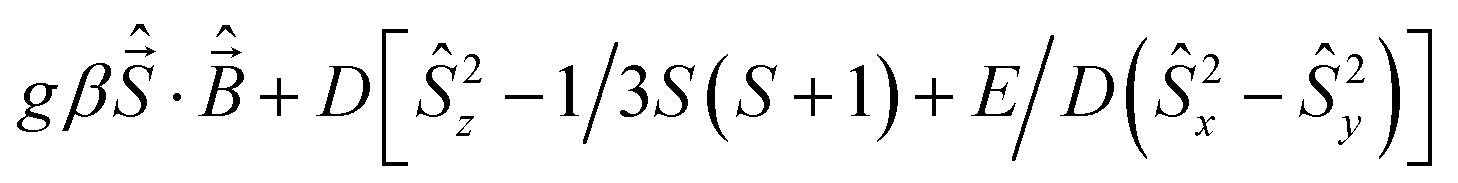DOI:
10.1039/C7RA09456F
(Paper)
RSC Adv., 2017,
7, 50713-50719
Synthesis and reactivity of a 4His enzyme model complex†
Received
25th August 2017
, Accepted 9th October 2017
First published on 31st October 2017
Abstract
A new iron(II) complex has been prepared and characterized. [Fe(TrIm)4(OTf)2] (1, TrIm = 1-tritylimidazole). The solid state structure of 1 has been determined by X-ray crystallography. Compound 1 crystallizes in triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with a = 13.342(7) Å, b = 13.5131(7) Å and c = 13.7025(7) Å. The iron center resides in distorted octahedral geometry coordinated to four equatorial imidazole groups and two axial triflate oxygens groups. The complex is high spin between 20 K and 300 K as indicated by variable field variable temperature magnetic measurements. A fit of the magnetic data yielded g = 2.24 and D = −0.80 cm−1. A large HOMO–LUMO gap energy (3.89 eV) exists for 1 indicating high stability. Addition of H2O2 or tBuOOH to 1 results in formation of an oxygenated intermediate which upon decomposition results in oxidation of the trityl substituent on the imidazole ligand.
, with a = 13.342(7) Å, b = 13.5131(7) Å and c = 13.7025(7) Å. The iron center resides in distorted octahedral geometry coordinated to four equatorial imidazole groups and two axial triflate oxygens groups. The complex is high spin between 20 K and 300 K as indicated by variable field variable temperature magnetic measurements. A fit of the magnetic data yielded g = 2.24 and D = −0.80 cm−1. A large HOMO–LUMO gap energy (3.89 eV) exists for 1 indicating high stability. Addition of H2O2 or tBuOOH to 1 results in formation of an oxygenated intermediate which upon decomposition results in oxidation of the trityl substituent on the imidazole ligand.
Introduction
Carotenoids are isoprene-containing compounds possessing conjugated π systems.1 In plants, carotenoids carry out a variety of critical functions. They operate as accessory light-harvesting components (antenna complex)2,3 of photosynthetic systems, light induced antioxidants (brought about by the low-lying triplet state poised to quench triplet chlorophyll and singlet oxygen4), membrane fluidity regulators, pigments (yellow to red-orange; important in attracting pollinators)2 for leaves, vegetables, and fruits.5 Along with carotenoids being essential in plants, carotenoid-derived products serve as important sources of bioactive compounds called apocarotenoids.6 Apocarotenoids serve as pigments, hormones (abscisic acid and strigolactones),7 aroma and scent compounds, regulatory compounds and molecules with yet to be determined functions. Carotenoids are also present in various fungi and heterotrophic bacteria as well as all photosynthetic organisms.6 In animals, carotenoids yield essential precursors necessary for vision, embryonic development, cellular homeostasis, and immunity (vitamin A).2,8,9 Carotenoid Cleavage Dioxygenases (CCDs) are enzymes possessing a nonheme site and are found in all life forms. CCD processes carotenoids and related substrates.10,11 CCDs generally catalyze the cleavage of carotenoid carbon–carbon double bonds to yield aldehydes and/or ketones. CCD must have appeared early in evolution since one of the products (retinal) is critical for photoreception by the ancient type 1 opsin protein family, a process critical in phototactic and phototrophic organisms.12
Synthesis of abscisic acid (ABA), occurs during the oxidative cleavage of the 11,12 carbon–carbon double bond of a 9-cis-epoxycarotenoid (9-cis-violaxanthin).13 This reaction is carried out by the enzyme viviparous14 (VP14) in maize (Zea mays). The X-ray structure of VP14 has been determined (PDB: 3NPE) and reveals an active site consisting of a nonheme iron coordinated to four histidine side chain residues. The remaining sites are occupied by water (or hydroxide) and dioxygen.13 The putative bound oxygen molecule is coordinated to iron in an end-on fashion. This type of iron coordination is also seen in photosystem II, superoxide reductase, and fumurate reductase.14–16 Apocarotenoid cleavage oxygenase (ACO), a CCD from cyanobacteria catalyzes the formation of 3-hydroxyretinal from 3-hydroxy-8′-apo-β-carotenol. The X-ray structure of ACO from Synechocystis was determined and also possesses iron ligated by four histidines.17 Retinal and its derivatives are involved in several mammalian biological processes and in particular the immune system and vision.18 CCDs which form retinal include β-carotene-15,15′-oxygenase (BCO1) generating retinal from symmetrical cleavage β-carotene. Retinal becomes reduced to retinol and is converted to all-trans retinyl esters by lecithin retinol acyltransferase.19
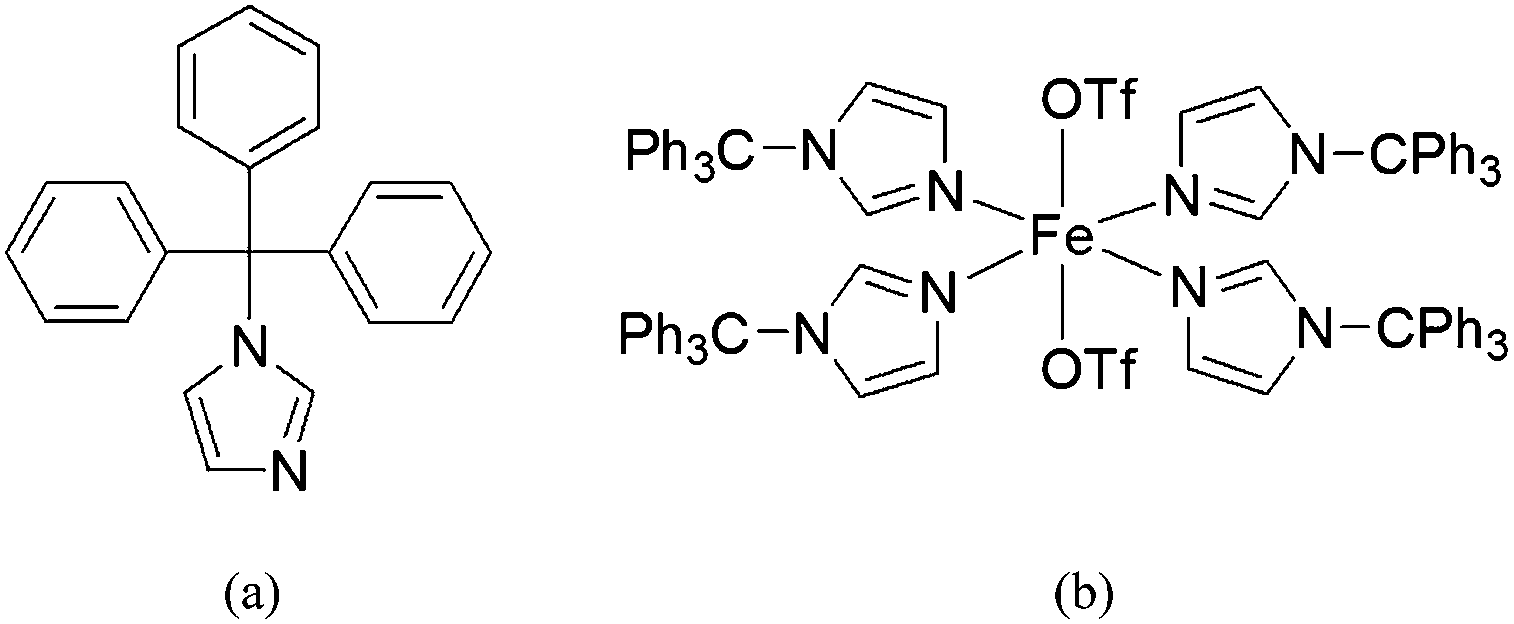
In our efforts to model the 4His active site of CCD, we have employed a bulky imidazole ligand (1-tritylimidazole, TrIm) to synthesize a 4His nonheme site ([Fe(TrIm)4(OTf)2] (1), Scheme 1). The synthesis, structure, and characterization (UV-vis and FTIR spectroscopy, and SQUID magnetometry) as well as theoretical studies (DFT) are presented. Reactivity studies are also presented.
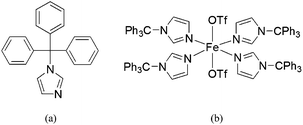 |
| | Scheme 1 (a) Structure of 1-tritylimidazole (TrIm) and (b) its iron(II) complex [Fe(TrIm)4(OTf)2] (1). | |
Experimental details
Materials
1-Tritylimidazole and tBuOOH (TBHP, 70% dried over anhydrous magnesium sulfate) were purchased from Sigma-Aldrich Chemical Co. H2O2 (30% dried over anhydrous magnesium sulfate to afford 78% as determined by density) was purchased from EMD Millipore. Fe(OTf)2·2CH3CN was synthesized according to a literature method.20 Pure dry solvents were afforded using an Innovative Technologies, Inc. Solvent Purification System. Elemental analysis was performed on pulverized crystalline samples that were heated under vacuum and sealed in a glass ampule prior to submission (Atlantic Microlabs, Inc., Norcross, GA).
Synthesis
[Fe(TrImA)4(OTf)2]·2Et2O (1·2Et2O). To a stirring suspension of 1-tritylimidazole (158.9 mg, 0.512 mmol) in dichloromethane (2 mL) was added a solution of Fe(OTf)2·2CH3CN (55.8 mg, 0.128 mmol) in dichloromethane (2 mL) under dry nitrogen atmosphere. After 1 h of stirring, the solution was filtered and placed in an ether diffusion chamber. After 24 h, colorless crystals were deposited. The crystals were collected and washed with ether. Yield: 155.7 mg (76%). Selected IR bands (KBr pellet, cm−1): 1489 (s), 1444 (s), 1308 (s), 1235 (s), 1216 (s), 1167 (s), 1112 (s), 1078 (s), 1025 (s), 932 (s), 845 (m), 747 (s), 663 (s), 633 (s) and 516 (m). UV-vis (CH3CN; λmax, nm (ε, M−1 cm−1)): 255 (8202, sh), 322 (2040, sh); (CH2Cl2; λmax, nm (ε, M−1 cm−1)): 256 (6775, sh), 294 (2320, sh). Anal. calcd for C90H72N8O6F6S2Fe (1): C, 67.75; H, 4.55; N, 7.02. Found: C, 67.96; H, 4.99; N, 6.63.
Physical characterization
A Varian 3100 Excalibur Series was used to collect the FTIR spectra. Optical spectra were collected using a Cary 50 UV-vis spectrophotometer. Variable temperature magnetic susceptibilities were measured using a Quantum Design MPMS3 SQUID magnetometer calibrated with a 765-Palladium standard purchased from NIST (formally NSB). A powdered sample of 1 was placed in a plastic bag. The sample was measured in the temperature range 2–300 K with H = 0.6 T. The sample was placed in a plastic drinking straw for the measurement. The magnetic contribution of the bag was determined between 2–300 K as well and subtracted from the sample. The molar magnetic susceptibility was corrected for the diamagnetism of the complex using tabulated values of Pascal's constants to obtain a corrected molar susceptibility. The program julX written by E. Bill was used for the simulation and analysis of magnetic susceptibility data.21 The Hamiltonian operator (Ĥ) was| |
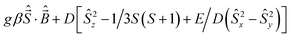 | (1) |
where g is the average electronic g value, D the axial zero-field splitting parameter, and E/D is the rhombicity parameter. Magnetic moments were obtained from numerically generated derivatives of the eigenvalues of eqn (1), and summed up over 16 field orientations along a 16-point Lebedev grid to account for the powder distribution of the sample. Intermolecular interactions were considered by using a Weiss temperature, θW, as perturbation of the temperature scale, kT′ = k(T − θW). GCMS experiments were performed on an HP 6890/5973 gas chromatograph. The products were identified and quantified by comparing the retention times along with mass spectra and peak areas with authentic standards.
Computational details
Quantum chemical calculations providing energy minimized molecular geometries, molecular orbitals (HOMO–LUMO), and vibrational spectra for compound 1 were carried out using density functional theory (DFT) as implemented in the GAUSSIAN09 (Rev. C.01) program package.22 We employed the hybrid functional PBE0.23 The basis set used was 6-31G(d).24 Full ground state geometry optimization was performed without any symmetry constraints. Only the default convergence criteria were used during the geometry optimizations. Initial geometry coordinates were taken from the crystal structure in the quintet state. Optimized structures were confirmed to be local minima (no imaginary frequencies for both cases). Selected theoretically derived metric parameters are shown in Table 2. Molecular orbitals were generated using Avogadro25 (an open-source molecular builder and visualization tool, Version1.1.0. http://avogadro.openmolecules.net/).
X-ray crystallography
Compound 1 was crystallized from a dichloromethane/ether solution. A colorless prism crystal with dimensions 0.224 × 0.185 × 0.064 mm was mounted on a Nylon loop using a very small amount of paratone oil. Data were collected using a Bruker CCD based diffractometer equipped with an Oxford Cryostream low-temperature apparatus operating at 173 K. Data were measured using omega and phi scans of 0.5° per frame for 10 s. The total number of images was based on results from the program COSMO26 where redundancy was expected to be 4.0 and completeness of 100% out to 0.83 Å. Cell parameters were retrieved using APEX II software27 and refined using SAINT on all observed reflections. Data reduction was performed using the SAINT software,28 which corrects for Lp. Scaling and absorption corrections were applied using SADABS29 multi-scan technique, supplied by George Sheldrick. The structures are solved by the direct method using the SHELXS-97 program and refined by least squares method on F2, SHELXL-2014,30 which are incorporated in OLEX2.31 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were calculated by geometrical methods and refined as a riding model. The crystal used for the diffraction study showed no decomposition during data collection. All drawings are done at 50% ellipsoids.
Results and discussion
Synthesis
[Fe(TrIm)4(OTf)2] (1) was synthesized in good yield at room temperature under dry nitrogen conditions by reacting 4 equiv. 1-tritylimidazole (TrIm) with Fe(OTf)2·2CH3CN in dichloromethane. Suitable crystals of 1 for X-ray studies were grown from dichloromethane/ether.
Crystallography
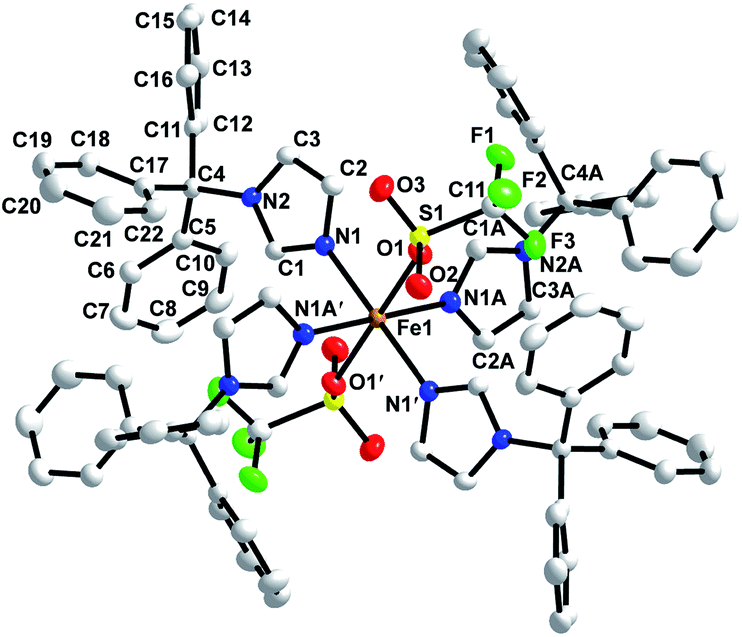
The crystal structure of 1 was determined. Crystallographic parameters are shown in Table 1 while selected bond distances and angles are given in Table 2. The structure of 1 was solved in the P space group. The structure reveals iron(II) in distorted octahedral geometry with ligands derived from two triflate oxygens and four 3-imidazole nitrogens (Fig. 1). The Fe–NIm distances are 2.1823(15) Å (Fe1–N1) and 2.1807(15) Å (Fe1–N1A) which is in the range of other known high spin FeII–NIm compounds (2.103–2.272 Å).32–38 The iron triflate Fe1–O1 distance is 2.1843(13) Å which is in the range of other known high spin FeII–Otriflate bond distances (2.025–2.211 Å).20,39–50 Hydrogen bonding (D⋯A: 2.995 Å) is observed between an imidazole hydrogen located on C1A and a coordinated triflate oxygen (O1). Close contacts between a triflate fluorines and phenyl carbons (F3⋯C7′: 3.320 Å; F3⋯C8′: 3.322 Å; F1⋯C19′ 3.133 Å; F1⋯C16′: 3.295 Å; F1⋯C18′: 3.120 Å) are observed. Each iron is well isolated from other irons with the closest distance between iron centers being 13.234 Å. No π–π stacking is observed among aromatic groups and all phenyl rings are approximately orthogonal within each trityl group.
Table 1 Crystallographic parameters for [Fe(TrIm)4(OTf)2]·2Et2O (1·2Et2O)
| |
1·2Et2O |
| Empirical formula |
C98H92N8O8F6S2Fe |
| Formula weight |
1743.76 |
| Temperature/K |
173.15 |
| Wavelength (Å) |
0.7103 |
| Crystal system |
Triclinic |
| Space group |
P |
| a/Å |
13.2342(7) |
| b Å |
13.5131(7) |
| c/Å |
13.7025(7) |
| α/° |
98.0060(10) |
| β/° |
116.1990(10) |
| γ/° |
93.3490(10) |
| Volume/Å3 |
2157.39(19) |
| Z |
1 |
| Dcalc/Mg m−3 |
1.342 |
| μ/mm−1 |
0.300 |
| Goodness-of-fit on F2 |
1.035 |
| Final R indexes [I > 2σ(I)] |
R1 = 0.0384, wR2 = 0.0948 |
| Final R indexes [all data] |
R1 = 0.0508, wR2 = 0.1032 |
Table 2 Selected bond lengths (Å) and angles (°) for [Fe(TrIm)4(OTf)2]·2Et2O (1·2Et2O). Calculated values are in brackets
| Distances |
| Fe1–O1 |
2.1843(13) [2.159] |
N1–C2 |
1.374(2) [1.372] |
| Fe1–N1 |
2.1823(15) [2.173] |
N2–C1 |
1.355(2) [1.358] |
| Fe1–N1 |
2.1807(15) [2.178] |
N2–C3 |
1.375(2) [1.381] |
| N1–C1 |
1.318(2) [1.319] |
N2–C4 |
1.503(2) [1.487] |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Angles |
| N1–Fe1–N1A |
93.13(6) [92.87] |
N1–Fe1–O1′ |
86.93(5) [91.56] |
| N1–Fe1–N1′ |
180.00(8) [180.00] |
N1A–Fe1–O1 |
95.24(6) [83.88] |
| N1–Fe1–N1A′ |
86.87(6) [87.13] |
N1A–Fe1–O1′ |
84.76(6) [86.12] |
| N1A–Fe1–N1′ |
86.87(6) [87.15] |
N1′–Fe1–O1 |
86.93(5) [91.56] |
| N1A–Fe1–N1A′ |
180.00(9) [180.00] |
N1′–Fe1–O1′ |
93.07(5) [88.44] |
| N1′–Fe1–N1A′ |
93.13(6) [92.85] |
N1A′–Fe1–O1 |
84.76(6) [86.12] |
| N1–Fe1–O1 |
93.07(5) [88.44] |
N1A′–Fe1–O1′ |
95.24(6) [83.88] |
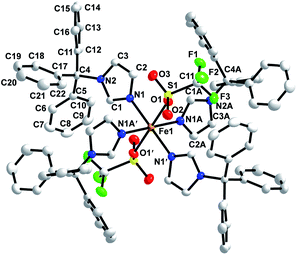 |
| | Fig. 1 X-ray crystal structure of [Fe(TrIm)4(OTf)2] (1) with thermal ellipsoids drawn at the 50% probability level. | |
Magnetic susceptibility
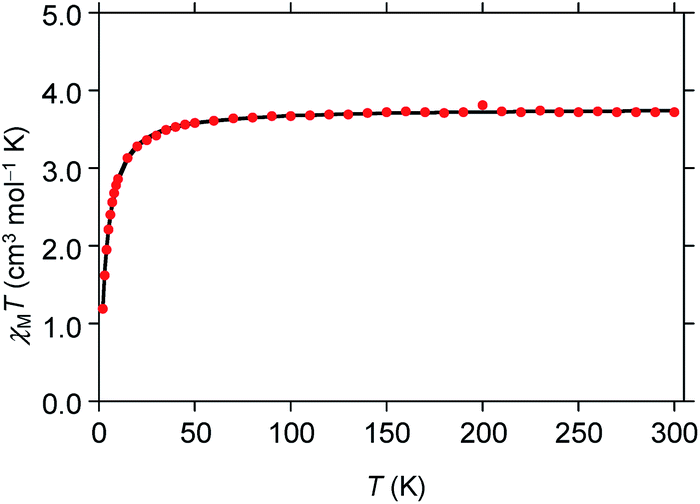
A powdered sample of 1 was characterized using variable temperature magnetic susceptibility (χM). The measurements were taken between 2–300 K with an applied field of 0.6 T. At 300 K χMT has a value of 3.73 cm3 mol−1 K (Fig. 2) consistent with high spin iron(II) (S = 2), 3.25–4.06 cm3 mol−1 K.51 As the temperature is lowered, the χMT value begins to decreases at 50 K quickly towards 1.17 cm3 mol−1 K at 2 K. This trend is likely due to zero-field splitting of high spin iron(II). This behavior is consistent with low symmetry at the iron center.52–54 The data was simulated using the program julX21 which gave a very good fit using g = 2.24 and D = −0.80 cm−1 which is typical for distorted octahedral geometry.55
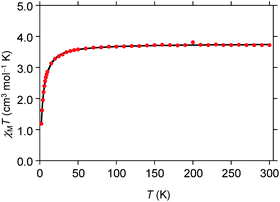 |
| | Fig. 2 Temperature-dependent molar magnetic susceptibility (χMT) for [Fe(TrIm)4(OTf)2] (1) at H = 0.6 T. | |
DFT studies
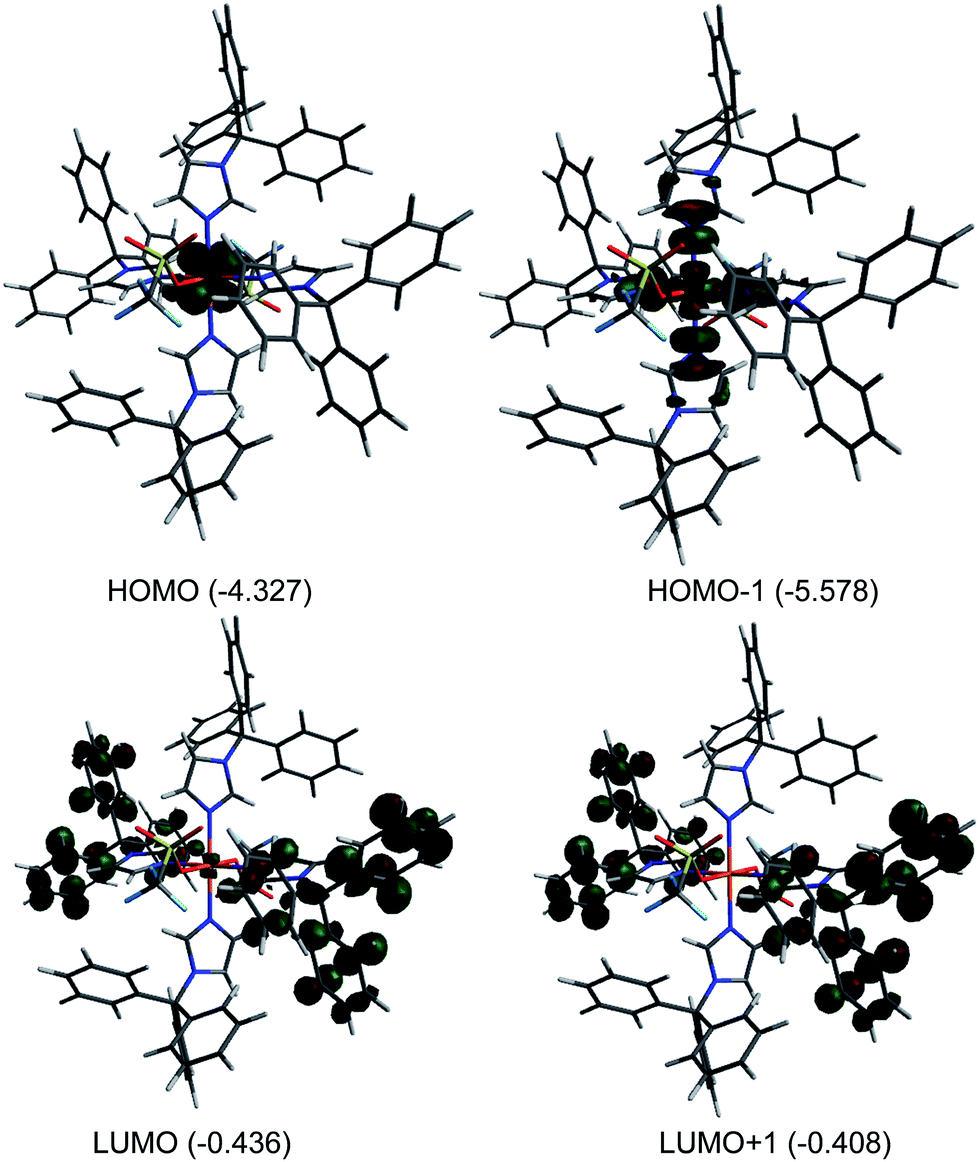
Theoretical calculation (DFT) were performed on 1 in the gas phase using X-ray coordinates as the starting point to optimize the ground state structure. The energies were obtained using PBE0/6-31G(d). The calculated metric parameters for the optimized structure were compared to those empirically determined (Table 2). The largest difference between the calculated and experimental bond distances is 0.025 Å (Fe1–O1), while the bond angles are in good agreement. Despite slight differences, the calculated structure is very close to the experimental structure and therefore the electronic properties can be confidently surmised. The calculated Mulliken charge value on the iron(II) of 1 is +1.35 consistent with high spin iron(II).56 Fig. 3 illustrates selected molecular orbitals: HOMO, HOMO−1, LUMO, and LUMO+1 for 1. It is found that the highest occupied molecular orbital (HOMO, β-412) is largely distributed over the dxz/dyz orbital while the HOMO−1 (α-416) is associated with the dx2−y2 and nitrogen sp2 orbital in an antibonding fashion. The lowest unoccupied molecular orbital (LUMO, β-413) is primarily distributed over ring unhybridized p orbitals and to a lesser extent the dxz/dyz orbital while the LUMO+1 (β-414) consists of unhybridized p orbitals. The HOMO–LUMO gap for 1 is large (3.891 eV) indicating high stability.57,58
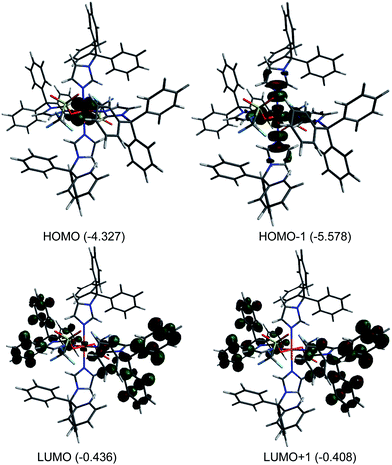 |
| | Fig. 3 Plots of molecular orbitals: HOMO−1, HOMO, LUMO, and LUMO+1 for [Fe(TrIm)4(OTf)2] (1). Orbital energies (eV) are indicated. | |
Reactivity studies
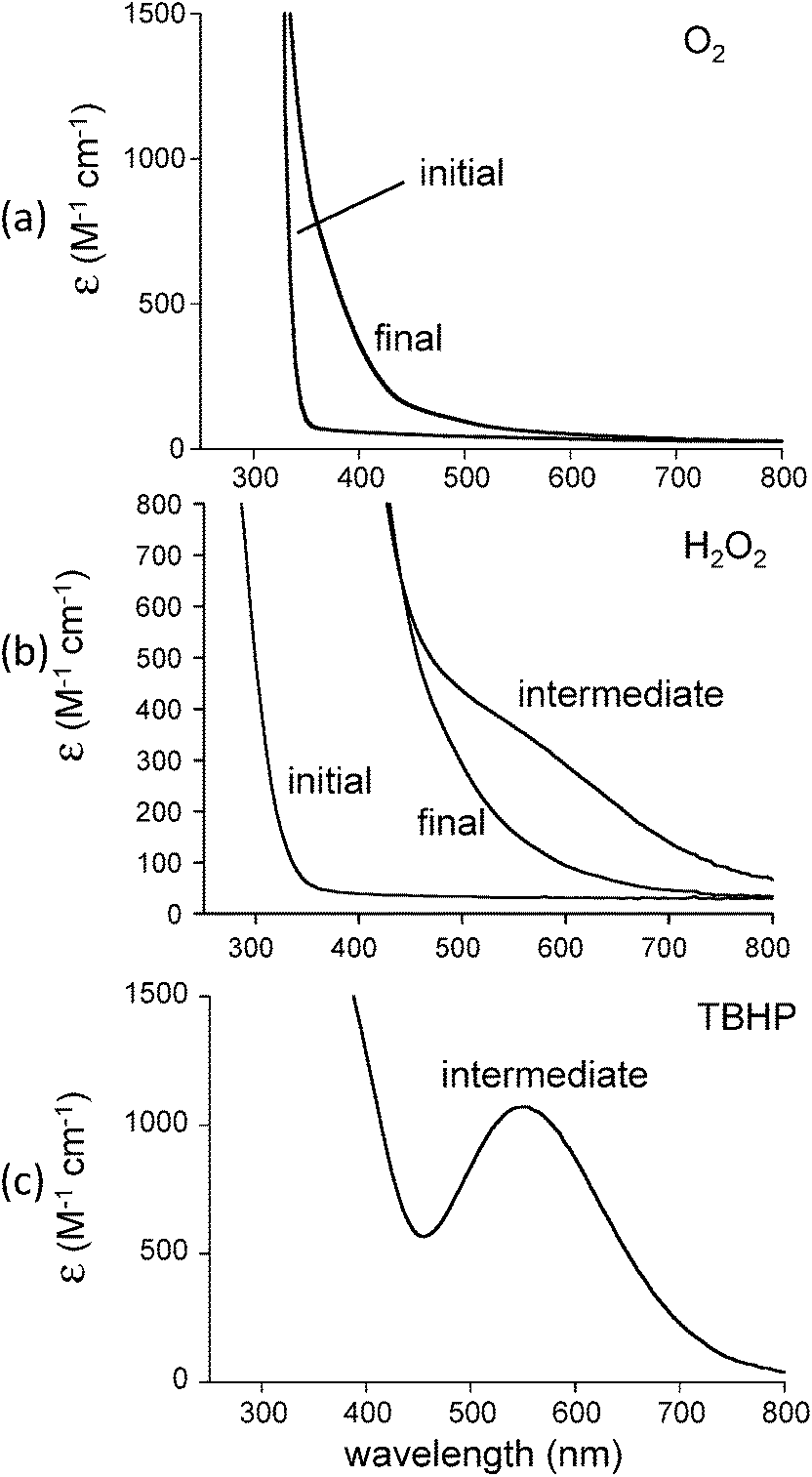
In our efforts to mimic CCD reactivity we reacted 1 with 1 equiv. cis-stilbene in the presence of dioxygen. It was anticipated that formation of benzaldehyde would be observed should biomimetic activity be present. The initial colorless solution immediately turned pale yellow when dioxygen was bubbled into the solution (Fig. 4(a)). No temperature sensitive intermediates were observed during oxygenation. Analysis of the reaction mixture (after addition of acid and extraction with CH2Cl2) by GCMS, however, indicated that the cis-stilbene was unaffected and that no other oxygenated products were detected. We then decided to use H2O2, a stronger oxidizing agent, for this reaction. Addition of 20 equiv. hydrogen peroxide (78%) to 1 in dichloromethane in the presence of cis-stilbene at 25 °C yielded a dark purple-brown solution which gradually turned orange. Analysis of the reaction mixture by GCMS in this case indicated that triphenylmethanol (0.5 equiv.) had formed with no cis-stilbene-derived products. A control reaction (lacking iron) resulted in no oxidation products. This result indicates the preference for ligand oxidation over cis-stilbene oxidation which is likely due to the close juxtapositioning of the ligand to the oxygenated intermediate compared to cis-stilbene. In our attempts to stabilize the purple-brown intermediate, we conducted a low temperature UV-vis experiment. A solution of 1 in dichloromethane was first cooled to −30 °C then 20 equiv. H2O2 was introduced to generate the intermediate species with a shoulder at ∼590 nm (Fig. 4(b)). The absorption intensity of shoulder began to decrease almost immediately and was gone after several minutes.
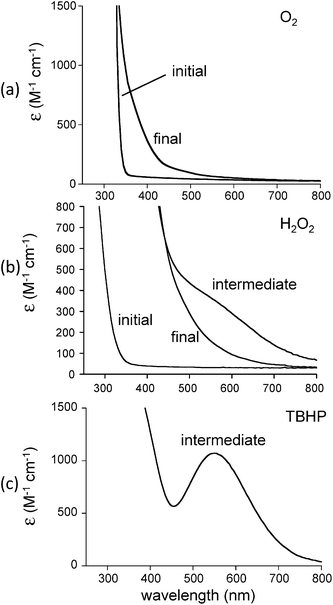 |
| | Fig. 4 Reaction of [Fe(TrIm)4(OTf)2] (1) in CH2Cl2 with (a) O2 in the presence of cis-stilbene at 25 °C, (b) 20 equiv. H2O2 at −30 °C, and (c) tBuOOH (TBHP) at −30 °C. | |
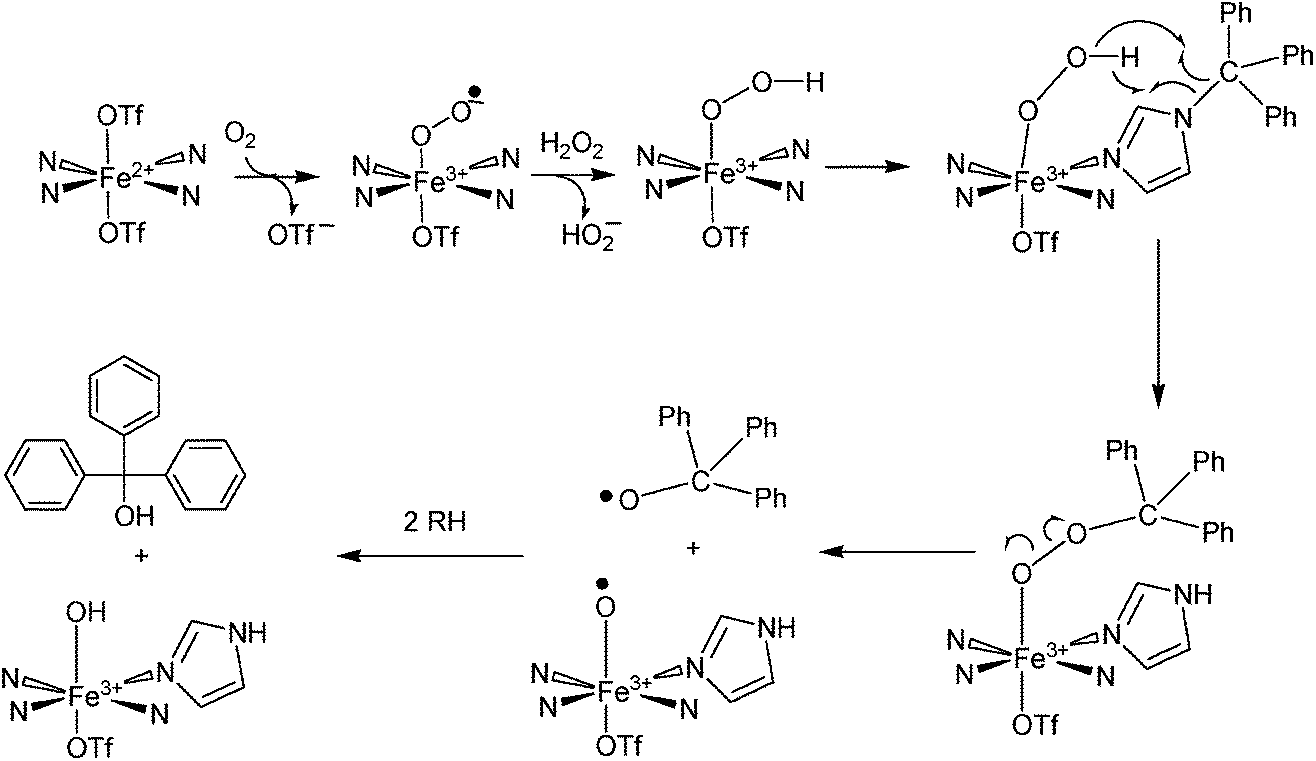
To explain these results, we have proposed a reaction mechanism which is shown in Scheme 2. In this mechanism, the iron(II) center in 1 first becomes oxidized to Fe(III) (due to the presence of ambient oxygen and possibly forming an LFeIII–O2− species59) and reacts with the hydrogen peroxide to form what is believed, based on spectroscopic characteristics, to be a LFeIII–OOH species.60–62 The protonated superoxide species would be displaced. Since triphenylmethyl is known to form the stable triphenylmethyl radical (Ph3C˙),63 we hypothesize that this species exchanges with the hydroperoxo group and forms LFeIII–OOCPh3 which along with LFeIII–OOH could account for the dark brown-purple intermediate species. Homolytic scission of the O–O bond in LFeIII–OOCPh3 could produce LFeIII–O˙ and ˙OCPh3 radicals. The ⋅OCPh3 species could abstract an H atom from an RH species (solvent or ligand) to form triphenylmethanol while the LFeIII–O⋅ species could additionally abstract an H atom to form LFeIII–OH. It should also be noted that a long-lived purple species (λmax = 550 nm, ε = 1070 M−1 cm−1) is generated by reacting 1 with tBuOOH at −30 °C (Fig. 4(c)). This intermediate is expected to be Fe–OOtBu.59–61 Upon decomposition, Ph3COCH3 is the primary product detected presumably formed by combination of ˙OCPh and ˙CH3 (derived from decomposition of tBuO˙64).
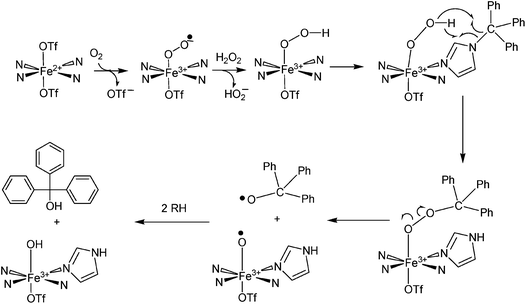 |
| | Scheme 2 Proposed mechanism for oxidation of the trityl group attached to [Fe(TrIm)4(OTf)2] (1) when H2O2 is added. | |
Conclusion
In this study we have utilized 1-tritylimidazole (TrIm) to model enzymatic 4His nonheme sites. Reaction of 4 equiv. TrIm with Fe(OTf)2·2MeCN yields [Fe(TrIm)4(OTf)2] (1) in good yield. The four imidazole ligands are bonded in a square plane while triflate oxygens are in the apical positions. Variable temperature magnetic susceptibility measurements revealed 1 to be high spin between 20 K and 300 K. simulations of the data yielded g = 2.24 and D = −0.80 cm−1. DFT studies reproduced metric parameters for 1 and the HOMO–LUMO gap energy (3.89 eV) indicated a high degree of stability. Reactivity studies of 1 with hydrogen peroxide or tBuOOH indicate that an oxygenated intermediate is formed which subsequently decomposes to yield oxidized trityl substituent products.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
FAC acknowledges the receipt of an OU-REF grant. JL acknowledges a graduate fellowship from OU. TAH acknowledges an OU Provost Award. We thank Prof. M. M. Szczęśniak for assistance with the DFT calculations. NIH Grant No. R15GM112395 and NSF Grant No. CHE-0748607 and CHE-0821487 are gratefully acknowledged.
Notes and references
- X. W. Sui, M. Golczak, J. Y. Zhang, K. A. Kleinberg, J. von Lintig, K. Palczewski and P. D. Kiser, J. Biol. Chem., 2015, 290, 30212–30223 CrossRef CAS PubMed.
- B. Zhang, C. Liu, Y. Q. Wang, X. Yao, F. Wang, J. S. Wu, G. J. King and K. D. Liu, New Phytol., 2015, 206, 1513–1526 CrossRef CAS PubMed.
- M. H. Walter and D. Strack, Nat. Prod. Rep., 2011, 28, 663–692 RSC.
- S. A. Baba, D. Jain, N. Abbas and N. Ashraf, J. Plant Physiol., 2015, 189, 114–125 CrossRef CAS PubMed.
- V. A. Tu, A. Kaga, K. H. Gericke, N. Watanabe, T. Narumi, M. Toda, B. Brueckner, S. Baldermann and N. Mase, J. Org. Chem., 2014, 79, 6808–6815 CrossRef CAS PubMed.
- M. Bruno, P. Beyer and S. Al-Babili, Arch. Biochem. Biophys., 2015, 572, 126–133 CrossRef CAS PubMed.
- L. H. Liu, Z. Y. Shao, M. Zhang and Q. M. Wang, Mol. Plant, 2015, 8, 28–39 CrossRef CAS PubMed.
- J. Amengual, M. A. K. Widjaja-Adhi, S. Rodriguez-Santiago, S. Hessel, M. Golczak, K. Palczewski and J. von Lintig, J. Biol. Chem., 2013, 288, 34081–34096 CrossRef CAS PubMed.
- S. Raghuvanshi, V. Reed, W. S. Blaner and E. H. Harrison, Arch. Biochem. Biophys., 2015, 572, 19–27 CrossRef CAS PubMed.
- M. Yahyaa, A. Berim, T. Isaacson, S. Marzouk, E. Bar, R. Davidoyich-Rikanati, E. Lewinsohn and M. Ibdah, J. Agric. Food Chem., 2015, 63, 8275–8282 CrossRef CAS PubMed.
- S. Holger, R. Kurtzer, W. Eisenreich and W. Schwab, J. Biol. Chem., 2006, 281, 9845–9851 CrossRef PubMed.
- O. P. Ernst, D. T. Lodowski, M. Elstner, P. Hegemann, L. S. Brown and H. Kandori, Chem. Rev., 2014, 114, 126–163 CrossRef CAS PubMed.
- S. A. J. Messing, S. B. Gabelli, I. Echeverria, J. T. Vogel, J. C. Guan, B. C. Tan, H. J. Klee, D. R. McCarty and L. M. Amzel, Plant Cell, 2010, 22, 2970–2980 CrossRef CAS PubMed.
- C. R. D. Lancaster, A. Kroger, M. Auer and H. Michel, Nature, 1999, 402, 377–385 CrossRef CAS PubMed.
- G. Katona, P. Carpentier, V. Niviere, P. Amara, V. Adam, J. Ohana, N. Tsanov and D. Bourgeois, Science, 2007, 316, 449–453 CrossRef CAS PubMed.
- J. P. McEvoy and G. W. Brudvig, Biochemistry, 2008, 47, 13394–13403 CrossRef CAS PubMed.
- X. W. Sui, P. D. Kiser, T. Che, P. R. Carey, M. Golczak, W. X. Shi, J. von Lintig and K. Palczewski, J. Biol. Chem., 2014, 289, 12286–12299 CrossRef CAS PubMed.
- D. P. Kloer, S. Ruch, S. Al-Babili, P. Beyer and G. E. Schulz, Science, 2005, 308, 267–269 CrossRef CAS PubMed.
- D. Babino, M. Golczak, P. D. Kiser, A. Wyss, K. Palczewski and J. von
Lintig, ACS Chem. Biol., 2016, 11, 1049–1057 CrossRef CAS PubMed.
- K. S. Hagen, Inorg. Chem., 2000, 39, 5867–5869 CrossRef CAS PubMed.
- julX, http://www.mpibac.mpg.de/bac/index_en.php/logins/bill/julX_en.php.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian09, Revision C.01, Gaussian Inc., Wallingford CT, 2010 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef CAS.
- M. Hanwell, D. Curtis, D. Lonie, T. Vandermeersch, E. Zurek and G. Hutchison, J. Cheminf., 2012, 4, 1–17 Search PubMed.
- COSMO V1.61, Software for the CCD Detector Systems for Determining Data Collection Parameters, Bruker Analytical X-ray Systems, Madison, WI, 2009 Search PubMed.
- APEX2 V2010.11-3. Software for the CCD Detector System, Bruker Analytical X-ray Systems, Madison, WI, 2010 Search PubMed.
- SAINT V 7.68A Software for the Integration of CCD Detector System, Bruker Analytical X-ray Systems, Madison, WI, 2010 Search PubMed.
- SADABS V2008/2 Program for absorption corrections using Bruker-AXS CCD based on the method of Robert Blessing; R. H. Blessing, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 33–38 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- N. Brefuel, C. Duhayon, S. Shova and J. P. Tuchagues, Chem. Commun., 2007, 5223–5225 RSC.
- S. W. Jin, W. Z. Chen and H. Y. Qiu, Cryst. Growth Des., 2007, 7, 2071–2079 CAS.
- I. Morgenstern-Badarau, F. Lambert, A. Deroche, M. Cesario, J. Guilhem, B. Keita and L. Nadjo, Inorg. Chim. Acta, 1998, 276, 234–241 CrossRef.
- A. K. Patra, K. S. Dube, G. C. Papaefthymiou, J. Conradie, A. Ghosh and T. C. Harrop, Inorg. Chem., 2010, 49, 2032–2034 CrossRef CAS PubMed.
- U. Ray, D. Banerjee, J. C. Liou, C. N. Lin, T. H. Lu and C. Sinha, Inorg. Chim. Acta, 2005, 358, 1019–1026 CrossRef CAS.
- W. Liu, G. Zhang, X. Li, B. L. Wu and H. Y. Zhang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, M938–U1085 CAS.
- E. Ohshima, K. Yoshida, K. Sugiyama and H. Uekusa, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, 1093–1094 Search PubMed.
- J. Li, A. Banerjee, P. L. Pawlak, W. W. Brennessel and F. A. Chavez, Inorg. Chem., 2014, 53, 5414–5416 CrossRef CAS PubMed.
- L. Benhamou, A. Thibon, L. Brelot, M. Lachkar and D. Mandon, Dalton Trans., 2012, 41, 14369–14380 RSC.
- M. Grau, A. Kyriacou, F. C. Martinez, I. M. de Wispelaere, A. J. P. White and G. J. P. Britovsek, Dalton Trans., 2014, 43, 17108–17119 RSC.
- G. Panchbhai, W. M. Singh, B. Das, R. T. Jane and A. Thapper, Eur. J. Inorg. Chem., 2016, 3262–3268 CrossRef CAS.
- K. Umehara, S. Kuwata and T. Ikariya, Inorg. Chim. Acta, 2014, 413, 136–142 CrossRef CAS.
- J. England, R. Gondhia, L. Bigorra-Lopez, A. R. Petersen, A. J. P. White and G. J. P. Britovsek, Dalton Trans., 2009, 5319–5334 RSC.
- S. Gosiewska, J. J. L. M. Cornelissen, M. Lutz, A. L. Spek, G. van Koten and R. J. M. K. Gebbink, Inorg. Chem., 2006, 45, 4214–4227 CrossRef CAS PubMed.
- G. J. P. Britovsek, J. England and A. J. P. White, Dalton Trans., 2006, 1399–1408 RSC.
- N. J. Hardman, X. G. Fang, B. L. Scott, R. J. Wright, R. L. Martin and G. J. Kubas, Inorg. Chem., 2005, 44, 8306–8316 CrossRef CAS PubMed.
- G. J. P. Britovsek, J. England and A. J. P. White, Inorg. Chem., 2005, 44, 8125–8134 CrossRef CAS PubMed.
- D. W. Blakesley, S. C. Payne and K. S. Hagen, Inorg. Chem., 2000, 39, 1979–1989 CrossRef CAS PubMed.
- J. Y. Chen and L. K. Woo, Polyhedron, 1999, 18, 825–830 CrossRef CAS.
- C. J. O'Connor, Prog. Inorg. Chem., 1982, 29, 203–283 CrossRef.
- B. N. Figgis and M. A. Hitchman, Ligand Field Theory and Its Applications, Wiley-VCH, New York, 2000 Search PubMed.
- T. Fujinami, K. Nishi, N. Matsumoto, S. Iijima, M. A. Halcrow, Y. Sunatsuki and M. Kojima, Dalton Trans., 2011, 40, 1230–12309 RSC.
- Y. Sunatsuki, R. Kawamoto, K. Fujita, H. Maruyama, T. Suzuki, H. Ishida, M. Kojima, S. Iijima and N. Matsumoto, Inorg. Chem., 2009, 48, 8784–8795 CrossRef CAS PubMed.
- R. Boca, Coord. Chem. Rev., 2004, 248, 757–815 CrossRef CAS.
- G. J. Long, S. Tanase, F. Remacle, G. Periyasamy and F. Grandjean, Inorg. Chem., 2009, 48, 8173–8179 CrossRef CAS PubMed.
- N. D. Sousa, R. B. de Lima, A. L. P. Silva, A. A. Tanaka, A. B. F. da Silva and J. D. G. Varela, Comput. Theor. Chem., 2015, 1054, 93–99 CrossRef.
- A. L. P. Silva, L. F. de Almeida, A. L. B. Marques, H. R. Costa, A. A. Tanaka, A. B. F. da Silva and J. D. G. Varela, J. Mol. Model., 2014, 20, 2131 CrossRef PubMed.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr, Chem. Rev., 2004, 104, 939–986 CrossRef CAS PubMed.
- F. Namuswe, G. D. Kasper, A. A. Sarjeant, T. Hayashi, C. M. Krest, M. T. Green, P. Moenne-Loccoz and D. P. Goldberg, J. Am. Chem. Soc., 2008, 130, 14189–14200 CrossRef CAS PubMed.
- M. P. Jensen, M. Costas, R. Y. Ho, J. Kaizer, A. Mairata i Payeras, E. Munck, L. Que Jr, J. U. Rohde and A. Stubna, J. Am. Chem. Soc., 2005, 127, 10512–10525 CrossRef CAS PubMed.
- G. Roelfes, V. Vrajmasu, K. Chen, R. Y. Ho, J. U. Rohde, C. Zondervan, R. M. La Crois, E. P. Schudde, M. Lutz, A. L. Spek, R. Hage, B. L. Feringa, E. Munck and L. Que Jr, Inorg. Chem., 2003, 42, 2639–2653 CrossRef CAS PubMed.
- W. P. Nuemann, W. Uzick and A. K. Zarkadis, J. Am. Chem. Soc., 1986, 108, 3762–3770 CrossRef.
- M. Finn, R. Friedline, N. K. Suleman, C. J. Wohl and J. M. Tanko, J. Am. Chem. Soc., 2004, 126, 7578–7584 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available:. CCDC 1548010. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra09456f |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  c,
Richard J. Staplesc and
Ferman A. Chavez
c,
Richard J. Staplesc and
Ferman A. Chavez