.88 |
| LMC10–8–16, V | 10 | 8 | 16 | 712 | 0.75 | 0.90 | 83.33 |
| LMC10–8–16, F | 10 | 8 | 16 | 672 | 0.56 | 0.72 | 77.78 |
| LMC10–8–24 | 10 | 8 | 24 | 642 | 0.77 | 0.91 | 84.62 |
| LMC10–8–32 | 10 | 8 | 32 | 643 | 0.77 | 0.91 | 84.62 |
| LMC10–12–16 | 10 | 12 | 16 | 628 | 0.55 | 0.68 | 80.88 |
| LMC10–16–16 | 10 | 16 | 16 | 572 | 0.46 | 0.61 | 75.41 |
| LMC10–20–16 | 10 | 20 | 16 | 487 | 0.45 | 0.56 | 80.36 |
The derivative plots from thermogravimetric analysis (DTG) in Fig. S3† shows that Mg(CH3COO)2·4H2O has total two weight loss states. The former one occurs below 200 °C, indicating the loss of its crystal water, and the latter one provides MgO nanoparticles from the decomposition of Mg(CH3COO)2·4H2O at the temperature ranging from 300 °C to 350 °C. Compared with pure Pluronic F127, the DTG peak of F127 mixed together with lignin appears a 5 °C shift to the higher temperature of 366 °C, which could signify the hydrogen bonding between F127 and lignin.24 After further adding Mg acetate, the DTG peak of the eventual precursor mixture appears at around 364 °C. Such a 2 °C shift to lower temperature of 364 °C can be attributed to the weak chelation between Mg2+ and lignin, and the dispersion of Mg acetate via the chelation with the O atoms of F127 in some extent.
To ascertain the relationship between MgO/lignin mixing ratio and the eventual corresponding pore texture properties, we remained the mass of F127 as an invariant constant and gradually increased the mixing ratio of MgO/lignin in the precursor mixture. As clearly shown in Fig. 2a, MgO/lignin mixing ratio is a crucial factor for the eventual porosity of LMCs. After adding MgO template, BET SSA and pore volume increased greatly and kept apparent growth trend at a lower dosage of Mg(CH3COO)2·4H2O, whereas they gradually decreased with the increasing MgO/lignin mixing mass ratio. The obtained LMC10–8–16, the sample synthesized with 8 g of MgO and 16 g of F127, got the richest porosity with a BET SSA of 642 m2 g−1 and a total pore volume of 0.86 cm3 g−1, including a mesopore volume of 0.72 cm3 g−1.
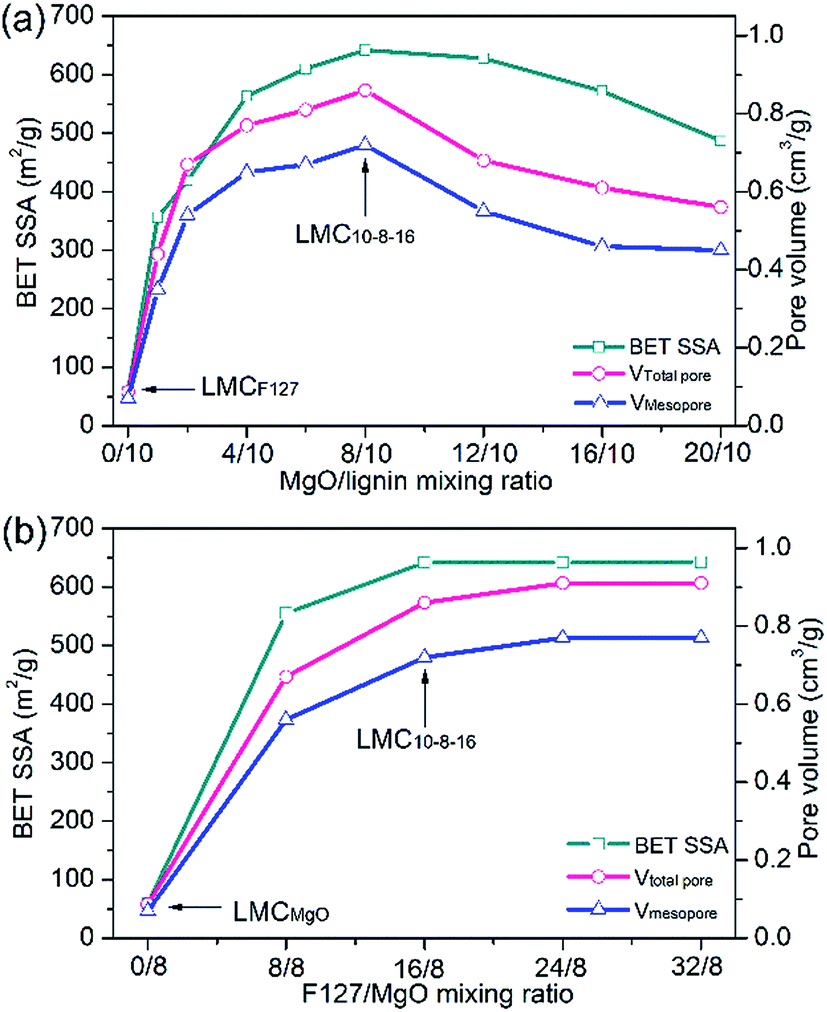 | ||
| Fig. 2 (a) Optimal level of MgO/lignin mixing ratio and (b) optimal level of F127/MgO mixing ratio as determined from BET SSA, mesopore and total pore volume. | ||
Pluronic F127 functions as dispersant to prevent the agglomeration of Mg acetate as well as soft template agent. Therefore, the dosage of F127 is another key factor affecting the eventual porosity of carbons. Fig. 2b depicts the relationship between F127/MgO mixing ratio and the corresponding BET SSA and pore volume of carbons. Compared with LMCMgO, the sample without F127, the using of F127 template substantially dispersed Mg acetate and enlarged the porosity of carbons. But on the whole, with F127/MgO mixing ratio increasing, the BET SSA and pore volume kept relatively steady and little growth.
In order to ensure Mg acetate can be dispersed well, vacuum drying and freeze-drying technologies were applied for the comparison with atmospheric pressure environment during solvent evaporation. The movement of Mg2+ in solution at higher temperature is much faster than that at lower temperature. During solvent evaporation, both vacuum drying and freeze-drying can significantly reduce the temperature, control the drying rate of solvent, and therefore restrain the fast movement of Mg2+ to prevent its agglomeration. Especially, freeze drying can preserve the skeleton of the entire composite of precursors and keep Mg2+ in its own space fixed. Two groups of samples were synthesized by the same precursors mixing mass ratio and experimental condition as LMC10–8–16, except that one of the two mixed precursors solutions was exsiccated by vacuum drying at 45 °C (labelled as LMC10–8–16, V) and the other by vacuum freeze-drying (labelled as LMC10–8–16, F), respectively. Results indicate that ways to exsiccate the precursor mixture liquid exert an implication to the eventual porosity of as-made carbons. LMC10–8–16, V exhibits a mesopore volume of 0.75 cm3 g−1, with BET SSA of 712 m2 g−1 and total pore volume of 0.90 cm3 g−1, which are larger than those of LMC10–8–16. The higher porosity of LMC10–8–16, V indicates that vacuum environment promoted the development of pores and enlarged the porosity of carbon. While freeze drying kept each component in its own space fixed, and significantly hindered the spontaneous agglomeration of template during solvent evaporation. However, the mesopore and total pore volume of obtained LMC10–8–16, F decreased to 0.56 cm3 g−1 and 0.72 cm3 g−1, respectively, which are both smaller than those of LMC10–8–16. The lower porosity of freeze-dried sample indicates that vacuum freezing environment may hinder the enlargement of porosity. As mentioned before, Saha et al.20 fabricated mesoporous carbon from solvent-extracted lower-molecular-weight fraction of lignin via surfactant template route. The corresponding optimal carbon exhibits a BET SSA of 418 m2 g−1 and a mesopore volume of 0.34 cm3 g−1, which are still much smaller than our best result via dual templates route.
Fig. 3a depicts the N2 adsorption–desorption isotherms of N2 at 77 K for selected LMCs. The obvious hysteresis loops, typical feature of type IV (according to IUPAC classification), confirm the presence of mesoporosity. Fig. 3b indicates the dominant well-developed mesopore structure. LMCF127 as well as LMCMgO has a broad pore size distributions, while LMC10–1–16 shows dominant peaks around 9 nm. Compared with LMC10–8–16 and LMC10–8–16, F, LMC10–8–16, V exhibits a narrower and smaller pore size distribution ranging from 6 nm to 11 nm due to the enhanced dispersibility under 45 °C vacuum environment during solvent evaporation.
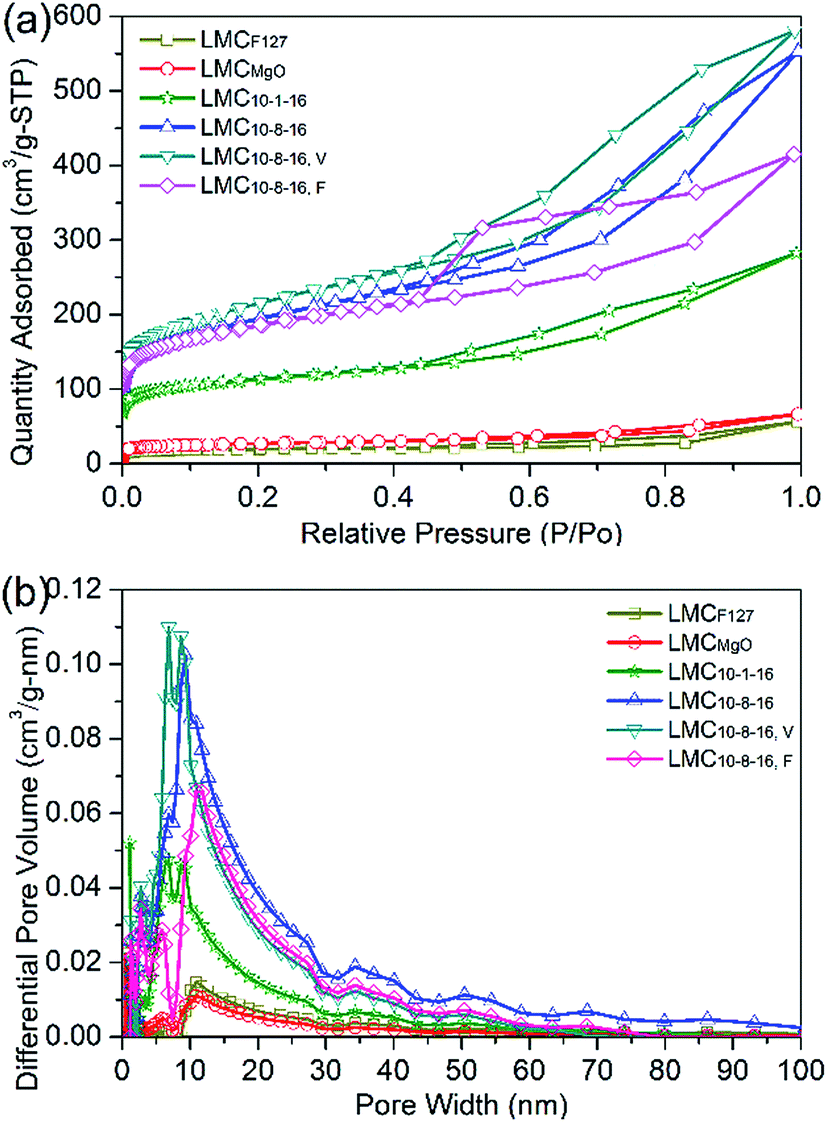 | ||
| Fig. 3 (a) N2 adsorption–desorption isotherms with (b) differential pore size distributions for the selected typical LMCs. | ||
As shown in Fig. 4a, layer-stacking surface structures with slit-like pores are visible clearly in LMCF127 because of the lack of MgO substrates to support carbon frameworks and partial pore structures shrinkage and collapse. LMCMgO also shows some rare random pores (Fig. S4a†) as LMCF127 due to the partial recrystallization and agglomeration of Mg acetate. Moreover, pores in LC (Fig. S4b†), the sample without any templating agent, are rarer than both LMCF127 and LMCMgO. However, LMC10–8–16 synthesized via dual templates route has a much more developed pore structures and a better pore structure connectivity (Fig. 4b). TEM image (Fig. 4e) depicts that LMC10–8–16 is abundant with worm-like disordered aperiodic pores. While pores in LMC10–8–16, V are relatively more regular and uniform (Fig. 4c and f), which indicates that vacuum environment during solvent evaporation can enhance the uniformity of templates and therefore induce a narrower pore size distribution. As shown in Fig. 4d and S4c,† pores in LMC10–8–16, F are much complex, and some open large pores larger than 100 nm are also noticeable. These large pores can be attributed to the inevitable change of liquid solvent into solid ice crystals during freezing and the further sublimation of certain ice crystals with particle size over 100 nm after drying completely.
 | ||
| Fig. 4 FESEM micrographs for (a) LMCF127, (b) LMC10–8–16, (c) LMC10–8–16, V and (d) LMC10–8–16, F; TEM micrographs for (e) LMC10–8–16 and (f) LMC10–8–16, V. | ||
Wide-angle XRD patterns for obtained LMCs are shown in Fig. 5a. Broad diffraction peaks can be obviously seen at 2θ = 24° and 44°, typical (002) and (100) diffraction of graphitic carbon.35 The XRD patterns of LMCF127 and LMCMgO are consistent with typical nanostructured mesoporous nongraphitized carbon black (NMCB) powders,36 exhibiting a strong and sharp peak at 2θ = 45°. However, there are no strong and sharp peaks in XRD patterns for other LMCs, indicating the dominating amorphous carbon structure of samples. Fig. 5b is the Raman spectra of all carbons. The G band at around 1590 cm−1 is derived from bond stretching of sp2 carbon pairs in both rings and chains, implying the presence of graphitic structure, and the D band at around 1350 cm−1 is induced by the breathing mode of aromatic rings, indicating the lattice defects due to structure distortion.19,37,38 Therefore, the peak intensity ratio of D band to G band usually acts as a measure for disorder degree or graphitization degree. As listed in Table S1,† the ID/IG values of all LMCs are very close to each other, and demonstrate that precursors mixing ratios as well as ways to exsiccate the precursor mixing liquids have little effect on the graphitization degree of obtained carbons. The overall XPS survey spectra in Fig. 5c as well as Table S2† reveals the presence of dominating carbon and significant presence of oxygen. A high surface oxygen content can improve surface wettability in aqueous solutions. However, after four times of measurements for each sample, the average oxygen contents are statistically similar and range from 16.05% to 20.29%, which indicates that under the same carbonization conditions, different preparation methods provided in this work have little effect on the oxygen contents of obtained carbons.
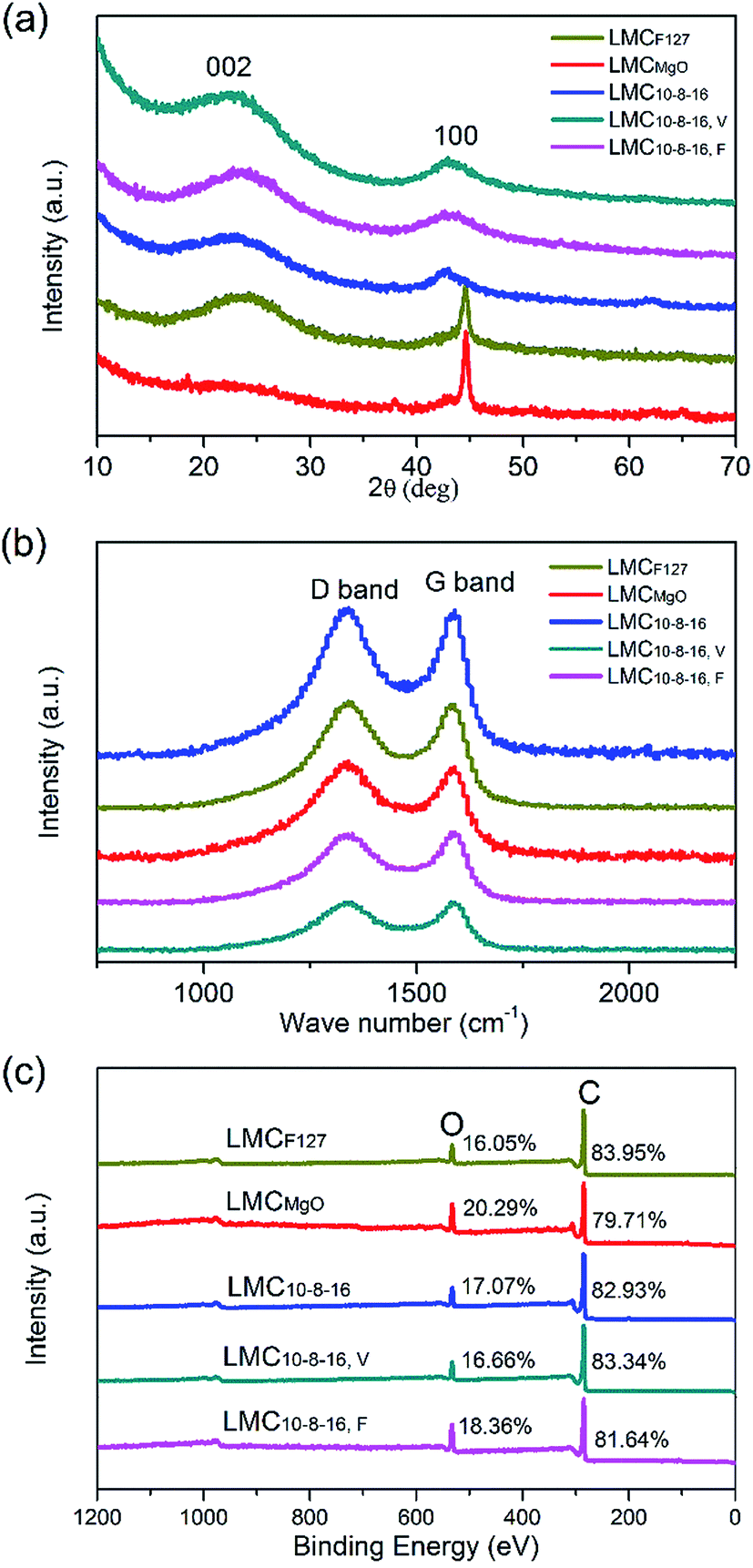 | ||
| Fig. 5 (a) XRD patterns, (b) Raman spectra, and (c) overall XPS survey spectra for LMCs. | ||
3.2 Electrochemical analysis
We further directly investigated the electrochemical performance in a three-electrode configuration by mixing carbon samples with acetylene black and PTFE together as electrode. Cyclic voltammetry (CV) measurements were carried out at different scan rates ranging from 5 to 100 mV s−1 within the potential window of 0 to 0.8 V. At a slow scan rate of 5 mV s−1, samples prepared via dual templates route exhibited ultimate capacitance behavior, evidenced by the nearly rectangular shaped plots (Fig. 6a), the typical performance of electric double-layer capacitor39 (EDLC), suggesting highly reversible charge–discharge responses. When shifting to a faster scan rate of 10 mV s−1, the CV plots shapes of LMC10–8–16, V and LMC10–8–16, F tended to deviate from rectangular and gradually form shuttle-shaped plots, especially at the scan rate of 20 mV s−1 (Fig. 6b). The CV curve for LMC10–8–16 still kept more rectangular shape before the scan rate increased to over 50 mV s−1, when it became shuttle shape (Fig. S5a†). The CV plots of selected carbons are shown in Fig. S5b–f.† LMCF127, LMCMgO and LMC10–8–16, F showed poor charge–discharge responses, evidenced by the triangle-shaped CV plots of LMCF127 at any scan rate ranging from 5 to 100 mV s−1 and by the shuttle-shaped CV plots of LMCMgO and LMC10–8–16, F at the faster scan rate. Besides, redox pseudocapacitance peaks were also observed clearly in the CV curves of LMCF127, which can be mainly related to surface oxygen-containing functionalities. LMCF127 is consisted of 83.95% C and 16.05% O on the surface. Such a high O content mainly induced the pseudocapacitance behavior of LMCF127. A gradual depression of the plateau values in the CV profiles of all carbons was observed with the increase of scan rate, caused by that the voltage signal couldn't reach the pores effectively and by the ohmic resistance difference between mesopores at the top and at the bottom of the electrolyte under increased scan rate.36 Fig. 6c shows the relationship between specific galvanostatic capacitance (Cg) and scan rate. Among the five selected typical samples, LMCMgO exhibited the smallest Cg value throughout all scan rates, decreasing from 105.6 F g−1 at 5 mV s−1 scan rate to 37.3 F g−1 at 100 mV s−1 scan rate. Though LMC10–8–16, F exhibited the largest specific capacitance of 151 F g−1 at 5 mV s−1 scan rate, the Cg value appeared an apparent sharp decline, and only maintained 54 F g−1 at 100 mV s−1 scan rate, which rarely reached 57.5% Cg value of LMC10–8–16. Saha et al.21 prepared activated mesoporous carbons from pre-cross-linked lignin via F127 templating route and further coupled with activation by KOH and CO2. Though the porosity of these carbons were largely enhanced after activation, the specific capacitance of their optimal carbon was only 100 F g−1 at 5 mV s−1, which was still smaller than LMCMgO, the sample with the smallest Cg value among our all samples.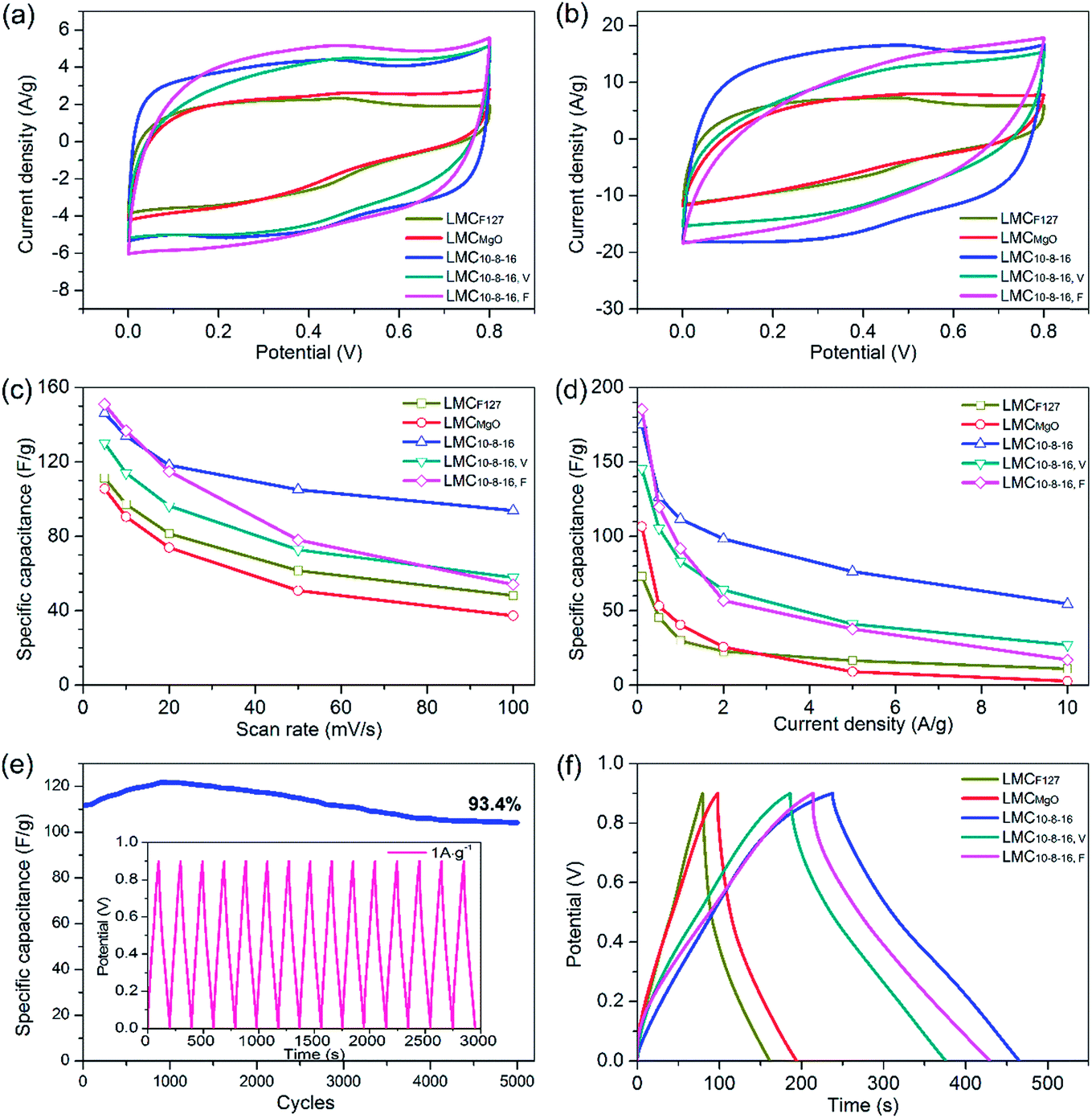 | ||
| Fig. 6 Cyclic voltammograms of carbons determined in a three-electrode configuration: (a) at 5 mV s−1 scan rate, and (b) at 20 mV s−1 scan rate; (c) specific capacitance vs. scan rate, and (d) specific capacitance vs. discharge current density; (e) cyclic performance of LMC10–8–16 at a current density of 1 A g−1 with the galvanostatic charge/discharge curve shown in inset; (f) galvanostatic charge/discharge curves for carbons at a current density of 0.5 A g−1. | ||
Fig. 6d shows the relationship between specific galvanostatic capacitance and increasing current density and depicts that the Cg value of LMCF127 and LMCMgO are still relatively small. At a current density of 0.5 A g−1, LMC10–8–16 showed a specific capacitance of 126.4 F g−1, about two times larger than that of LMCF127 and LMCMgO. Such poor electrochemical behavior for LMCF127 and LMCMgO can be undoubtedly attributed to the small surface area and the lack of capacity for charge storage. Besides, as shown in Fig. 6e, after 5000 cycles at 1 A g−1, LMC10–8–16 displayed good electrochemical stability for maintaining 93.4% capacitance retention from the initial Cg value of 111.6 F g−1. The Cg value of LMC10–8–16, F still exhibited a sharp decline trend and decreased from 186.3 F g−1 at a lower current density of 0.1 A g−1 to mere 17 F g−1 at the high current density of 10 A g−1. Fig. 6f is the typical triangle-like charge/discharge profiles of obtained carbons at the current density of 0.5 A g−1. Obviously, LMC10–8–16 exhibited a longer charge/discharge time for a complete cycle and a better reversible capacitive behavior than the other four samples. Galvanostatic charge/discharge curves at various of current densities of LMC10–8–16 were also provided in Fig. S6.† Although sample LMC10–8–16, V possesses the largest BET SSA, mesopore and total pore volume, it didn't exhibit a better electrochemical performance than LMC10–8–16 through our study. Generally, for an example of the electrochemical behavior of LMC10–8–16 and LMCF127, a large specific surface area has a positive impact for storing more charge and therefore performs a better electrochemical behavior. However, not all surface could establish electric double layer. Pore structure connectivity and surface functionalities also have effect on eventual electrochemical performance. As mentioned above, MgO nanoparticles just function as substrates by space occupying effect, and the vacuum environment during solvent evaporation can enhance the uniformity of templates. Pores in LMC10–8–16, V are much regular and isolated, and LMC10–8–16, V may have a much poorer pore structure connectivity than LMC10–8–16. It is relatively more difficult for LMC10–8–16, V to meet the rapid ions transportation, leading to a higher ion-transport resistance and more insufficient ionic diffusion. Furthermore, XPS data indicates the small differences of surface oxygen content in all as-made carbons. Therefore, the better pore connectivity may be the major factor that induced the better electrochemical performance of LMC10–8–16 than that of LMC10–8–16, V.
4. Conclusions
Lignin-derived mesoporous carbons with dominating pore width of 9 nm have been successfully synthesized, the BET SSA and total pore volume of which reached 712 m2 g−1 and 0.90 cm3 g−1, respectively. F127 functions mainly to disperse MgO sufficiently and the amount of MgO nanoparticles determines the porosity, which was significantly enlarged in comparison with carbons via single template route. The mixing mass ratio of lignin to nano-sized MgO as well as the dosage of F127 plays a major role to enrich the porosity of carbons. Vacuum environment during solvent evaporation enhanced porosity and pore uniformity, but didn't enhance the electrochemical performance. The carbon with atmospheric pressure environment, on contrast, exhibited a better electrochemical behavior. While vacuum freeze drying preserved the skeleton of the entire precursors composite and also significantly hindered the agglomeration of template. The corresponding carbon exhibited a much smaller mesopore and total pore volume but a larger specific capacitance of 186.3 F g−1 than those of carbon with atmospheric pressure environment.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially sponsored by the National Science & Technology Pillar Program during the Twelfth Five-year Plan Period (Grant No. 2015BAD14B06).References
- S. Solomon, G. K. Plattner, R. Knutti and P. Friedlingstein, Irreversible climate change due to carbon dioxide emissions, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(106), 1704–1709 CrossRef CAS PubMed.
- R. J. White, V. Budarin, R. Luque, J. H. Clark and D. J. Macquarrie, Tuneable porous carbonaceous materials from renewable resources, Chem. Soc. Rev., 2009, 38(12), 3401–3418 RSC.
- J. Lorenz, K. Panagiotou and A. Steger, Catalytic Carbonization of Lignin for Production of Electrically Conductive Charcoal, J. Biobased Mater. Bioenergy, 2012, 6(1), 69–74 CrossRef.
- W. Boerjan, J. Ralph and M. Baucher, Lignin biosynthesis, Annu. Rev. Plant Biol., 2003, 54(1), 519–546 CrossRef CAS PubMed.
- M. M. Titirici, R. J. White, N. Brun, V. L. Budarin, D. S. Su and F. D. Monte, et al., Sustainable carbon materials, Chem. Soc. Rev., 2015, 44(1), 250–290 RSC.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Valorization of biomass: deriving more value from waste, Science, 2012, 337(6095), 695–699 CrossRef CAS PubMed.
- J. D. Nguyen, B. S. Matsuura and C. R. J. Stephenson, ChemInform Abstract: A Photochemical Strategy for Lignin Degradation at Room Temperature, J. Am. Chem. Soc., 2014, 136(4), 1218–1221 CrossRef CAS PubMed.
- H. N. Lyckeskog, C. Mattsson, L. E. Amand, L. Olausson, S. I. Andersson and L. Vamling, et al., The storage stability of bio-oils derived from the catalytic conversion of softwood Kraft lignin in subcritical water, Energy Fuels, 2016, 30(4), 3097–3106 CrossRef.
- A. Rahimi, A. Ulbrich, J. J. Coon and S. S. Stahl, Formic-acid-induced depolymerization of oxidized lignin to aromatics, Nature, 2014, 515(7526), 249–252 CrossRef CAS PubMed.
- W. Sangchoom and R. Mokaya, Valorization of Lignin Waste: Carbons from Hydrothermal Carbonization of Renewable Lignin as Superior Sorbents for CO2 and Hydrogen Storage, ACS Sustainable Chem. Eng., 2015, 3(7), 1658–1667 CrossRef CAS.
- D. Montané, V. Torné-Fernández and V. Fierro, Activated carbons from lignin: kinetic modeling of the pyrolysis of Kraft lignin activated with phosphoric acid, Chem. Eng. J., 2005, 106(1), 1–12 CrossRef.
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, 3D aperiodic hierarchical porous graphitic carbon material for high-rate electrochemical capacitive energy storage, Angew. Chem., Int. Ed., 2008, 47(2), 373–376 CrossRef CAS PubMed.
- W. Zhang, Z. H. Huang, Z. Guo, C. Li and F. Kang, Porous carbons prepared from deoiled asphalt and their electrochemical properties for supercapacitors, Mater. Lett., 2010, 64(17), 1868–1870 CrossRef CAS.
- C. Zheng, Q. Li, M. Yoshio and H. Wang, Cooperation of micro- and meso-porous carbon electrode materials in electric double-layer capacitors, J. Power Sources, 2010, 195(13), 4406–4409 CrossRef CAS.
- X. J. He, N. Zhao, J. S. Qiu, N. Xiao, M. X. Yu and C. Yu, et al., Synthesis of hierarchical porous carbons for supercapacitors from coal tar pitch with nano-Fe2O3 as template and activation agent coupled with KOH activation, J. Mater. Chem. A, 2013, 1(33), 9440–9448 CAS.
- M. J. Valero-Romero, E. M. Márquez-Franco, J. Bedia, J. Rodriguez-Mirasol and T. Cordero, Hierarchical porous carbons by liquid phase impregnation of zeolite templates with lignin solution, Microporous Mesoporous Mater., 2014, 196(196), 68–78 CrossRef CAS.
- R. Ruiz-Rosas, M. J. Valero-Romero, D. Salinas-Torres, J. Rodriguez-Mirasol, T. Cordero and E. Morallon, et al., Electrochemical performance of hierarchical porous carbon materials obtained from the infiltration of lignin into zeolite templates, ChemSusChem, 2014, 7(5), 1458–1467 CrossRef CAS PubMed.
- D. Salinas-Torres, R. Ruiz-Rosas, M. J. Valero-Romero, J. Rodriguez-Mirasol, T. Cordero and E. Morallon, et al., Asymmetric capacitors using lignin-based hierarchical porous carbons, J. Power Sources, 2016, 326, 641–651 CrossRef CAS.
- H. Li, D. Yuan, C. H. Tang, S. X. Wang, J. T. Sun and Z. B. Li, et al., Lignin-derived interconnected hierarchical porous carbon monolith with large areal/volumetric capacitances for supercapacitor, Carbon, 2016, 100, 151–157 CrossRef CAS.
- D. Saha, E. A. Payzant, A. S. Kumbhar and A. M. Naskar, Sustainable mesoporous carbons as storage and controlled-delivery media for functional molecules, ACS Appl. Mater. Interfaces, 2013, 5(5), 5868–5874 CAS.
- D. Saha, Y. C. Li, Z. H. Bi, J. H. Chen, J. K. Keum and D. K. Hensley, et al., Studies on Supercapacitor Electrode Material from Activated Lignin-Derived Mesoporous Carbon, Langmuir, 2014, 30(3), 900–910 CrossRef CAS PubMed.
- D. Saha, K. E. Warren and A. K. Naskar, Soft-templated mesoporous carbons as potential materials for oral drug delivery, Carbon, 2014, 71(7), 47–57 CrossRef CAS.
- W. Libbrecht, A. Verberckmoes, J. W. Thybaut, P. V. D. Voort and J. D. Clercq, Soft templated mesoporous carbons: tuning the porosity for the adsorption of large organic pollutants, Carbon, 2017, 116, 528–546 CrossRef CAS.
- D. Saha, R. Zacharia and A. K. Naskar, Soft-Templated Mesoporous Carbons: Chemistry and Structural Characteristics, in Polymer Precursor-Derived Carbon, American Chemistry Society, Washington, DC, 2014, pp. 61–83 Search PubMed.
- M. Inagaki, M. Kato, T. Morishita, K. Morita and K. Mizuuchi, Direct preparation of mesoporous carbon from a coal tar pitch, Carbon, 2007, 45(5), 1121–1124 CrossRef CAS.
- T. Morishita, Y. Soneda, T. Tsumura and M. Inagaki, Preparation of porous carbons from thermoplastic precursors and their performance for electric double layer capacitors, Carbon, 2006, 44(12), 2360–2367 CrossRef CAS.
- W. Zhang, Z. H. Huang, G. Cao, F. Kang and Y. Yang, Coal tar pitch-based porous carbon by one dimensional nano-sized MgO template, J. Phys. Chem. Solids, 2012, 73(12), 1428–1431 CrossRef CAS.
- He, R. C. Li, J. S. Qiu, K. Xie, P. H. Ling and M. X. Yu, et al., Synthesis of mesoporous carbons for supercapacitors from coal tar pitch by coupling microwave-assisted KOH activation with a MgO template, Carbon, 2012, 50(13), 4911–4921 CrossRef CAS.
- W. Zhang, Z. H. Huang, G. Cao, F. Kang and Y. Yang, A novel mesoporous carbon with straight tunnel-like pore structure for high rate electrochemical capacitors, J. Power Sources, 2012, 204(204), 230–235 CrossRef CAS.
- T. Morishita, K. Ishihara, M. Kato and M. Inagaki, Preparation of a carbon with a 2nm pore size and of a carbon with a bi-modal pore size distribution, Carbon, 2007, 45(1), 209–211 CrossRef CAS.
- B. Wang, J. S. Chen, H. B. Wu, Z. Wang and X. W. Lou, Quasiemulsion-templated formation of α-Fe2O3 hollow spheres with enhanced lithium storage properties, J. Am. Chem. Soc., 2011, 133(43), 17146 CrossRef CAS PubMed.
- Z. Wang, D. Luan, S. Madhavi, Y. Hu and X. W. Lou, Assembling carbon-coated α-Fe2O3 hollow nanohorns on the CNT backbone for superior lithium storage capability, Energy Environ. Sci., 2012, 5(1), 5252–5256 CAS.
- C. R. Zhao, W. K. Wang, Z. B. Yu, A. B. Wang and Y. S. Yang, Nano-CaCO3 as template for preparation of disordered large mesoporous carbon with hierarchical porosities, J. Mater. Chem., 2010, 20(5), 976–980 RSC.
- G. Yang, H. Han, T. Li and C. Du, Synthesis of nitrogen-doped porous graphitic carbons using nano-CaCO3 as template, graphitization catalyst, and activating agent, Carbon, 2012, 50(10), 3753–3765 CrossRef CAS.
- S. Tomita, A. Burian, J. C. Dore, D. LeBolloch, M. Fujii and S. Hayashi, Diamond nanoparticles to carbon onions transformation: X-ray diffraction studies, Carbon, 2002, 40(9), 1469–1474 CrossRef CAS.
- S. Prabaharan, R. Vimala and Z. Zainal, Nanostructured mesoporous carbon as electrodes for supercapacitors, J. Power Sources, 2006, 161(1), 730–736 CrossRef CAS.
- Y. Fang, Y. Y. Lv, R. C. Che, H. Y. Wu, X. H. Zhang and D. Gu, et al., Two-dimensional mesoporous carbon nanosheets and their derived graphene nanosheets: synthesis and efficient lithium ion storage, J. Am. Chem. Soc., 2013, 135(4), 1524–1530 CrossRef CAS PubMed.
- G. Eda and M. Chhowalla, Chemically Derived Graphene Oxide: Towards Large-Area Thin-Film Electronics and Optoelectronics, Adv. Mater., 2010, 22(22), 2392–2415 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Capacitive Energy Storage in Nanostructured Carbon–Electrolyte Systems, Acc. Chem. Res., 2012, 46(5), 1094–1103 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09464g |
| This journal is © The Royal Society of Chemistry 2017 |