
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the existence of AgM9(VO4)6I (M = Ba, Pb)†
E. V.
Johnstone
 a,
D. J.
Bailey
a,
M. C.
Stennett
a,
J.
Heo
bc and
N. C.
Hyatt
*a
a,
D. J.
Bailey
a,
M. C.
Stennett
a,
J.
Heo
bc and
N. C.
Hyatt
*a
aUniversity of Sheffield, Materials Science and Engineering Department, Sheffield, S10 2TN, UK. E-mail: erikvjohnstone@gmail.com
bDepartment of Materials Science and Engineering, Pohang University of Science and Technology (POSTECH), Pohang, Gyeongbuk 790-784, South Korea
cDivision of Advanced Nuclear Engineering, Pohang University of Science and Technology (POSTECH), Pohang, Gyeongbuk 790-784, South Korea
First published on 17th October 2017
Abstract
The syntheses of the reported compounds AgM9(VO4)6I (M = Ba, Pb) were reinvestigated. Stoichiometric amounts of AgI with either M3(VO4)2 (M = Ba, Pb) or PbO and V2O5 were reacted in the solid-state at elevated temperatures in air or in flame-sealed quartz vessels. The resulting products were characterized by X-ray diffraction, scanning electron microscopy with energy dispersive X-ray analysis, and thermal analyses. Results show that, for all reaction conditions, the target AgM9(VO4)6I (M = Ba, Pb) phases could not be isolated. Instead, heterogeneous phase distributions of primarily M3(VO4)2 (M = Ba, Pb) and AgI were obtained. These findings demonstrate that AgI incorporation into single phase, iodine-deficient apatite derivatives for the immobilization of iodine-129 are not feasible under such conditions. This conclusion is important for the conditioning of iodine-129 in advanced reprocessing flowsheets, where iodine is typically sequestered as AgI.
Introduction
A myriad of apatite (A10(BO4)6X2) and apatite supergroup phases, such as hedyphane, belovite, britholite, and ellestadite groups, are found as naturally occurring minerals, a testament to their chemically robust character.2 Because the apatite phase and those similar to it are capable of structurally substituting a host of different cation (A and B sites) and anion variants (X site), they are ideal synthetic templates for the immobilization of chemically complex radioactive wastes. Previous studies have already shown that an assortment of fission products (e.g., Cs+, Sr2+, Ag+, Eu2+, Ln3+, I−),3,4 actinide components (e.g., Th4+, U4+, U6+),5 and other process additives or corrosion products (e.g., Na+, Co2+, Pb2+, F−, Cl−) can be integrated into the apatite structure for purposes of nuclear waste disposal.6,7Due to its volatile nature and radiotoxicity, radioiodine, e.g., 129I and 131I, has been a long-standing problem in terms of its containment during reprocessing and disposal in nuclear fuel cycles.8 In particular, because of its long half-life, 129I (t1/2 = 15.7 × 106 y) must be properly treated in order to minimize its long-term accumulation and effect on the environment.9 Radioiodine is of concern due to its environmental dispersal during nuclear reprocessing,10 accidents,11,12 and weapons testing.13 Current regulatory practices overseeing reprocessing utilize dispersion and dilution of 129I with natural 127I to reduce its impact on the biosphere.14 However, it is possible that future reprocessing and waste schemes will focus on its capture and disposal, where a range of robust treatments and waste forms will be necessary for its management.8
Specifically, of these materials, iodoapatite phases, such as M10(VO4)6I2 (M = Ba, Pb), have been suggested as possible waste forms for radioiodine, and, in particular, the Pb variant has been the topic of many recent studies.15–23 Using a variation of these M10(VO4)6I2 host phases, the synthesis and characterization of the compounds AgBa9(VO4)6I and AgPb9(VO4)6I were reported using AgI as an iodine source.1 The use of AgI as the iodine carrier is attractive because this compound is formed in the spent nuclear fuel and occurs in various stages of reprocessing, such as during fuel dissolution, where it comprises a part of the undissolved solids (UDS),24–26 and in the dissolver off-gas capture of gaseous iodine with solid Ag or AgNO3-loaded sorbents (e.g., zeolite, alumina, or silica).8
The aim of this study was to synthesize compounds AgM9(VO4)6I (M = Ba, Pb) and investigate the feasibility of AgI incorporation and retention into these matrices. Batched powders corresponding to AgM9(VO4)6I (M = Ba, Pb) were reacted using standard solid-state ceramic syntheses in air and in sealed quartz vessels at elevated temperatures. The resulting materials were studied using X-ray diffraction, microscopy, and thermal analysis techniques. However, as reported herein, our attempts to synthesize “AgBa9(VO4)6I” and “AgPb9(VO4)6I” for further investigation proved unsuccessful, with no evidence for the formation of compounds of this stoichiometry incorporating AgI. Nevertheless, our X-ray diffraction data are in good agreement with those published previously for these hypothecated compounds, which on careful inspection, show the phase assemblage to comprise the M3(VO4)2 and AgI starting materials, as also evidenced by scanning electron microscopy/energy dispersive X-ray analysis.
Experimental
Synthesis of AgM9(VO4)6I (M = Ba, Pb)
Stoichiometric amounts of M3(VO4)2 (M = Ba, Pb) and AgI, were batched to yield a final reacted composition of AgM9(VO4)6I (M = Ba, Pb). The batched powders were pulverized and mixed as a slurry with isopropanol in an agate mortar and pestle. After drying, the resulting powders were pressed at 2 tonnes into ∼250 mg, 6 mm disk pellets. Pellets were reacted in air in a box furnace or under vacuum in flame-sealed quartz tubes (10 mm O.D., 8 mm I.D., 180 length) in an alumina tube furnace at 700 °C for 5 h. Because preparations of Pb10(VO4)6I2 have also been reported starting with the component oxides, additional reactions for the Pb series were also performed using the same synthetic procedure with PbO, V2O5, and AgI as starting materials; characterization of these materials are reported in the ESI.†Characterization techniques
Powder X-ray diffraction (PXRD) was performed on a Bruker D2 Phaser system operating with Ni filtered Cu Kα radiation and a position sensitive detector. Samples were pulverized with an agate mortar and pestle and dispersed on a low-background silicon holder. Measurements were made from 10° < 2θ < 70° with a step size of 0.02 increments at scan rate of 1.0 min−1. Phase analysis was completed using Diffrac.Suite Eva V.3 (Bruker) and Rietveld refinements were performed using Topas V.4.2 (Bruker). Scanning electron microscopy (SEM) (Hitachi-TM3030) and energy-dispersive X-ray spectroscopy (EDX) (Quantax 70) measurements were performed at an accelerating voltage of 15 keV with backscattered electron (BSE) detection. Pieces of reacted pellets were cold-mounted in an epoxy resin and polished to 1 μm using SiC grit paper and diamond polishing paste with a polishing cloth. Thermal gravimetric (TG) and differential thermal analysis (DTA) measurements (Netzsch Jupiter STA 449F3) of powdered samples were performed under flowing Ar(g) in an alumina crucible with a scan rate of 10 °C min−1 from 30 to 1150 °C.Results and discussion
Synthesis and formation of “AgPb9(VO4)6I”
Powders of Pb3(VO4)2 and AgI were batched and reacted using standard solid-state ceramic techniques. The combined starting materials produced yellow-white powders before reacting. After pressing into pellets and reacting, the resulting pellets were dark grey-blue and produced pale yellow to yellow-brown powders upon grinding. For reactions performed in quartz tubes, no apparent sublimation of iodine from the reacted pellet was observed at the cooler end of the tube.The PXRD patterns of “AgPb9(VO4)6I” are shown in Fig. 1. The PXRD patterns of the resulting products were similar, irrespective of sealed tube or open reaction conditions. Phase analysis of each pattern identified β-Pb3(VO4)2 (monoclinic P121/c1) and β/γ-AgI (hexagonal P63mc/cubic F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m, respectively) as the primary phases. Additionally, the Pb9.85(VO4)6I1.7 (hexagonal P63/m) phase was identified, as a minor component, in the “AgPb9(VO4)6I” sample reacted in a sealed tube. The formation of Pb9.85(VO4)6I1.7 is hypothesised to be a result of the decomposition of AgI to yield Ag metal and I2, and subsequent reaction of the latter with β-Pb3(VO4)2.
3m, respectively) as the primary phases. Additionally, the Pb9.85(VO4)6I1.7 (hexagonal P63/m) phase was identified, as a minor component, in the “AgPb9(VO4)6I” sample reacted in a sealed tube. The formation of Pb9.85(VO4)6I1.7 is hypothesised to be a result of the decomposition of AgI to yield Ag metal and I2, and subsequent reaction of the latter with β-Pb3(VO4)2.
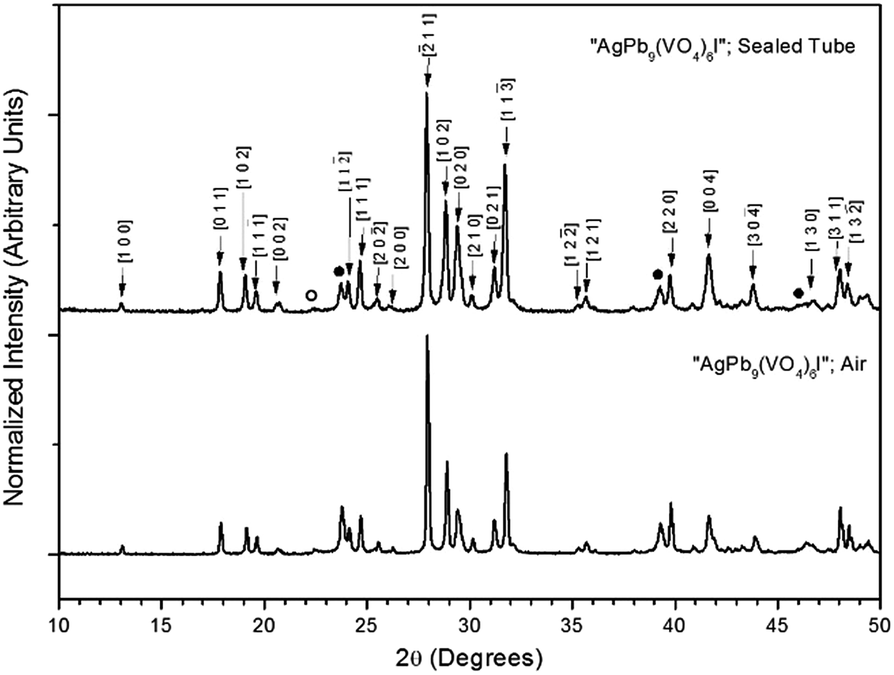 | ||
| Fig. 1 PXRD patterns of “AgPb9(VO4)6I” treated at 700 °C for 5 h in either air or a sealed quartz tube. Markers indicate peaks assigned to the β-Pb3(VO4)2 (labelled (hkl) Miller indices), γ-AgI (●), and β-AgI (○) phases. | ||
Rietveld fitting of X-ray diffraction data of “AgPb9(VO4)6I” was effectively achieved assuming a mixture of β-Pb3(VO4)2, γ-AgI, and Pb9.85(VO4)6I1.7 as shown in Fig. 2; the refined phase fractions were 70 ± 5%, 8 ± 1%, and 22 ± 5%, respectively. The phase fraction of β-AgI was determined to be < 1%. Although the crystal structure of “AgPb9(VO4)6I” was stated to be of monoclinic symmetry, no space group was determined, but the compound was reported not to adopt an apatite related structure.1 Inspection of Table 1 shows the reported unit cell parameters of “AgPb9(VO4)6I” to be close to those determined for β-Pb3(VO4)2, the major phase identified in our reaction product, which adopts the monoclinic space group P121/c1. The PXRD patterns acquired here for “AgPb9(VO4)6I” in both open and closed systems are comparable to those reported by Uno et al. for “AgPb9(VO4)6I”.1 In these PXRD patterns, reflections are evident at 2θ = 23.7°, 39.3° and 46.4°, which may be attributed to γ-AgI (indexed (hkl) as (111), (022) and (113)), as highlighted in Fig. 1 here. These same reflections are apparent, but unindexed, in Fig. 1 of Uno et al.1 These data suggest that the compound identified by Uno et al. as “AgPb9(VO4)6I” was, in fact, β-Pb3(VO4)2.
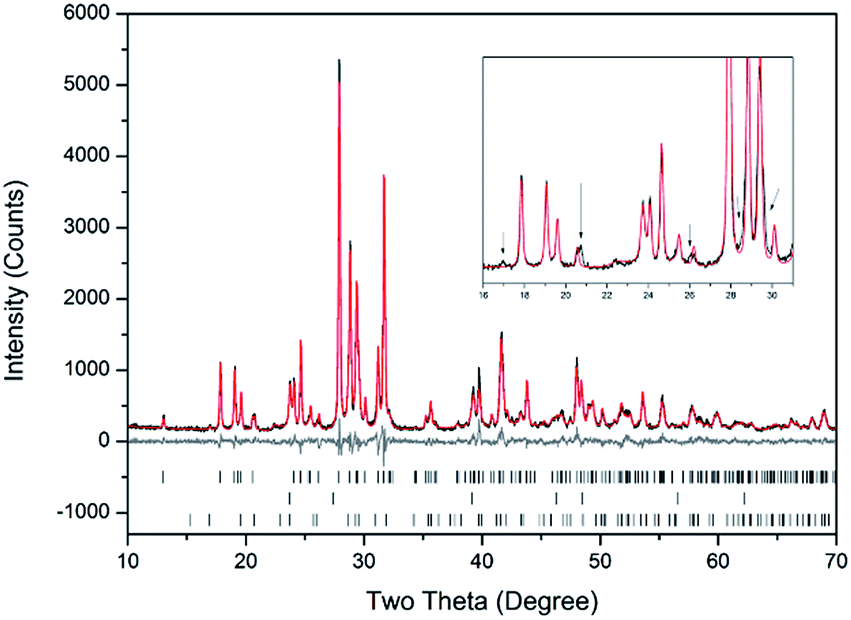 | ||
| Fig. 2 Rietveld analysis of PXRD data of “AgPb9(VO4)6I” reacted at 700 °C in a sealed quartz tube; the data are shown in black, the fit is shown in red, and the difference plot in grey. Tick marks show allowed reflections of β-Pb3(VO4)2 (upper ticks), γ-AgI (middle ticks), and Pb9.85(VO4)6I1.7 (bottom ticks). Inset: Rietveld fit without the Pb9.85(VO4)6I1.7 phase showing consequent reflection misfits, highlighted by black arrows. | ||
| “AgPb9(VO4)6I”1 | β-Pb3(VO4)2 in “AgPb9(VO4)6I” this study | β-Pb3(VO4)2 [ref. 27] | Pb9.85(VO4)6I1.7 [ref. 17] | |
|---|---|---|---|---|
| a Not reported. | ||||
| Crystal system | Monoclinic (NRa) | Monoclinic P121/c1 | Monoclinic P121/c1 | Hexagonal P63/m |
| a (Å) | 7.509 | 7.518 (1) | 7.514 | 10.422 |
| b (Å) | 6.097 | 6.072 (1) | 6.107 | — |
| c (Å) | 9.268 | 9.555 (1) | 9.526 | 7.467 |
| α (°) | 90 | 90 | 90 | 90 |
| β (°) | 111.8 | 115.3 (3) | 115.2 | 90 |
| γ (°) | 90 | 90 | 90 | 120 |
In order to further investigate the product phase assemblage and element distribution, sectioned pieces of the reacted pellets were mounted into an epoxy resin, polished, and characterized using SEM/EDX. In Fig. 3, the SEM image and corresponding EDX element distribution maps for V, Ag, I, and Pb are shown for “AgPb9(VO4)6I” reacted at 700 °C in a sealed quartz tube. From the BSE imaging, at least two primary phases were identified, distinguished according to contrast. Element distribution maps further support this conclusion, with association of Ag and I in one phase, and association of Pb and V in a second phase. In accordance with the PXRD data, these two phases can be interpreted as the AgI and β-Pb3(VO4)2 phases, respectively. EDX analyses (Fig. S3†) of individual β-Pb3(VO4)2 grains demonstrated the absence of measurable iodine concentration. The apparent weak iodine signal associated with β-Pb3(VO4)2 grains in Fig. 3 is attributed to the background X-ray emission in the I Lα energy window (i.e., no discernible I Lα emission line was observed in the corresponding EDX spectra). Small inclusions of Ag metal were also identified in the microstructure of this “AgPb9(VO4)6I” sample (Fig. S4†); this phase was not clearly evident in the associated PXRD data due to reflection overlap from compounds with more complex and lower symmetry structures. The presence of Ag metal supports a hypothesised mechanism of Pb9.85(VO4)6I1.7via decomposition of AgI, liberating I2 which subsequently reacts with β-Pb3(VO4)2. It was not possible to conclusively identify the minor Pb9.85(VO4)6I1.7 phase in the microstructure by BSE imaging. Using well-established formulations,28 the BSE coefficients for β-Pb3(VO4)2 and Pb10(VO4)6I2 were estimated as η = 0.2903 and η = 0.3064, respectively, yielding a difference in contrast of 5%. Such a small difference in contrast, which would be further reduced by non-stoichiometry in the apatite phase, is acknowledged to make differentiation of the two phases challenging.28 Nevertheless, by forming a Red – Green – Blue colour map from the EDX signals associated with the Pb Mα, I Lα and V Kα signals, it was possible to identify small white regions that afforded EDX spectra exhibiting X-ray emission lines characteristic of all three elements, but excluding Ag Lα emission lines (Fig. S5†). These regions were identified as the Pb9.85(VO4)6I1.7 component required to adequately fit X-ray diffraction data in Fig. 1. The elemental analysis of the whole field of view in Fig. 3 is displayed in Table 2; the determined average composition is comparable with the expected and reported composition for the “AgPb9(VO4)6I” phase.
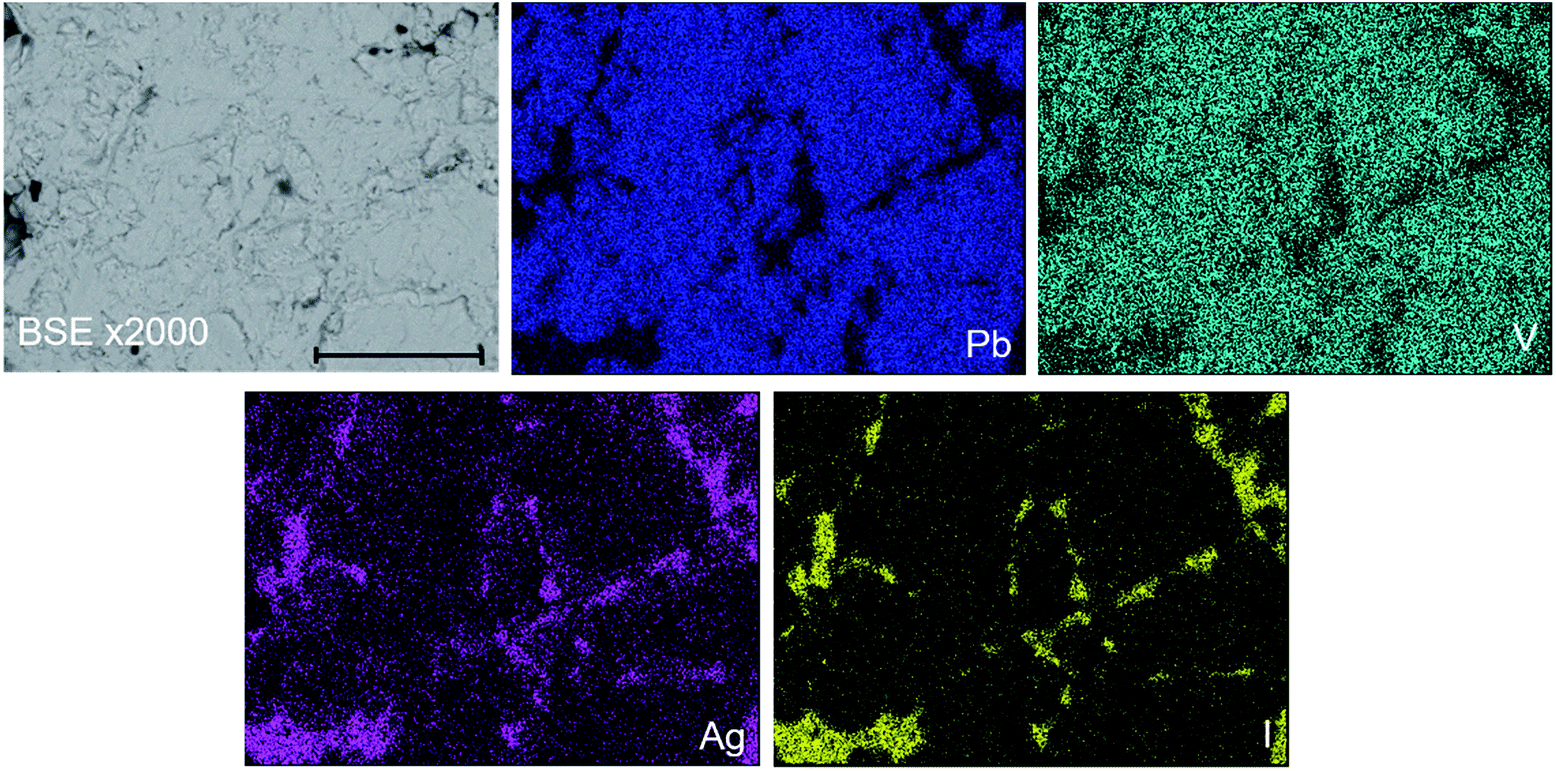 | ||
| Fig. 3 BSE SEM image at ×2000 magnification (scale bar length = 30 μm) and EDX map of Pb (blue), V (teal), I (yellow), and Ag (pink) present in “AgPb9(VO4)6I” reacted at 700 °C for 5 h in a sealed quartz tube. | ||
| Element | “AgPb9(VO4)I” | “AgBa9(VO4)6I” | ||||
|---|---|---|---|---|---|---|
| Elemental wt% | Elemental wt% | |||||
| Stoich. | Lit.1 | Meas. | Stoich. | Lit.1 | Meas. | |
| Ag | 3.87 | 4.33 | 5.3 ± 0.2 | 4.99 | 3.30 | 5.7 ± 0.2 |
| Pb | 66.86 | 64.14 | 62 ± 2 | — | — | — |
| Ba | — | — | — | 57.21 | 62.67 | 56 ± 1 |
| V | 10.96 | 15.02 | 10.8 ± 0.3 | 14.15 | 15.83 | 14.2 ± 0.4 |
| O | 13.77 | 11.48 | 14 ± 2 | 17.77 | 12.24 | 18 ± 2 |
| I | 4.55 | 5.03 | 7.9 ± 0.3 | 5.87 | 4.61 | 6.1 ± 0.2 |
TG-DTA analysis (Fig. S6†) of “AgPb9(VO4)6I” reacted at 700 °C for 5 h, in a sealed tube, is characterized with significant weight loss shown in the TG curve beginning at ∼630 °C continuing up to ∼1100 °C. Uno et al. reported “AgPb9(VO4)6I” to be stable up to ∼677 °C,1 which is broadly consistent with the weight loss data obtained in this study. In accordance with the SEM and XRD data, this thermal behaviour can be associated with the melting and iodine release/decomposition of AgI and Pb9.85(VO4)6I1.7 within the matrix. Additionally, a DTA signal was observed at ∼150 °C, which was indicative of the β/γ → α phase transformation of AgI,29 confirming the presence of this compound.
Synthesis and formation of “AgBa9(VO4)6I”
Powders of Ba3(VO4)2 and AgI were batched and reacted using standard solid-state ceramic techniques. The combined starting materials produced pale yellow powders, before reacting. After pressing into pellets and reacting, the resulting pellets were pale yellow with small dark inclusions, and yielded pale and canary yellow powders for those reacted in sealed tubes or air, respectively. For reactions performed in quartz tubes, no apparent sublimation of iodine from the reacted pellet was observed at the cooler end of the tube. The PXRD patterns of “AgBa9(VO4)6I” reacted in air and a sealed tube are shown in Fig. 4. The PXRD patterns of the resulting products were similar, irrespective of sealed tube or open reaction conditions. Phase analysis of each PXRD pattern identified major phases: Ba3(VO4)2 (trigonal R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) and β/γ-AgI (hexagonal P63mc/cubic F3m, respectively). Rietveld fitting of X-ray diffraction data of “AgBa9(VO4)6I” was effectively achieved assuming a mixture of Ba3(VO4)2, γ-AgI, and β-AgI, as shown in Fig. 5; the refined phase fractions were 92 ± 1%, 7 ± 1%, and 1.0 ± 0.1%, respectively.
m) and β/γ-AgI (hexagonal P63mc/cubic F3m, respectively). Rietveld fitting of X-ray diffraction data of “AgBa9(VO4)6I” was effectively achieved assuming a mixture of Ba3(VO4)2, γ-AgI, and β-AgI, as shown in Fig. 5; the refined phase fractions were 92 ± 1%, 7 ± 1%, and 1.0 ± 0.1%, respectively.
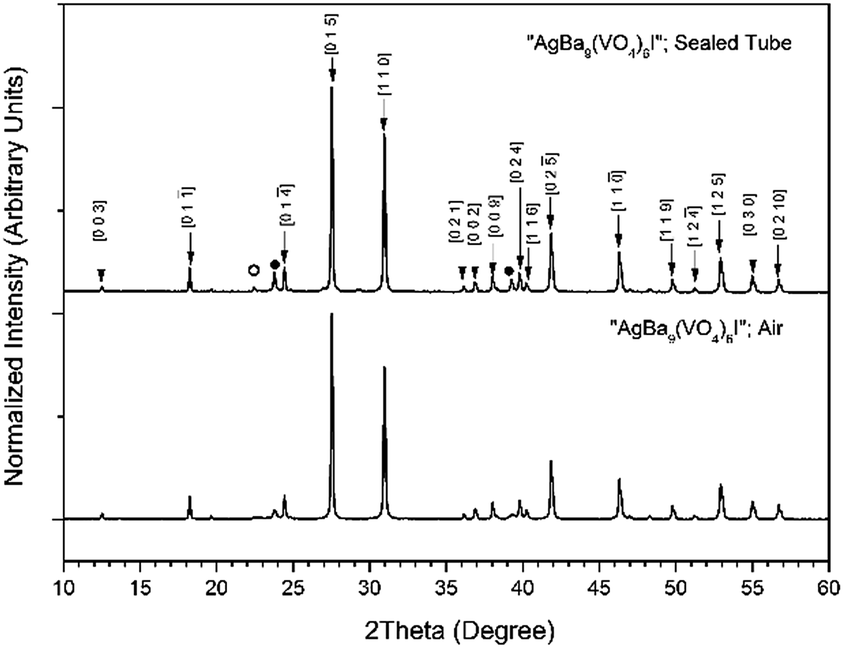 | ||
| Fig. 4 PXRD patterns of “AgBa9(VO4)6I” treated at 700 °C for 5 h in either air or a sealed quartz tube. Markers indicate peaks assigned to the Ba3(VO4)2 (labelled hkl Miller indices), β-AgI (○), and γ-AgI (●) phases. | ||
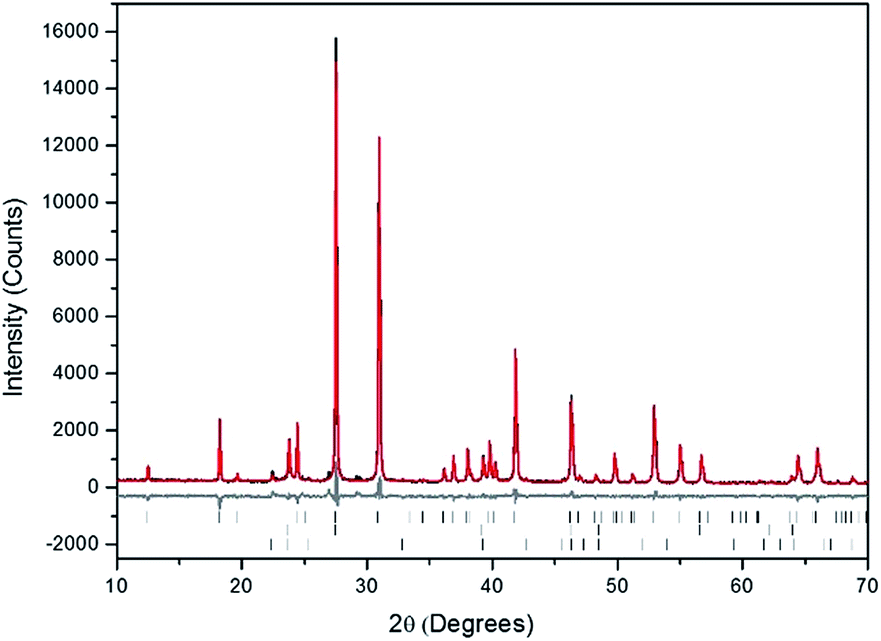 | ||
| Fig. 5 Rietveld analysis of PXRD data of “AgBa9(VO4)6I” (black) fit reacted at 700 °C in a sealed quartz tube; the data are shown in black, the fit is shown in red, and the difference plot in grey. Tick marks show allowed reflections of Ba3(VO4)2 (upper ticks) and γ-AgI (middle ticks), and β-AgI (lower ticks). | ||
Note, that although the crystal structure of “AgBa9(VO4)6I” was stated to be of rhombohedral symmetry, no space group was determined, and the compound was reported not to adopt an apatite related structure. Inspection of Table 3 shows the reported unit cell parameters of “AgBa9(VO4)6I” to be close to those determined for Ba3(VO4)2, the major phase in our reaction product, which adopts the trigonal space group R/m.30 The PXRD patterns acquired here for “AgBa9(VO4)6I” in both open and closed systems are comparable to those reported in the by Uno et al. for this purported phase.1 In these PXRD patterns, reflections are evident at 2θ = 23.7° and 39.3°, which may be attributed to γ-AgI (indexed as (111) and (022)) and 2θ = 22.4° to β-AgI (indexed as (010)), as highlighted in Fig. 5 here. These reflections are also apparent but unindexed in Fig. 1 of Uno et al.1 These data suggest that the compound identified by Uno et al. as “AgBa9(VO4)6I” was, in fact, Ba3(VO4)2.
The phase assemblage and element distribution for “AgBa9(VO4)6I” was similar to that observed in the Pb counterpart. In Fig. 6, the BSE-SEM image and corresponding EDX maps for V, Ba, I, and Ag are presented for “AgBa9(VO4)6I” reacted at 700 °C in a sealed quartz tube. From the BSE image, two major phases were identified, based on their differentiated contrasts. Element distribution maps were consistent with the BSE images, with a notable association of Ag and I in one phase and Ba and V in a second phase (Fig. S7†). These two phases can be interpreted as the AgI and Ba3(VO4)2 phases, respectively, identified by PXRD. Elemental analysis (Table 2) of the entire field of view in Fig. 6, yielded an elemental composition consistent with that of “AgBa9(VO4)6I” as reported in the literature and determined by the calculated stoichiometry.1 Once again, this information indicates that AgI incorporation into an apatite or other single phase product was not achieved. Small areas of Ag metal were also identified, indicative of decomposition of AgI but there was no evidence for the presence of an iodoapatite phase, as in the counterpart Pb system.
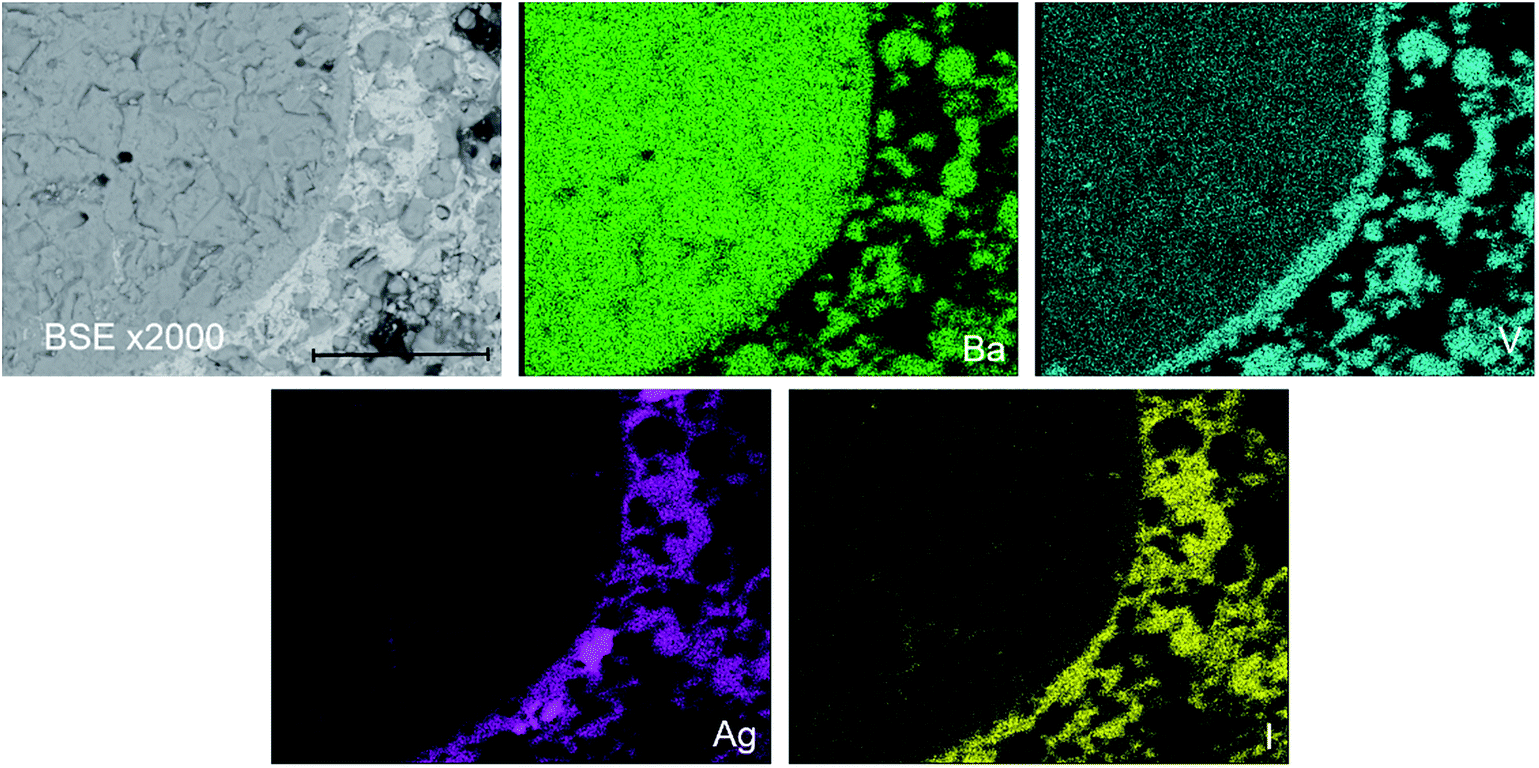 | ||
| Fig. 6 BSE-SEM image ×2000 magnification (scale bar length = 30 μm) and distribution of V (teal), Ba (green), I (yellow), and Ag (pink) in a sectioned pellet of “AgBa9(VO4)6I” reacted at 700 °C for 5 h in a sealed quartz tube. | ||
The TG-DTA analysis (Fig. S8†) of “AgBa9(VO4)6I” reacted at 700 °C for 5 h in a sealed quartz tube was characterized by a gradual weight loss, beginning at ∼680 °C in the TG curve. This is consistent with the reported thermal stability of purported “AgBa9(VO4)6I”, which was stated to decompose above ∼677 °C.1 The β/γ → α phase transformation of AgI was also identified in the DTA signal occurring at ∼152 °C,29 a further indication of the presence of unreacted AgI in the sample.
Conclusions
Syntheses of lead/barium vanadium iodoapatite and Ag–I bearing single phases, i.e., “AgM9(VO4)6I” (M = Ba, Pb) were attempted, but proved unsuccessful, contrary to previous literature reports.1 Reactions were carried out in either sealed quartz tubes or in air at elevated temperatures, which yielded similar results for each system. Notably, for each system investigated, AgI incorporation into an apatite, apatite-like, or alternative ternary phase was not observed.In these reactions, AgI was mostly unreactive and found as a secondary phase, responsible for the sequestration of the majority of iodine in the sample. However, lead iodovanadinite was identified in the Pb system from reactions performed in closed environments, most likely due to decomposition of AgI and reaction of liberated iodine with the β-Pb3(VO4)2. These results indicate that the stability and formation of AgI is favoured over the formation of a single phase apatite incorporating both Ag and I. Nevertheless, the observed formation of Pb9.85(VO4)6I1.7 suggests that AgI may be a suitable substrate for the synthesis of this phase as a sacrificial iodine source with stoichiometric quantities of PbO, V2O5 and Pb3(VO4)2. This hypothesis will be the subject of a future communication.
Furthermore, excluding Ag(IO3) and iodargyrite, the terrestrial mineral form of AgI, no other Ag/I-bearing monolith phases have been reported.8,31 However, Ag/lanthanide-containing Pb fluoro- and chloroapatites (i.e., Pb8AgLn (PO4)6X2 and Pb6Ag2Ln2(PO4)6X2; Ln = La, Nd, Eu and X = F, Cl) have been prepared,3 although no bromo- or iodo-analogues have been reported. Likewise, the endmember compound Ag2Pb8(PO4)6 and its analogue Ag2Pb8(AsO4)6, which exhibit the apatite structure with fully unoccupied halide channels, have also been synthesized;32,33 the vanadate-derivative Ag2Pb8(VO4)6 has yet to be discovered and could give further insight into the nature of these materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The corresponding author (EVJ) would like to thank the Engineering and Physical Sciences Research Council (EPSRC) for the financial support under grant ref. EP/M026566/1. Jong Heo was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT: Ministry of Science and ICT) (NRF-2015M2A7A1000191). Additionally, the authors would like to show gratitude for the financial support concerning the operation of MIDAS facilities at the University of Sheffield by the Department of Energy and Climate Change.References
- M. Uno, A. Kosuga, S. Masuo, M. Imamura and S. Yamanaka, J. Alloys Compd., 2004, 384, 300–302 CrossRef CAS.
- M. Pasero, A. R. Kampf, C. Ferraris, I. V. Pekov, J. Rakovan and T. J. White, Eur. J. Mineral., 2010, 22, 163–179 CrossRef CAS.
- I. Mayer, A. Semadja and V. J. Weiss, Solid State Chem., 1980, 34, 223–229 CrossRef CAS.
- I. Mayer, E. Fischbein and S. J. Cohen, Solid State Chem., 1975, 14, 307–312 CrossRef CAS.
- O. Terra, F. Audubert, N. Dacheux, C. Guy and R. J. Podor, J. Nucl. Mater., 2007, 366, 70–86 CrossRef CAS.
- J. Wang, Front. Earth Sci., 2015, 3, 1–11 CAS.
- E. E. Jay, P. C. M. Fossati, M. J. D. Rushton and R. W. Grimes, J. Mater. Chem. A, 2015, 3, 1164–1173 CAS.
- B. J. Riley, J. D. Vienna, D. M. Strachan, J. S. McCloy and J. L. Jerden Jr, J. Nucl. Mater., 2016, 470, 307–326 CrossRef CAS.
- X. Hou, V. Hansen, A. Aldahan, G. Possnert, O. C. Lind and G. Lujaniene, Anal. Chim. Acta, 2009, 26, 181–196 CrossRef PubMed.
- J. E. Moran, S. Oktay, P. H. Santschi and D. R. Schink, Environ. Sci. Technol., 1999, 33, 2536–2542 CrossRef CAS.
- Y. Koo, Y. Yang and K. Song, Prog. Nucl. Energy, 2014, 74, 61–70 CrossRef CAS.
- T. Imanaka and H. Koide, J. Environ. Radioact., 1986, 4, 149–153 CrossRef CAS.
- E. S. Gilbert, R. Tarone and E. J. Ron, J. Natl. Cancer Inst., 1998, 90, 1654–1660 CrossRef CAS PubMed.
- P. D. Wilson, The Nuclear Fuel Cycle from Ore to Waste, Oxford University Press, Oxford, 1996 Search PubMed.
- M. Uno, M. Shinohara, K. Kurosaki and S. J. Yamanaka, J. Nucl. Mater., 2001, 294, 119–122 CrossRef CAS.
- F. Lu, T. Yao, Y. Danon, J. Zhou, R. C. Ewing and J. Lian, J. Am. Ceram. Soc., 2015, 98, 2261–3366 Search PubMed.
- F. Audubert, J. M. Savariault and J. L. Lacout, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 271–273 Search PubMed.
- F. Lu, T. Yao, J. Xu, S. Scott, Z. Dong, R. C. Ewing and J. Lian, RSC Adv., 2014, 4, 38718–38725 RSC.
- Y. J. Suetsugu, J. Nucl. Mater., 2014, 454, 223–229 CrossRef CAS.
- M. Zhang, E. R. Maddrell, P. K. Abraitis and E. K. H. Salje, Mater. Sci. Eng., B, 2007, 137, 149–155 CrossRef CAS.
- M. Stennett, I. J. Pinnock and N. C. Hyatt, J. Nucl. Mater., 2011, 414, 352–359 CrossRef CAS.
- T. Yao, S. Scott, G. Xin, F. Lu and J. Lian, J. Am. Ceram. Soc., 2015, 98, 3733–3739 CrossRef CAS.
- T. Yao, L. Fengyuan, H. Sun, J. Wang, R. C. Ewing and J. Lian, J. Am. Ceram. Soc., 2014, 97, 2409–2412 CrossRef CAS.
- T. Sakurai, A. Takahashi, N. Ishikawa, Y. Komaki, M. Ohnuki and T. Adachi, Nucl. Technol., 1989, 85, 206–212 CrossRef CAS.
- T. Sakurai, A. Takahashi, N. Ishikawa, Y. Komaki, M. Ohnuki and T. Adachi, Nucl. Technol., 1992, 99, 70–79 CrossRef CAS.
- E. C. Buck, E. J. Mausolf, B. K. McNamara, C. Z. Soderquist and J. M. Schwantes, J. Nucl. Mater., 2016, 482, 229–235 CrossRef CAS.
- P. Garnier, G. Calvarin, J. F. Berar and D. Weigel, Mater. Res. Bull., 1984, 19, 407–414 CrossRef CAS.
- J. Goldstein, D. E. Newbury, P. Echlin, D. C. Joy, A. D. Romig Jr, C. E. Lyman, C. Fiori and E. Lifshin, Scanning Electron Microscopy and X-Ray Microanalysis, Plenum Press, New York and London, 2nd edn, 1992 Search PubMed.
- W. A. Bassett and T. Takahashi, Am. Mineral., 1965, 50, 1576–1594 CAS.
- P. Süsse and M. J. Buerger, Z. Kristallogr., 1970, 131, 161–174 CrossRef.
- L. L. Burger, R. D. Scheele and K. D. Wiemers, Selection of a Form for Fixation of Iodine-129, PNL-4045, Battelle Pacific Northwest Laboratory, Richland, WA, 1981 Search PubMed.
- G. Engel and H. Jäckle, Z. Anorg. Allg. Chem., 1979, 448, 71–78 CrossRef CAS.
- R. Ternane, M. Ferid, N. Kbir-Ariguib and M. Trabelsi-Ayedi, J. Alloys Compd., 2000, 308, 83–86 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, PXRD, and SEM/EDX of “AgPb9(VO4)6I” from PbO, V2O5, and AgI. Additional PXRD figures, SEM images, EDX spectra, and TG-DTA measurements for “AgM9(VO4)6I (M = Pb, Ba)”. See DOI: 10.1039/c7ra09313f |
| This journal is © The Royal Society of Chemistry 2017 |