
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePromotion effect of water in catalytic fireplace soot oxidation over silver and platinum†
O. A. Shromova,
N. M. Kinnunen*,
T. A. Pakkanen and
M. Suvanto *
*
Department of Chemistry, University of Eastern Finland, P. O. Box 111, FI-80100, Joensuu, Finland. E-mail: mika.suvanto@uef.fi; niko.kinnunen@uef.fi
First published on 28th September 2017
Abstract
Fireplaces are one of the largest sources of soot emissions. One possible approach to reduce wood-based soot emission is to use catalysts that have been designed for fireplace applications. This study compares the activities of silver and platinum catalysts in the oxidation of real fireplace soot in different gas atmospheres. Another goal is to examine the effects of water on the catalyst activity depending on the moisture content in the reaction gas. The study shows that the gas feed composition affects the performance of the catalyst. Unlike platinum, the silver catalyst directly oxidizes the soot in the presence of air and NO. The addition of water into the air promotes the soot oxidation activity of both the silver and platinum catalysts. The catalysts improve their activity with the increase in water content. For both the silver and platinum catalysts, the partial dissociation of water with oxygen and the formation of hydroxyls are promoted, which oxidize soot. Thermal ageing shows that silver has a higher thermal stability and retains its oxidation activity for longer than platinum under all of the applied conditions. Silver is a more suitable option for the catalytic oxidation of soot emission of fireplaces.
1. Introduction
Over the last few decades, the issue of soot emissions has bothered many researchers because soot contributes to global warming1,2 and harmfully affects human health.3–9 To eliminate soot emissions, one possible solution is the use of catalysts. However, in the development of efficient soot oxidation catalysts, various sources of soot must be considered because the structure and composition of soot strongly depend on the type of fuel.5,10–15 For example, there is a clear difference between wood- and diesel-originated soot. Specific compounds in wood, such as lignin and its derivatives, which are produced during burning, provide a high quantity of oxygenated species in the soot structure.12–15 Wood soot has more amorphous structures than diesel soot.5,11,13 Furthermore, wood soot significantly differs in the composition of polycyclic aromatics and other hydrocarbons adsorbed on the soot surface.10Currently, platinum and silver are known components of catalysts used to eliminate particulate matter (PM) emissions from diesel engines. In particular, platinum is a widely used component of commercial catalysts in diesel particulate filters.16 Platinum indirectly oxidizes soot through the formation of nitrogen dioxide from nitrogen monoxide, which is present in considerable amounts in diesel exhaust gases.16–18 However, platinum also assists soot oxidation in air through the “spillover effect”.19,20 Uchisawa et al.21,22 reported that the addition of water promoted soot oxidation over platinum only in the presence of nitrogen monoxide in the gas feed mixture.
As an alternative candidate, silver has attracted the attention of many researchers because of its low cost and comparably high activity in oxidation processes. Most investigations on silver focus on diesel soot combustion in air,23–26 in which silver exhibits an even higher activity than platinum and palladium.27 However, in the presence of NO in the gas mixture, silver catalysts show comparably high activity only when the silver species are presented as nitrates.28 Despite significant development in the field of PM emission reduction in the car industry, the elimination of soot emissions from fireplaces and residential wood combustors requires thorough investigation. Only a few researchers have attempted to apply catalysts to reduce the emissions of wood soot and other products of incomplete combustion. For example, a metal wire mesh covered with platinum and palladium29 and a catalytic monolith covered with different La–Cr perovskite catalysts doped with gold30 have been reported.
The lack of information on wood-based soot emission removal catalysis motivates us to examine silver and platinum catalysts, which can be subsequently applied to oxidize soot in actual fireplaces. We aim to compare silver and platinum catalysts in different reaction gas mixtures to mimic realistic conditions. More precisely, the study answers the following questions. Are there any differences between the reaction pathways of silver and platinum with a change in the gas feed composition? How does water affect the catalyst activity, and what is the influencing mechanism of water? Which catalyst is more thermally durable? And finally, what type of catalyst is the best option for application in fireplaces?
2. Experimental
2.1. Soot material
Real soot, which was produced during wood burning in a fireplace, was used for all of the activity experiments. Tulikivi Oyj (Finland) provided the soot for the experiments. Elemental analysis showed that the soot contained ∼50 wt% of carbon, ∼4 wt% of hydrogen, ∼2.2 wt% of nitrogen and ∼0.7 wt% of sulfur. The fireplace soot also contained approximately 30 wt% of inorganic residues, which mainly consisted of K, Na, Ca, Mg, Zn, Si and O. The last 13 wt% could be attributed to oxygen of the organic matter in the soot.2.2. Catalyst preparation and treatment
The catalysts were prepared via impregnation by mixing a lanthanum-stabilized aluminium oxide support material with an aqueous solution of a AgNO3 precursor (63 wt% Ag, Alfa Aesar, Germany) or with a 30 vol% propionic acid–water solution of the C10H14O4Pt precursor (48 wt% Pt, Alfa Aesar, Germany). The loadings of active components were 1 and 2 wt% for both the silver and platinum catalysts. The catalyst components were impregnated by mixing for 24 hours at room temperature. Then the solvent was evaporated by heating in a sand bath. Finally, the catalysts were dried in an oven by slow heating (heating rate: 2.5 °C min−1) to avoid damage inside the pores. The catalysts were calcined at 600 °C for 4 hours in static air. After calcination, half of the prepared catalyst was aged at 1000 °C for 4 hours in static air to study the effect of time and temperature on the catalytic properties. An additional series of silver catalysts was prepared by an identical procedure. The loadings of the active component for this series were 2, 5 and 7 wt%.2.3. Catalyst characterization and catalytic activity evaluation
Powder X-ray measurements were performed to identify the present phases of the catalyst components and determine the Pt and Ag crystallite sizes using a Bruker-AXD D8 Advance device with Cu Kα radiation. The diffraction patterns were recorded at 2θ values of 25–85° with a scanning step of 0.08°.The specific surface areas were calculated from the adsorption of nitrogen and analysis of the nitrogen adsorption/desorption curves, measured with a Quantachrome autosorb iQ/ASiQwin device. The specific surface areas were evaluated based on the Brunauer–Emmett–Teller theory (multipoint BET). The pore volume and pore size distributions were determined by the NLDFT method. The total pore volume was determined from the adsorption curve at a relative pressure of P/P0 = 0.995. The average pore size was estimated based on the Kelvin equation. Prior treatment of the samples included heating them at 350 °C under a vacuum for 150 min to remove moisture.
The morphology of the catalyst surface was characterized using a Hitachi S4800 FE-SEM. Measurements were carried out at a working distance of 15 mm and an accelerating voltage of 20 kV. Detailed chemical analysis of the catalyst surface was performed with an energy dispersive X-ray spectrometer (Thermo Electron EDS).
The soot oxidation activities of the fresh and aged catalysts were measured using a batch flow reactor. The experiments were performed with tight contact between the catalyst and the soot. The contact was reached using a ball mill. The mixture of the soot and catalyst in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 was packed into a reactor in two layers, which were separated by quartz wool. Four different compositions of gas feed mixture were used to estimate the catalyst activity under various conditions (Table 1). The effect of water on soot oxidation was also examined. The gas feed compositions for these experiments are shown in Table 2. The flow rates for each gas were controlled by the corresponding mass flow controllers, and water was supplied using a peristaltic pump. High flow rates were applied to approach realistic conditions. The addition of water was initiated at a temperature of 100 °C. Temperature-programmed heating was performed from room temperature to 600 °C with a heating rate of 7 °C min−1. The reaction products were analysed with high accuracy using an FTIR gas analyser (Gasmet™) with an analysis frequency of 20 seconds.
:20 was packed into a reactor in two layers, which were separated by quartz wool. Four different compositions of gas feed mixture were used to estimate the catalyst activity under various conditions (Table 1). The effect of water on soot oxidation was also examined. The gas feed compositions for these experiments are shown in Table 2. The flow rates for each gas were controlled by the corresponding mass flow controllers, and water was supplied using a peristaltic pump. High flow rates were applied to approach realistic conditions. The addition of water was initiated at a temperature of 100 °C. Temperature-programmed heating was performed from room temperature to 600 °C with a heating rate of 7 °C min−1. The reaction products were analysed with high accuracy using an FTIR gas analyser (Gasmet™) with an analysis frequency of 20 seconds.
| Air | Air + NO | Air + H2O | Air + NO + H2O | |
|---|---|---|---|---|
| O2 | 21% | 20.9% | 17.9% | 17.8% |
| NO | — | 500 ppm | — | 500 ppm |
| H2O | — | — | 15% | 15% |
| N2 | Balance | Balance | Balance | Balance |
| Total flow rate | 1200 ml min−1 | 1200 ml min−1 | 1412 ml min−1 | 1412 ml min−1 |
| Regime | I | II | III |
|---|---|---|---|
| O2 | 13% | 13% | 13% |
| H2O | 0% | 7.5% | 15% |
| N2 | Balance | Balance | Balance |
| Total flow rate | 1200 ml min−1 | 1200 ml min−1 | 1200 ml min−1 |
The effect of water on the CO oxidation was examined using the described setup. The catalyst with a mass of 400 mg was heated to 120 °C under the flow of 4167 ppm CO/13 vol% O2 with N2 as a balance gas. The total flow rate was 1200 ml min−1. After the temperature stabilization, 7.5 vol% of water was added to the gas mixture for 20 minutes, and then the water addition was stopped. The reaction products were analysed using an FTIR gas analyser (Gasmet™).
The chemical state of the active component was determined based on the CO reduction. A sample of 400 mg was heated from room temperature to 400 °C with a heating rate of 7 °C min−1 under the flow of 5000 ppm CO in N2. The total flow rate was 400 ml min−1. The concentrations of consumed carbon monoxide were analysed using an FTIR gas analyser (Gasmet™). No pretreatment was performed prior to the experiments.
3. Results and discussion
3.1. Effect of H2O and NO on the catalyst activity
Fig. 1 shows the soot oxidation activity of the Ag- and Pt-based catalysts under different conditions. The figure is divided into two parts: soot oxidation without (left side) and with (right side) the addition of water. In air, all of the catalysts promoted soot oxidation, and the differences between the maxima of the oxidation curves for the Ag- and Pt-based catalysts were negligible. However, soot oxidation for the high-loaded catalysts occurred in a lower temperature region than that for the low-loaded catalysts. Thus, an increase in the active-component loading has a positive effect on the activity of the catalysts.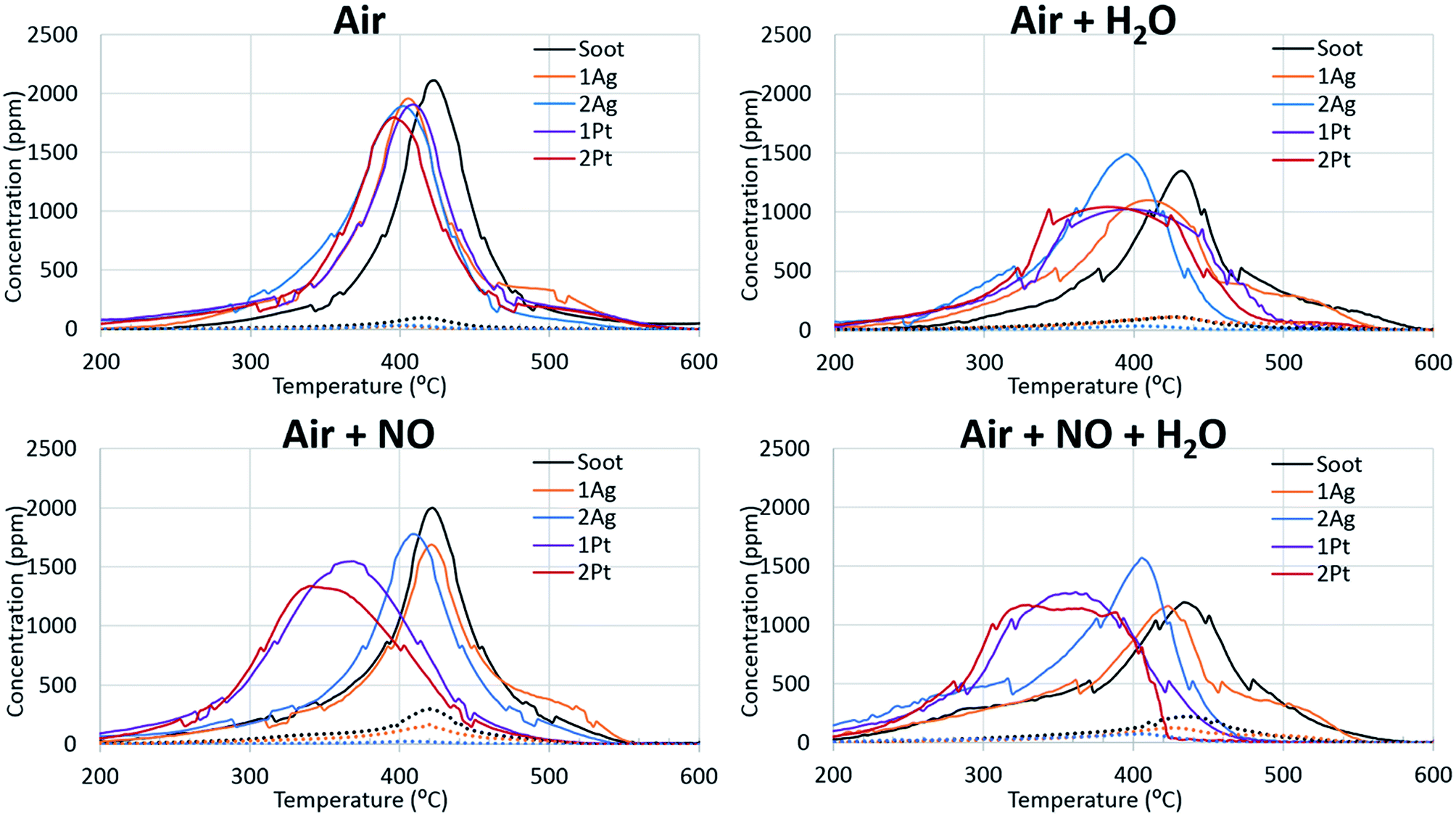 | ||
| Fig. 1 Silver and platinum activity in fireplace soot oxidation under the applied conditions. The solid line indicates CO2 evolution and the dotted line indicates CO evolution. | ||
For CO emission during fireplace soot combustion (dotted line), silver catalysts have a minor disadvantage compared to platinum catalysts. Carbon monoxide was released during the soot oxidation over the silver catalysts. However, the amount of CO emission decreased with an increase in the silver loading. This evidence leads to the assumption that complete cessation of carbon monoxide release is only a result of the increase in silver loading.
Non-catalytic soot oxidation. The addition of water to the reaction gas mixture is an important issue for the development of catalysts for fireplace soot combustion because the water content in flue gases can be high, particularly in the case of non-dried wood. The comparison of the non-catalysed soot oxidation curves with and without water illustrates that water in the gas feed composition affected the soot combustion (Fig. 1). The addition of water increased the total flow rate, which can cause a concentration dilution effect. However, the replacement of the water with nitrogen showed negligible changes in the position of the CO2 release peak and insignificant concentration deviation (ESI Fig. S1†). The addition of water increased the temperature of the soot oxidation and changed the ratio of the CO and CO2. This effect of water can be explained by a direct reaction of water with soot, which can decrease the reaction rate. A change in the ratio of CO/CO2 can be caused by the effect of water on the stability of oxygen complexes on the soot surface.31
Catalytic soot oxidation. Fig. 1 demonstrates that water also affects the catalytic soot oxidation. The significant difference between the CO2 evolution peaks for soot combustion with and without catalysts shows that water promotes the activity of the catalysts. A shift in the CO2 evolution towards lower temperatures was observed with the presence of water, which is the opposite of that observed in the soot oxidation without the catalyst. Moreover, in the case of the platinum catalysts, the addition of water decreased the initiation temperature of the soot combustion. In the case of the silver catalysts, the soot oxidation over the 2Ag catalyst ended at a lower temperature than the oxidation over the same catalyst without water. The highly-loaded silver catalyst oxidized soot in a narrower temperature range in the lower temperature region than the other catalysts and showed the highest activity under these conditions. In addition, an increase in the active-component loading improved the catalyst performance in the presence of water.
The behaviours of the silver and platinum catalysts significantly differed with the presence of nitrogen monoxide in the reaction gas mixture. Fig. 2 shows the differences in the routes of the soot oxidation reaction over the silver and platinum catalysts under the effect of NO. Specifically, the graphs for the platinum catalysts show that nitrogen monoxide was converted into dioxide in the entire soot combustion process. The nitrogen dioxide concentration was maximized before the intensive soot oxidation began. This evidence indicates that platinum affords the formation of NO2, which is a strong soot oxidant.7 Meanwhile, the silver catalysts behaved differently from the corresponding platinum catalysts. Initially, the silver oxidized NO like platinum. However, when the soot combustion began, the catalyst focused only on the reaction with soot; the conversion of NO to NO2 tended to zero and reached its minimum approximately at the point of the maximum soot oxidation rate. Therefore, the silver-based catalysts directly oxidized the soot. Direct oxidation requires interaction between the soot and highly mobile silver particles.35,36 However, the formation of silver nitrates/nitrites hindered the mobility of silver particles and impeded the soot oxidation. Thus, the changes in the activity of the silver-based catalysts after the addition of the nitrogen oxides into the reaction gas also confirm the direct oxidation of the soot over the silver catalysts.
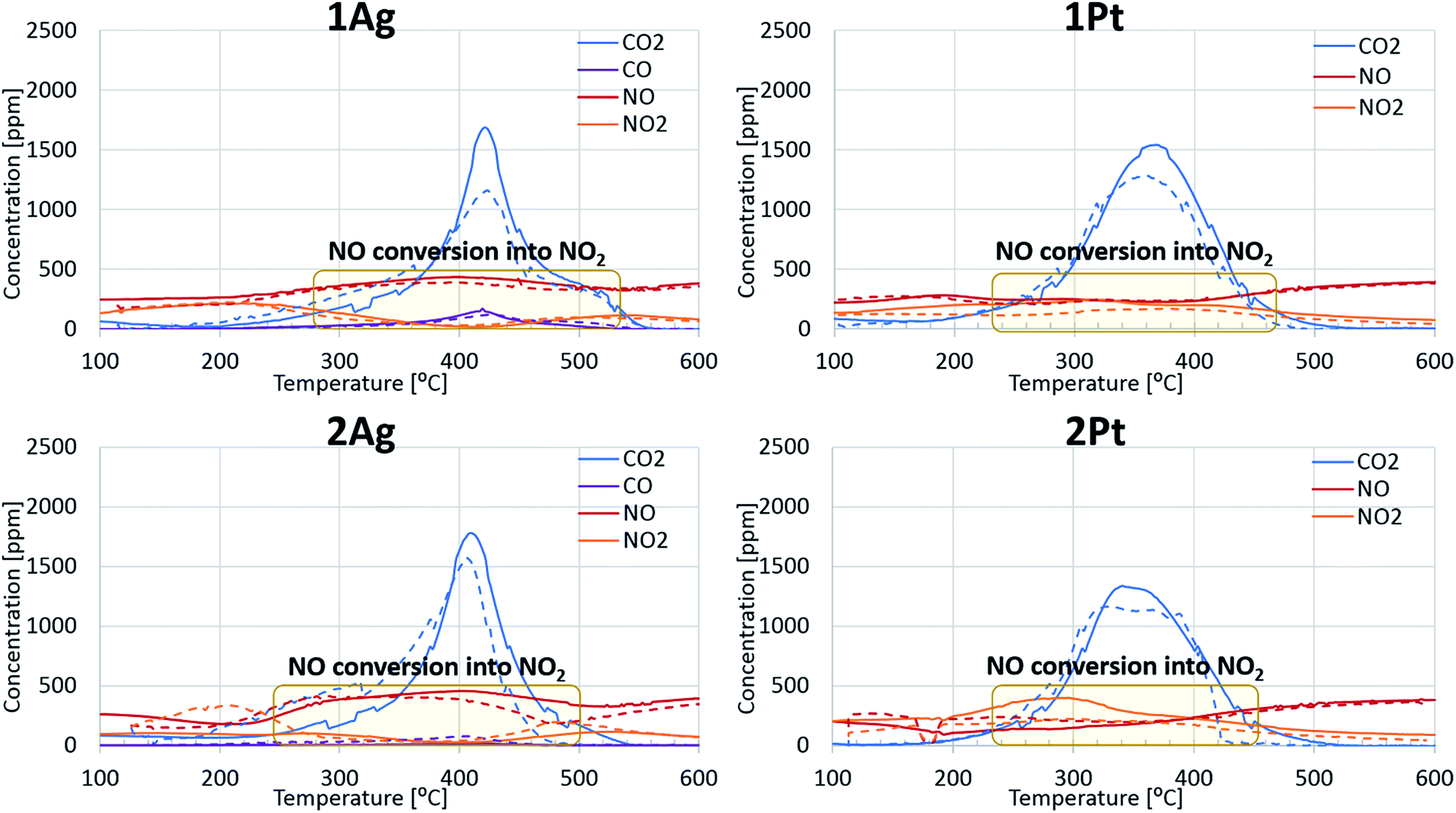 | ||
| Fig. 2 Differences in the reaction pathway of soot oxidation over silver and platinum catalysts in the presence of NO. The solid line indicates oxidation without water and the dashed line indicates oxidation with water. | ||
The addition of water to the mixture of technical air and nitrogen monoxide improved the activity of both the silver- and platinum-based catalysts, as observed from Fig. 1 and 2. Particularly, the soot oxidation under these conditions began and ended at lower temperatures than that of the oxidation without water. The combined effect of water and nitrogen monoxide for Pt-based catalysts during fireplace soot combustion is analogous to the platinum catalytic behaviour in carbon black oxidation under similar conditions.22,37 This can be explained by the interaction of water with nitrogen monoxide and dioxide, which results in the formation of nitric and nitrous acids as intermediates and nitronium ions (NO2+) in acids, which easily react with soot and cause the hydrolysis of oxygenated species formed on the soot surface during oxidation.22,37–39
3.2. Water promotion mechanism
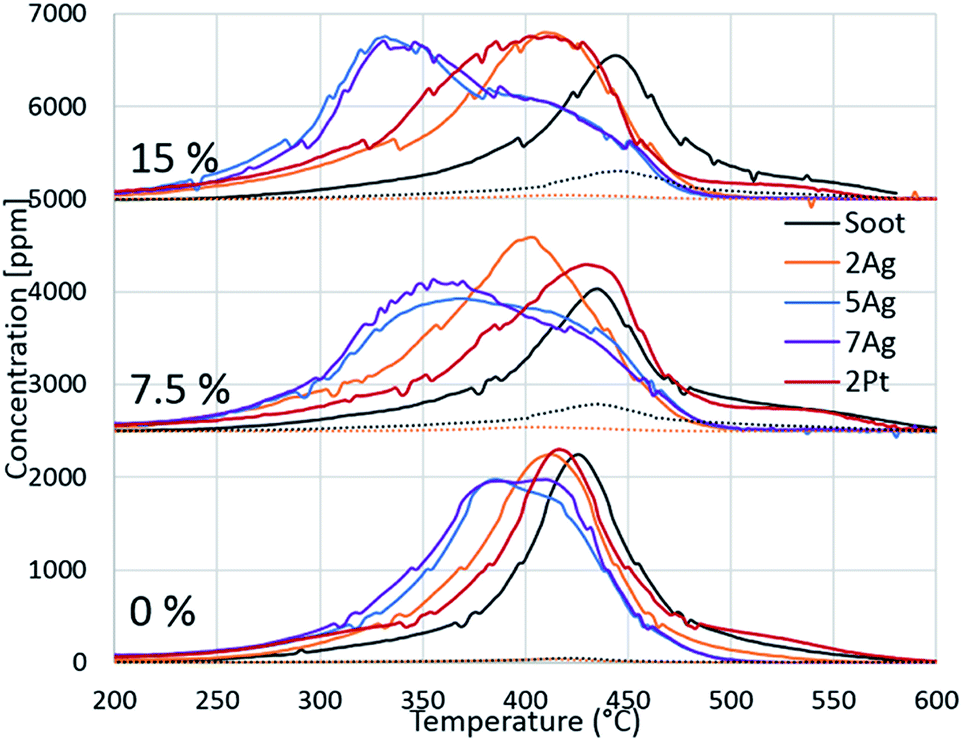
Because of the evident effect of water on fireplace soot oxidation in our study and the reported water promotion effect on diesel soot oxidation,40,41 we decided to investigate the phenomenon more thoroughly. The silver and platinum catalysts with 2 wt% loadings were used, and two additional catalysts with increased silver loading (5 and 7 wt%) were synthesized for the experiments. The catalysts were tested in soot oxidation in three different regimes with different water concentrations in the gas feed. The oxygen concentration was 13 vol%, which is the average concentration in flue gases in fireplaces.42 To avoid the dilution effect, the total flow rates for all of the experiments were constant. The results are shown in Fig. 3. As previously discussed, water inhibits the oxidation of pure soot and changes the CO/CO2 ratio. Identical behaviour was observed in the comparison of the oxidation curves for all of the regimes with different water concentrations. Fig. 3 shows that the oxidation temperature increased with the increasing water content in the gas feed. In the case of catalytic soot oxidation, water positively affected the soot oxidation. Despite a small inhibitory effect of water on the 2Ag catalyst, water promoted all of the other catalysts. Fig. 3 shows the dependence of the oxidation temperature on the water concentration in the gas mixture and of the loading effect on the performance of the silver catalysts. However, this evidence leads to the question: why does water promote soot combustion?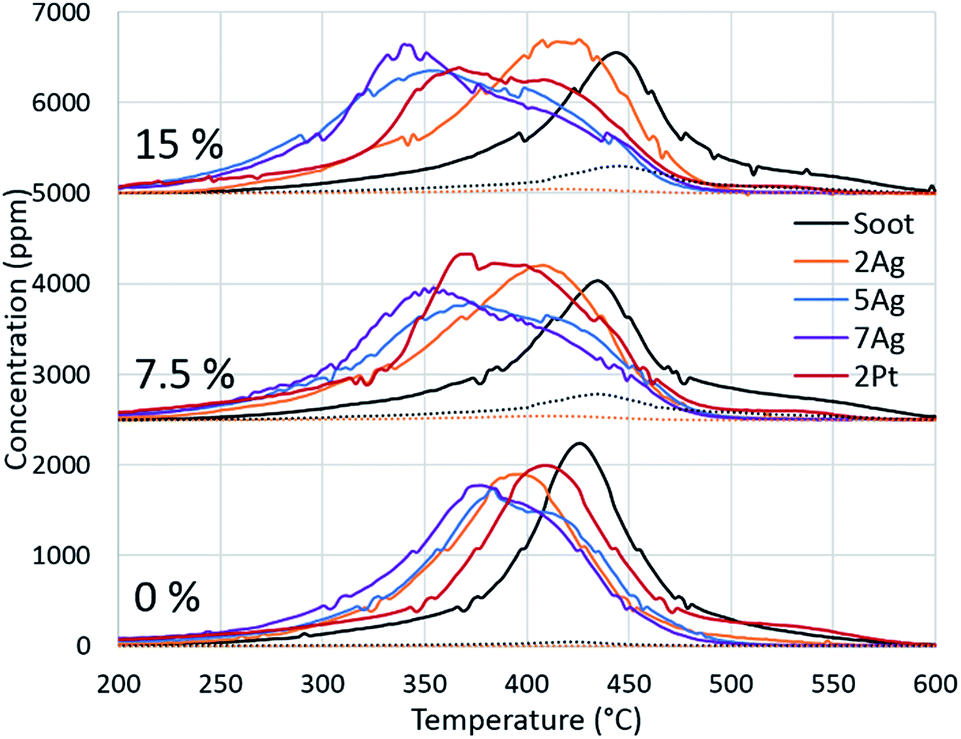 | ||
| Fig. 3 Dependence of the catalyst activity on the water concentration in the gas feed. | ||
We suggest the following water promotion mechanism:
• Water adsorbs onto the metallic surface of the active component with the aid of chemisorbed oxygen;
• The adsorbed water partially dissociates;
• The formed hydroxyl groups react with soot and oxidize it.
To confirm our hypothesis, the chemical state of the active component via CO reduction was determined, and the ability of the catalysts to oxidize carbon monoxide in the presence of water was tested. The reduction of the catalysts (Fig. 4) with CO shows that platinum existed in the metallic form: the absence of consumption of carbon monoxide indicates the absence of oxides on the platinum surface. For the silver catalysts, the consumption of carbon monoxide increased depending on the silver loading. Thus, we can assume that silver existed in the oxide form on the catalyst surface. However, the comparison of the CO reduction experiments for the silver catalyst and bulk silver oxide (Ag2O) with mass, which is equivalent to silver on the catalyst, shows that oxide consumed much more CO (ESI Fig. S3†). In addition, the XRD diffractograms indicate the presence of metallic silver in the highly-loaded catalysts (Fig. 5). Thus, silver partially existed in the oxide form; in particular, metallic silver particles were covered by oxygen.
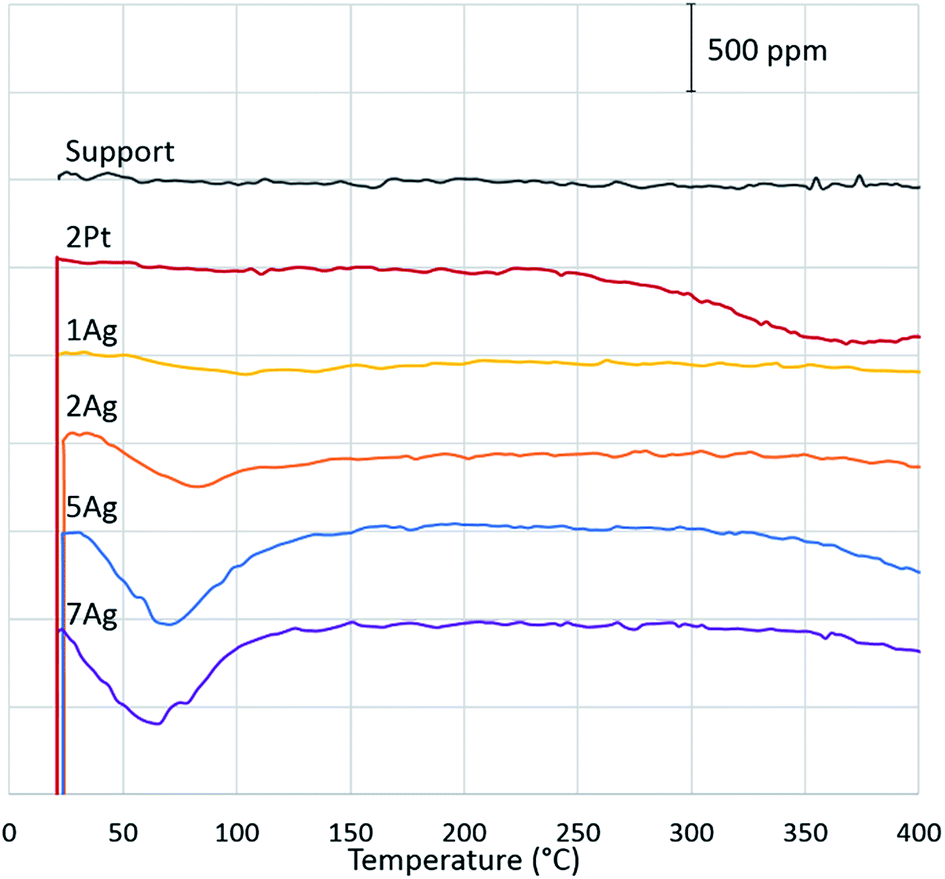 | ||
| Fig. 4 Determination of the chemical state of the active component with CO reduction. | ||
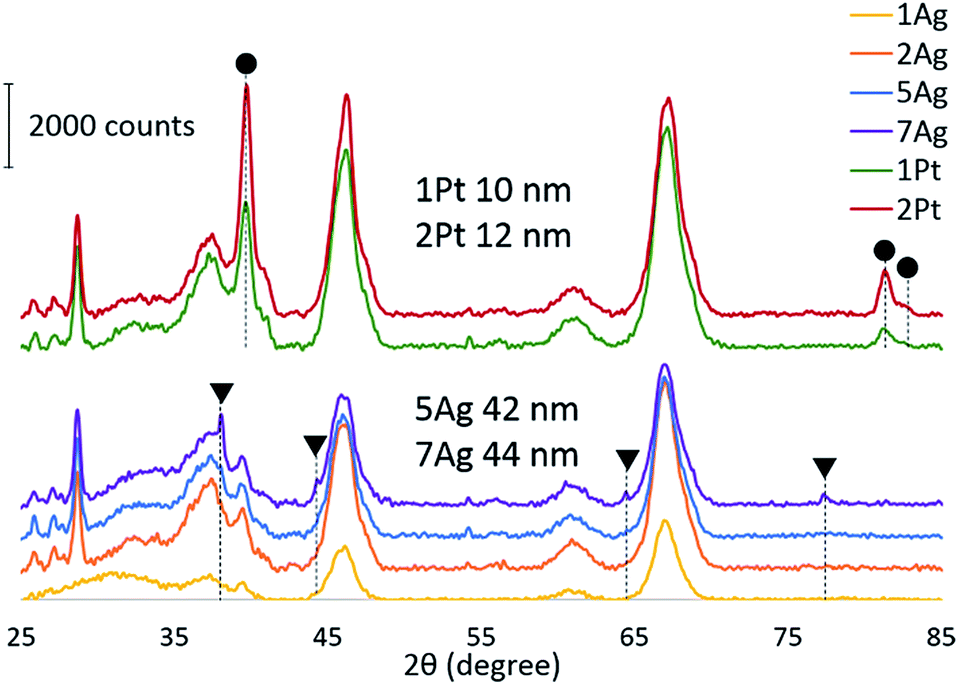 | ||
| Fig. 5 Powder XRD patterns for fresh Ag and Pt catalysts: metallic Pt ●; metallic Ag ▼. | ||
Water can dissociate on the metallic surfaces of oxygen-pretreated silver and platinum.43–46 Water interacted with the chemisorbed oxygen and dissociated into two adsorbed hydroxyl groups. Examination of the effect of water on CO oxidation confirms the partial dissociation of water on the silver and platinum surfaces (Fig. 6). In the case of platinum, the addition of water increased the conversion of carbon monoxide; hence, platinum assisted the water gas shift reaction. According to the mechanism, CO was converted through a carboxyl intermediate, in whose formation the adsorbed hydroxyl and carbon monoxide participated.47 Thus, water partially dissociated on the platinum during soot oxidation. For silver, the effect of water on CO oxidation was the opposite. The addition of water caused a significant decrease in carbon monoxide conversion. However, unlike platinum, CO did not adsorb on the metallic silver, and CO was oxidized through a reaction with the chemisorbed oxygen on the surface.48 Thus, the changes in conversion indicate that water interacted with the available oxygen on the silver surface, which confirms the formation of hydroxyls. In addition, water hindered the carbon monoxide oxidation regardless of the silver loading. Even at a higher temperature, the addition of water decreased the conversion (ESI Fig. S4†). The presence of oxygen in the reaction gas mixture was also important. Without oxygen, the active component did not interact with water, so the soot could not be oxidized (ESI Fig. S5†). Overall, both the platinum and silver partially dissociated water and formed hydroxyls on the surface. According to modelling and experimental studies, hydroxyls can oxidize soot that requires low activation energies.49–51 Based on experiments and the literature, we conclude that water promotes soot oxidation through the partial dissociation and formation of hydroxyls on silver and platinum surfaces.
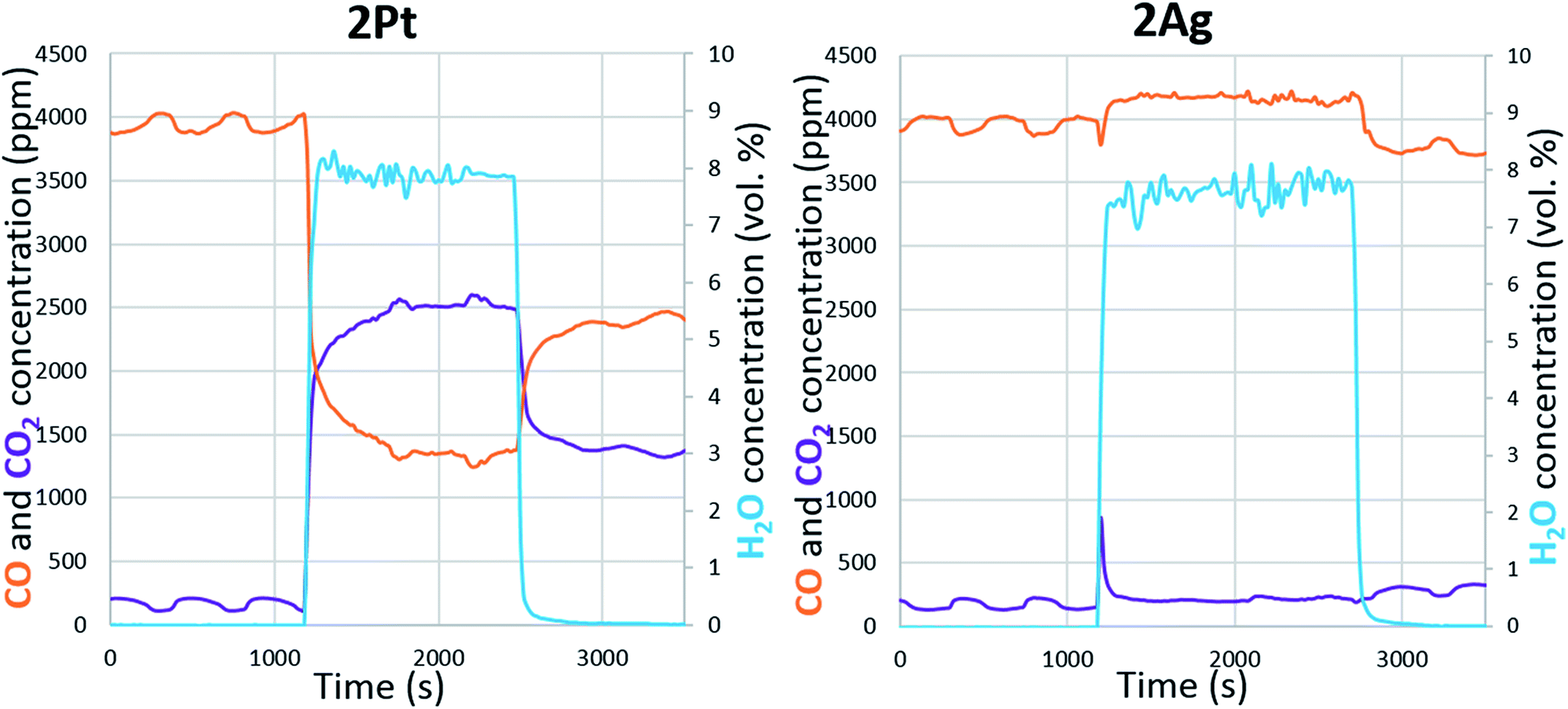 | ||
| Fig. 6 Water effect on the oxidation of carbon monoxide. | ||
3.3. Thermal ageing effect
Since soot combustion is an exothermic reaction, local overheating of the catalyst can occur, which causes agglomeration and sintering. Thus, thermal ageing is a demonstrative procedure to estimate the thermal resistance and oxidation capability of catalysts after treatment at high temperatures. Fig. 7 shows the effect of ageing at high temperature on the activity of the catalyst for soot oxidation. The platinum catalyst lost its activity under all conditions, even when NO was present in the gas feed. The main reason for this activity decrease is the agglomeration of platinum at high ageing temperatures. In spite of negligable changes of the specific surface areas of the catalysts after soot oxidation (Table 3), the comparison of the platinum particle sizes of the catalysts before and after the reaction (ESI Fig. S11†) showed partial agglomeration of the platinum. After one cycle of the reaction, there were still small platinum particles, however, particles with larger sizes started emerging. We assume that after several cycles of soot oxidation the platinum particles would be of the same size as those in the case of aged catalysts. In the case of the silver catalysts, the catalyst with high silver loading retained its activity for soot oxidation even after ageing in all conditions. The high activity of the silver catalysts after thermal treatment can be due to the ability of large sintered silver particles supported on alumina to redisperse easily at sufficient oxygen concentrations.52 Silver particles of catalysts before and after reaction are approximately the same size, as is the case for the aged catalysts (ESI Fig. S10†). Thus, the silver catalysts showed high thermal durability, which further gives a significant advantage for possible applications in fireplaces.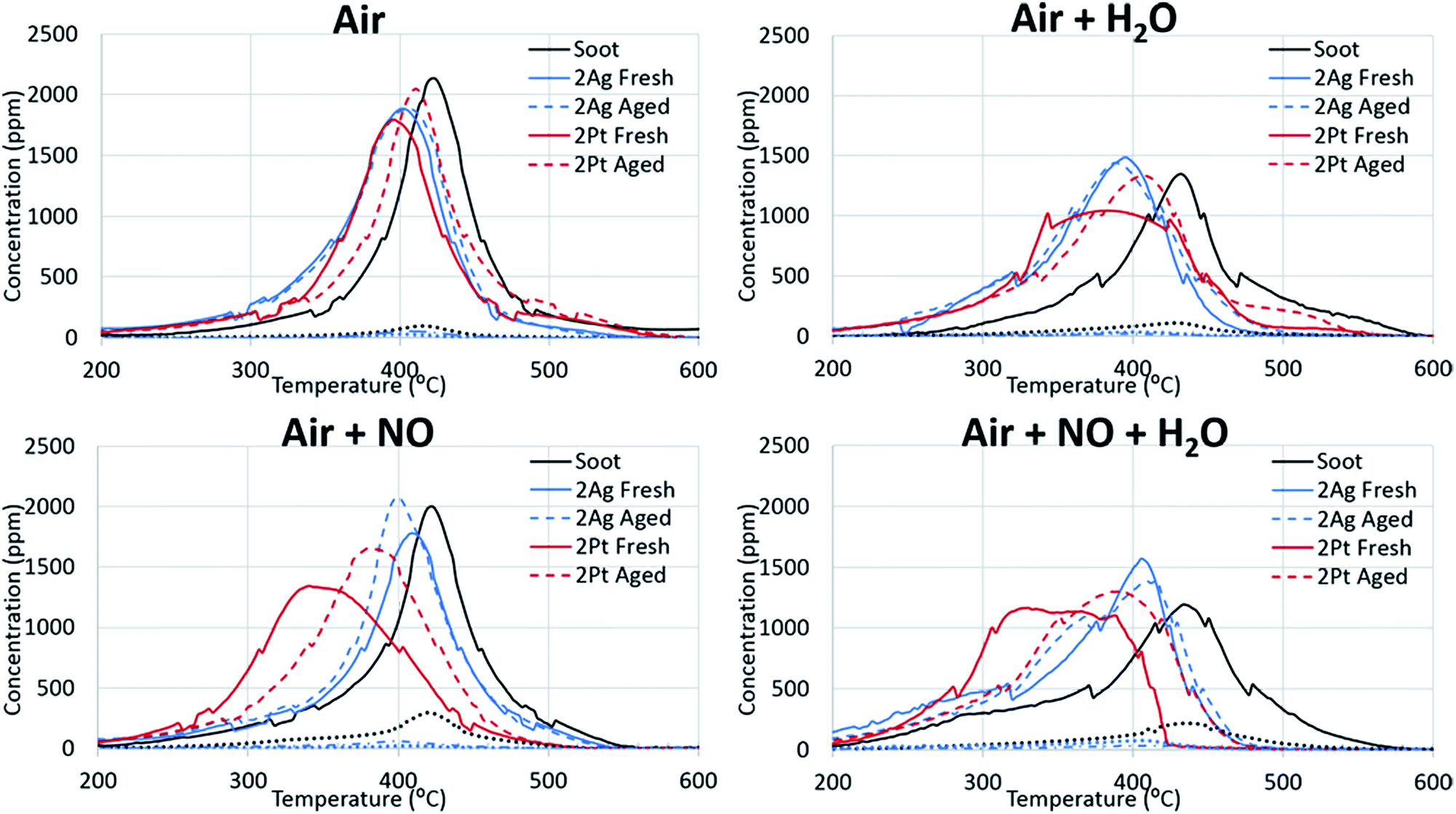 | ||
| Fig. 7 The thermal ageing effect on the activity of silver and platinum catalysts in fireplace soot combustion. The solid and dotted lines show the evolution of CO2 and CO, respectively, for fresh catalysts; the dashed and dash-dotted lines show the evolution of CO2 and CO, respectively, for the aged catalysts. | ||
| Specific surface area, m2 g−1 | Total pore volume, cm3 g−1 | Average pore diameter, nm | |
|---|---|---|---|
| a 1 Indicates catalysts after the reaction of soot oxidation in air; 2 indicates catalysts after the reaction of soot oxidation in air with the addition of NO; 3 indicates catalysts after the reaction of soot oxidation in air with the addition of water; 4 indicates catalysts after the reaction of soot oxidation in air with the addition of NO and water. | |||
| Support | 164 | 0.5900 | 14.4 |
|
|||
| 2Ag: | |||
| Fresh | 144 | 0.5337 | 14.8 |
| Spent1 | 153 | 0.5539 | 14.5 |
| Spent2 | 141 | 0.5166 | 14.7 |
| Spent3 | 139 | 0.5191 | 14.9 |
| Spent4 | 141 | 0.5074 | 14.4 |
|
|||
| 2Pt: | |||
| Fresh | 151 | 0.6914 | 18.3 |
| Spent1 | 136 | 0.6023 | 17.7 |
| Spent2 | 155 | 0.6709 | 17.3 |
| Spent3 | 131 | 0.6035 | 18.4 |
| Spent4 | 145 | 0.6440 | 17.8 |
Identical catalyst behaviour was observed in the case where the effect of water was examined (Fig. 8). The platinum catalysts had lower activity than the fresh ones. However, water slightly promoted the soot oxidation over platinum. The silver catalysts retained their activity for soot oxidation after ageing. The addition of water also improved the catalyst activity to comparable values to those of the fresh catalysts.
 | ||
| Fig. 8 Dependence of the activity of the aged catalysts on the water concentration in the feed gas. | ||
4. Conclusions
Our study shows that the catalytic behaviour of the silver and platinum catalysts changes with the applied conditions. In air, where oxygen is a main oxidant in the feed gas, the silver and platinum catalysts have similar reaction pathways and comparable activities in fireplace soot oxidation. The addition of NO into the reaction gas mixture causes different reaction routes for the silver and platinum. This result indicates that the silver-based catalysts follow direct oxidation, whereas the platinum catalysts follow NO2-assisted oxidation. However, the flue gases of real fireplaces contain extremely low quantities of NO and high amounts of water, so the effect of water addition is important to the catalytic performance of the active components. Although water addition increases the temperature of pure soot oxidation, in the case of catalytic soot combustion, water promotes the activity of the catalyst. The increase in water content in the gas feed improves the performance of both the silver and platinum catalysts. The promotion effect is based on the partial water dissociation on the active component surface in the presence of oxygen with the following formation of hydroxyls. The highly-loaded silver catalysts exhibit the highest activity among all of the catalysts in fireplace soot combustion under the effect of water.Thermal ageing at high temperatures causes essential changes in the catalyst activity. The aged platinum catalysts oxidize soot at higher temperatures than the fresh catalysts. The main reason for the activity loss is the agglomeration of platinum particles. However, the silver catalyst retains its activity, which demonstrates its high thermal stability and resistance to high temperatures and its potential opportunity for application in real fireplaces.
Conflicts of interest
There are no conflict to declare.Acknowledgements
The authors are grateful for the financial support from the Finnish Funding Agency of Technology and Innovation (TEKES, EVIM project) and the European Regional Development Fund (ERDF). We are also grateful to Tulikivi Oyj (Finland) and Uunisepät Oy (Finland). We would also like to thank the laboratory technician Taina Nivajärvi for conducting the elemental analysis.References
- Q. Li, J. Shang and T. Zhu, Atmos. Environ., 2013, 81, 68–75 CrossRef CAS.
- T. C. Bond, S. J. Doherty, D. W. Fahey, P. M. Forster, T. Berntsen, B. J. DeAngelo, M. G. Flanner, S. Ghan, B. Kärcher, D. Koch, S. Kinne, Y. Kondo, P. K. Quinn, M. C. Sarofim, M. G. Schultz, M. Schulz, C. Venkataraman, H. Zhang, S. Zhang, N. Bellouin, S. K. Guttikunda, P. K. Hopke, M. Z. Jacobson, J. W. Kaiser, Z. Klimont, U. Lohmann, J. P. Schwarz, D. Shindell, T. Storelvmo, S. G. Warren and C. S. Zender, J. Geophys. Res.: Atmos., 2013, 118, 5380–5552 CAS.
- Z. Cong, S. Kang, S. Gao, Y. Zhang, Q. Li and K. Kawamura, Environ. Sci. Technol., 2013, 47, 2579–2586 CrossRef CAS PubMed.
- C. A. Pope III, R. T. Burnett, M. J. Thun, E. E. Calle, D. Krfewski, K. Ito and G. D. Thurston, JAMA, J. Am. Med. Assoc., 2002, 287, 1132–1141 CrossRef.
- E. M. Fitzpatrick, A. B. Ross, J. Bates, G. Andrews, J. M. Jones, H. Phylaktou, M. Pourkashanian and A. Williams, Process Saf. Environ. Prot., 2007, 85, 430–440 CrossRef CAS.
- A. M. Nienow and J. T. Roberts, Annu. Rev. Phys. Chem., 2006, 57, 105–128 CrossRef CAS PubMed.
- B. R. Stanmore, J. F. Brilhac and P. Gilot, Carbon, 2001, 39, 2247–2268 CrossRef CAS.
- A. Raj, G. R. da Silva and S. H. Chung, Combust. Flame, 2012, 159, 3423–3436 CrossRef CAS.
- P. I. Jalava, R. O. Salonen, K. Nuutinen, A. S. Pennanen, M. S. Happo, J. Tissari, A. Frey, R. Hillamo, J. Jokiniemi and M. Hirvonen, Atmos. Environ., 2010, 44, 1691–1698 CrossRef CAS.
- A. Kocbach, Y. Li, K. E. Yttri, K. R. Cassee, P. E. Schwarze and E. Namork, Part. Fibre Toxicol., 2006, 3, 1–10 CrossRef PubMed.
- A. Braun, F. E. Huggins, A. Kubatova, S. Wirick, M. M. Maricq, B. S. Mun, J. D. McDonald, K. E. Kelly, N. Shah and G. P. Huffman, Environ. Sci. Technol., 2008, 42, 374–380 CrossRef CAS PubMed.
- A. B. Ross, S. Junyapoon, J. M. Jones, A. Williams and K. D. Bartle, J. Anal. Appl. Pyrolysis, 2005, 74, 494–501 CrossRef CAS.
- E. M. Fitzpatrick, J. M. Jones, M. Pourkashanian, A. B. Ross, A. Williams and K. D. Bartle, Energy Fuels, 2008, 22, 3771–3778 CrossRef CAS.
- J. M. Wilson, M. T. Baeza-Romero, J. M. Jones, M. Pourkashanian, A. Williams, A. B. Lea-Langton, A. B. Ross and K. D. Bartle, Energy Fuels, 2013, 27, 1668–1678 CrossRef CAS.
- R. L. V. Wal and C. J. Mueller, Energy Fuels, 2006, 20, 2364–2369 CrossRef.
- B. J. Cooper, H. J. Jung and J. E. Toss, F01N 3/023(20060101), United States, 1990, patent issued after 1st publication within TVPP.
- H. J. Stein, Appl. Catal., B, 1996, 10, 69–82 CrossRef CAS.
- M. Hosoya and M. Shimoda, Appl. Catal., B, 1996, 10, 83–97 CrossRef CAS.
- A. Bueno-López, K. Krishna, B. van der Linden, G. Mul, J. A. Moulijn and M. Makkee, Catal. Today, 2007, 121, 237–245 CrossRef.
- G. Neri, L. Bonaccorsi, A. Donato, C. Milone, M. G. Musolino and A. M. Visco, Appl. Catal., B, 1997, 11, 217–231 CrossRef CAS.
- J. Oi Uchisawa, A. Obuchi, Z. Zhao and S. Kushiyama, Appl. Catal., B, 1998, 18, L183–L187 CrossRef CAS.
- J. Oi-Uchisawa, A. Obuchi, A. Ogata, R. Enomoto and S. Kushiyama, Appl. Catal., B, 1999, 21, 9–17 CrossRef CAS.
- E. Aneggi, J. Llorca, C. de Leitenburg, G. Dolcetti and A. Trovarelli, Appl. Catal., B, 2009, 91, 489–498 CrossRef CAS.
- G. Corro, U. Pal, E. Ayala, E. Vidal and E. Guilleminot, Top. Catal., 2013, 56, 467–472 CrossRef CAS.
- G. Corro, U. Pal, E. Ayala and E. Vidal, Catal. Today, 2013, 212, 63–69 CrossRef CAS.
- T. Nanba, S. Masukawa, A. Abe, J. Uchisawa and A. Obuchi, Catal. Sci. Technol., 2012, 2, 1961–1966 CAS.
- C. Lim, H. Kusaba, H. Einaga and Y. Teraoka, Catal. Today, 2011, 175, 106–111 CrossRef CAS.
- K. Villani, R. Brosius and J. A. Martens, J. Catal., 2005, 236, 172–175 CrossRef CAS.
- A. Hukkanen, T. Kaivosoja, O. Sippula, K. Nuutinen, J. Jokiniemi and J. Tissari, Atmos. Environ., 2012, 50, 16–23 CrossRef CAS.
- S. Bensaid, F. A. Deorsola, D. Fino and N. Russo, Catal. Today, 2012, 197, 76–89 CrossRef CAS.
- J. P. A. Neeft, X. T. Nijhuis, E. Smakman, M. Makkee and J. A. Moulijn, Fuel, 1997, 76, 1129–1136 CrossRef CAS.
- J. Kašpar, P. Fornasiero and N. Hickey, Catal. Today, 2003, 77, 419–449 CrossRef.
- R. Brosius, K. Arve, M. H. Groothaert and J. A. Martens, J. Catal., 2005, 231, 344–353 CrossRef CAS.
- J. Tissari, K. Hytönen, J. Lyyränen and J. Jokiniemi, Atmos. Environ., 2007, 41, 8330–8344 CrossRef CAS.
- D. Gardini, J. M. Christensen, C. D. Damsgaard, A. D. Jensen and J. B. Wagner, Appl. Catal., B, 2016, 183, 28–36 CrossRef CAS.
- K. Kamatani, K. Higuchi, Y. Yamamoto, S. Arai, N. Tanaka and M. Ogura, Sci. Rep., 2015, 5, 10161–10166 CrossRef PubMed.
- S. Liu, X. Wu, H. Luo, D. Weng and R. Ran, J. Phys. Chem. C, 2015, 119, 17218–17227 CAS.
- M. Mehring, M. Elsener, L. Bächli and O. Kröcher, Fuel, 2012, 50, 2100–2109 CAS.
- M. Jeguirim, V. Tschamber, J. F. Brilhac and P. Ehrburger, Fuel, 2005, 84, 1949–1956 CrossRef CAS.
- C. Lee, Y. Jeon, S. Hata, J. Park, R. Akiyoshi, H. Saito, Y. Teraoka, Y. Shul and H. Einaga, Appl. Catal., B, 2016, 191, 157–164 CrossRef CAS.
- Y. Gao, A. Duan, S. Liu, X. Wu, W. Liu, M. Li, S. Chen, X. Wang and D. Weng, Appl. Catal., B, 2017, 203, 116–126 CrossRef CAS.
- J. Tissari, K. Hytönen, J. Lyyränen and J. Jokiniemi, Atmos. Environ., 2007, 41, 8330–8344 CrossRef CAS.
- P. A. Thiel and T. E. Madey, Surf. Sci. Rep., 1987, 7, 211–385 CrossRef CAS.
- G. Wang, S. Tao and X. Bu, J. Catal., 2006, 244, 10–16 CrossRef CAS.
- X. Bao, M. Muhler, B. Pettinger, Y. Uchida, G. Lehmpfuhl, R. Schlögl and G. Ertl, Catal. Lett., 1995, 32, 171–183 CrossRef CAS.
- C. Wang, G. Deo and I. E. Wachs, J. Phys. Chem. B, 1999, 103, 5645–5656 CrossRef CAS.
- L. C. Grabow, A. A. Gokhale, S. T. Evans, J. A. Dumesic and M. Mavrikakis, J. Phys. Chem. C, 2008, 112, 4608–4617 CAS.
- G. W. Keulks and C. C. Chang, J. Phys. Chem., 1970, 74, 2590–2595 CrossRef CAS.
- P. Roth, T. Eckhardt, B. Franz and J. Patschull, Combust. Flame, 1998, 115, 28–37 CrossRef CAS.
- D. E. Edwards, D. Y. Zubarev, W. A. Lester and M. Frenklach, J. Phys. Chem. A, 2014, 118, 8606–8613 CrossRef CAS PubMed.
- C. P. Fenimore and G. W. Jones, J. Phys. Chem., 1967, 71, 593–597 CrossRef CAS.
- Y. Gao, X. Wu, S. Liu, M. Ogura, M. Liu and D. Weng, Catal. Sci. Technol., 2017, 7, 3524–3529 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09291a |
| This journal is © The Royal Society of Chemistry 2017 |