
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Density functional theoretical studies on the ring-opening polymerization mechanism of oxetane cation series compounds†
Hai-Xia Wanga,
Min Pu *b and
Yu-Cheng Dingc
*b and
Yu-Cheng Dingc
aSchool of Science, Xi'an Jiaotong University, Xi'an 700049, P. R. China
bState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: pumin@mail.buct.edu.cn
cSchool of Mechanic Engineering, Xi'an Jiaotong University, Xi'an 700049, P. R. China
First published on 25th October 2017
Abstract
The mechanism of ring-opening polymerization of oxetane cation series compounds was investigated using the B3LYP and MP2 methods of density functional theory and ab initio methods, at the basis set levels of 6-31G(d,p) and 6-311++G(d,p). The geometrical parameters of the reactant, transition state, intermediate and product of a series of multi-polymer species in the reaction pathway were fully optimized. The structural changes of species in the reaction pathway are explained herein. The computing results show that the polymerization of oxetane is performed by the O atom of oxetane continuously attacking the C atom of the oxetane cation. The energy analysis of the reaction process shows that the acidized oxetane can easily polymerize with other oxetane molecules to form a copolymer, and the activation energy in the initial step is very low. The equilibrium and transition state characteristics of every stationary point in the reaction pathway were determined through vibrational analysis. The corresponding reactant and product of each transition state were verified according to the intrinsic reaction coordinates traced from the transition state of different hierarchical polymers. Finally, the solvent effects of tetrahydrofuran and dichloromethane are discussed herein based on the self-consistent reaction field theory.
1. Introduction
Oxetane is an important precursor for light polymerization in organic chemistry. It can be used as a monomer for UV cationic polymerization,1,2 a polymer electrolyte supporter,3 and can be used in coatings, sealants, adhesives and advanced ink products.4–6 Oxetane exhibits strong alkaline properties, and can be combined with an acidic hydrogen ion to initiate cationic ring-opening polymerization by photo-excitation. The polymerization shows a high reaction speed, low viscosity, high adhesive strength, small curing shrinkage rate, and low toxicity and volatility. The polymer product has good chemical resistance, heat stability and excellent mechanical properties. Therefore, research into the structure and properties of oxetane and its cation has attracted much attention in recent years.7–11Normally, a series of oxetane derivatives can be used as monomers of polymerization. The experimental processes are typically monitored using UV, FTIR and other means. The reactions are normally initiated using lasers of different wavelengths. Boron trifluoride hydrate, hexafluorophosphate, hexa-fluoroantimonate, aromatic diazonium salts, and aryl iodonium salts are common initiators.12–14 The reaction speeds are typically fast and it is often difficult to inhibit the reaction with oxygen once polymerization has started. Because the entire reaction speed in a continuous reaction process is determined by the slowest reaction step, if the light source is strong enough and the quantum yield of the reaction is fixed, the reaction speed depends on the reactivity of the monomer and the end site of the growing chain. Therefore, research into the reaction mechanism and the structure of the monomers in the light-cation ring-opening polymerization is particularly important in organic synthesis.
There has been some theoretical research carried out on the electronic structure of oxetane. Most work has been focused on calculating the structural properties of monomers and complexes,15–18 or the reaction mechanisms of pyrolysis19–21 and photolysis22–24 of oxetane. Ferreira et al.15 predicted the structural stability of a complex of the oxetane monomer with hydrogen halide and discussed the special nature of the weak interaction using density functional theory calculations at the B3LYP/6-311++G(d,p) level. Ottaviani et al.16 computed the electronic structure of oxetane hydrate and gave the geometry and main bond parameters by combining the jet millimeter wave spectrum with quantum chemical calculations. Earlier, Chen et al.19 explored the pyrolysis reaction mechanism of oxetane using a semi-empirical molecular orbital method, and indicated that the biradical reaction was the most interesting process in the variety of primitive reaction pathways. Recently, Tahan20 and Shiroudi21 determined that formaldehyde and an olefin were the main products of oxetane single molecule pyrolysis, and gave a high-pressure limit of the reaction rate constants according to computations using B3LYP/6-311+G**, B3PW91/6-311+G** and MPW1PW91/6-311+G**. Studies investigating the photolysis reaction mechanism in oxetane polymerization have gradually increased. Most research has been based on the Paterno–Buchi reaction. Palmer22 studied the photolysis reaction mechanism of oxetane using the MC-SCF method. Lee et al.23 reported UV photolysis molecular beam experiments with oxetane at 193.3 nm. Yang, et al.24 illustrated the phenomenon that azetidin is generated faster than oxetane in the (6–4) photolysis of 5-methyluracil using theoretical calculations.
The most important uses of oxetane and its derivatives are in the field of cationic photo-polymerization,25–29 since oxetane is the simplest molecule in the series of oxygen hetero-ring compounds. Understanding the single molecular polymerization mechanism of oxetane is very significant for explaining the more complex polymerization mechanisms of oxetane derivatives. In this paper, density functional theory and ab initio methods of quantum chemistry were used to study the mechanism of monomer polymerization of oxetane with its cation, and to determine the transition state of each step of the reaction. The aim of this work was to analyze and speculate the changes in atomic charges, frontier molecular orbitals and the interactions between different species, and finally, to reveal the microscopic mechanism of the series polymerization of oxetane.
2. Computational methods
Structure models of oxetane, its acidified cation and the polymer were established according to the reported experimental data.30 The geometries of the reactants, transition state and products were fully optimized by using the B3LYP and MP2 methods at the basis levels of 6-31G(d,p) and 6-311++G(d,p). The characteristics of the equilibrium and transition states were confirmed by the number of imaginary frequencies based on the vibrational analysis of the geometries; the zero-point energy (ZPE) correction was obtained at the same time. The intrinsic reaction coordinates (IRC) of the oxetane polymerization were calculated from the transition state to determine what the corresponding reactants and products were. The quantum chemical calculations of the oxetane molecular geometry and electronic structure were completed using the Gaussian 09 program,31 Mulliken atomic charge was taken from the Gaussian program results, and Becke, Hirshfeld and ADCH (Hirshfeld population included atomic dipole correction) atomic charge calculations were computed using Multiwfn program.32 The solvent effects on the mechanism were discussed based on the polarizable continuum model (PCM) of self-consistent reaction field (SCRF) theory. The molecular structure, molecular orbitals and transition state imaginary vibration modes were drawn using the HyperChem program,33 and the data from the Gaussian computation results were converted to HyperChem data format using the GTH program.343. Results and discussion
3.1 The analysis of structural features and reaction tendency of oxetane and its acidified cation
Oxetane (Ox) is a four-membered heterocyclic molecule with a saturated chemical bond. Fig. 1 gives the structural models of Ox and its protonated cation (Ox-H+). The tension of small molecule cyclic compounds is usually large, and the four-membered ring of oxetane obviously possesses this feature. The thermodynamic parameters observed when adding water to 1,3-propanediol may be explained by this. The C–C bond length of Ox remains constant at about 0.152–0.153 nm before and after the addition, while the C–C–C bond angle changes from 83° to 113°. The hydration heat of Ox is about −83 kJ mol−1. These results suggest that the ring-opening of oxetane takes place easily. The main bond parameters of oxetane were computed using the B3LYP/6-31G(d,p) and MP2/6-31G(d,p) methods. The main optimized geometric parameters of Ox and Ox-H+ are listed in the ESI.† It can be seen that the differences are small between the different computing levels, and the parameters of Ox are also close to those of the experimental data.30 The results also show that both density functional theory and ab initio methods are suitable for calculating the electronic structure of the Ox system. Because the atom numbers in the Ox polymer system increase as the reaction progresses, in order to explain the problem of inconsistencies in the calculation data, the B3LYP/6-31G(d,p) results were mainly used to discuss the polymerization changes in this paper.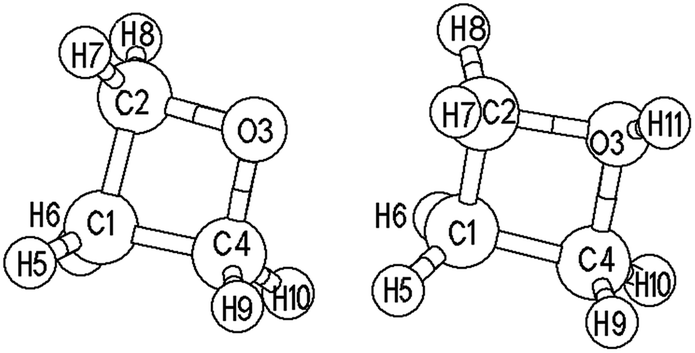 | ||
| Fig. 1 The structural models of oxetane (Ox) and its protonated ion (Ox-H+). | ||
One feature of the chemical reaction was the migration of electrons between different atoms, resulting in the rupture of old bonds and the building of new bonds. The atomic charge data, obtained using several different theoretical calculation methods using the basis set of 6-31G(d,p), are shown in the ESI.† The adaptability of the atomic charge calculation method can be evaluated according to the calculation results. The relative changes in several atomic charge values were basically fitted using the electronic gains and losses rule; however, the absolute values of the atomic charge showed large differences. Generally, it was considered that C and O atoms mostly have a negative charge, while the H atom has a positive charge. Comparing the calculation results of different methods, the absolute values of the Becke and ADCH atomic charge were better fitted to the chemical characteristics than those of the Mulliken and Hirshfeld atomic charge, which was also observed by Lu et al.35 in their atomic charge values. The changes in atomic charge value could explain the interaction of oxetane with its protonated ion to a certain extent; however, the scale and significance of the atomic charges may also need further investigation. The ADCH charge calculations showed that the O atom charge changes from −0.287 to −0.221 when Ox is combined with H+ to form Ox-H+, while the ADCH charge of the adjacent C and O atoms changes from −0.026 to −0.013. This could explain why O atoms of Ox would attack C atoms of Ox-H+ in the initial stage of the nucleophilic reaction. The reduction of the negative charge of the C atom of Ox or the electrostatic attraction may be due to the power of polymerization.
The regular geometries of oxetane and its protonated ion may possess C2v and Cs symmetry; however, the calculated self-consistent field energy of an unsymmetrical molecular structure is lower than that of a symmetrical structure. The symmetry restrictions of Ox and Ox-H+ were canceled in the subsequent computation. Graphics of the two highest occupied molecular orbitals (MO 15 and 16) and the two lowest unoccupied molecular orbitals (MO 17 and 18) of Ox and Ox-H+, drawn with the HyperChem program, are shown in the ESI.† Since the polymerization reaction of oxetane involves mainly the O atom of Ox attacking the C atom of Ox-H+, when an O atom and a C atom are in close proximity, the O atom HOMO should interact with the C atom LUMO. It can be obviously seen that the symmetry of MO 15 of the O atom in Ox is matched with MO 17 of the C atom in Ox-H+ for the formation of an O–C bond. In addition, if the symmetry matching of MO 16 of Ox with MO 18 of Ox-H+ took place, it would produce an OH2C bond. However, this possibility may be very low. When the four-membered ring planes of Ox and Ox-H+ are almost perpendicular, an O atom of Ox would approach a C atom of Ox-H+ along the C–C bond axis direction, and the symmetry matching can meet the principle of orbital maximum overlap. This is also consistent with the changes in the geometric parameters of the transition state in the polymerization of Ox with Ox-H+.
3.2 The structural characteristics of equilibrium and transition state on the polymerization pathways
The experimental results show that the polymerization of the oxetane derivative with its cation is usually classified as a cationic ring-opening reaction. This kind of process normally includes three processes, that is, photoinitiation, chain growth and termination, based on the principles of polymerization. Under the conditions of photoinitiation, the reaction system firstly produces a proton due to the interaction of the photoinitiator, and then an oxonium ion is formed, thereby attracting oxetane molecules to form a complex. Fig. 2 shows the structural model of the reactant, transition state and product in the polymerization reaction process of oxetane with its cation. The data for 4- and 5-polymerization are put into the ESI.† As can be seen from Fig. 2, the structures of the intermediates and products of the polymerization are close to those of polyalcohol or polyether. There is a four-membered heterocyclic structure containing oxygen in the side chain, which undergoes a ring-opening reaction to finally terminate the polymerization process. Table 1 lists the main geometric parameters of different species in the reaction pathway of the oxetane 2- to 6-polymerization, which were obtained using the B3LYP/6-31G(d,p) method. It is shown that the transition state structures of each step are closer to the reactant (complex of Ox with Ox-H+). Taking the dipolymer reaction as an example, while the polymerization reaction is progressing and the reactant interaction is increasing, the distance between the O atom of Ox and the C atom of Ox-H+ becomes 0.255 nm. This decreases to 0.222 nm in the transition state, and finally to 0.150 nm in the polymer intermediate. The changes in the O and C atom positions fully reflect the structural changes of polymerization. The C–C–C bond angle also changes to 94.30° in the transition state from 90.75° for the complex reactants, and finally increases to 109.63° in the ring-opening products. It can be seen that the changes in the bond parameters of the polymer system (R, TS, P) change little when the degree of polymerization is 3 to 6 (the data are listed in the ESI†). This means that when the degree of polymerization reaches a certain value, the polymerization of oxetane favors a repeated process.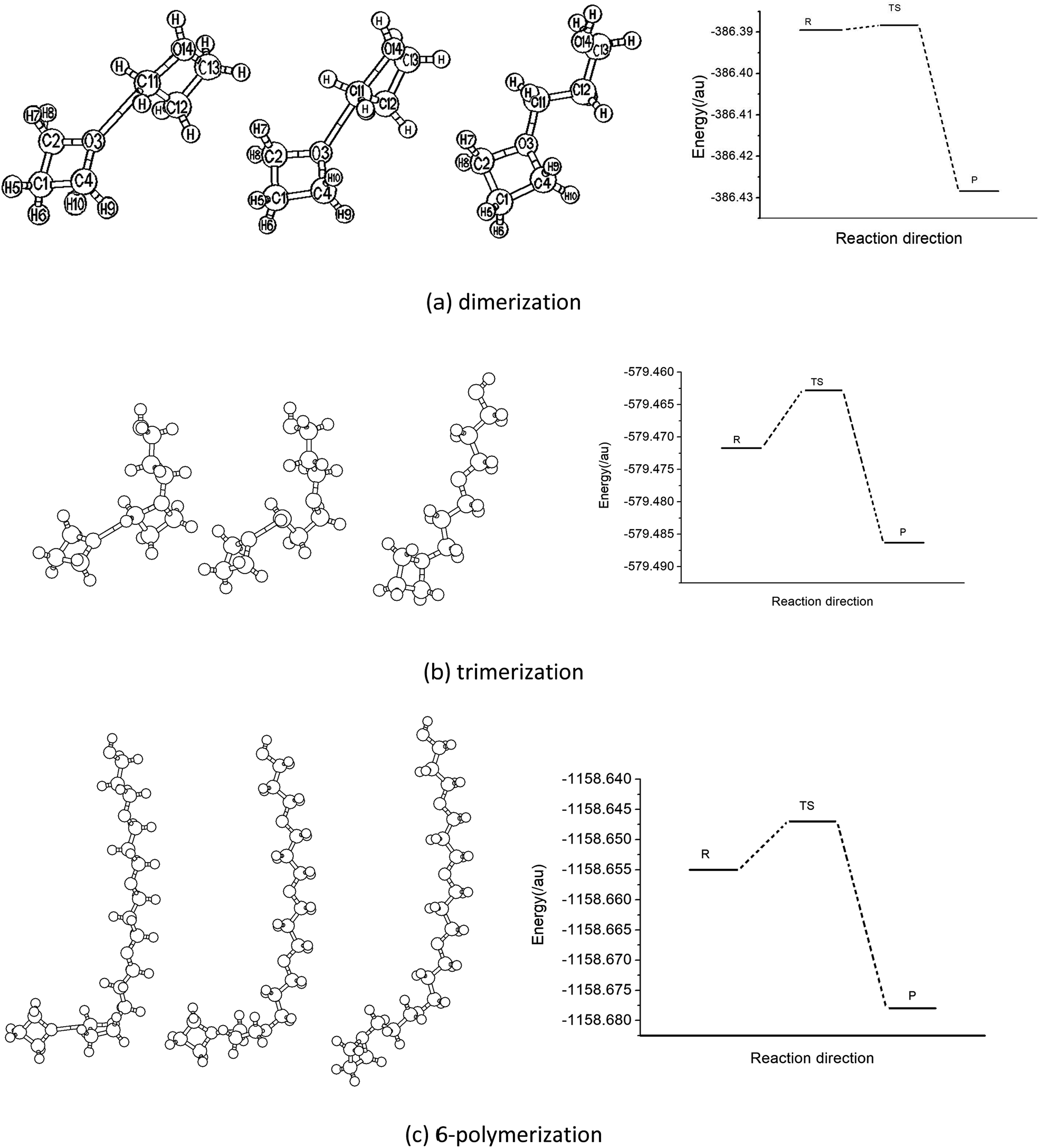 | ||
| Fig. 2 Structural models of the reactant, transition state and product of oxetane polymerization and the potential energy profiles of the reaction. | ||
| n | 2 | 3 | 4 | 5 | 6 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| O–C | CCC | CCO | O–C | CCC | CCO | O–C | CCC | CCO | O–C | CCC | CCO | O–C | CCC | CCO | |
| a Bond length: nm; bond angle: °. | |||||||||||||||
| R | 0.255 | 90.75 | 90.40 | 0.267 | 89.20 | 89.89 | 0.268 | 89.40 | 89.40 | 0.268 | 89.39 | 89.41 | 0.266 | 89.59 | 88.30 |
| TS | 0.222 | 94.30 | 93.85 | 0.208 | 95.72 | 95.60 | 0.211 | 95.86 | 94.99 | 0.211 | 95.84 | 95.03 | 0.211 | 95.85 | 94.99 |
| P | 0.150 | 109.63 | 106.43 | 0.149 | 111.74 | 105.91 | 0.150 | 108.67 | 106.56 | 0.150 | 108.69 | 106.54 | 0.150 | 108.71 | 106.74 |
By analyzing the structural parameters of the reactants, transition states and products, the polymerization of the Ox monomer in the initial process of the reaction was found to represent mainly the O atoms of oxetane attacking the C atom of Ox-H+ adjacent to the O atom along the direction of the C–O axis of Ox-H+. This is consistent with the symmetrical changes of the frontier molecular orbitals and may result in the charge attraction of Ox and Ox-H+ in the reaction process. The positions of the two carbon atoms of Ox adjacent to OH groups are equivalent, and they can accept the Ox monomer to carry out polymerization. This means that the products would have some isomers. The transition state geometries of oxetane dimerization are very close to the reactant complex and the product geometries are similar to that of polyether according to the structural parameters of the polymer chain.
3.3 The reaction barrier of polymerization and the pathway of intrinsic reaction coordinates
Bulut36 described that the reaction process of oxetane proceeds with an increase of the polymer chain, activation energy and exothermic heat of the reaction. Based on the transition state structure computed theoretically, the intrinsic reaction coordinates of each step in the polymerization reaction confirm that the reactants and products are the oxetane cation and its polymers, corresponding to the configuration of the transition state. Table 2 lists the self-consistent field energy of the polymerization system (ER, ETS, EP), forward activation energy (Ea) and backward activation energy (Eb), and reaction heat (ΔHr). It also shows that the transition state geometry of every step in the polymerization of oxetane is very close to that of its corresponding reactant. The activation barrier of dimerization in the initial steps is very low, at only about 3 kJ mol−1. The activation energy of the corresponding inverse reaction (decomposition) is relatively high, at about 105 kJ mol−1. This is consistent with the experimental characteristics of a high reaction rate and difficult polymerization-inhibition by oxygen. With the polymer chain growth, the activation energy of polymerization increases, and the reaction rate becomes gradually smoother.| n | ER | ETS | EP | Ea | Eb | ΔHr | |
|---|---|---|---|---|---|---|---|
| 2 | 6-31G(d,p) | −386.390 | −386.388 | −386.428 | 2.888 | 105.282 | −102.394 |
| 6-311++G(d,p) | −386.484 | −386.483 | −386.525 | 3.151 | 111.059 | −107.908 | |
| 3 | 6-31G(d,p) | −579.472 | −579.463 | −579.486 | 23.367 | 61.699 | −38.332 |
| 6-311++G(d,p) | −579.618 | −579.609 | −579.633 | 24.417 | 64.850 | −40.433 | |
| 4 | 6-31G(d,p) | −772.536 | −772.520 | −772.556 | 40.170 | 93.993 | −53.823 |
| 6-311++G(d,p) | −772.732 | −772.716 | −772.754 | 39.908 | 98.981 | −59.074 | |
| 5 | 6-31G(d,p) | −965.599 | −965.583 | −965.6193 | 40.483 | 94.342 | −53.860 |
| 6-311++G(d,p) | −965.844 | −965.826 | −965.8658 | 37.581 | 95.311 | −57.729 | |
| 6 | 6-31G(d,p) | −1158.655 | −1158.647 | −1158.678 | 21.004 | 82.814 | −61.810 |
| 6-311++G(d,p) | −1158.951 | −1158.943 | −1158.974 | 22.361 | 83.504 | −61.143 | |
The intrinsic reaction coordinates of the single step 2- to 6-polymerization of oxetane with its cation were calculated using the B3LYP/6-31G(d,p) method. Fig. 3 shows the combination of several groups of intrinsic reaction coordinates in the polymerization pathways of Ox and Ox-H+. The energy of the complex of Ox and Ox-H+ was selected as the zero point of relative energy. The potential energy profile in the reaction path shows a change in the ladder shape. It shows the total energy of the system reduces with the increase of polymerization degree, and the activation energy of each step in the reaction becomes gradually constant. Such a low energy barrier of the initial step means that the reaction of Ox with Ox-H+ takes place so easily that the monomers of Ox and Ox-H+ would almost not exist in a container at the same time. The high backward energy barrier of polymerization means that some dimers or multi-polymers would be stable and difficult to be dissociated in the reaction system. This explanation of the polymerization of Ox with Ox-H+ is in accordance with the experimental description of oxetane.
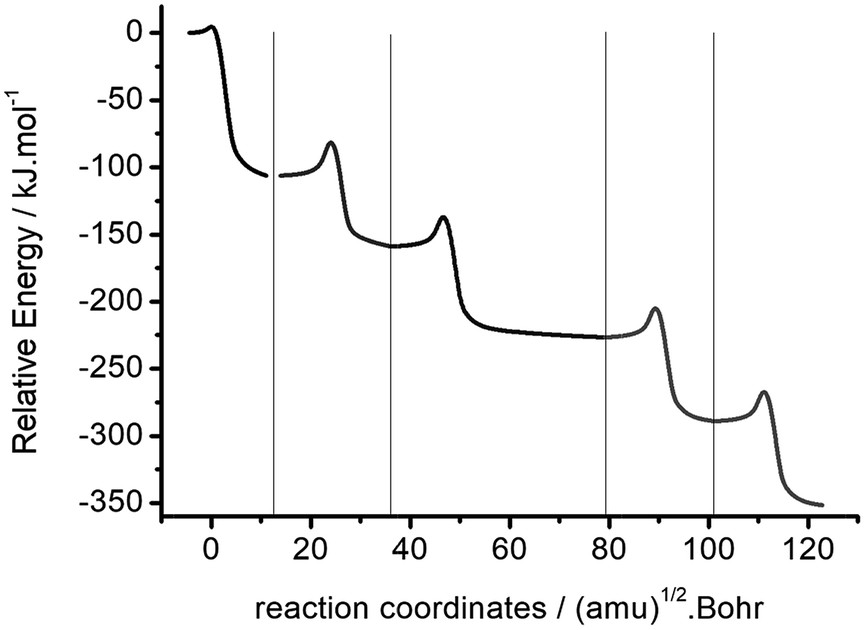 | ||
| Fig. 3 Combination of intrinsic reaction coordinates of the polymerization pathway of Ox with Ox-H+. | ||
3.4 Solvent effects
Normally, the polymerization reaction of oxetane derivatives with its cation is carried out in an organic solvent such as tetrahydrofuran (THF) or dichloromethane (DCM), according to the experimental reports. Here, two kinds of solvent effects of the polymerization were computed using the polarizable continuum model. Table 3 lists the geometric parameters of each species on the polymerization pathways, which were fully optimized at the level of B3LYP/6-31G(d,p), based on the PCM model. The computational results show that the solvent effects of THF and DCM have a small effect on the geometry, causing an increase of about 0.01 nm in the O(Ox)–C(Ox-H+) inter-atomic distance, compared to the situation with no solvent. This means that the different molecules that are not bonded present a certain relaxation and move away from each other under the solvent effects. Table 4 lists the energies of the reactants (ER), transition state (ETS) and products (EP), at each step of the 2- to 6-polymerization in the solvent. Comparing Tables 2 and 4, the solvent effects of THF and DCM obviously affect the system energy and balance the activation energy of each step in the reaction. However, the geometries of the species on the reaction path show no large changes. The forward activation energy of the initial step is still small, while the backward activation energy is large. This also means that the reaction speed of the initial process should be fast, and dimers or multi-polymers of Ox and Ox-H+ may exist stably in the polymerization process.| n | THF | DCM | |||||
|---|---|---|---|---|---|---|---|
| R | TS | P | R | TS | P | ||
| a Bond length: nm; bond angle: °. | |||||||
| 2 | O–C | 0.268 | 0.215 | 0.149 | 0.269 | 0.216 | 0.149 |
| CCC | 89.85 | 95.06 | 110.47 | 89.75 | 94.81 | 110.50 | |
| CCO | 89.97 | 95.29 | 107.52 | 89.83 | 94.96 | 107.57 | |
| 3 | O–C | 0.273 | 0.211 | 0.148 | 0.273 | 0.211 | 0.148 |
| CCC | 89.13 | 95.41 | 111.44 | 89.13 | 95.39 | 111.41 | |
| CCO | 89.71 | 95.36 | 106.77 | 89.72 | 95.34 | 106.82 | |
| 4 | O–C | 0.274 | 0.213 | 0.149 | 0.274 | 0.213 | 0.149 |
| O–C | 89.32 | 95.69 | 109.41 | 89.39 | 95.70 | 109.66 | |
| CCC | 89.17 | 94.79 | 107.53 | 89.25 | 94.81 | 107.72 | |
| 5 | O–C | 0.274 | 0.213 | 0.149 | 0.274 | 0.213 | 0.149 |
| CCC | 109.65 | 95.76 | 109.53 | 89.47 | 95.76 | 109.79 | |
| CCO | 108.06 | 94.98 | 107.53 | 89.32 | 94.98 | 107.67 | |
| 6 | O–C | 0.274 | 0.149 | 0.274 | 0.149 | ||
| CCC | 89.47 | 109.40 | 89.48 | 109.46 | |||
| CCO | 89.19 | 107.55 | 89.20 | 107.58 | |||
| Solvent | n | ER (au) | ETS (au) | EP (au) | Ea (kJ mol−1) | Eb (kJ mol−1) | ΔH (kJ mol−1) |
|---|---|---|---|---|---|---|---|
| THF | 2 | −386.458961 | −386.454075 | −386.496147 | 12.828 | 110.460 | −97.632 |
| 3 | −579.531281 | −579.522109 | −579.558345 | 24.081 | 95.138 | −71.057 | |
| 4 | −772.594669 | −772.582167 | −772.623358 | 32.824 | 108.147 | −75.323 | |
| 5 | −965.659637 | −965.647255 | −965.688565 | 32.509 | 108.459 | −75.950 | |
| 6 | −1158.721140 | −1158.712398 | −1158.751291 | 22.952 | 102.114 | −79.161 | |
| DCM | 2 | −386.460639 | −386.456134 | −386.498024 | 11.829 | 109.982 | −98.154 |
| 3 | −579.532984 | −579.523776 | −579.560470 | 24.176 | 96.340 | −72.164 | |
| 4 | −772.596227 | −772.584023 | −772.625201 | 32.042 | 108.113 | −76.071 | |
| 5 | −965.661470 | −965.649171 | −965.690427 | 32.291 | 108.318 | −76.027 | |
| 6 | −1158.723142 | −1158.714419 | −1158.753550 | 22.902 | 102.738 | −79.836 |
4. Conclusions
The mechanism of the ring-opening polymerization of oxetane with its cation series compounds was revealed, through theoretical calculations using density functional theory and ab initio methods of quantum chemistry.The computational results show that the polymerization process of oxetane with its cation mainly proceeds through the O atom of the oxetane molecule attacking α-carbon atoms of the oxetane cation after light initiation. Each step of the polymerization process is an elementary reaction. The geometries of the transition state of each elementary reaction approach the structure of a complex of oxetane and its cation. The forward activation energy of polymerization in the initial step was found to be very small, while the backward energy barrier of polymerization was found to be high. Therefore, the reaction speed should be very fast and dimers or multi-polymers may be stable in the initial process and the reaction process, respectively. With an increase in the polymerization degree of the reactant, the activation energy of the reaction process becomes large and constant. All the polymerization reaction steps would be easy to perform. The solvent effects of THF and DCM obtained from the PCM model show that they may cause the relaxation of the polymerization system, and the effects on the geometry of the reaction system would be low. In summary, the polymerization of oxetane with its cation would be easy and fast. The results of our theoretical quantum chemistry calculations are in accordance with experiments.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by the National Natural Science Foundation of China (90923040, 21173019).References
- M. Sangermano, R. Bongiovanni and G. Malucelli, Polymer, 2004, 45, 2133–2139 CrossRef CAS.
- S. Feser and K. Meerholz, Chem. Mater., 2011, 23, 5001–5005 CrossRef CAS.
- Y. Shintani and H. Tsutsumi, Electrochemistry, 2010, 78, 387–389 CrossRef CAS.
- H. Kazuhiro, CN1930213, US57102905A, 2007 03 14.
- H. Sasaki, Abstr. Pap. Am. Chem. Soc., 2001, 222, U311 Search PubMed.
- H. Liu, J. Mo and H. Liu, J. Huazhong Univ. Sci. Technol., 2008, 36, 129–132 CAS.
- M. Alves, B. Grignard, A. Boyaval, R. Mereau, J. L. Winter, P. Gerbaux, C. Detrembleur, T. Tassaing and C. Jerome, ChemSusChem, 2017, 10, 1128–1138 CrossRef CAS PubMed.
- B. J. Li, Y. J. Luo and J. Zheng, Polym. Sci., Ser. A, 2017, 59, 301–309 CrossRef CAS.
- P. Champagne and K. N. Houk, J. Am. Chem. Soc., 2016, 138, 12356–12359 CrossRef CAS PubMed.
- R. Perez-Ruiz, S. Gil and M. A. Miranda, J. Org. Chem., 2005, 1376–1381 CrossRef CAS PubMed.
- B. Guichard, V. Poirier and C. Noel, Macromol. Chem. Phys., 1996, 197, 3631–3656 CrossRef CAS.
- J. X. Feng, L. J. Zhu, C. M. Lu, S. L. Teng, M. W. Young and C. G. Gogos, Polym. Eng. Sci., 2009, 49, 1107–1116 CAS.
- F. Zhan, X. E. Cheng and W. F. Shi, J. Appl. Polym. Sci., 2012, 123, 717–724 CrossRef CAS.
- B. Falk, S. M. Vallinas and J. V. Crivello, PMSE Prepr., 2003, 89, 279 CAS.
- F. C. Ferreira, B. G. Oliveiraa, E. Ventura, S. A. Monte, C. F. Braga, R. C. Araujo and M. N. Ramos, Spectrochim. Acta, Part A, 2006, 64, 156–160 CrossRef CAS PubMed.
- P. Ottaviani, M. Giuliano and B. Velino, et al., Chem.–Eur. J., 2004, 10, 538–543 CrossRef CAS PubMed.
- M. E. Sanz, V. M. Sanz, J. C. Lopez and J. L. Alonso, Chem. Phys. Lett., 2001, 342, 31–33 CrossRef CAS.
- M. A. Izquierdo, L. R. Domingo and M. A. Miranda, J. Phys. Chem. A, 2005, 109, 2602–2607 CrossRef CAS PubMed.
- G. J. Chen, D. C. Fang and X. Y. Fu, Acta Phys.-Chim. Sin., 1990, 6, 177–182 Search PubMed.
- A. Tahan and A. Shiroudi, Chin. J. Struct. Chem., 2011, 30, 697–705 CAS.
- A. Shiroudi, A. Tahan and E. Zahedi, Russ. J. Phys. Chem. A, 2012, 86, 1245–1249 CrossRef CAS.
- I. J. Palmer, I. N. Ragazos, F. Bernardi, M. Otivucci and M. A. Robb, J. Am. Chem. Soc., 1994, 116, 2121–2132 CrossRef CAS.
- S. H. Lee, J. Chem. Phys., 2009, 131, 224309 CrossRef PubMed.
- Z. B. Yang, L. A. Eriksson and R. B. Zhang, J. Phys. Chem. B, 2011, 115, 9681–9686 CrossRef CAS PubMed.
- H. L. Wu, J. L. Yang, M. Luo, R. Y. Wang, J. T. Xu, X. H. Zhang and D. J. Darensbourg, Macromolecules, 2016, 49, 8863–8868 CrossRef CAS.
- K. J. Zheng, X. Q. Zhu, X. C. Qian, J. Li, J. L. Yang and J. Nie, Polym. Int., 2016, 65, 1486–1492 CrossRef CAS.
- E. M. Christ, D. Hobernik, M. Bros, M. Wagner and H. Frey, Biomacromolecules, 2015, 16, 3297–3307 CrossRef CAS PubMed.
- A. Harrane, N. Naar and M. Belbachir, Mater. Lett., 2007, 61, 3555–3558 CrossRef CAS.
- J. A. Ivie, D. J. Darensbourg and C. E. Webster, Abstr. Pap. Am. Chem. Soc., 2011, 241, 520 Search PubMed.
- R. A. Creswell, Mol. Phys., 1975, 30, 217–222 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- T. Lu and F. W. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- HyperChem(TM) Professional 8.0, Hypercube, Inc., 1115 NW 4th Street, Gainesville, Florida 32601, USA, February, 2011, http://www.hyper.com/, assessed Search PubMed.
- H. X. Wang and M. Pu, Comp. Appl. Chem., 2003, 20, 354–356 CAS.
- T. Lu and F. W. Chen, Acta Phys.-Chim. Sin., 1990, 6, 177–182 Search PubMed.
- U. Bulut and J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3205–3220 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09317a |
| This journal is © The Royal Society of Chemistry 2017 |