
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of Cu–Mg/ZnO catalysts and catalysis in dimethyl oxalate hydrogenation to ethylene glycol: enhanced catalytic behavior in the presence of a Mg2+ dopant
Xiangpeng Kong ab,
Zheng Chenbc,
Yuehuan Wua,
Ruihong Wanga,
Jiangang Chen*b and
Lifeng Dinga
ab,
Zheng Chenbc,
Yuehuan Wua,
Ruihong Wanga,
Jiangang Chen*b and
Lifeng Dinga
aDepartment of Chemistry and Chemical Engineering, Taiyuan Institute of Technology, Taiyuan 030008, PR China
bState Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, PR China. E-mail: chenjg19@163.com; chenjg@sxicc.ac.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, PR China
First published on 24th October 2017
Abstract
Mg2+ doped nanoscale Cu–Mg/ZnO catalysts prepared by the co-precipitation method have been systematically characterized focusing on the amount of Mg2+ ions incorporated. The amount of Mg2+ dopant was demonstrated to have profound influence on the evolution of textural and structural properties, the functionality of active phases and the catalytic behavior of the as-synthesized ternary catalysts (Cu, ZnO and Mg2+). The Cu–1Mg/ZnO catalyst with 1 wt% MgO loading was found to be helpful for enhanced Cu dispersion and an increased amount of active surface Cu0 sites, which promoted catalytic activity in dimethyl oxalate (DMO) hydrogenation to ethylene glycol (EG) effectively. Further increasing the Mg2+ concentration results in the aggregation of surface metal Cu nano-particles (NPs), and thus causes the reduction in the number of surface active Cu0 sites and the activity of the Cu/ZnO based catalyst. However, the high density of the surface Cu+ sites and O2− centers generated in the Cu–4Mg/ZnO catalyst with 4.0 wt% MgO loading facilitates superb hydrogenation activity. Under the optimized reaction conditions, the Cu–4Mg/ZnO catalyst shows 100% DMO conversion and an EG yield of 95% for longer than 300 h. During the DMO hydrogenation process, Cu0 sites are assumed to afford atomic hydrogen by dissociative adsorption and spillover. The reaction rate greatly depends on the dissociative adsorption of DMO molecules by the surface Cu+ and oxygen vacancies, originating from tight contact between the Cu NP ZnO matrix and Mg2+ dopant. Additionally, the strengthened metal-support interaction (MSI) originating from the enhanced chemical interaction between the Mg2+ modified ZnO substrate and the Cu NPs leads to excellent stability.
1. Introduction
Ethylene glycol (EG), as an all-round chemical in the chemical industry, is widely used in energy, plastics, automobiles, and chemicals.1 To date, EG has been mainly produced from the traditional petrochemical derived ethylene oxide hydrolysis route, wherein the productivity is limited by shrinking oil resources and high energy consumption.2 Converting syngas to fine chemicals has been deemed a promising strategy to offset the insufficient capacity of the traditional technology, owing to the virtues of adequate feedstocks, high efficiency and environmental friendliness.3 In the past few decades, considerable effort has been put into conducting the direct homogeneous synthesis of EG from syngas using noble metal catalysts such as Co, Ru and Rh.4,5 Nevertheless, the drawbacks of high cost, low efficiency, excessive energy consumption, and subsequent separation issues hinder its industrial prospects. Alternatively, most researchers have focused on the indirect synthesis of EG from the hydrogenation of dimethyl oxalate (DMO) and the DMO synthesis from syngas has been industrialized successfully.6 Thus, a catalyst for the DMO selective vapor-phase hydrogenation to EG has become a key topic in achieving the indirect synthesis of EG from syngas.Ester selective hydrogenation to the corresponding alcohol is mostly carried out on Cu nano-particles (NPs), owing to their essential feature of the selective activation of C–O/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds.7 However, the low Hüttig temperature (134 °C, the temperature at which defective atoms will diffuse) and Tamman temperature (405 °C, the temperature at which bulk atoms will be mobile) generate the issue of Cu NP agglomeration during the highly exothermic DMO hydrogenation process, hindering the service efficiency of the Cu-based catalysts.8 The metal support interaction (MSI) has shown great potential in guaranteeing the catalytic activity and stability of the Cu-based catalysts. For this reason, it would be worthwhile elucidating and fully utilizing the support components, affording both adequate active sites and strengthened MSIs. It is well known that ZnO is a wide band gap semiconductor (3.37 eV) with a high surface-to-volume ratio, intrinsic properties and a large exciton binding energy (60 meV).9 The unique properties of the ZnO material render its potential application in many fields, such as in gas sensors,10 photocatalysts,11 optoelectronic applications12 and so on. Also, ZnO can provide active sites for hydrogen spillover, or act as a structure-directing support to control the dispersion of metallic Cu NPs.13 In particular, Cu NPs supported on a ZnO material for CO/C–O bond selective hydrogenation reactions has been attracting lots of interest from researchers.14 Nonetheless, the Cu/ZnO catalyst exhibits poor activity and stability in the hydrogenation reaction. Partial substitution of group IIIA elements in Cu/ZnO based catalysts can improve their catalytic performance effectively; moreover, the co-precipitation method was effective for the preparation of an active Cu/ZnO based catalyst.15 For instance, Zhang et al.16 and Wen et al.17 prepared Cu/ZnO based catalysts with an Al3+ promoter, achieving enhanced Cu dispersion, adequate surface Cu0 sites and balanced Cu0–Cu+ sites. Accordingly, enhanced activity for the EG synthesis from DMO hydrogenation was obtained over the ZnO–Al2O3 mixed oxide supported Cu NPs. Li et al.18 synthesized CuZnGa catalysts by a pH-controlled co-precipitation method for use as catalysts in the DMO hydrogenation reaction. They discerned that the addition of Ga3+ into nano-beta phase CuZn favors enhanced catalytic activity and thermal stability. Apart from Al3+ and Ga3+, Mg2+ can be easily doped in the lattice of ZnO, due to its similar radius and electronic shell to a Zn atom, favoring an increased band gap.19 Moreover, Mg2+ dopants encourage electronic interaction between the ZnO material and metallic NPs, producing enhanced MSIs and an improved metal-support interface.20,21 In addition, the tight contact between ZnO and the Cu NPs is believed to affect the in situ formation of catalytically active sites,22 wherein distinct ZnO morphology with a porous structure, surface morphology and optical properties can be regenerated with the assistance of Mg2+ dopants, and they are critically dependent on the amount of Mg2+.20,21 In any case, surface Mg2+ additives tend to produce more abundant surface oxygen vacancies (Mg–O2− pairs and O2− ions) in the catalysts.23 Additionally, the synergistic effect between oxygen vacancies and Cu NPs could produce defective sites, which is helpful for negatively charged C–O/CO bonds that have been proposed to benefit C–O/CO selective hydrogenation.24 Recently, Behrens and coworkers25 have reported that defect sites in catalysts improve the hydrogenation behavior. What is more, previous studies revealed that Mg2+ additives are prone to promote surface Cu+ sites formation, which can improve the activity of the water–gas shift reaction by lowering the activation energy, while their influence on C–O/CO selective hydrogenation has been rarely reported.26 Thus, it is inferred that the possible synergistic effect among Cu, Zn and Mg species tends to endow the Cu–Mg/ZnO composites with distinct performance in the DMO hydrogenation to EG.27 Nonetheless, limited studies have been reported on Mg2+ doped ZnO dispersed Cu NPs for C–O/CO selective hydrogenation.
O bonds.7 However, the low Hüttig temperature (134 °C, the temperature at which defective atoms will diffuse) and Tamman temperature (405 °C, the temperature at which bulk atoms will be mobile) generate the issue of Cu NP agglomeration during the highly exothermic DMO hydrogenation process, hindering the service efficiency of the Cu-based catalysts.8 The metal support interaction (MSI) has shown great potential in guaranteeing the catalytic activity and stability of the Cu-based catalysts. For this reason, it would be worthwhile elucidating and fully utilizing the support components, affording both adequate active sites and strengthened MSIs. It is well known that ZnO is a wide band gap semiconductor (3.37 eV) with a high surface-to-volume ratio, intrinsic properties and a large exciton binding energy (60 meV).9 The unique properties of the ZnO material render its potential application in many fields, such as in gas sensors,10 photocatalysts,11 optoelectronic applications12 and so on. Also, ZnO can provide active sites for hydrogen spillover, or act as a structure-directing support to control the dispersion of metallic Cu NPs.13 In particular, Cu NPs supported on a ZnO material for CO/C–O bond selective hydrogenation reactions has been attracting lots of interest from researchers.14 Nonetheless, the Cu/ZnO catalyst exhibits poor activity and stability in the hydrogenation reaction. Partial substitution of group IIIA elements in Cu/ZnO based catalysts can improve their catalytic performance effectively; moreover, the co-precipitation method was effective for the preparation of an active Cu/ZnO based catalyst.15 For instance, Zhang et al.16 and Wen et al.17 prepared Cu/ZnO based catalysts with an Al3+ promoter, achieving enhanced Cu dispersion, adequate surface Cu0 sites and balanced Cu0–Cu+ sites. Accordingly, enhanced activity for the EG synthesis from DMO hydrogenation was obtained over the ZnO–Al2O3 mixed oxide supported Cu NPs. Li et al.18 synthesized CuZnGa catalysts by a pH-controlled co-precipitation method for use as catalysts in the DMO hydrogenation reaction. They discerned that the addition of Ga3+ into nano-beta phase CuZn favors enhanced catalytic activity and thermal stability. Apart from Al3+ and Ga3+, Mg2+ can be easily doped in the lattice of ZnO, due to its similar radius and electronic shell to a Zn atom, favoring an increased band gap.19 Moreover, Mg2+ dopants encourage electronic interaction between the ZnO material and metallic NPs, producing enhanced MSIs and an improved metal-support interface.20,21 In addition, the tight contact between ZnO and the Cu NPs is believed to affect the in situ formation of catalytically active sites,22 wherein distinct ZnO morphology with a porous structure, surface morphology and optical properties can be regenerated with the assistance of Mg2+ dopants, and they are critically dependent on the amount of Mg2+.20,21 In any case, surface Mg2+ additives tend to produce more abundant surface oxygen vacancies (Mg–O2− pairs and O2− ions) in the catalysts.23 Additionally, the synergistic effect between oxygen vacancies and Cu NPs could produce defective sites, which is helpful for negatively charged C–O/CO bonds that have been proposed to benefit C–O/CO selective hydrogenation.24 Recently, Behrens and coworkers25 have reported that defect sites in catalysts improve the hydrogenation behavior. What is more, previous studies revealed that Mg2+ additives are prone to promote surface Cu+ sites formation, which can improve the activity of the water–gas shift reaction by lowering the activation energy, while their influence on C–O/CO selective hydrogenation has been rarely reported.26 Thus, it is inferred that the possible synergistic effect among Cu, Zn and Mg species tends to endow the Cu–Mg/ZnO composites with distinct performance in the DMO hydrogenation to EG.27 Nonetheless, limited studies have been reported on Mg2+ doped ZnO dispersed Cu NPs for C–O/CO selective hydrogenation.
In the present work, a series of porous Cu–xMg/ZnO catalysts modified by different amounts of Mg2+ dopant are synthesized using a facile co-precipitation method, and the structural evolution and catalytic behavior of the Cu–Mg/ZnO catalysts in the DMO hydrogenation were investigated systematically by changing the amount of Mg2+ dopant.
2. Experimental
2.1 Catalyst preparation
The Cu–xMg/ZnO catalysts with different amounts of Mg2+ dopant (the mole ratio of Cu/Zn is fixed at 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) were prepared using the continuous co-precipitation method with Na2CO3 as the precipitant. Typically, 200 mL of mixed aqueous solution (1 M) of the starting materials with given atomic ratios (Cu:Zn mole ratio = 4:5, and the starting precursors were Cu(NO3)2·3H2O, Zn(NO3)2·6H2O, and Mg(NO3)2·6H2O) was used as a precursor solution, and 200 mL of Na2CO3 solution (1 M) was used as a precipitating agent. Co-precipitation was conducted at 75 °C in a water bath, and the flow rates of the two solutions were adjusted to give a constant pH value of ca. 8.0. The resultant suspension was aged for 24 h at room temperature. Then, the precipitate was filtrated and washed using distilled water until the conductivity of the filtrate was less than 2 mS m−1. After being dried in air at 120 °C for 12 h, the precursors were calcined at 400 °C for 6 h. The as-synthesized precursors from the co-precipitation process were denoted as CZMx-p and the catalysts after calcination were labeled as CuO–xMg/ZnO. The corresponding reduced catalysts, which were reduced at 300 °C under a flow of H2 for 4 h, were cooled to room temperature. Finally, the samples were transferred into a glass bottle without air and denoted as Cu–xMg/ZnO (C, Z and M stand for, Cu2+, Zn2+ and Mg2+ precipitate, respectively, and x denotes the MgO loading).
:5) were prepared using the continuous co-precipitation method with Na2CO3 as the precipitant. Typically, 200 mL of mixed aqueous solution (1 M) of the starting materials with given atomic ratios (Cu:Zn mole ratio = 4:5, and the starting precursors were Cu(NO3)2·3H2O, Zn(NO3)2·6H2O, and Mg(NO3)2·6H2O) was used as a precursor solution, and 200 mL of Na2CO3 solution (1 M) was used as a precipitating agent. Co-precipitation was conducted at 75 °C in a water bath, and the flow rates of the two solutions were adjusted to give a constant pH value of ca. 8.0. The resultant suspension was aged for 24 h at room temperature. Then, the precipitate was filtrated and washed using distilled water until the conductivity of the filtrate was less than 2 mS m−1. After being dried in air at 120 °C for 12 h, the precursors were calcined at 400 °C for 6 h. The as-synthesized precursors from the co-precipitation process were denoted as CZMx-p and the catalysts after calcination were labeled as CuO–xMg/ZnO. The corresponding reduced catalysts, which were reduced at 300 °C under a flow of H2 for 4 h, were cooled to room temperature. Finally, the samples were transferred into a glass bottle without air and denoted as Cu–xMg/ZnO (C, Z and M stand for, Cu2+, Zn2+ and Mg2+ precipitate, respectively, and x denotes the MgO loading).
2.2 Characterization
The textural properties of the samples were determined by a nitrogen adsorption method using a Micromeritics Tristar II 3000 Analyzer at −196 °C, and the BET surface areas were calculated from the isotherms using the Brunauer–Emmett–Teller (BET) method.The powder X-ray diffraction (XRD) patterns were conducted on a Shimadzu XRD-6000 diffractometer with a graphite-filtered Cu Kα source. The average crystallite size (Dhkl) was calculated using the Scherrer equation.
The number of surface metallic Cu0 sites exposed was determined by N2O titration; first they were oxidized by N2O, followed by H2 titration using the procedure described in the report.28
Thermal decomposition of the catalyst precursors was studied by the thermogravimetric (TG) method using a NETZSCH TG209F1 thermal analyzer. The measurements were performed in the temperature range 100–700 °C with a linear temperature program with b = 10 °C min−1 in a continuous flow of synthetic air (30 mL min−1).
The morphology of the catalysts was investigated using scanning electron microscopy (SEM) (JSM-6701F, Japan).
Transmission electron microscopy (TEM) images of the catalysts were obtained using a JEM 2010 microscope operated at 200 kV.
H2-temperature programmed reduction (H2-TPR) was carried out using a dynamic analyzer (Micromeritics, Model 2920). About 30 mg of catalyst was treated in 5% H2/95% Ar (v/v) (with a flow rate of 50 mL min−1), and the reduction temperature was increased from room temperature to 600 °C at a heating rate of 10 °C min−1.
In CO2 temperature programmed desorption (CO2-TPD), the catalyst was first reduced at 300 °C in a H2 flow of 30 mL min−1 for 2 h. After cooling to room temperature, the catalyst was saturated with pure CO2 (30 mL min−1) at 50 °C for 60 min and then flushed with an Ar flow (40 mL min−1) to remove all physically adsorbed molecules. Afterwards, the TPD experiment was performed with a heating rate of 10 °C min−1 under an Ar flow (40 mL min−1), and the desorbed CO2 was detected using an AMETEK mass spectrometer. The CO2 peak area was quantitatively calibrated by injecting CO2 pulses.
FT-IR characterization of the catalysts was performed using a Bruker Vector 22 spectrometer equipped with a DTGS detector. 2 mg of the as-prepared calcined samples was finely grounded, dispersed in 200 mg KBr, and pelletized.
X-ray photoelectron spectroscopy (XPS) was carried out on a Quantum 2000 Scanning ESCA Microprobe instrument (Physical Electronics) equipped with an Al Kα X-ray radiation source (hm = 1486.6 eV).
2.3 Catalytic behavior test
The catalytic behavior of the as-synthesized samples was tested on a fixed-bed tubular reactor with an inner diameter of 10 mm. The calcined CuO–xMg/ZnO samples (2.0 mL) were packed and sandwiched by quartz powders (20–40 meshes) in a tubular reactor, and then were reduced in a 5% H2/Ar atmosphere at 300 °C for 12 h at a heating rate of 2 °C min−1. After cooling to the reaction temperature, 12.5 wt% DMO (purity >99%) in methanol and H2 were fed into the reactor at a H2/DMO molar ratio of 100:1 and a system temperature of 220 °C. During the hydrogenation process, the total pressure was kept at 2.5 MPa, and the room-temperature liquid space velocity of DMO was 2.0 h−1. The products were collected and analyzed using a gas chromatograph (Finnigan Trace GC ultra) fixed with a 30 m HP-5 capillary column and a flame ionization detector (FID).
3. Results
3.1 Evolution of crystalline phase and morphology
Fig. 1 shows the XRD patterns of the as-synthesized precursors after drying at 120 °C in air (A), the CuO–xMg/ZnO catalysts calcined at 400 °C in air (B) and the Cu–xMg/ZnO catalysts reduced at 300 °C in a H2 atmosphere (C) with different amounts of Mg2+. The XRD pattern in Fig. 1(A) of CZM0-p shows a series of characteristic diffraction peaks assigned to aurichalcite [(Cu, Zn)5(CO3)2(OH)6 (JCPDS 11-0287)]. However, Harding et al.29 identified that obtaining pure aurichalcite is difficult, due to the occasional insertion of the hydrozincite layer into the aurichalcite structure which facilitates twinning in the aurichalcite structure. Inspection of the relevant JCPDS database revealed that the unassigned peaks were also characteristic of the malachite [Cu2CO3(OH)2] structure.20 Unexpectedly, the crystallinity decreased when more Mg2+ dopants were incorporated in the precursors. This is probably due to large distortions in the precursor as a result of the replacement of Zn2+ (ionic radius = 0.057 nm) with Mg2+ (ionic radius = 0.060 nm), which facilitates an amorphous precipitate (such as hydroxides and hydroxyl carbonates).30 Additionally, no reflection line corresponding to the chemical phase containing Mg species was detected, indicating that the promoters do not form crystalline segregated biphases. After annealing at 400 °C, all of the peaks in the XRD patterns in Fig. 1(B) could be well indexed to crystalline ZnO and CuO for all of the catalysts, implying the as-synthesized precursor transformed to the CuO and ZnO nanostructure by thermally decomposing. The crystallite sizes of CuO calculated using the Scherrer equation are shown in Table 1. After being modified, the CuO NP size increases dramatically from 11.42 to 12.81 nm at first with 1.0 wt% MgO loading, however, it decreases gradually when the dopant amount is increased from 1.0 wt% to 4.0 wt%. Nevertheless, the CuO peaks became adversely strengthened with excessive 5.0 wt% MgO additive, which is indicative of the formation of larger CuO NPs. The above results suggest that only sufficient Mg2+ doping favors the enhanced dispersion of the CuO species. Except for the CuO and ZnO characteristic diffraction peaks, there are another two diffraction peaks which are attributed to calcite and CuMgO3 (JCPDS 41-1365) phases in the CuO–5Mg/ZnO sample, demonstrating crystalline calcite and CuMgO3 formation.31 Upon reduction, the XRD patterns in Fig. 1(C) of the reduced samples display three distinct weak diffraction peaks located at 43.5°, 50.4° and 74.1° that are attributed to face-centered cubic (fcc) Cu (JCPDS 04-0836). The absence of CuO characteristic diffraction peaks shows that the almost crystalline CuO was completely reduced under the present reduction conditions. Additionally, the intensity of the Cu diffraction peaks when adjusting the Mg2+ content exhibits a similar trend to the CuO NPs in the calcined CuO–xMg/ZnO catalysts. What is more, MgO loading of less than 4.0 wt% shows indistinctive influence on the crystalline ZnO NP sizes of the resultant catalysts, as shown in Table 1. In contrast, 5.0 wt% MgO loading leads to a great increase in ZnO NP size, which illustrates that a trace amount of Mg2+ additive shows unobvious influence on the crystalline ZnO NP size, but excess would generate an impurity phase instead of entering into the intracellular of hierarchical ZnO nanostructures.32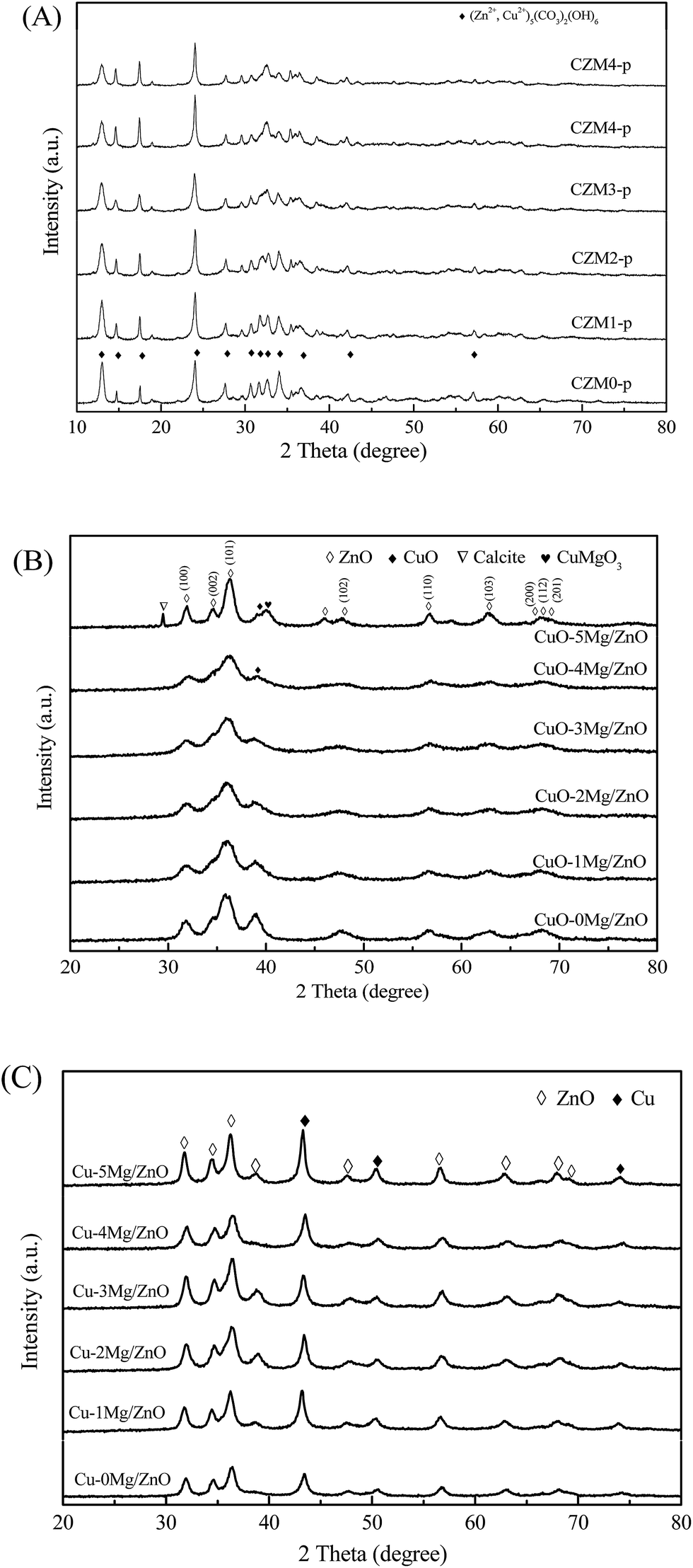 | ||
| Fig. 1 XRD patterns of the as-synthesized precursors (A), the calcined CuO–xMg/ZnO catalysts (B) and the reduced Cu–xMg/ZnO catalysts (C) with different amounts of Mg2+. | ||
| Catalyst | SBET (m2 g−1) | Vp (cm3 g−1) | DCuOb | DCub | DZnOb | SCua (m2 g−1) | Cu dispersiona (%) | Surface atomic Cuc (wt%) | Surface atomic Mgc (wt%) |
|---|---|---|---|---|---|---|---|---|---|
| a SCu and copper dispersion were measured using N2O adsorption.b DCuO in calcined C–xMg/ZnO catalysts, DCu, and DZnO in reduced Cu–xMg/ZnO were calculated from the reflection planes in the XRD patterns using the Scherrer equation.c Surface atomic Cu and Mg in the resultant sample was analyzed using SEM-EDS. | |||||||||
| Cu–0Mg/ZnO | 50.3 | 0.21 | 11.4 | 11.3 | 11.5 | 2.40 | 13.0 | 23.8 | 0 |
| Cu–1Mg/ZnO | 74.7 | 0.19 | 12.8 | 12.8 | 11.6 | 3.59 | 19.1 | 24.4 | 0.056 |
| Cu–2Mg/ZnO | 83.8 | 0.22 | 12.3 | 12.4 | 11.7 | 3.20 | 17.5 | 26.6 | 0.93 |
| Cu–3Mg/ZnO | 90.2 | 0.24 | 11.8 | 11.6 | 11.8 | 2.79 | 15.2 | 27.5 | 1.41 |
| Cu–4Mg/ZnO | 93.1 | 0.27 | 11.0 | 11.1 | 11.6 | 2.45 | 13.4 | 28.3 | 2.42 |
| Cu–5Mg/ZnO | 27.2 | 0.17 | 13.4 | 13.3 | 12.6 | 2.27 | 12.3 | 29.4 | 3.50 |
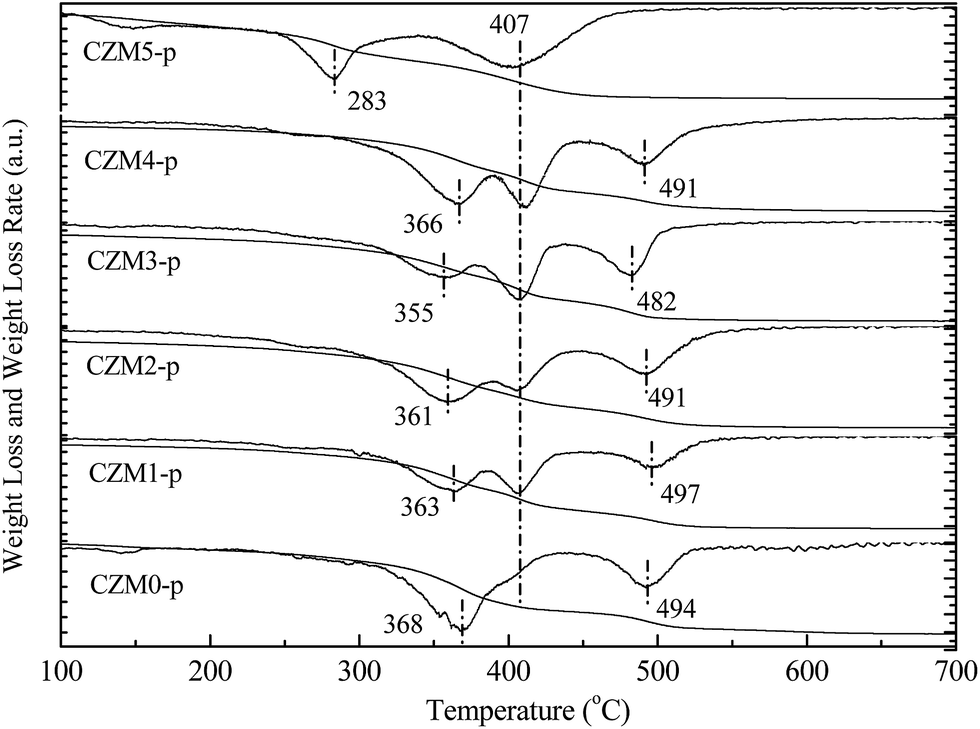 | ||
| Fig. 2 Thermogravimetry and differential thermogravimetry (TG-DTG) profiles of CZMx-p precursors in an Ar atmosphere. | ||
Fig. 2 presents the TG-DTG profiles of the CZMx-p precursors in an Ar atmosphere. The DTG profile of CZM0-p exhibits three weight losses at 368, 407 (weak shoulder) and 494 °C. The first weight loss at 368 °C was assigned to the decomposition of the hydroxycarbonate structure to produce an anion modified oxide, i.e. an oxide in which some oxygen ions are replaced by either OH− or CO32− species. The weak shoulder at 407 °C should correspond to the phase decomposition of zincian malachite.33 The third weight loss at 494 °C should be ascribed to the decomposition of Cu oxocarbonates formed during the former steps of the thermal decomposition of the aurichalcite structure.34 Compared to CZM0-p, the weight loss corresponding to the phase decomposition of zincian malachite became distinct with the introduction of a small amount of Mg2+ dopants, suggesting that Mg2+ ions favor zincian malachite phase formation during co-precipitation. However, CZM5-p contained only two distinct losses at 277 and 412 °C, which can be explained by the fact that the synthesis of the aurichalcite structure was suppressed, but the zincian malachite phase has only been synthesized when a excessive amount of Mg2+ ions was introduced.33 These assignments were supported by the relevant XRD pattern in Fig. 1(A), which again showed that the mole ratio of aurichalcite and malachite in the precipitate precursors depends greatly on the Mg2+ concentration.
Mg2+ dopant induced effects on morphology of the as-synthesized samples have been examined using SEM and SEM-EDS, and the results are presented in Fig. 3. For the Cu–0Mg/ZnO catalyst, Fig. 3(a) shows the formation of agglomerated nanosheets, which self-aggregate into large particles surrounded by a developed porous structure. Fig. 3(b)–(d) depict the SEM micrographs of the Mg2+ modified samples with varying MgO loading levels from 1.0 wt% to 5.0 wt%. The particle sizes are still small and each particle is probably composed of nanoscale CuO and ZnO crystallites detected by XRD. With the increase in Mg2+ percentage, the dispersion of particles become more average, and the particle sizes become smaller. Interestingly, it is found that the as-prepared samples undergo significant morphological changes induced by changing the Mg2+ content doped in the Cu/ZnO structure. After being modified by the Mg2+ dopant, the crystal growth along the sideways direction was suppressed and the particle shape of the ZnO powder changed to agglomerated and dense-stacking nanorods, suggesting enormous Mg2+ ion induced effects on ZnO growth. It is deduced that the chemical morphology of the precursors are influenced by the Mg2+ dopant and further determine the growth mechanisms of the Cu–xMg/ZnO nanocomposites. Klubnuan et al.35 proposed that pure ZnO had a narrower energy band gap (Eg) value compared to that of the MgO/ZnO nanocomposites because of the presence of higher defect concentrations. The Eg values of the Mg/ZnO nanocomposites slightly increased as a function of the amount of Mg2+. Nevertheless, there appeared to be a lot of bulky sheet-crystals on the surface of the Cu–5Mg/ZnO catalyst (Fig. 3(f)), which might have congregated due to the excessive amount of Mg2+ dopant.36
 | ||
| Fig. 3 SEM and SEM-EDS mapping images of the reduced Cu–xMg/ZnO samples: (a) Cu–0Mg/ZnO, (b) Cu–1Mg/ZnO, (c) Cu–2Mg/ZnO, (d) Cu–3Mg/ZnO, (e) Cu–4Mg/ZnO and (f) Cu–5Mg/ZnO. (g) SEM-EDS mapping and EDS analysis images of the Cu–4Mg/ZnO sample. | ||
The chemical compositions of the as-prepared catalysts were analyzed using energy-dispersive spectroscopy (SEM-EDS). Fig. 3(g) shows the chemical composition of the Cu–4Mg/ZnO catalyst, with Zn, Cu, Mg and O present in different positions of the nanocrystals. The inner surface is considerably depleted of Cu and Mg species, but is rich in Zn species. In contrast, the Mg and Cu species accumulated preferentially on the aggregated nano-plates surface. The varying of the surface chemical composition via the quantity of Mg2+ dopant further confirmed that the resultant catalysts mainly consist of two different crystalline phases originating from different precursors decomposing upon calcination.
TEM images of the reduced Cu–xMg/ZnO catalysts with different amounts of Mg2+ dopant are shown in Fig. 4. It is found from Fig. 4(a) that light gray spherical ZnO NPs are identified along with obvious copper aggregation in the Cu–0Mg/ZnO catalyst. Notably, a trace amount of Mg2+ dopant can effectively promote Cu dispersion, which improves gradually when the Mg2+ concentration increases until 4.0 wt% MgO loading. Nonetheless, excessive 5.0 wt% MgO loading adversely causes serious sintering of Cu species, as shown in Fig. 4(f). Additionally, Cu metal and ZnO are quite separately distributed, while the Mg species is rather uniformly distributed on both Cu metal and ZnO. The calculated results from the line width in XRD (vide infra) showed that the average size of the Cu metal particles on the catalysts is less than 15.0 nm, whereas that of ZnO is 10.0–15.0 nm in all of the samples tested. Therefore, it is likely that small-sized Cu metal particles are located on nanoscale ZnO crystals and Mg species are distributed on or in both Cu metal and ZnO particles.
 | ||
| Fig. 4 TEM images of the reduced Cu–xMg/ZnO catalysts: (a) Cu–0Mg/ZnO, (b) Cu–1Mg/ZnO, (c) Cu–2Mg/ZnO, (d) Cu–3Mg/ZnO, (e) Cu–4Mg/ZnO and (f) Cu–5Mg/ZnO. | ||
3.2 Chemical composition and porosity
The amount of Cu and Mg in the as-synthesized samples is summarized in Table 1. The actual Cu content is slightly lower than the preset value of the designed composition, indicating the incomplete precipitation of the Cu species. The BET surface area and pore volume is summarized in Table 1. It is found that the Cu–0Mg/ZnO possesses a relatively low BET surface area of 50.3 m2 g−1 and pore volume of 0.21 cm3 g−1. It is worth noting that doping Mg2+ ions corresponding to a 1.0 wt% MgO loading gives rise to an increased BET surface area of 74.7 m2 g−1, but an indistinctive loss of pore volume to 0.19 cm3 g−1. However, they increased gradually with more Mg2+ dopants, and reached the maximum values of 93.1 m2 g−1 and 0.27 cm3 g−1, respectively, with 4.0 wt% MgO loading. The reason for the increase in surface area should be closely related to formation of a ZnO framework with the Mg2+ dopant, as shown in Fig. 3.37 However, excessive 5.0 wt% MgO loading causes the BET surface area and pore volume to dramatically drop. From the N2 adsorption–desorption isotherms (Fig. 5(A)), it is found that all of the samples display the type IV with a H4-type hysteresis loop, suggesting mesoporous structures in the as-synthesized samples. Additionally, a sudden steep increase in nitrogen uptake in the characteristic relative pressure (P/P0) range of 0.60–0.95 for all of the samples is indicative of the formation of a typical mesoporous structure with uniform pore diameters.38 Fig. 5(B) illustrates the pore size distribution of the samples. The Cu–0Mg/ZnO sample exhibits the highest peak intensity at about 14.5 nm, while that of the Cu–xMg/ZnO samples drops gradually to 8.4 nm when the MgO loading increases from 1.0 wt% to 4.0 wt%. Nevertheless, a 5.0 wt% MgO loading results in a larger pores structure generated at the expense of partial micropores, indicating that excessive Mg2+ doping over-promotes the agglomeration of composite NPs. As a result, the BET surface area of the Cu–5Mg/ZnO dropped sharply, but the pore volumes became enlarged. The reason should be that Mg2+ ions, which have the same valence state and similar ionic radius to the Zn2+ species, tend to interact with ZnO to modulate the arrangement of the composite NPs. The Cu dispersion measured using the N2O titration method is shown in Table 1. Although a trace amount of Mg2+ dopant contributes to the enhanced BET surface area, the Cu dispersion still declined from 17.5% to 12.3% with MgO loading increasing from 1.0 wt% to 5.0 wt%. Based on the above results, it is deduced that a trace amount of Mg2+ dopant in the Cu/ZnO system can induce a significant impact on the structure evolution of the resultant catalysts.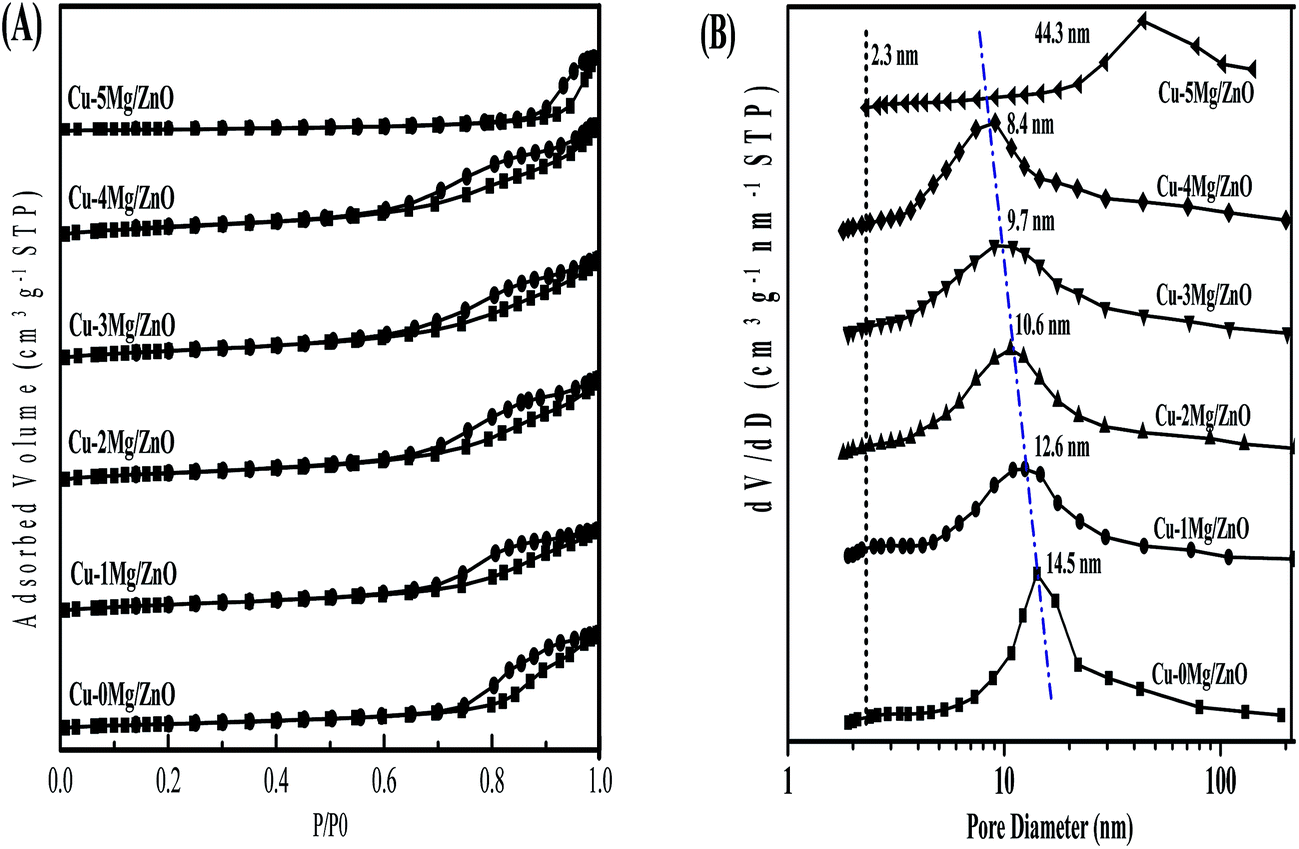 | ||
| Fig. 5 N2 adsorption–desorption isotherms (A) and pore size distribution curves (B) calculated using the BJH equation in the desorption branch of the samples. | ||
3.3 Reduction behavior of the calcined samples
H2 temperature-programmed reduction (H2-TPR), a very useful tool for exploring metal–metal and metal–support interactions, is used to gain insight into the promotion effect of the Mg2+ dopant on the reducibility of the copper species. From Fig. 6, it is found that the CuO–0Mg/ZnO catalyst features a weak tailing at the low-temperature side of the symmetrical main peak located at 230 °C, corresponding to the reducing of highly dispersed isolated and anchored CuO NPs.39 After being modified by 1.0 wt% MgO additives, the hydrogen consumption peak at 230 °C became weakened with a new distinct reduction peak emerging at 262 °C, indicating two types of component in the CuO–xMg/ZnO catalysts. Further increasing the MgO loading caused the main hydrogen consumption peak to shift to the lower temperature, due to more surface CuO species being accessible to reduce gas promoted by Mg2+ dopant.40 Nonetheless, the initial reduction temperature of the CuO–5Mg/ZnO is observed at 356 °C, which is probably attributed to the strengthened MSI and limited CuO surface area exposed to H2. Based on the above results, it is deduced that the Mg2+ dopant can not only act as structure promoter to affect the Cu dispersion, but also can regulate the chemical interaction between the Cu species and Zn–Mg substrate.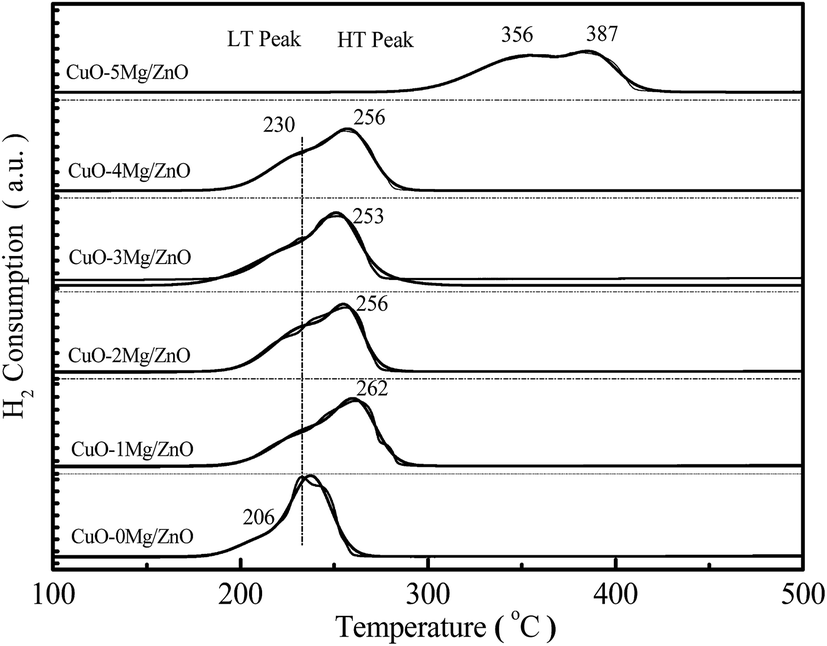 | ||
| Fig. 6 H2-TPR profiles of the calcined CuO–xMg/ZnO catalysts. | ||
3.4 Surface basicity of the pre-reduced samples
To gain more insight into the influence of the Mg2+ dopant on the surface basicity of the as-synthesized samples, CO2-TPD experiments were conducted. As shown in Fig. 7, all of the samples display two distinct CO2 desorption peaks positioned in different temperatures regions: α peak (T ≤ 400 °C) and β peak (T ≥ 400 °C). The α peaks should correspond to desorption of CO2 adsorbed on the surface Cu0 sites,41 and the β peaks should be associated with CO2 desorption from the basic sites afforded by the low-coordination surface O2− (Zn–O) sites.42,43 Obviously, the intensity of β peaks became gradually strengthened with an increase in the amount of Mg2+ ions until 4.0 wt% MgO loading, and then reduced. The surface Mg2+ ions should be responsible for the increased surface O2− sites, but an excessive amount of Mg2+ ions led to partial surface O2− sites being blocked and a decreased specific surface area.44 Additionally, the intensity of the α peaks exhibit a similar trend to that of the β peaks. Most of all, the Cu–4Mg/ZnO exhibits the strongest β desorption peaks. Thus, it is deduced that the Mg2+ dopant can regulate both the strength and density of the surface basic sites in Cu–xMg/ZnO.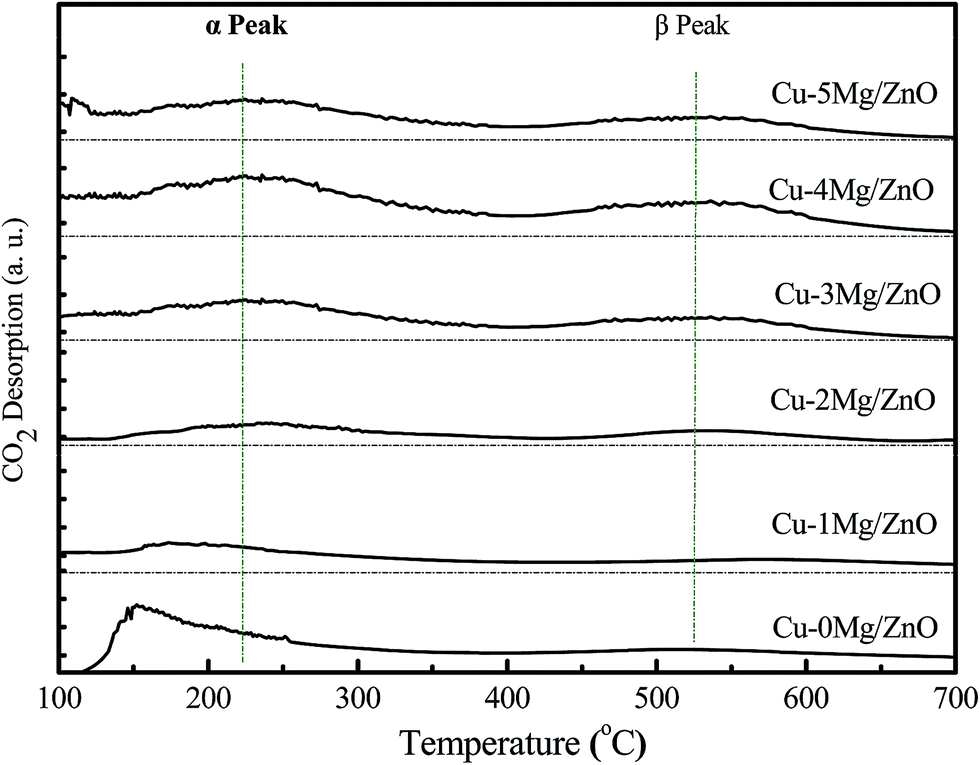 | ||
| Fig. 7 CO2-TPD profiles of the reduced Cu–xMg/ZnO catalysts. | ||
3.5 The FT-IR spectra of the pre-reduced catalysts
FT-IR is a technique used to explore the chemical bonding in a material. The band positions and numbers of the absorption peaks are dependent on crystalline structure, chemical composition and also on morphology.45 The characteristic peaks exhibited by the FT-IR spectra of the Cu–xMg/ZnO catalysts are shown in Fig. 8(A). The main, broad absorption bands around 2700–3500 and 1092 cm−1 are attributed to normal polymeric O–H stretching vibrations of H2O in the Cu–Zn–O lattice.46 Another absorption band at 1630 cm−1 is assigned to the H–O–H bending vibration, originating from H2O molecules absorbed by the nanocomposite. The three absorption bands at around 492, 1388 and 1458 cm−1 should correspond to the Zn–O bond.47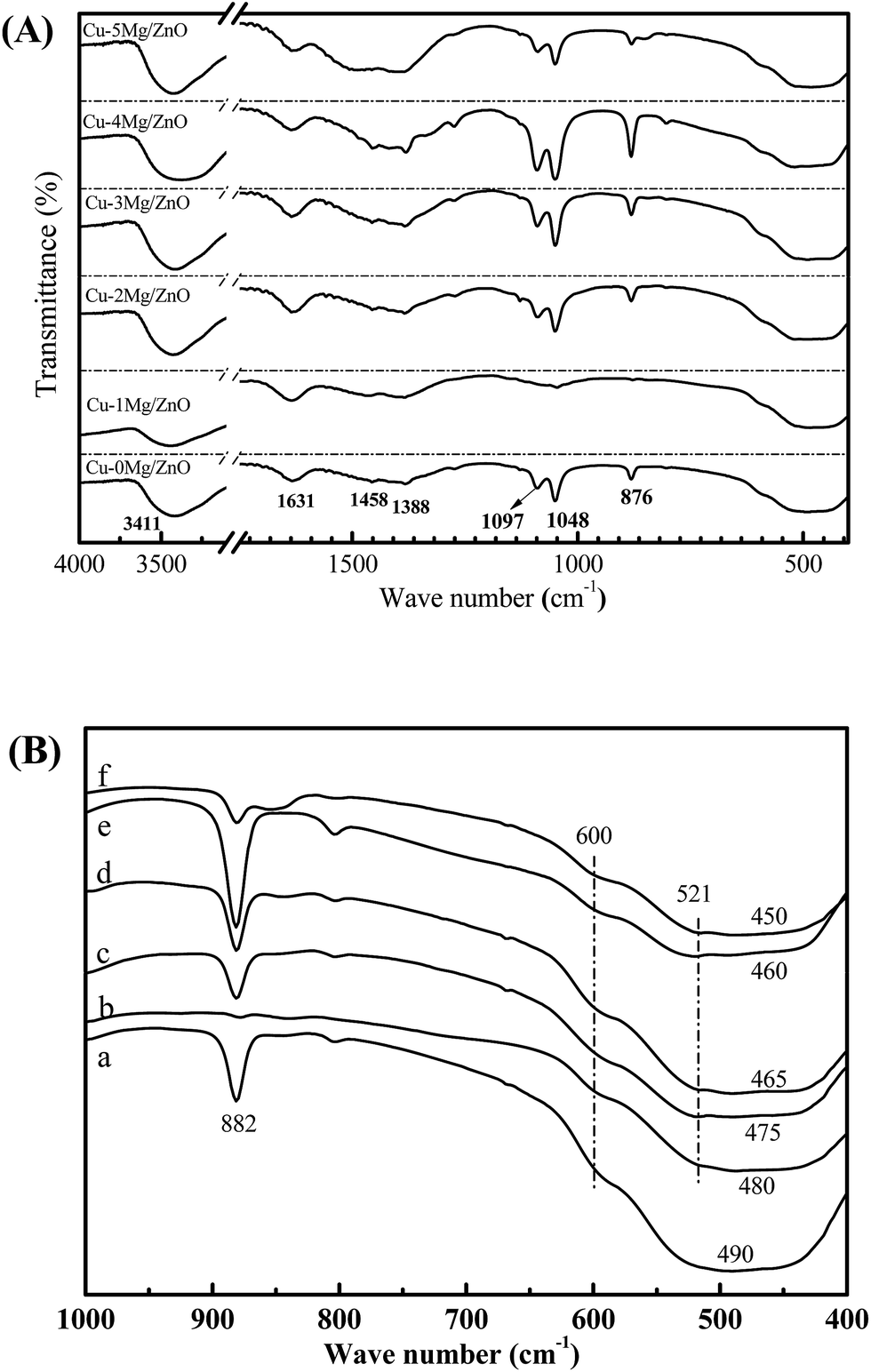 | ||
| Fig. 8 The FT-IR spectra of the reduced Cu–xMg/ZnO samples: (a) Cu–0Mg/ZnO, (b) Cu–1Mg/ZnO, (c) Cu–2Mg/ZnO, (d) Cu–3Mg/ZnO, (e) Cu–4Mg/ZnO and (f) Cu–5Mg/ZnO. | ||
The characteristic FT-IR peaks below 1000 cm−1 are very important to study the presence or absence of Zn–O/Cu–O/Mg–O bonds and the functional groups, and the results are given in Fig. 8(B). Absorption bands observed in the range 420–680 cm−1 are attributed to the stretching modes of the Zn–Cu–O bond. A weak absorption peak at around 500 cm−1 corresponding to the octahedral co-ordination and a strong peak at around 600 cm−1 assigned to the tetrahedral co-ordination reveal the strong tetrahedral orientation of the Zn–Cu–O bond.48 With Mg2+ doped into the Cu/ZnO catalysts, it is noted that an additional weak band appeared at 521.4 cm−1, which can be ascribed to the Mg–O–Zn bond. Therefore, the evolution of the diagnostic bands of ZnO should be apparently associated with the substitution of Mg2+ for Zn2+.49 Additionally, a medium weak band at 882 cm−1 assigned to defects vanished in the presence of the Mg2+ dopant, but became gradually strengthened with more Mg2+ ions embedded, suggesting the creation of more defects with a sufficient amount of Mg2+ dopant.48 The results are well supported by CO2-TPD and optical studies. Therefore, the evolution of the diagnostic bands of ZnO in our work is apparently associated with the substitution of Mg2+ for Zn2+.
3.6 Surface chemical states of the activated composites
In order to explore the surface chemical states of the Cu, Zn and Mg species in the reduced catalysts, XPS spectra of the reduced catalysts are illustrated in Fig. 9. As shown in Fig. 9(A), the intensive photoelectron peaks of the samples at 932.6 eV and 952.3 eV correspond to the binding energy (BE) of Cu 2p3/2 and Cu 2p1/2, respectively. There are not any satellite peaks between 942 and 944 eV ascribed to the Cu2+ species, implying the complete reduction of Cu2+ to Cu+ or/and Cu0.1 In terms of the Zn species, it is from Fig. 9(B) that the binding energies of Zn (2p2/3) for the Cu–xMg/ZnO catalysts are identified at 1021.7 and 1019 eV, respectively. In addition, the peak positions of the Zn 2p3/2 and Zn 2p1/2 states match closely with the standard values for ZnO, implying that Zn atoms are in the +2 oxidation state.49 What is more, the shift of the Zn 2p3/2 and Zn 2p1/2 peak further confirms that the Zn2+ species is partially substituted by Mg2+, being in the form of a Zn–Mg oxide solid solution.49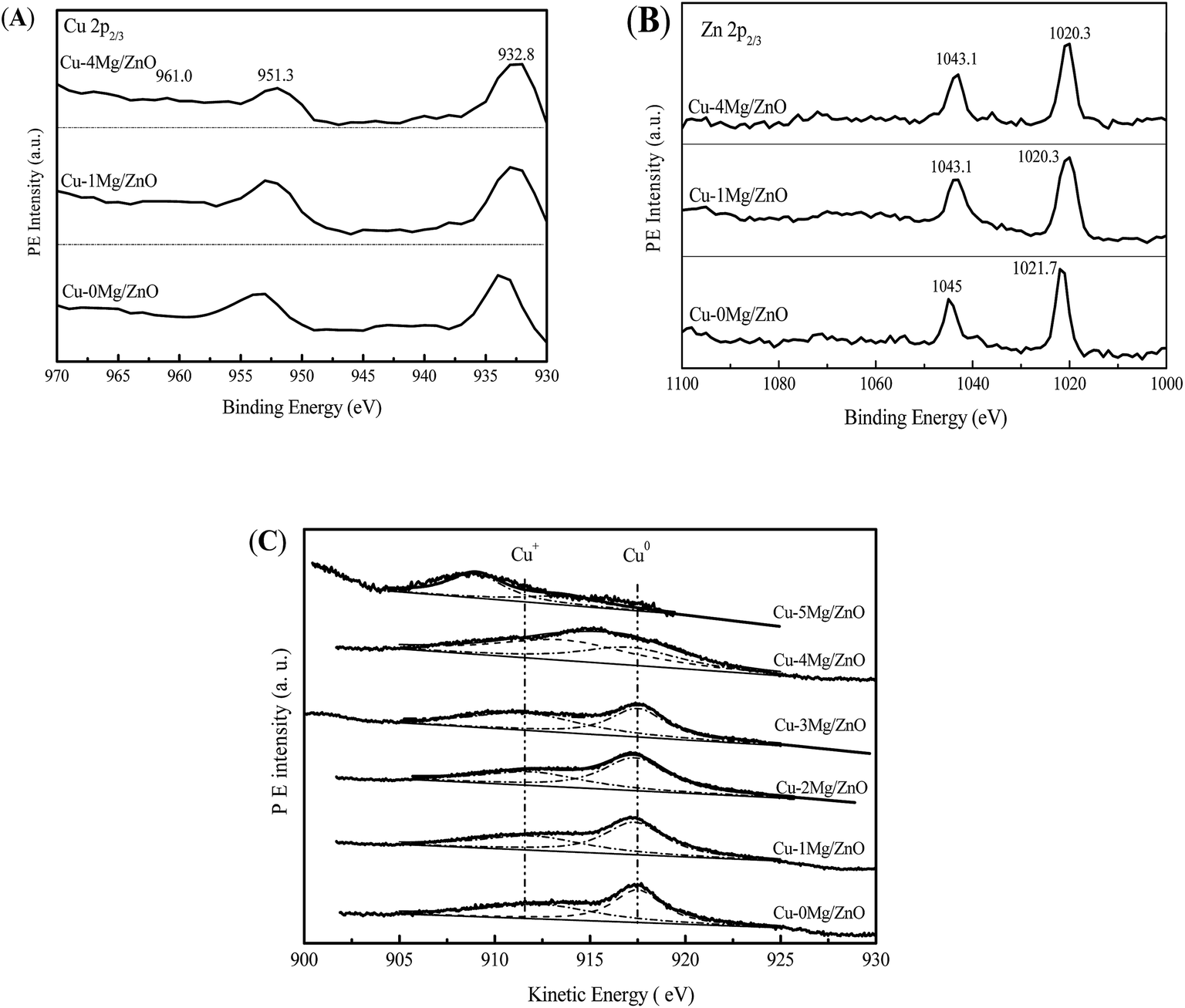 | ||
| Fig. 9 Cu 2p (A) and Zn 2p (B) XPS spectra and Cu LMM spectra (C) of the pre-reduced catalysts. | ||
Furthermore, it is quite difficult to distinguish the Cu+ and Cu0 only from the Cu 2p3/2 spectra because of the overlapping BE ranges for these two states. Generally, asymmetric and broad Auger kinetic energy peaks are observed and devolved into two symmetrical peaks located at around 916 and 918 eV in the Cu LMM spectra corresponding to Cu+ and Cu0 species, respectively. The simultaneous existence of Cu+ and Cu0 species in the Cu/ZnO based catalysts has been extensively discussed.1 It is obvious that the Cu+/Cu0 ratio of the Cu species increases with the Mg2+ concentration increasing from 1.0 wt% to 4.0 wt%, but is reduced with an excessive amount of Mg2+ dopant (5.0 wt%). Most of all, Cu–4Mg/ZnO possesses the maximum Cu+/Cu0 ratio of 55.2%, which is higher than that of other catalysts, as shown in Fig. 9(C) and Table 2. These results can be explained in terms of a relatively high affinity of the Mg–Zn solid solution for Cu NPs, and this interaction leads to a lower degree of reduction of surface copper and a partial positive charge on the copper surfaces.
| Catalysts | K.E.a (eV) | B.E. of Cu 2p3/2b (eV) | Cu+c (%) | SCu+ (m2 g−1) | |
|---|---|---|---|---|---|
| Cu+ | Cu0 | ||||
| a Kinetic energy.b Binding Energy.c Intensity ratio between Cu+ and (Cu+ + Cu0) by deconvolution of Cu LMM spectra. | |||||
| Cu–0Mg/ZnO | 916.2 | 918.6 | 933.9 | 38.6 | 1.51 |
| Cu–1Mg/ZnO | 915.7 | 918.1 | 932.8 | 42.5 | 1.81 |
| Cu–2Mg/ZnO | 915.2 | 917.7 | 932.8 | 46.4 | 2.35 |
| Cu–3Mg/ZnO | 914.5 | 917.0 | 932.8 | 49.6 | 2.51 |
| Cu–4Mg/ZnO | 913.7 | 916.1 | 932.8 | 55.2 | 2.70 |
| Cu–5Mg/ZnO | 909.1 | 914.0 | 932.0 | 40.4 | 1.82 |
3.7 Catalytic behavior
It is well known that the DMO vapor-phase hydrogenation process comprises several continuous reactions, including DMO hydrogenation to methyl glycolate (MG), MG sequential hydrogenation to EG, and EG deep hydrogenation to ethanol. Meanwhile, the byproducts of 1,2-butanediol (1,2-BDO) and 1,2-propanediol (1,2-PDO) can be synthesized by the dehydration reaction between EG and ethanol or methanol, however, this decreases the EG yield.1 The catalytic activity of the Cu–0Mg/ZnO and Mg2+-modified Cu–xMg/ZnO catalysts for the DMO hydrogenation reaction was evaluated, and the results are shown in Fig. 10(A). The Cu–0Mg/ZnO catalyst exhibits a 90% DMO conversion and a relatively low selectivity of 75% to EG. Notably, doping a trace amount of Mg species into Cu/ZnO can effectively improve the catalytic activity, which shows inverse S-type catalytic behavior in terms of EG selectivity in the range of 1.0–5.0 wt% MgO loading. Among the as-synthesized samples, the Cu–4Mg/ZnO sample affords the highest EG selectivity of 95.0% under the identical reaction conditions. Meanwhile, a trace amount of diol byproducts (1,2-propanediol and 1,2-butanediol) was also detected, due to the Guerbet reaction on the surface basic sites.38 What is more, the catalytic activity of the Cu–4Mg/ZnO catalysts is comparable with that of CuZnAl16,17 and CuZnGa18 catalysts reported in previous papers.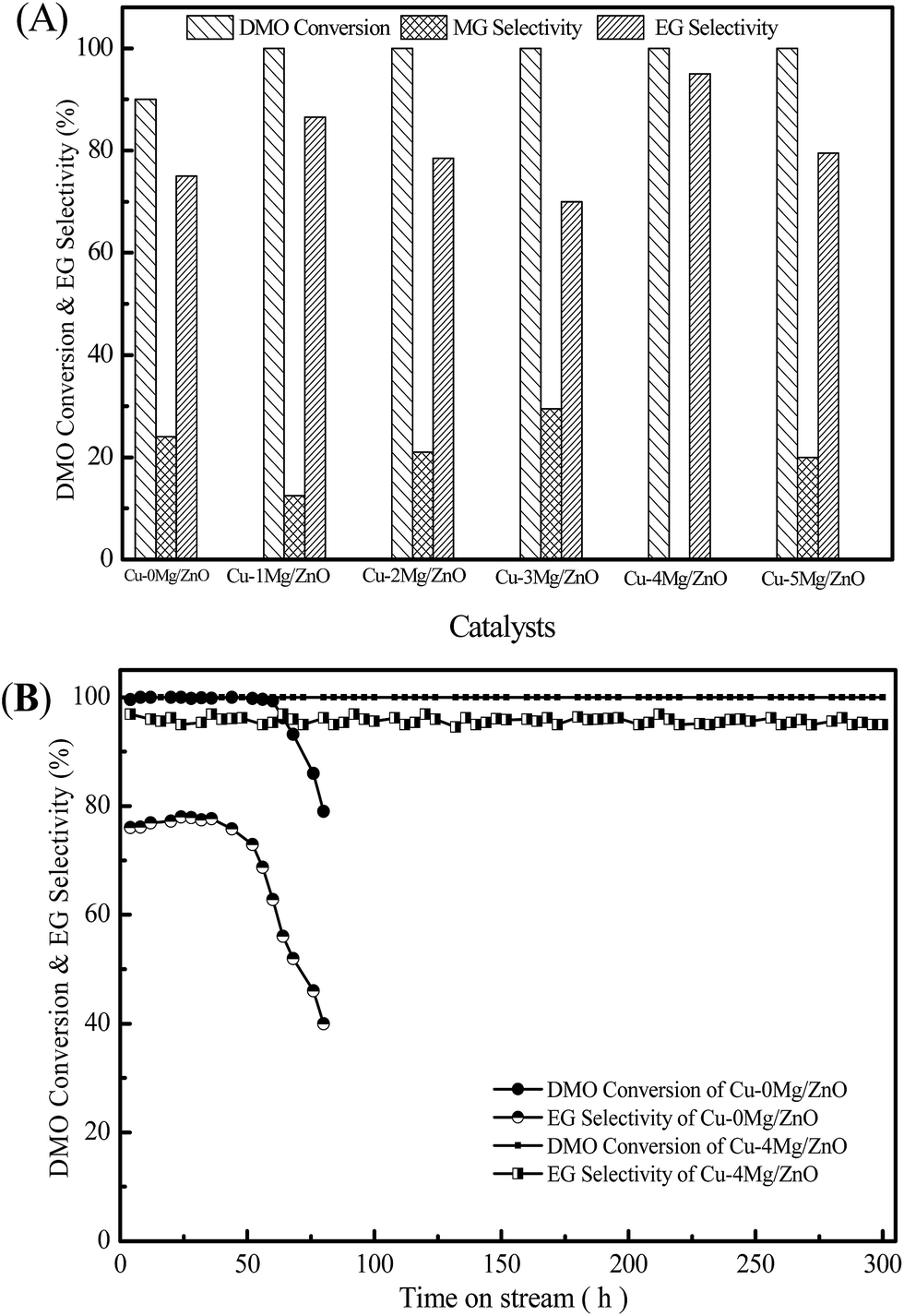 | ||
| Fig. 10 The catalytic activity of the Cu–xMg/ZnO catalysts (A), and the stability test of the Cu–0Mg/ZnO and Cu–4Mg/ZnO samples (B). Reaction conditions: reaction temperature = 220 °C, H2/DMO molar ratio = 100, P = 2.5 MPa, and LHSV = 2.0 h−1. | ||
Fig. 10(B) shows the stability test of the Cu–0Mg/ZnO and Cu–4Mg/ZnO catalysts as a function of reaction time. The activity of the Cu–4Mg/ZnO sample can be maintained for at least 300 h, demonstrating its excellent stability. In contrast, there is obvious activity loss of the Cu–0Mg/ZnO catalyst within 30 h under the identical reaction conditions.
4. Discussion
4.1 The evolution of structural and surface properties
The above characterizations depicted the evolution of microstructures and the physicochemical and surface properties of the construction of ternary functional components upon co-precipitation, calcination and reduction. In particular, these characteristics describe the critical dependency of the final precursor chemistry on the proportion of Mg2+ dopant. In the thermal process, the compounds in the precursors are transformed to the monoxides, Cu, and Zn species, but their different crystal structures mean that they are very poorly dissolvable in each other.50 The nano-particles of the oxides, mostly CuO and ZnO, maintain their porous nano-structures by “chemical memory” during heat thermal treatment.51 Scheme 1 summarizes the main structural evolutions of the ternary active phases (CuO, ZnO and Mg2+) via the Mg2+ content. The CZM0-p precursor mainly consists of a crystalline aurichalcite structure. Nanoscale CuO particles were formed upon the decomposition of crystalline aurichalcite and they are highly dispersed, thus, the reduction temperature is relatively low, but the amount of surface active Cu sites is relatively limited due to the Cu NPs being partially covered or encapsulated by the Zn–Mg oxide solid solution, as shown in Fig. 3 and 4. With 1.0 wt% MgO loading introduced, the Mg2+ ions not only can effectively improve the aurichalcite dispersion, but also promote the synthesis of a zincian malachite structure. After being calcined, the Mg2+ dopant induced the surface enrichment of the Cu species in the resultant catalysts. The assembling of the Cu species over the surface caused the growth of the Cu NPs and retarded the reduction process, but the surface active Cu species still increased. The increased surface Cu area derived from a higher proportion of the zincian malachite structure confirms the idea that a highly Zn-substituted form of zincian malachite is the relevant precursor phase for the preparation of Cu/ZnO catalysts with large Cu surface areas.52 Further increasing the Mg2+ concentration gives rise to the improved dispersion of the nano-compounds in the precursor, which should be helpful for the dispersion of the Cu species. This is possibly because the introduced Mg2+ dopants are easily absorbed on the surface of nano-compounds, which contributes to an enhanced BET surface area and the easy reduction of CuO NPs. Nonetheless, the monophase zincian malachite phase with excessive 5.0 wt% MgO loading leads to the Cu NP agglomeration dominating over the catalysts, adversely leading to a decreased amount of surface active Cu sites. Among the catalysts, the Cu–1Mg/ZnO catalyst has the maximum surface active Cu0 sites. What is more, Fig. 3 shows that there are at least two distinct types of Cu metal on the Mg2+ modified Cu–xMg/ZnO catalyst surface, presenting different morphologies and elemental compositions. The regime of higher Cu concentration may originate from the Cu-rich zincian malachite precursor, since the highest incorporation of Cu into ZnO is observed in the precursors containing the highest concentration of the Cu species.53 Multitudinous Cu species tend to agglomerate into coarser particles, which are a less efficient barrier against grain growth, resulting in a decrease of surface active Cu sites with an excessive amount of Mg2+ dopant.54 The synergistic effect of the ternary functional components (the Cu2+, Zn2+ and Mg2+ species) during the precipitation process should be responsible for the influence of Mg2+ on the surface Cu distribution and the decreased number of surface Cu sites exposed. The ZnO phase transformed in the sequence, single plates phase → agglomerated rods → biphase, and contained overlapped plates and agglomerated nano-rods when the MgO loading increased from 1.0 to 5.0 wt%. It should be noted that the formation and transformation of crystalline ZnO relies heavily on the amount of Mg2+ dopant in the catalysts. This is because the resultant form of ZnO is closely related to the disparate precursor and the strong interactions between the Cu NPs and Zn–Mg oxide solid solution.20 Thereby, a distribution within the common cationic sub-lattice of a single phase will be much more effective than the intermixing of individual Cu-rich and Zn-rich precursor phases, displaying a distribution on a microscopic level, but not on an atomic level.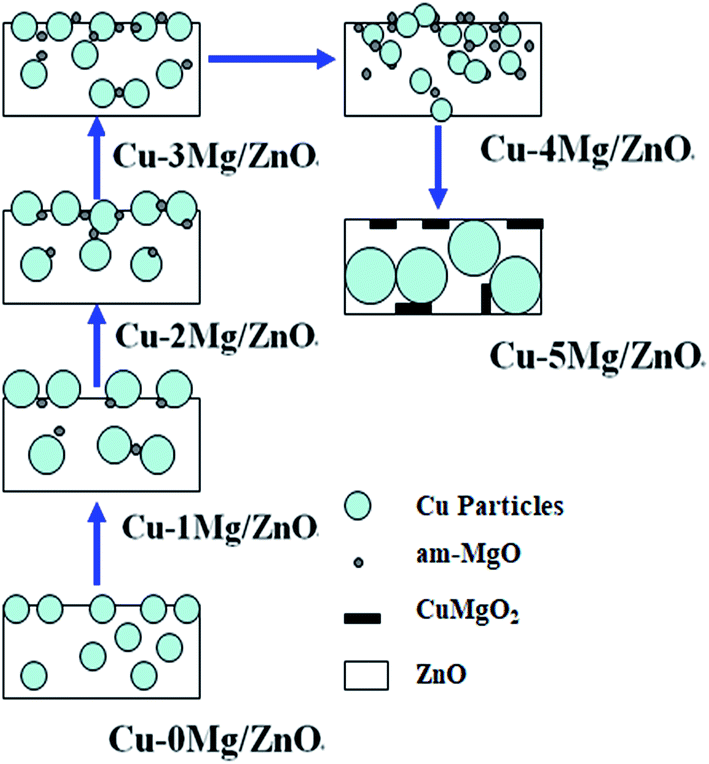 | ||
| Scheme 1 Structural evolutions of the ternary composites upon increasing the MgO loading. | ||
The surface basic properties are also associated with the Mg2+ concentration, as evidenced by the CO2-TPD results (Fig. 7). The density of surface basic sites increases when the MgO loading increases from 1.0 wt% to 4.0 wt%. The previous report identified that the 3-fold and 4-fold coordinated O2− anions on the ZnO surface facilitate the formation of surface Lewis basicity of the Cu/ZnO based catalysts.20 However, an excessive amount of Mg2+ ions caused the surface Lewis basicity of the ternary functional components to decrease adversely, which goes against the argument that the Mg2+ dopant is the main factor in determining the surface basic properties. Additionally, an increase in the amount of Mg2+ dopant led to reduced Cu particle sizes, but its contribution to the amount of surface active Cu sites is limited. Even with the smallest Cu NPs of 11.1 nm in the Cu–4Mg/ZnO catalyst, a small Cu surface area of about 2.45 m2 g−1, and consequently a poor degree of dispersion, was achieved. Presumably most of the Cu nanoparticles are located within the Zn2+ and Mg2+ matrix or are encapsulated due to the preparation by co-precipitation, which can be confirmed by Fig. 3 and 4.55 This conjecture is supported by the Cu NP sizes in the range from 11.1 nm to 13.3 nm, as derived from the Scherrer equation, which should lead to substantially larger specific Cu surface areas. Nonetheless, the Cu species in close proximity to ZnOx on the surface can also afford strong adsorption sites for oxygen-bound intermediates, leading to the partial coverage of the metal particles on ZnOx.56 So, it is inferred that the enhanced Cu–ZnO interface in presence of the Mg2+ dopant would be another reason for the increase in the amount of basic sites for the CO bonds. However, an excessive amount of Mg2+ dopants promote the formation of the CuMgO2 solid solution, inhibiting the synergistic effect of the ternary functional components.
Besides the effect of Mg2+ concentration on the surface basic properties, the surface chemical states of the Cu species were also changed with the variation in structural parameters. Notably, it can be argued from the Cu LMM spectra of the Cu–0Mg/ZnO samples that the Cu–ZnO interface originating from the Cu NPs located on the ZnO substrate can also promote Cu+ formation, as shown in Fig. 9(C) and Table 2. During the reduction process, partial Cu+ derived from Cu2+ would dissolve in the surface ZnO because Cu+ is isoelectronic with Zn2+, which benefits surface Cu+ formation, however, the solubility of Cu+ in ZnO is limited based on the requirement of electroneutrality.57,58 Also, no distinct binding energies of Cu 2p2/3 for the Cu–xMg/ZnO catalyst shift compared to those for the Cu–0Mg/ZnO catalyst until the concentration of the Mg2+ dopant corresponds to 4.0 wt% MgO loading. This suggests that no obvious electron transfer occurred in the Mg2+ doped Cu–xMg/ZnO (x = 1, 2, 3, 4). However, excessive 5.0 wt% MgO loading accelerates the shift of the energies of Cu 2p2/3 to the lower value of 914.0 eV, demonstrating the affinity for the high energy electrons of the Mg2+ dopant.59 As a result, the increase in the Cu–ZnO interface area and enhanced affinity for the electrons of the Zn–Mg solid solution with an increase in the amount of Mg2+ dopant incorporated promotes an increase in surface Cu+ formation, which is in accordance with previous reports.26,60,61 Furthermore, both the increased Cu–ZnO interface area and enhanced affinity for the electrons of the Zn–Mg substrate tend to strengthen the MSI, which can induce the reduction of surface Cu2+ species, as evidenced by the H2-TPR profiles (Fig. 6). Moreover, the strengthened MSI can stabilize the Cu NPs and effectively restrain the sintering.
From the above, the Mg2+ dopant tends to rearrange the dispersion of the Cu species, further regulating the density of the surface O2− centers located at the exposed Cu–ZnO interface. On the other hand, the chemical states and concentration of surface elements can be modulated by the Mg2+ dopant. Thus, the synergistic effect of the ternary functional components should be responsible for the structural evolution of the resultant catalysts.
4.2 The structure–performance relationships
The evolution of the microstructural and surface properties can be greatly modulated by the synergy of the Cu, Zn and Mg species, and thus, change the catalytic behaviors. The Cu–0Mg/ZnO catalyst had fewer surface active Cu0 sites exposed. After being modified by the Mg2+ dopant, it is obvious from Table 1 that the surface Cu0 sites increased significantly, while the surface Cu0 sites decreased gradually with more Mg2+ dopant introduced. Most of all, the Cu–1Mg/ZnO catalyst showed the maximum number of surface active Cu0 sites exposed, but Cu–4Mg/ZnO catalyst possesses the highest number of surface O2− sites and the highest Cu+ concentration (Fig. 7 and Table 2). In the DMO hydrogenation process, the abundance of exposed surface active surface Cu0 sites was proposed to be essential for the catalytic activity.1 Thus, the reactivity of the Cu–xMg/ZnO catalyst should decrease gradually when more Mg2+ species are incorporated, as should the yield of EG. As shown in Fig. 10, the EG selectivity of Cu–xMg/ZnO displayed a decreasing trend with MgO loading increasing from 1.0 wt% to 3.0 wt%, indicating that the activity is really well in line with the amount of surface active Cu0 sites exposed. However, Cu–4Mg/ZnO and Cu–5Mg/ZnO exhibited 95.0% and 80.0% yields of EG respectively, which is unexpectedly higher than the theoretical value corresponding to the active Cu0 sites. In particular, only pure copper or the ZnO phase is inactive in the C–O/CO selective hydrogenation.62 It is reasonable to conclude that the cascade reaction of DMO-to-EG is not only governed by a single parameter (surface Cu+/Cu0 sites), but rather by the synergistic concurrence of other factors. Thus, it is inferred that the synergistic effects between the surface Cu+/Cu0 sites and O2− centers should be responsible for the resultant catalytic activity of the Cu–xMg/ZnO catalysts in DMO hydrogenation.
In particular, the role of ZnO is of importance and seems to go beyond its function as a physical support or geometrical spacer. The strengthened MSI was discussed to contribute to the so-called synergy in the Cu–ZnO system. General mechanisms were accordingly proposed on the basis of the results, as shown in Scheme 2. During the DMO hydrogenation process, adsorbed H2 molecules can be dissociated on the Cu0 sites.63 Cu+ sites can act as electrophilic sites to polarize and activate the acyl group of the DMO reactant, and thus co-operate with the Cu0 sites to facilitate DMO selective hydrogenation to EG by the Cu–xMg/ZnO catalysts.64 Moreover, the dissociative adsorption of the ester molecules was proposed to be the rate-controlling step in the hydrogenation of DMO over the Cu based catalyst.65 Although a higher proportion of the Mg2+ dopant induces a detrimental effect on the concentration of surface Cu0 sites, the XPS data indicated that Mg2+ embed in the ZnO lattice can be helpful for polarizing the Cu species and facilitating the production of more surface Cu+ sites. On the other hand, increasing the amount of Mg2+ ions submitted in the Cu/ZnO system gives rise to an increased Cu/ZnO interface area and more surface O2− sites in the Cu–4Mg/ZnO and Cu–5Mg/ZnO catalysts. Moreover, the surface O2− sites, presumably located at the interface region between the Cu and ZnO species, can also act as active sites for substrate dissociation in the ester hydrogenation process.66 If the Cu NPs are near the surface O2− sites or the Cu–ZnO interface, then the activated substrate on the defects can spill over on to the nearby copper particles to react with the activated hydrogen on the Cu0 sites, leading to enhanced activity.67 Frost et al.68 established that the active sites of CO hydrogenation over ZrO2 or Y2O3-doped ZrO2 could result from oxygen anion vacancies, in which the higher mobility of O2− vacancies represents a higher reaction rate. On the condition of the absence of copper, a catalyst promoted by silver and gold oxides also possessed a high activity for CO activation, in which the higher mobility of oxygen vacancies represents a higher action ability. These studies discerned that the active site situated in the boundary between the Cu cluster and the support is another vital parameter for CO activation.69
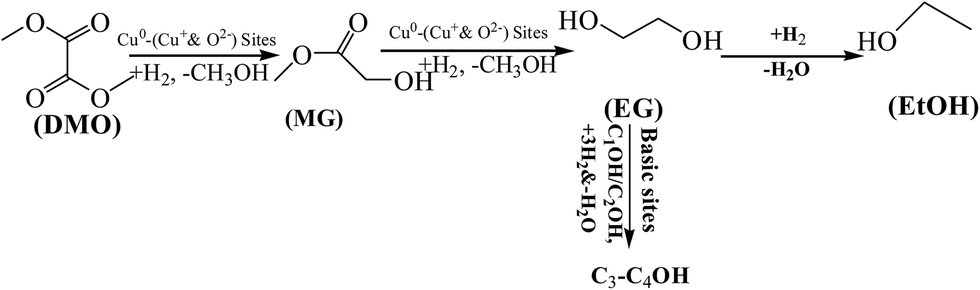 | ||
| Scheme 2 Reaction pathways for the conversion of DMO over the Cu–xMg/ZnO catalysts in H2 flow. | ||
The strengthened MSI in the presence of Mg2+ dopants is discussed to contribute to the so-called synergy in the Cu/ZnO system.52 The analysis of the fresh catalysts and used catalysts after lifetime evaluation (Cu–0Mg/ZnO and Cu–4Mg/ZnO) was conducted. The results show that there is no obvious particle size change of the Cu NPs in the used Cu–4Mg/ZnO compared to that of the fresh catalyst, confirming that the Mg2+ dopant can effectively stabilize Cu NPs owing to the strengthened MSI between the Cu0 and Mg–Zn spacer.70 Nonetheless, the Cu species in the used Cu–0Mg/ZnO catalyst existed only in the form of Cu2O NPs, revealing that the agglomeration of the Cu2O particles occurred during the hydrogenation process. Additionally, serious leaching of Cu0 occurred for the used Cu–0Mg/ZnO, which should be caused by the weak MSI. Furthermore, the Mg2+ dopant was proposed to effectively hinder the dissolution of the ZnO NPs by restricting the phase transfer reaction, which contributes to the excellent stability.
5. Conclusions
In conclusion, porous nanoscale Cu–Mg/ZnO catalysts were successfully prepared using the facile and controllable co-precipitation method. The Mg2+ dopant was found to promote both aurichalcite and zincian malachite structures as the relevant precursor phases, further affecting the physicochemical properties of the resultant catalysts by the chemical memory of the system. In the gas-phase hydrogenation of DMO to EG, doping Mg2+ into the Cu/ZnO system can significantly improve its activity and stability. Over the Cu–4Mg/ZnO catalyst, 100% DMO conversion and 95% EG yield was obtained for longer than 300 h under the optimized hydrogenation conditions. In the Cu–Mg/ZnO system, Mg2+ ions are highly dispersive and successfully incorporated into the lattice of ZnO, facilitating the synergistic effect of the Cu, Zn and Mg species. The excellent catalytic behavior is mainly attributed to the appropriate surface Cu0 sites and abundant surface Cu+ and O2− sites. What is more, the strengthened MSI originating from the enhanced Cu/ZnO interface in the ternary assembly (Cu, Zn, Mg species) contributed to the enhanced stability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the State Key Laboratory of Coal Conversion, National Natural Science Foundation of China (Grant No. 21373254, No. 21503256 and No 51604180), the Research Project supported by the Shanxi Scholarship Council of China (Grant No. 2016-114) and the Primary Research & Developement Plan of Shanxi Province (No. 201603D421038).References
- Y. Zhao, S. Li, Y. Wang, B. Shan, J. Zhang, S. Wang and X. Ma, Chem. Eng. J., 2017, 313, 759–768 CrossRef CAS.
- R. P. Ye, L. Lin, J. X. Yang, M. L. Sun, F. Li, B. Li and Y. G. Yao, J. Catal., 2017, 350, 122–132 CrossRef CAS.
- Y. Zhao, Y. Zhang, Y. Wang, J. Zhang, Y. Xu, S. Wang and X. Ma, Appl. Catal., A, 2017, 539, 59–69 CrossRef CAS.
- H. D. Dombek, J. Chem. Educ., 1986, 63, 210–212 CrossRef.
- Y. Tomotake, T. Matsuzaki, K. Murayama, E. Watanabe, K. Wada and T. Onoda, J. Organomet. Chem., 1987, 320, 239–247 CrossRef CAS.
- J. He, Y. Zhao, Y. Wang, J. Wang, J. Zheng, H. Zhang, G. Zhou, C. Wang, S. Wang and X. Ma, Chem. Commun., 2017, 53, 5376–5379 RSC.
- Y. Wang, Y. Shen, Y. Zhao, J. Lv, S. Wang and X. Ma, ACS Catal., 2015, 5, 6200–6208 CrossRef CAS.
- P. Ai, M. Tan, Y. Ishikuro, Y. Hosoi, G. Yang, Y. Yoneyama and N. Tsubaki, ChemCatChem, 2017, 9, 1–10 CrossRef.
- L. H. Yu, W. Chen, D. Z. Li, J. B. Wang, Y. Shao, M. He, P. Wang and X. Z. Zheng, Appl. Catal., B, 2015, 164, 453–461 CrossRef CAS.
- C. H. Kwak, H. S. Woo, F. Abdel-Hady, A. A. Wazzan and J. H. Lee, Sens. Actuators, B, 2016, 223, 527–534 CrossRef CAS.
- X. Bai, L. Wang, R. Zong, Y. Lv, Y. Sun and Y. Zhu, Langmuir, 2013, 29, 3097–3105 CrossRef CAS PubMed.
- Ü. Özgür, Y. I. Alivov, C. Liu, A. Teke, M. A. Reshchikov, S. Doğan, V. Avrutin, S. J. Cho and H. Morkoç, J. Appl. Phys., 2005, 98, 041301 CrossRef.
- H. Ren, C. H. Xu, H. Y. Zhao, Y. X. Wang, J. Liu and J. Y. Liu, J. Ind. Eng. Chem., 2015, 28, 261–267 CrossRef CAS.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS PubMed.
- Z. Lu, H. Yin, A. Wang, J. Hu, W. Xue, H. Yin and S. Liu, J. Ind. Eng. Chem., 2016, 37, 208–215 CrossRef CAS.
- S. Zhang, Q. Liu, G. Fan and F. Li, Catal. Lett., 2012, 142, 1121–1127 CrossRef CAS.
- C. Wen, F. Li, Y. Cui, W. L. Dai and K. Fan, Catal. Today, 2014, 233(13), 117–126 CrossRef CAS.
- M. M. J. Li, J. Zheng, J. Qu, F. Liao, E. Raine, W. C. H. Kuo, S. S. Su, P. Po, Y. Yuan and S. C. E. Tsang, Sci. Rep., 2016, 6, 20527–20534 CrossRef PubMed.
- J. Yang, Y. Wang, J. Kong and M. Yu, J. Alloys Compd., 2016, 657, 261–267 CrossRef CAS.
- S. Y. Peng, Z. N. Xu, Q. S. Chen, Z. Q. Wang, D. M. Lv, J. Sun, Y. Chen and G. C. Guo, ACS Catal., 2015, 5(7), 4410–4417 CrossRef CAS.
- B. M. Nagaraja, A. H. Padmasri, B. David Raju and K. S. Rama Rao, J. Mol. Catal. A: Chem., 2007, 265, 90–97 CrossRef CAS.
- S. Asthana, C. Samanta, A. Bhaumik, B. Banerjee, R. K. Voolapalli and B. Saha, J. Catal., 2016, 334, 89–101 CrossRef CAS.
- H. Yin, C. Zhang, H. Yin, D. Gao, L. Shen and A. Wang, Chem. Eng. J., 2016, 288, 332–343 CrossRef CAS.
- B. M. Nagaraja, V. Siva Kumar, V. Shasikala, A. H. Padmasri, B. Sreedhar, B. David Raju and K. S. Rama Rao, Catal. Commun., 2003, 4, 287–293 CrossRef CAS.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B. L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- T. Shishido, M. Yamamoto, I. Atake, D. Li, Y. Tian, H. Morioka, M. Honda, T. Sano and K. Takehira, J. Mol. Catal. A: Chem., 2006, 253, 270–278 CrossRef CAS.
- G. Zeng, Q. Liu, R. Gu, L. Zhang and Y. Li, Catal. Today, 2011, 178, 206–213 CrossRef CAS.
- S. Xia, Z. Yuan, L. Wang, P. Chen and Z. Hou, Appl. Catal., A, 2011, 403, 173–182 CrossRef CAS.
- M. M. Harding, B. M. Kariuki, R. Cernik and G. Cressey, Acta Crystallogr., Sect. B: Struct. Sci., 1994, 50, 673–676 Search PubMed.
- A. N. Mallika, A. R. Reddy, K. S. Babu, Ch. Sujatha and K. V. Reddy, Opt. Mater., 2014, 36, 879–884 CrossRef.
- J. B. Ries, Paleobiology, 2005, 31, 445–458 CrossRef.
- J. Yang, Y. Wang, J. Kong, M. Yu and H. Jin, J. Alloys Compd., 2016, 657, 261–267 CrossRef CAS.
- G. J. Millar, I. H. Holm, P. J. R. Uwins and J. Drennan, J. Chem. Soc., Faraday Trans., 1998, 94, 593–600 RSC.
- T. M. Yurieva, React. Kinet. Catal. Lett., 1995, 55(2), 513–521 CrossRef CAS.
- S. Klubnuan, P. Amornpitoksuk and S. Suwanboo, Mater. Sci. Semicond. Process., 2015, 39, 515–520 CrossRef CAS.
- B. Hu and K. Fujimoto, Appl. Catal., A, 2008, 346, 174–178 CrossRef CAS.
- R. K. Singha, A. Yadav, A. Agrawal, A. Shukla, S. Adak, T. Sasaki and R. Bal, Appl. Catal., B, 2016, 191, 165–178 CrossRef CAS.
- A. Yin, C. Wen, X. Guo, W. L. Dai and K. Fan, J. Catal., 2011, 280, 77–88 CrossRef CAS.
- Z. Lu, H. Yin, A. Wang, J. Hu, W. Xue, H. Yin and S. Liu, J. Ind. Eng. Chem., 2016, 37, 208–215 CrossRef CAS.
- S. Asthana, C. Samanta, A. Bhaumik, B. Banerjee, R. K. Voolapalli and B. Saha, J. Catal., 2016, 334, 89–101 CrossRef CAS.
- K. W. Jun, W. J. Shen, K. S. R. Rao and K. W. Lee, Appl. Catal., A, 1998, 174, 231–238 CrossRef CAS.
- L. J. I. Coleman, W. Epling, P. R. Hudgins and E. Croiset, Appl. Catal., A, 2009, 363, 52–63 CrossRef CAS.
- H. F. Wang, H. Ariga, R. Dowler, M. Sterrer and H. J. Freund, J. Catal., 2012, 286, 1–5 CrossRef CAS.
- T. M. Brenner, T. A. Flores, P. F. Ndione, E. P. Meinig, G. Chen, D. C. Olson, T. E. Furtak and R. T. Collins, J. Phys. Chem. C, 2014, 118, 12599–12607 CAS.
- Z. Yang, Z. Ye, Z. Xu and B. Zhao, Phys. E, 2009, 42(2), 116–119 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Parts-A and B, John Wiley & Sons, New York, 1997 Search PubMed.
- Y. Yang, Y. Yang, H. Wu and S. Guo, CrystEngComm, 2013, 15, 2608–2615 RSC.
- M. Ashokkumar and S. Muthukumaran, Opt. Mater., 2014, 37, 671–678 CrossRef CAS.
- X. Qiu, L. Li, J. Zheng, J. Liu, X. Sun and G. Li, J. Phys. Chem. C, 2008, 112, 12242–12248 CAS.
- D. M. Whittle, A. A. Mirzaei, J. S. J. Hargreaves, R. W. Joyner, C. J. Kiely, S. H. Taylor and G. J. Hutchings, Phys. Chem. Chem. Phys., 2002, 4, 5915–5920 RSC.
- P. Podbrscek, Z. C. Orel and J. Macek, Mater. Res. Bull., 2009, 44, 1642–1646 CrossRef CAS.
- M. Behrens, J. Catal., 2009, 267, 24–29 CrossRef CAS.
- B. Bems, M. Schur, A. Dassenoy, H. Junkes, D. Herein and R. Schlögl, Chem.–Eur. J., 2003, 9(9), 2039–2052 CrossRef CAS PubMed.
- J. Ďurišin, K. Ďurišinová, M. Orolínová and K. Saksl, Mater. Lett., 2004, 58(29), 3796–3801 CrossRef.
- J. Schittkowski, K. Tölle, S. Anke, S. Stürmer and M. Muhler, J. Catal., 2017, 352, 120–129 CrossRef CAS.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B. L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- M. Popovaa, M. Dimitrova, V. DalSantob, N. Ravasiob and N. Scottic, Catal. Commun., 2012, 17, 150–153 CrossRef.
- S. Zhang, Q. Hu, G. Fan and F. Li, Catal. Commun., 2013, 39, 96–101 CrossRef CAS.
- Z. He, H. Lin, P. He and Y. Yuan, J. Catal., 2011, 277, 54–63 CrossRef CAS.
- C. Liu, X. Guo, Q. Guo, D. Mao, J. Yu and G. Lu, J. Mol. Catal. A: Chem., 2016, 425, 86–93 CrossRef CAS.
- K. Nishida, D. Li, Y. Zhan, T. Shishido, Y. Oumi, T. Sano and K. Takehira, Appl. Clay Sci., 2009, 44, 211–217 CrossRef CAS.
- A. Karelovic, A. Bargibant, C. Fernández and P. Ruiz, Catal. Today, 2012, 197, 109–118 CrossRef CAS.
- D. J. Thomas, J. T. Wehrli, M. S. Wainwright and D. L. Trimm, Appl. Catal., A, 1992, 86, 101–114 CrossRef CAS.
- J. Gong, H. Yue, Y. Zhao, S. Zhao, L. Zhao, J. Lv, S. Wang and X. Ma, J. Am. Chem. Soc., 2012, 134, 13922–13925 CrossRef CAS PubMed.
- S. Li, Y. Wang, J. Zhang, S. Wang, Y. Xu, Y. Zhao and X. Ma, Ind. Eng. Chem. Res., 2015, 54, 1243–1250 CrossRef CAS.
- E. Strunk, K. Kähler, X. Xia, M. Comotti, F. Schüth, T. Reinecke and M. Muhler, Appl. Catal., A, 2009, 359, 121–128 CrossRef.
- A. Yin, X. Guo, W. L. Dai and K. Fan, J. Phys. Chem. C, 2010, 114, 8523–8532 CAS.
- J. C. Frost, Nature, 1988, 334, 577–590 CrossRef CAS.
- S. Zhang, J. Yu, H. Li, D. Mao and G. Lu, Sci. Rep., 2016, 6, 20527–20541 CrossRef PubMed.
- M. M. Villaverde, N. M. Bertero, T. F. Garetto and A. J. Marchi, Catal. Today, 2013, 213, 87–92 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |