
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilica film deposited on diamond-structured polymer microlattices by dip coating
Fang Wei ab,
Yongfeng Zhouab,
Qinglin Yang*ab,
Lin Guoab and
Lei Jiangab
ab,
Yongfeng Zhouab,
Qinglin Yang*ab,
Lin Guoab and
Lei Jiangab
aKey Laboratory of Bio-inspired Smart Interfacial Science and Technology of Ministry of Education, Beijing, China. E-mail: yangql@buaa.edu.cn
bSchool of Chemistry and Environment, Beihang University, Beijing 100191, China
First published on 29th November 2017
Abstract
The specific strength of lightweight cellular materials mainly depends on their structure and composition. In this study, polymer microlattices with diamond structures were fabricated by 3D printing. Then, silica materials with high strength were covered on the polymer microlattices using a dip-coating method, and we obtained polymer/SiO2 microlattices, which had the properties of low density, high compressive strength, and good thermostability. Moreover, three types of silica films with different thickness, i.e., 10.1 μm, 18.3 μm, and 23.6 μm, were deposited on the microlattices. The compressive strength of the polymer/SiO2 microlattices can be improved upon increasing the thickness of the silica coating. The thermostability of the polymer can be enhanced after coating with the silica materials and was improved more dramatically with a thicker coating. Light hollow-tube SiO2 microlattices with a density of 14.7 kg m−3 were also fabricated by calcining the polymer template at high temperatures.
Introduction
As a type of lightweight cellular material, microlattice materials have periodicity and ordered structures and have recently attracted significant attention in the field of lightweight materials. Lightweight materials have properties of good storage,1,2 excellent noise reduction,3,4 prominent absorption and shielding,5 ideal permeability and adsorption,6 shape memory,7,8 effective thermal conductivity,9 and strong energy absorption,10,11 and they have a wide range of applications in aviation, aerospace, and architecture. Existing lightweight materials can be divided into approximately three categories according to the structure of the materials,12 i.e., aerogels,13–15 foams,16–18 and microlattices.9,19–27 When compared with that of other lightweight cellular materials, the pore distribution of microlattice materials is very ordered, and the greatest advantage of microlattice materials lies in the periodicity of their structures as well as their manual design. As a result, the strength of the materials will be higher. By researching the structure of the lattice, we can know that the density and mechanical performance of the lattice depends strongly on the cell geometry including shape, size, and structural hierarchy.28–31 Among the common topologies used to produce structured materials, the diamond structure is preferred for attaining material with high strength and low density due to its special configuration, which exhibits high spatial symmetry and strong stability to resist deformation.11,32,33The strength of lattice materials not only depends on the architecture but also on the constituent materials. The materials that make up the architecture are polymers. There were three reasons to select polymers over other materials. First, polymers have a wide range of applications in research. Thus, we wanted to investigate that the changes in their mechanical properties and thermal stability after being coated using a low-cost and simple method, to expand their applications. Second, polymers can be calcined, and we aimed to construct a perfect hollow tube structure to investigate its functionality to obtain materials with a perfect combination of mechanical structure and function. Ceramics are materials with high strength in inorganic materials. Third, the polymer materials that we used to fabricate the architecture by 3D printing have better precision,34,35 which is beneficial for the subsequent coating process, and it is easier to obtain a smooth coating. Silica is an important inorganic material, which belongs to atomic crystal and possesses excellent mechanical and thermal properties due to the diamond-like structure formed by the Si–O covalent bonds.36 To protect the surface of the polymeric materials and improve other properties of the materials, a ceramic film can be coated on the surface of polymeric materials to obtain new types of composite, which have attracted significant attention; moreover, some meaningful research has been carried out in this regard recently.37–41 A biomimetic study has shown that hybrid materials consisting of an inorganic shell and a polymer core exhibit higher mechanical performance than the individual constituent materials. Therefore, by fabricating polymer/ceramic composites, the materials can significantly improve their mechanical properties.25,42–46
Surface coating technology is an important way to modify the surface of materials, which plays an important role in improving material usability, expanding the scope of a material and realizing the functional combination of materials. There are many ways, such as electroless plating,1,5,22,24,26,29,47 electroplating,1,5,47 atomic layer deposition,23,48 plasma vapor deposition,25 and dip-coating,27 to deposit films on microlattice materials. As a kind of mature technology with low cost, the dip-coating method can be used to produce high quality films over large areas, and thus, it was chosen as the preferred coating method in this study. Based on previous studies,12,25,26 in this study, silica films were deposited on the surface of diamond-structured polymer microlattices via a dip-coating method, and the resulting lightweight diamond-structured microlattice polymer/silica thin film composite materials with certain mechanical strength and thermal stability were successfully fabricated.
Experimental
Fabrication of the 3D polymer microlattices
3D polymer microlattices with a diamond structure were designed using computer aided design (CAD) software (Solid Works v2011 sp4.0) and manufactured by 3D printing. A 3D printer (MiiCraft, Young Optics Inc) based on stereolithography was used in this study. The stereolithography resin used in this study was acrylate-based EX-200 resin (Yiwu Realmaker Electronic Technolgy Co. Ltd). The size of the resulting 3D microlattice was approximately 3 × 2 × 1 cm3.The process began with a photosensitive polymer resin bath. The 3D model was first converted into an STL file format and digitally sliced into a sequence of 2D layers, which were projected to solidify the liquid photosensitive resin. After one layer was solidified on the bottom of the tank, the object being built was recoated with a second layer of fresh resin, and then, solidification of the next layer began. These steps were repeated until the whole object was fabricated.
Fabrication of the polymer/SiO2 microlattices
The polymer scaffolds were conformally coated with SiO2 layers via dipping–withdrawing cycles under ambient conditions. The precursor was a model glass of SiO2–Na2O (purity > 95%, Haihua Co., Shandong, China) with a Na2O concentration of 29 mol% and SiO2 concentration of 71 mol%. It has been denoted silica in the study since its main ingredient is silica. The withdrawal speed was 10 mm min−1, and the dipping time was 15 min. The polymers coated with SiO2 were dried at 80 °C using an oven for 60 min in air after each coating was finished. The thickness of the SiO2 layers was controlled by repeating the cycle from dipping to heat treatment. In this study, to ensure that the densities of the microlattice materials are within the density range of lightweight materials, we coated the material with 1–3 layers.Fabrication of the hollow-tube SiO2 microlattices
Hollow-tube SiO2 microlattices were obtained by calcining the polymer/SiO2 microlattices to remove the polymer template using a tube furnace (RT 50-250, Nabertherm, Germany). The polymer/SiO2 microlattices were heated to 500 °C at a heating rate of 10 °C min−1 and kept at this temperature for 120 min to completely remove the polymer template under a nitrogen atmosphere. The calcination temperature depends on the thermal decomposition temperature of the template.Characterization and performance testing of the microlattice materials
Environmental scanning electron microscopy (ESEM, FEI Quanta-250 FEG) was conducted to investigate the surface morphology and measure layer thickness of the resultant microlattices at an acceleration voltage of 10 kV.X-ray diffraction (XRD) (D/MAX-1200, Rigaku Denki Co. Ltd, Japan) was used to identify the phase composition present in the silica layer using Cu Kα radiation (λ = 1.5406 Å) at 40 kV and 30 mA in the 2θ range of 10–80°.
Energy-dispersive X-ray spectrometry (EDS) was also used to analyze the composition of the inorganic SiO2 coating of the polymer/SiO2 microlattices.
TGA measurements were used to characterize the thermal stability of the polymer/silica composites in terms of their thermal degradation under a nitrogen atmosphere at a heating rate of 10 °C min−1 and to evaluate the effect of the silica films on the degradation of the matrix.
Compression tests were performed using a servo-electric INSTRONR 5565 frame with a 5 kN load cell controlled using the Bluehill 2.0 software. All the microlattices were tested between the compression plates at a loading displacement rate of 0.5 mm min−1, and the loading area was 3 × 2 cm2.
The compressive strength was determined using the initial peak strength.
The apparent density was calculated by measuring the weight and dimensions of the microlattices.
Results and discussion
The 3D polymer/SiO2 microlattice fabricated in this study is illustrated in Fig. 1. The microlattice was designed using a series of regular diamond cell connected at their vertices. Each diamond cell was made up of four 2 mm-long and 0.6 mm diameter struts intersecting at the angle of 109° 28' (Fig. 1a–c). The length of a strut was determined as node-to-node distance (L) and the diameter of a strut was defined as the average diameter (D). The strut diameter of the polymer/SiO2 was in accordance with that of the design (Fig. 1e). Fig. 1f shows a coating thickness (t) of 10.1 μm. The coating was uniform, as observed in Fig. 1e and f. Fig. 1g shows the hollow-tube SiO2 microlattice, whose density is only 14.7 kg m−3, which belongs to ultralight class of materials and can be fully supported on a dandelion. The average diameter of the hollow-tube SiO2 was about 547 μm, which was lower than that of solid SiO2, and the SiO2 film was not as uniform as solid SiO2 due to the slight shrinkage that occurred after the thermal treatment. The thickness was unchanged for the hollow-tube SiO2 microlattice when compared with that of the solid SiO2 microlattice (Fig. 1i). | ||
| Fig. 1 The architecture of the polymer/SiO2 microlattices: (a–e) CAD images of the diamond structure design: (a) the diamond-structured microlattice; (b) a magnified section of (a); (c) the unit cell; (d) a digital image of the 3D printed polymer microlattice coated with SiO2 via dip-coating; and (e) an SEM image of a strut in (d). (f) An SEM image of the layer thickness of a strut in (d). (g) A digital image of the hollow-tube SiO2 microlattice (ρ = 14.7 kg m−3) supported on a dandelion. (h) An SEM image of a strut of (g). (i) An SEM image of the layer thickness of a strut in (h). | ||
The XRD patterns show that all the samples exhibit the same peak at around 24°, which corresponds to amorphous silica, indicating that the main constituent of the coating layers is amorphous SiO2 (Fig. 2a).49 EDS shows that the layer composition is 71% SiO2 and 29% Na2O (Fig. 2b). A small amount of the element C present in the EDS (Fig. 2b) may be derived from the polymer core of the polymer/SiO2 microlattices. The element C presented in Fig. 2c was higher than that observed in Fig. 2b, which might be derived from the calcined products of the polymer template.
 | ||
| Fig. 2 (a) The XRD patterns obtained for SiO2 gel at 25 °C, the SiO2 coating dried at 80 °C and the SiO2 coating calcined at 500 °C. (b) The EDS patterns of the polymer/SiO2 microlattices. (c) The EDS patterns of the hollow-tube SiO2 microlattices. | ||
The stress–strain curves of the pure polymer, polymer/silica composites, and hollow-tube silica microlattice material are shown in Fig. 3. It can be seen that the compression strength of the pure polymer microlattice is 205 kPa, and the compression stress of the polymer/silica composites with different coating thicknesses of silica film (10.1 μm, 18.3 μm, and 23.6 μm) is higher than that found for the pure microlattice and far higher than that of the hollow-tube silica microlattice material. The compressive strengths of the coated microlattices are higher than that of the pure polymer microlattice because silica is stiffer than EX-200 polymer and carries the bulk of the externally applied load. The compressive strength of the polymer/SiO2 microlattices increases significantly upon increasing the thickness of the coating. When the coating thickness was 10.1 μm, the maximum compressive strength was 286 kPa, which was 1.40 times that of the pure polymer microlattice. When the coating thickness was 18.3 m, the maximum compressive strength was 339 kPa, which was 1.65 times that of the pure polymer microlattice. The compressive strength of the hybrid microlattices increased to 402.6 kPa, which was 1.97 times that of the pure polymer microlattice, when the coating thickness was 23 μm. In addition, it was demonstrated that the mechanical properties of the microlattices could be significantly enhanced at a lower density by introducing a SiO2 coating. The reinforcement mechanism can be considered as the stiff SiO2 coating carrying the compressive forces, whereas the light polymeric core serves to resist early face cracking and improve the toughness. The compressive strength of the hybrid structures primarily depends on the thickness of the coating, which is the only variable factor, because the lattices possess the same structure including geometric configuration and elemental size. However, the strength of the polymer/SiO2 composites increases to just less than twice than that of the pure polymer; if we optimize the related parameters, we can obtain a higher mechanical strength. This is one of our goals in our further research to use an ideal oxide precursor with better mechanical properties and obtain composites with obvious mechanical properties. Furthermore, the hierarchy has an important effect on the strength; thus, the increased thickness of the SiO2 coating leads to an increase in the strength of the polymer/SiO2 microlattices.
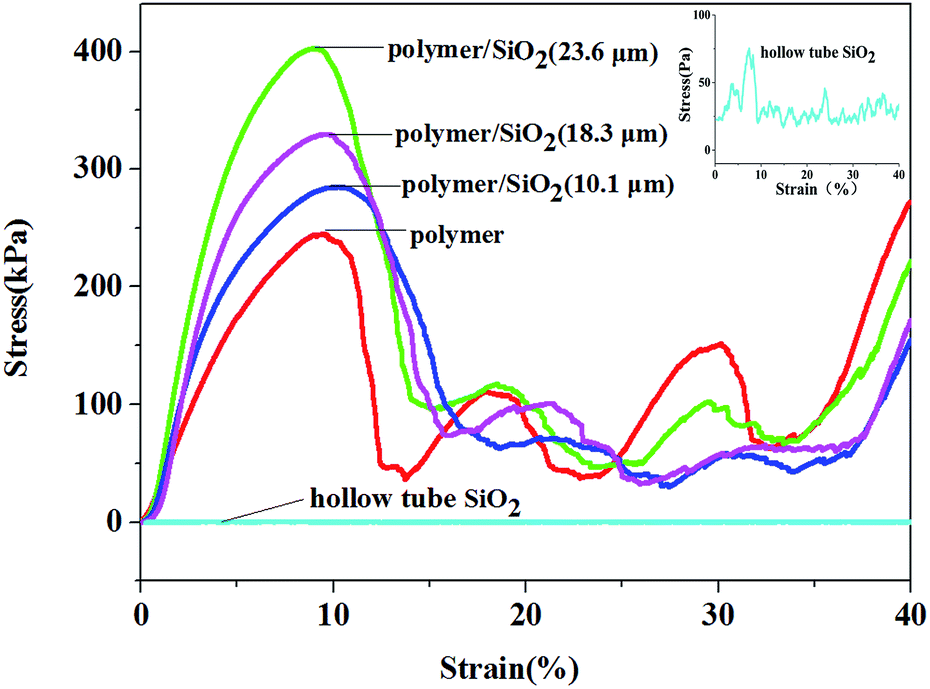 | ||
| Fig. 3 The stress–strain curves obtained for the polymer, polymer/SiO2 composites, and hollow tube silica microlattices. The magnification curve obtained for the hollow tube silica microlattice is in the top right corner. | ||
All samples exhibit a similar mechanical response to compression, as shown in Fig. 3, due to the chosen geometry and architecture. Generally, the samples deformed elastically over the first 3–8% strain due to the low densities and long slender trusses of the diamond structures, followed by a peak of stress between 8% and 10% strain, which corresponds to buckling of the top and bottom layers of the cells. After this peak, the stress declines rapidly before it increases again. This suggested that the structure was fully collapsed sequentially as the unit cells buckled layer by layer. Then, the structure of the polymer and polymer/SiO2 composites was totally destroyed, and they just simply stacked together; thus, the stress strength of the polymer and polymer/SiO2 composites had no discipline, and there was no comparability between them after their collapse.
The compressive strength of the hollow-tube SiO2 microlattice was about 90 Pa, which was far lower than that found for the polymer/SiO2 microlattices. Therefore, the polymer/SiO2 composite microlattices exhibit better mechanical performance than the pure polymer lattice and hollow-tube SiO2 microlattice. The parameters and apparent densities are provided in Table 1.
| Samples | SiO2 thickness [μm] | Density [kg m−3] | Compressive strength [kPa] | Specific compressive strength [kPa m3 kg−1] |
|---|---|---|---|---|
| Polymer | 0 | 85.5 | 203.8 | 2.384 |
| Polymer/SiO2 | 10.1 | 103.1 | 285.2 | 2.766 |
| Polymer/SiO2 | 18.3 | 115.09 | 336.5 | 2.924 |
| Polymer/SiO2 | 23.6 | 122.46 | 403.2 | 3.293 |
| Hollow-tube SiO2 | 10.1 | 14.7 | 0.090 | 0.006 |
To investigate the thermal stability of the polymer and polymer/SiO2 composites with different coating thicknesses of the silica film (10.1 μm, 18.3 μm, and 23.6 μm), TGA and DTG were conducted. The materials were heated to 800 °C from room temperature. It was obvious that all the samples degraded between 300 °C and 550 °C. This temperature range was chosen for analysis. The TGA and DTG curves obtained for all the samples are shown in Fig. 4, and the corresponding TGA parameters are given in Table 2. For the polymer/silica composites, as shown in Fig. 4 and Table 2, the thermal degradation temperatures, such as the initial temperature of degradation (Tonset), the degradation temperature at 50% weight loss (T0.5), and the degradation temperature at maximum weight loss rate (Tmax), increase continuously and dramatically as a function of the silica coating thickness. The maximum improvement in Tonset, T0.5, and Tmax was 59.58 °C, 43.23 °C, and 37.72 °C, respectively. The maximum weight loss rate (Vmax) given in Table 2 remained hardly unchanged for polymer/SiO2 (10.1 μm) and polymer/SiO2 (18.3 μm) when compared with the case of the pure polymer, but increased obviously for polymer/SiO2 (23.6 μm); this might indicate that the thermal degradation of the polymer was accelerated by the thicker coating of SiO2. The residue at 800 °C increased from 4.53% to 29.38% and remained unchangeable above 500 °C; this suggested that the polymer was almost completely decomposed in the atmosphere at 500 °C and the residue was mainly silica. In view of these results, the thermal stability of the polymer/SiO2 composites increased, and the thermal stability was enhanced upon increasing the thickness of the silica coating.
 | ||
| Fig. 4 The TGA and DTG curves obtained for the polymer/SiO2 composites compared with the pure polymer. | ||
| Sample | TGA results | ||||
|---|---|---|---|---|---|
| Tonset (°C) | T0.5 (°C) | Tmax (°C) | Vmax (% per °C) | % Residue at 800 °C | |
| Polymer | 307.6 | 402.6 | 410.1 | 1.59 | 4.5 |
| Polymer/SiO2 (10.1 μm) | 332.9 | 411.1 | 418.6 | 1.54 | 12.8 |
| Polymer/SiO2 (18.3 μm) | 363.1 | 426.1 | 428.6 | 1.58 | 14.8 |
| Polymer/SiO2 (23.6 μm) | 367.1 | 445.8 | 445.8 | 1.91 | 29.4 |
In summary, we have demonstrated a feasible method for the synthesis of lightweight diamond-structured microlattice polymer/silica thin film composite materials by combining the methods of 3D printing and dip-coating. With regard to the thickness of the coating, the polymer/silica lattices exhibits higher strength than the pure polymer lattice and hollow-tube SiO2 lattices. The mechanical properties of the obtained composite microlattices improved upon increasing the thickness of the SiO2 coating. When the coating thickness was 23.6 μm, the compressive strength of the hybrid microlattices approached 402.6 kPa, which was 1.97 times that of the pure polymer microlattice. The corresponding density was 122.46 kg m−3, and the specific compressive strength was 3.293 kPa m3 kg−1. This study enables the mechanical properties of the polymer microlattices to be improved significantly within relativity low densities. Moreover, the thermal stability of the polymers increased with the silica coating, and the thermal stability of the polymer/silica thin film composites enhanced with an increase in the thickness of the SiO2 coating. When the coating thickness was 23.6 μm, the maximum improvement for Tonset, T0.5, and Tmax were 59.6 °C, 43.2 °C, and 37.7 °C, respectively. Hollow-tube SiO2 microlattices were also fabricated by calcining the polymer template at high temperatures, and the mechanical property test was carried out. The polymer/silica composites microlattices are expected to have a wide range of applications in the weight reduction of space vehicles, sound insulation materials, and so on.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support provided by the National Natural Science Foundation of China (Project Number: 51372010).Notes and references
- L. Salari-Sharif, T. A. Schaedler and L. Valdevit, J. Mater. Res., 2014, 29, 1755 CrossRef CAS.
- L. Wu, W. Li, P. Li, S. Liao, S. Qiu, M. Chen, Y. Guo, Q. Li, C. Zhu and L. Liu, Small, 2014, 10, 1421 CrossRef CAS PubMed.
- J. Bian, Prog. Mater. Sci., 2001, 46, 559 CrossRef.
- Z. Xu, H. Sun and C. Gao, APL Mater., 2013, 1, 030901 CrossRef.
- T. A. Schaedler, C. J. Ro, A. E. Sorensen, Z. Eckel, S. S. Yang, W. B. Carter and A. J. Jacobsen, Adv. Eng. Mater., 2014, 16, 276 CrossRef CAS.
- Y. Wei, X. Li, Z. Xu, H. Sun, Y. Zheng, L. Peng, Z. Liu, C. Gao and M. Gao, Polym. Chem., 2015, 6, 973 RSC.
- V. P. Shastri, I. Martin and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 1970 CrossRef CAS.
- P. Singhal, J. N. Rodriguez, W. Small, S. Eagleston, J. V. Water, D. J. Maitland and T. S. Wilson, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 724 CrossRef CAS PubMed.
- N. T. Trifale, E. A. Nauman and K. Yazawa, ACS Appl. Mater. Interfaces, 2016, 8, 35534 CAS.
- Z. Liu, W. Chen, J. Carstensen, J. Ketkaew, R. M. O. Mota, J. K. Guest and J. Schroers, Acta Mater., 2016, 105, 35 CrossRef CAS.
- F. Côté, V. S. Deshpande, N. A. Fleck and A. G. Evans, Int. J. Solids Struct., 2006, 43, 6220 CrossRef.
- Y. Gao, Y. F. Zhou, Q. L. Yang, L. Guo and L. Jiang, Progress in Chemistry, 2015, 27, 1714 Search PubMed.
- M. Mecklenburg, A. Schuchardt, Y. K. Mishra, S. Kaps, R. Adelung, A. Lotnyk, L. Kienle and K. Schulte, Adv. Mater., 2012, 24, 3486 CrossRef CAS PubMed.
- H. Sun, Z. Xu and C. Gao, Adv. Mater., 2013, 25, 2554 CrossRef CAS PubMed.
- G. Nystrom, M. P. Fernandez-Ronco, S. Bolisetty, M. Mazzotti and R. Mezzenga, Adv. Mater., 2016, 28, 472 CrossRef PubMed.
- B. C. Tappan, M. H. Huynh, M. A. Hiskey, D. E. Chavez, E. P. Luther, J. T. Mang and S. F. Son, J. Am. Chem. Soc., 2006, 128, 6589 CrossRef CAS PubMed.
- J. Yin, X. Li, J. Zhou and W. Guo, Nano Lett., 2013, 13, 3232 CrossRef CAS PubMed.
- N. Chen and Q. Pan, ACS Nano, 2013, 7, 6875 CrossRef CAS PubMed.
- J. Xiong, R. Mines, R. Ghosh, A. Vaziri, L. Ma, A. Ohrndorf, H. J. Christ and L. Wu, Adv. Eng. Mater., 2015, 17, 1253 CrossRef CAS.
- M. G. Rashed, M. Ashraf, R. A. W. Mines and P. J. Hazell, Mater. Des., 2016, 95, 518 CrossRef CAS.
- X. Li and H. Gao, Nat. Mater., 2016, 15, 373 CrossRef CAS PubMed.
- X. Wang, Q. Guo, X. Cai, S. Zhou, B. Kobe and J. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 2583 CAS.
- L. R. Meza, S. Das and J. R. Greer, Science, 2014, 345, 1322 CrossRef CAS PubMed.
- T. A. Schaedler, A. J. Jacobsen, A. Torrents, A. E. Sorensen, J. Lian, G. R. Greer, L. Valdevit and W. B. Carter, Science, 2011, 334, 962 CrossRef CAS PubMed.
- Y. F. Zhou, C. Z. Yao, Q. L. Yang, L. Guo and L. Jiang, Adv. Eng. Mater., 2016, 18, 236 CrossRef CAS.
- J. Xu, Y. Gao, H. Huang, Q. L. Yang, L. Guo and L. Jiang, Sci. China: Chem., 2016, 59, 1632 CrossRef CAS.
- A. J. Jacobsen, S. Mahoney, W. B. Carter and S. Nutt, Carbon, 2011, 49, 1025 CrossRef CAS.
- T. Tancogne-Dejean, A. B. Spierings and D. Mohr, Acta Mater., 2016, 116, 14 CrossRef CAS.
- J. Rys, L. Valdevit, T. A. Schaedler, A. J. Jacobsen, W. B. Carter and J. R. Greer, Adv. Eng. Mater., 2014, 16, 889 CrossRef CAS.
- H. Wadley, Compos. Sci. Technol., 2003, 63, 2331 CrossRef CAS.
- T. A. Schaedler and W. B. Carter, Annu. Rev. Mater. Res., 2016, 46, 187 CrossRef CAS.
- S. Castilloramírez, J. Corander, P. Marttinen and M. Wang, Rapid Prototyp. J., 2012, 18, 49 CrossRef.
- Q. Zheng, S. Ju and D. Jiang, Compos. Struct., 2014, 109, 23 CrossRef.
- D. A. Türk, F. Brenni, M. Zogg and M. Meboldt, Mater. Des., 2017, 118, 256 CrossRef.
- J. Shin, R. S. Sandhu and G. Shih, J. Digit. Imag., 2017, 30, 572 CrossRef PubMed.
- W. Chen, K. Soshu and M. Yoshinari, J. Am. Ceram. Soc., 2007, 90, 2078 CrossRef CAS.
- K. Sun, P. Xie, Z. Wang, T. Su, Q. Shao, J. Ryu, X. Zhang, J. Guo, A. Shankar, J. Li, R. Fan, D. Cao and Z. Guo, Polymer, 2017, 125, 50 CrossRef CAS.
- K. Sun, Z. Zhang, R. Fan, M. Chen, C. Cheng, Q. Hou, X. Zhang and Y. Liu, RSC Adv., 2015, 5, 61155 RSC.
- K. Sun, R. Fan, Y. Yin, J. Guo, X. Li, Y. Lei, L. An, C. Cheng and Z. Guo, J. Phys. Chem. C, 2017, 121, 7564 CAS.
- P. Xie, K. Sun, Z. Wang, Y. Liu, R. Fan, Z. Zhang and G. Schumacher, J. Alloys Compd., 2017, 725, 1259 CrossRef CAS.
- K. Sun, Z. D. Zhang, L. Qian, F. Dang, X. H. Zhang and R. H. Fan, Appl. Phys. Lett., 2016, 108, 061903 CrossRef.
- J. Bauer, S. Hengsbach, I. Tesari, R. Schwaiger and O. Kraft, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2453 CrossRef CAS PubMed.
- K. Tushtev, M. Gonsior, M. Murck, G. Grathwohl and K. Rezwan, Adv. Eng. Mater., 2014, 16, 156 CrossRef CAS.
- C. A. Wang, Y. Huang, Q. Zan, H. Guo and S. Cai, Mater. Sci. Eng., C, 2000, 11, 9 CrossRef.
- M. Ashby, J. Am. Ceram. Soc., 2011, 94, s3 CrossRef CAS.
- L. J. Bonderer, A. R. Studart and L. J. Gauckler, Science, 2008, 319, 1069 CrossRef CAS PubMed.
- A. Torrents, T. A. Schaedler, A. J. Jacobsen, W. B. Carter and L. Valdevit, Acta Mater., 2012, 60, 3511 CrossRef CAS.
- X. Zheng, H. Lee, T. H. Weisgraber, M. Shusteff, J. DeOtte, E. B. Duoss, J. D. Kuntz, M. M. Biener, Q. Ge, J. A. Jackson, S. O. Kucheyev, N. X. Fang and C. M. Spadaccini, Science, 2014, 344, 1373 CrossRef CAS PubMed.
- V. Malinovschi, A. Marin, M. Mihalache and I. Iosub, Surf. Coat. Technol., 2016, 296, 96 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |