
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Relative quantification of neuronal polar lipids by UPLC-MS reveals the brain protection mechanism of Danhong injection†
Jing Xu‡
 ab,
Liying Tang‡a,
Qiong Zhang‡b,
Junying Weia,
Minghua Xiana,
Ye Zhaoa,
Qiang Jiaa,
Xin Lib,
Yi Zhanga,
Yan Zhaoc,
Hongwei Wu*a and
Hongjun Yang*a
ab,
Liying Tang‡a,
Qiong Zhang‡b,
Junying Weia,
Minghua Xiana,
Ye Zhaoa,
Qiang Jiaa,
Xin Lib,
Yi Zhanga,
Yan Zhaoc,
Hongwei Wu*a and
Hongjun Yang*a
aInstitute of Chinese Materia Medica, China Academy of Chinese Medical Sciences, Dong Nei Nan Xiao Jie 16, Beijing 100700, China. E-mail: whw9905012@163.com; hongjun0420@vip.sina.com; Fax: +86-64062692; Tel: +86-13611325156
bDepartment of Pharmaceutical Science, Shanxi Medical University, No. 56 Xinjian South Road, Taiyuan 030001, Shanxi, China
cBeijing Omics Biological Technology Co., LTD., China
First published on 25th September 2017
Abstract
Promising results from clinical trials have fueled a growing acceptance of Danhong injection (DHI) as a Chinese Materia Medica standardized product for the treatment of ischemic stroke. However, little information is available on the underlying mechanisms of DHI especially in lipidomics. In this study, experiments on permanent middle cerebral artery occlusion (MCAO) in mice were carried out to confirm the protective effect of DHI. Furthermore, primary mouse cortical neurons subjected to oxygen-glucose-deprivation (OGD) were used to investigate the protective mechanisms of DHI. A UPLC-MS profiling analysis for neuronal polar lipids including phosphatidylcholines (PCs), sphingomyelins (SMs) and ceramides (Cers) was carried out in order to study the potential biomarkers in this OGD-induced neuron injury model. The results showed that pretreatment with DHI resulted in a significantly smaller infarct volume and better neurological scores than pretreatment with saline in MCAO mice. In an OGD-induced neuron injury model, DHI exhibited remarkable neuroprotection by reducing the neuronal damage and excessive accumulation of intracellular reactive oxygen species, suppressing intracellular free calcium influx and apoptosis. Meanwhile, 28 biomarkers of PCs, SMs and Cers sub-species of neuronal injury induced by OGD have been identified for the first time. The perturbations could be partly reversed by DHI intervention such as PC (17:0,0:0), PC (18:0,0:0), PC (16:0,0:0), PC (P-16:0,0:0) and SM (18:0,16:0). The results specifically provide information on the relationships between PCs, SMs, Cers sub-species and neuronal damage mechanisms during an ischemic stroke. Overall, we found that the therapeutic effects of DHI on cerebral ischemia are partially due to interferences with the PC and SM metabolisms.
1. Introduction
Stroke represents the second leading cause of death worldwide, with a high incidence of morbidity often observed in surviving victims. Stroke results in approximately 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 deaths annually, with ischemic strokes accounting for about 90%.1,2 Ischemic stroke results in a series of complex and multifaceted pathological and physiological alterations, including reactive oxygen species (ROS) outburst, inflammatory mediator overproduction, lipid peroxidation, [Ca2+] overload, and brain blood barrier (BBB) disruption.3–5 However, the mechanisms of ischemic stroke are largely unclear and clinical trials have failed to show positive effects in patients with ischemic stroke. Therefore, systematic investigations of the complex pathological cascades during ischemic brain injury may help in the development of effective treatments and novel therapeutic tools against cerebral ischemia.
000000 deaths annually, with ischemic strokes accounting for about 90%.1,2 Ischemic stroke results in a series of complex and multifaceted pathological and physiological alterations, including reactive oxygen species (ROS) outburst, inflammatory mediator overproduction, lipid peroxidation, [Ca2+] overload, and brain blood barrier (BBB) disruption.3–5 However, the mechanisms of ischemic stroke are largely unclear and clinical trials have failed to show positive effects in patients with ischemic stroke. Therefore, systematic investigations of the complex pathological cascades during ischemic brain injury may help in the development of effective treatments and novel therapeutic tools against cerebral ischemia.
Traditional Chinese Medicine (TCM) has been practiced for millennia using multiple components to treat as well as prevent many complex and refractory disease states. Danhong injection (DHI), a Chinese Materia Medica standardized product, attracted a considerable amount of attention to find more agents and treatment options for ischemic brain vascular diseases. The raw materials of DHI are Radix Salviae miltiorrhizae and Flos Carthami tinctoria. Chemically, a number of phytochemical constituents, including catechols derived from Radix Salviae miltiorrhizae and quinochalcones, flavonoids derived from Flos Carthami tinctoria, have been identified in DHI.6 Ten main components of DHI, including procatechuic aldehyde, ferulic acid, salvianolic acid B, etc., have been quantitatively analyzed for quality control using HPLC-UV.7 Potential mechanisms of DHI have been reported by various research groups, involving antifibrinolytic and antioxidant,8 transcription factors mediator,9 activating Nrf2 signaling and NF-κB signaling pathways,10,11 as well as brain and heart co-protection effects by reinstating the arginine vasopressin level.12 In a recent study, DHI showed a strong ameliorative effect on cerebral ischemia-reperfusion damages in rats due to its protective effect on the blood–brain barrier and the reversal of neutrophil infiltration by suppressing the upregulation of matrix metallopeptidase-9 expression.13 Although investigations on the mechanism of DHI have resulted in significant breakthroughs, investigations on the mechanism of DHI from a lipidomics perspective are extremely rare.
It has already been described in the literature that brain tissue contains a high concentration of phospholipids and sphingolipids.14,15 Ischemic injury characterized by low oxygen and insufficient glucose supply may induce changes in the composition of membrane phospholipids and sphingolipids and may further initiate the production of second messengers for cellular signal transduction. Lipidomics, a rapidly expanding research field in metabonomics, offers powerful tools to understand the pathogenesis of ischemic injury and may further help uncover the therapeutic mechanisms involved in TCM. In neuron or brain tissue, polar lipids, particularly phosphatidylcholines (PCs), sphingomyelins (SMs) and ceramides (Cers), play a crucial role in the physiological function and pathological processes. PCs, the most abundant phospholipid on the outer surface of cellular membranes, are important for maintaining the cellular structure and exhibit important functions in signal transduction and membrane trafficking.16,17 The metabolism of SMs creates a variety of byproducts that plays a significant role in cell homeostasis. When ischemia occurs, PCs and SMs are susceptible to extensive hydrolysis.18,19 SMs hydrolysis results in accumulation of Cers which activates diverse signal pathways and leads to cell damage and death.20,21 PCs hydrolysis usually causes functional cell membrane fluidity disorders and may further result in messenger transduction of surface-to-nucleus phenomena in neural cells.22 Although changes in the amount of PCs, SMs and Cers may be highly significant to ischemic stroke, the relationships between these polar lipid changes, especially for sub-species and neuron injury, remain largely unclear.
In this study, a UPLC-MS profiling analysis for neuronal polar lipids, including PCs, SMs and Cers, was carried out on neurons subjected to oxygen-glucose-deprivation as a method to investigate the therapeutic mechanisms of DHI. In an effort to obtain reliable results and improve the accuracy of lipid analysis, a target database including the identified 98 PCs, 28 SMs and 41 Cers was constructed based on a LC-MS platform using internal standard standards as well as the relative data of accurate masses and MS/MS fragments of the organic samples. The corresponding checklist can be found in the ESI.† Before lipidomics analysis, permanent middle cerebral artery occlusion (MCAO) mice were used to confirm the protective effect of DHI in vivo. Subsequently, primary mouse cortical neurons subjected to oxygen-glucose-deprivation (OGD) were used to investigate the pathogenesis of acute ischemic stroke along with all underlying mechanisms of DHI.
2. Materials and methods
2.1. Chemicals and reagents
Danhong injection (DHI) with batch number 17011023 was provided by Shandong Danhong Pharmaceutical Co., Ltd (Shandong, China). Ginaton with batch number 1B166 was provided by Chi Sheng Pharma & Biotech Co., Ltd (Taiwan, China), and Edaravone with batch number H20131342 was obtained from Simcere Pharmaceutical Group Co., Ltd (Nanjing, China). Minimum essential medium (MEM), D-glucose and 0.25% trypsin–EDTA were purchased from Life Technologies (LA, USA). Donor Equine Serum (DES) was obtained from Hyclone (Logan, UT, US). L-Glutamine, Ca2+- and Mg2+-free Hank's balanced salt solution (HBSS) were purchased from Gibco BRL (Carlsbad, NY, USA). 0.5% 2,3,5-triphenyltetrazolium chloride, poly-D-lysine and 5-fluoro-20-deoxyuridine (5-FU) were obtained from Sigma-Aldrich (St Louis, MO, USA). A cytotoxicity LDH assay kit-WST (LDH) was purchased from Dojindo (Kumamoto, Japan). A fluo-3/AM assay kit was purchased from Molecular Probes (Eugene, OR, USA). The fluorescein isothiocyanate (FITC)-Annexin V/propidium iodide (PI) apoptosis assay kit and reactive oxygen species (ROS) assay kit were purchased from Nanjing Jiancheng Biotech Co, Ltd. (Nanjing, China). The internal standard mixtures including PCs, SMs and Cers were purchased from Avanti Polar Lipids. All organic solvents used throughout the studies were of HPLC-grade and purchased from Fisher Scientific (Shanghai, China). All other chemicals were of analytical grade and purchased from Sigma-Aldrich (Shanghai, China) unless stated otherwise.2.2. MCAO surgery and drug administration
Male C57 BL/6 mouse were obtained from the Animal Breeding Centre of Beijing Vital River Laboratories Company. The project identification code was 20162003. All experimental procedures were approved by the Academy of Chinese Medical Science's Administrative Panel on Laboratory Animal Care and performed in accordance with institutional guidelines and ethics of the committee as part of the China Academy of Chinese Medical Sciences (February 1st, 2016).In this study, a permanent middle cerebral artery occlusion (MCAO) mouse model was applied. According to the reported methods described previously, MCAO surgery was carried out by intraluminal occlusion using a monofilament.23 Twenty four male C57 BL/6 mice were randomly divided into 4 groups: sham operation (Sham), MCAO group with water treatment (Model), MCAO with DHI-treated group (5 mL kg−1) and MCAO with Ginaton (positive drug) treated group (5 mL kg−1). The drugs were intraperitoneally injected twice a day in the morning and evening for a total of 3 days. On the third day and after the last injection in the morning, stroke was induced in the mice by MCAO. Sham mice were subjected to the same procedures with the exception of a nylon filament insertion into the common carotid artery.
2.3. Evaluation of neurological defects and infarct volume measurement
To evaluate the protective effects of DHI against ischemic stroke, the neurologic function and infarct area were measured. Six hours after MCAO-operation, the neurological function was blindly evaluated by Longa's Neurological Severity Score.24 Then, all mice were euthanized with a lethal dose of isoflurane. Five coronal sections of the brain (1 mm thickness) were immediately cut and the slices were stained with 0.5% 2,3,5-triphenyltetrazolium chloride (Sigma, St. Louis, MO, USA) for 15 minutes at 37 °C. Finally, numeric images were captured for the quantification of the infarct volume. The infarct volume of each slice was calculated as the infarct area × thickness (1 mm). The summation of the infarct volumes for all brain slices was defined as the total infarct volume.2.4. Primary mouse cortical neuron culture and drug administration
Primary mouse cortical neurons from embryos of timed pregnant (14 days) C57 BL/6 mice were isolated and cultivated by methods as described previously.25 After isolation of cortical neurons, the cells were counted and planted into poly-L-lysine-coated culture dishes with equal cell numbers (1 × 106 cells per well in 6-well plates, 3 × 104 cells per well in 96-well plates). The neurons were maintained at 37 °C in a humidified incubator with 5% CO2 atmosphere. The culture medium supplemented with 10% DES, 200 mM L-glutamine, 1 M D-glucose and 15 nM 5-FU was half changed with fresh culture medium every 2 days. 8 d after planting, the purity of the cells was confirmed by the use of mouse anti-MAP2 staining (Ab11267; 1200; Abcam, Cambridge, UK). As the purity was calculated to be over 87%, this stage was deemed best for further studies (results of MAP2 immunoreactivity can be found in the ESI†).Neurons in the OGD group were washed with HBSS and the culture media were then replaced with glucose-free HBSS and incubated for 6 h in oxygen-free N2/CO2 (95%/5%) atmosphere at 37 °C. To determine the effect of DHI, cultured cortical neurons were treated with DHI (1–0.01 μL mL−1) and 0.01 μM EDA for 6 h of OGD. EDA was used as a positive control during OGD. The cortical neurons undergoing neither drug treatment nor OGD served as control. At the end of cell treatments, different tests were carried out as described below. 6-well plates and 96-well plates were used for lipidomics analysis and all other evaluations, respectively.
2.5. Measurement of the cell viability, reactive oxygen species, [Ca2+] levels and apoptosis
The cell viability was quantitatively assessed by measurement of LDH released into the bathing medium. Cortical neuron supernatants were collected and LDH activity in the medium was determined according to the manufacturer's protocol of the LDH assay kit (Dojindo, Kumamoto, Japan).26The levels of intracellular free calcium were determined by loading the cells with fluo-3/AM. The neurons were washed and subsequently incubated with 1 mM of fluo-3/AM in the dark at 37 °C for 30 min. The fluo-3 fluorescence was excited at 488 nm and measured at 520 nm with a microplate reader (SpectraMax M5, USA).27
Formation of ROS was determined by using the fluorescent probe 2,7-dichlorofluorescein diacetate (DCFH-DA). Cell-permeant nonfluorescent DCFH-DA has been shown to be oxidized to the highly fluorescent species 2,7-dichlorofluorescein in the presence of ROS. Neurons were washed with PBS and were incubated with 10 μM DCF-DA for 1 h at 37 °C in the dark. The fluorescence intensity was measured using a microplate reader (SpectraMax M5, USA) at an excitation wavelength of 488 nm and an emission wavelength of 525 nm.28
The apoptosis rate was measured using an annexin V-FITC/PI apoptosis detection kit (Nanjing Jiancheng Biotech Co, Ltd., Nanjing, China) and flow cytometry (BD, CA, USA). Neurons were washed with PBS and subjected to Annexin V-FITC and propidium iodide (PI) double staining as described in the manufacturer's instructions. After incubation for 30 minutes at 37 °C, the stained neurons were analyzed by flow cytometry and the rate of cell apoptosis was determined.29
2.6. Lipid profiling analysis of cortical neurons
The cells (1 × 106 cells per well) were gently scraped to dislodge them from the plate and then transferred using a 3 mL Eppendorf® tube. After centrifugation for 5 minutes at 100g at 4 °C, the supernatant was removed. Then, 1.5 mL of chloroform/methanol/water (3:1:1, v/v) was added and the mixture was ultrasonicated in an ice-water bath for 1 h. The mixture was then centrifuged for 5 minutes at 100g at 4 °C and the liquid phase was transferred and evaporated using a steam of nitrogen. Subsequently, the residue was dissolved in 200 μL of isopropanol/acetonitrile (1:1, v/v), followed by centrifugation at 4 °C (13000 × g for 10 min). The supernatant was then analyzed by LC-MS.
Separation for relative quantification of PCs, Cers, and SMs was performed by UltiMate™ 3000 Rapid Separation LC (RSLC) system (Thermo Scientific, USA). As stationary phase, an ACQUITY UPLC HSS T3 C18 column (1.8 μM, 100 × 2.1 mm, Waters™, USA) was used suited for polar compound retention. The mobile phase consisted of solvent A (0.1% formic acid–water containing 2 mM ammonium formate) and solvent B (methanol) with a gradient elution (0–2 min, 20–30% B; 2–5 min, 30–45% B; 5–6.5 min, 45–55% B; 6.5–12 min, 55–65% B; 12–14 min, 65–85% B; 14–17.5 min, 85–100% B; 17.5–18 min, 100–100% B). The re-equilibration was 2 minutes with 20% B. The flow rate of the mobile phase was 0.3 mL min−1. The column temperature was maintained at 45 °C and the sample manager temperature was set at 4 °C.
A Thermo Scientific™ Q Exactive hybrid quadrupole Orbitrap mass spectrometer equipped with a HESI-II probe was used in the positive electrospray ionization mode. The pos HESI-II spray voltages were 3.7 kV, the heated capillary temperature was 320 °C, the sheath gas pressure was 30 psi, the auxiliary gas setting was 10 psi, and the heated vaporizer temperature was 300 °C. The parameters of the full mass scan were as follows: resolution of 70000, auto gain control target under 1 × 106, maximum isolation time of 50 ms, and m/z range of 150–1500.
2.7. Data processing and statistical analysis
The raw LC-MS data was imported to the Skyline software (http://skyline.gs.washington.edu/) for the relative quantification of the lipid species according to the retention time and accurate mass in the constructed polar lipids database. Briefly, the workflow for using Skyline for the analysis of 165 targeted polar lipids consisted of the following steps: (1) flat file containing the 165 molecules; (2) import the small molecule list into the Skyline, building an analysis template; (3) import the raw data. The chromatographic data for each lipid were manually analyzed to determine the quality of the signal and peak shape. Before chemometrics analysis, all of the detected ion signals in each sample were normalized to the obtained total ion count value.Multivariate analysis was performed using SIMCA-P 12.0 software. A Principal Components Analysis (PCA) was first used as an unsupervised method to visualize the differences for all groups. Supervised regression modeling was then performed on the data set by using Partial Least Squares Discriminant Analysis (PLS-DA) to identify the potential biomarkers. The biomarkers were then filtered and confirmed by combining the results of the VIP values (VIP > 1) and t-test (P < 0.05).
All values measured in vivo and in vitro are presented as means ± standard error of the mean. Statistical significance was determined by one-way ANOVA followed by Tukey multiple comparison test or Student's t-tests. A value of P < 0.05 was considered to be statistically significant.
3. Results
3.1. DHI reduces MCAO-induced infarct size and improves neurologic function
In order to investigate the protective effects of DHI against MCAO in mice, the neurologic function and infarct area were determined. Six hours after MCAO-operation, the mean neurological score in the model group (2.45 ± 0.19) was found to be significantly (P < 0.001) greater than that of the sham group, indicating a neurological defect after MCAO. In the DHI and Ginaton (positive drug) groups, the neurological defect was determined to be significantly improved compared to the model group (P < 0.01, cf. Fig. 1A). | ||
| Fig. 1 Neurological scores and infarct area by DHI pre-treatment: neurobehavioral score (A), infarct area rate (B) and TTC staining of brain (C). #P < 0.05, ##P < 0.01, ###P < 0.001 the model group versus the sham group; *P < 0.05, **P < 0.01, ***P < 0.001 the DHI group (or positive group) versus the model group. Histograms represent mean ± SD, n = 6. | ||
A similar phenomenon also appeared in the cerebral infarct area in the serial coronal brain sections. As shown in Fig. 1B and C, MCAO-induced ischemia produced a marked infarct area in the serial coronal brain sections. TTC staining of the relevant mouse tissue after DHI treatment showed a significantly lower degree of ischemic injury compared to the MCAO mice. Moreover, the corresponding infarct volumes also demonstrated that both DHI and Ginaton exhibited significant protective effects against MCAO-induced ischemic injury. All of the experimental results taken in concert suggest a crucial protective effect of DHI on ischemic stroke in vivo.
3.2. DHI protects the OGD-induced neuron injury
LDH is a stable cytoplasmic enzyme present in most cells and is often found to be rapidly released into the cell culture supernatant upon plasma membrane damages. As shown in Fig. 2A, the leakage rate of LDH in the OGD group dramatically increased compared to the control group, indicating cortical neuron injury. Treatment with DHI (3 μL mL−1, 1 μL mL−1, 0.3 μL mL−1, 0.1 μL mL−1, 0.03 μL mL−1, 0.01 μL mL−1) and EDA (0.01 μM, positive drug) showed a remarkable reduction of the leakage rate of LDH compared to the OGD group. These results confirmed that DHI exhibits a degree of neuroprotection against an OGD-induced cortical neuron injury. | ||
| Fig. 2 Protective effects of DHI on primary cortical neuronal cell under oxygen-glucose deprivation for 6 h. Cell viability (A), [Ca2+] fluorescence intensity (B), ROS levels (C), apoptosis (D). #P < 0.05, ##P < 0.01, ###P < 0.001 the OGD group versus the control group; *P < 0.05, **P < 0.01, ***P < 0.001 the DHI group (or positive group) versus the OGD group. Histograms represent mean ± SD, n > 3. | ||
A calcium surge represents a crucial step in brain damage after a stroke. As shown in Fig. 2B, the levels of calcium in the OGD group were all found to be significantly increased compared to the control group. Compared to the OGD-group, upon treatment with DHI (1 μL mL−1, 0.3 μL mL−1, 0.1 μL mL−1, 0.03 μL mL−1, 0.01 μL mL−1) and EDA (0.01 μM), the levels of calcium were found to be remarkably reduced. These results suggested that DHI can reduce the intracellular calcium influx to prevent further damages.
Fig. 2C shows the changes in the ROS-dependent fluorescence intensity detected in cortical neurons of each group. As shown here, the DCF fluorescent intensity significantly increased when compared to the control group, indicating that OGD induced an obvious elevation in the ROS level (P < 0.001). However, the overproduction of intracellular ROS was found to be significantly reduced by following pretreatments with DHI (1 μL mL−1, 0.3 μL mL−1, 0.1 μL mL−1, 0.03 μL mL−1, 0.01 μL mL−1) and EDA (0.01 μM), further indicating that DHI reduced intracellular ROS levels in OGD treated cortical neurons (P < 0.01).
An Annexin V-FITC and PI double-stain were used to evaluate the apoptosis percentage. As shown in Fig. 2D, under normal conditions, a very low level (10%) of neuronal apoptosis could be detected, but the apoptosis percentage was significantly increased to 60.7% after OGD (P < 0.001). After DHI and EDA treatment, the apoptosis percentage was reversed to 27.2%, 24.1%, 26.7%, 29.2%, 32.2% and 24.8%, respectively, upon treatment with 1 μL mL−1, 0.3 μL mL−1, 0.1 μL mL−1, 0.03 μL mL−1, 0.01 μL mL−1 DHI and 0.01 μM EDA during OGD (P < 0.01).
Taken in concert, all experimental results demonstrated cerebral protective effects of DHI on ischemic stroke. DHI could reduce the LDH release and the excess generation of ROS. Furthermore, the intracellular calcium influx and apoptosis were found to be suppressed upon DHI treatment. In the doses of DHI-treated OGD groups, a DHI dose of 0.3 μL mL−1 exhibited the best neuroprotective efficacy. Therefore, this dose was selected for the subsequent lipidomics analysis.
3.3. Lipid profiling analysis of cortical neurons
Fig. 3 represents the corresponding base peak chromatogram (BPC) of cortical neuron samples in ESI+ mode. Based on the generated targeted database and according to the retention time and accurate mass values, all compounds in the database were extracted. Finally, 117 polar lipids including 93 PC, 21 SM and 3 Cer in all the cell samples were identified and the peak area values of the identified lipids were aligned for further statistical analysis using the Skyline software. | ||
| Fig. 3 Representative base peak chromatogram (BPC) obtained from cortical neuron in ESI+ mode. The 28 potential lipids biomarkers of OGD-induced cortical neuron damage were encoded as following PC1–PC18 represent PC (14:0/0:0), PC (20:5/0:0), PC (18:2/0:0), PC (16:0/0:0), PC (0:0/22:4), PC (22:4/0:0), PC (0:0/17:0), PC (17:0/0:0), PC (20:2/0:0), PC (P-16:0/0:0), PC (O-16:0/0:0), PC (0:0/18:0), PC (P-18:0/0:0), PC (18:0/0:0), PC (O-18:0/0:0), PC (22:6/14:0), PC (P-18:0/18:2), PC (18:0/22:5) respectively; SM1–SM9 represent SM (d16:1/16:0), SM (d16:1/17:0), SM (d18:1/16:0), SM (d18:0/16:0), SM (d18:1/17:0), SM (d17:1/24:1), SM (d18:2/23:0), SM (d18:1/24:1), SM (d18:2/24:0), respectively; Cer represents Cer (d14:1/18:0). | ||
QC samples were used to demonstrate the stability of the LC-MS system. Five quality control (QC) samples of cell blends collected at the beginning of the sequence were run and the QC samples were run at regular intervals (every ten samples) throughout the entire sequence. The RSDs of the peak areas and retention times of all identified lipids in the QC samples were calculated and we determined that more than 90% of RSDs were less than 20% for the QC samples. The values of RSD > 20% in the QC group were excluded. Therefore, the repeatability and stability of the global experimental performances were high and suitable for this study. Furthermore, the cluster of the QC samples in the PCA scores scatter plot also demonstrated a satisfactory stability and repeatability of this lipidomics profiling analysis method (cf. Fig. 4).
 | ||
Fig. 4 The PCA 3D scores plot using the first four components.  : QC group, : QC group,  : control group, : control group,  : OGD group, : OGD group,  : DHI group, : DHI group,  : positive group. : positive group. | ||
3.4. Identification of potential biomarkers
To investigate the global lipidomics metabolism variations, PCA was used to analyze all observations acquired. PCA, an unsupervised pattern recognition method for handling metabolomics data, can classify the lipid metabolic phenotypes based on all imported samples. As shown in the PCA score 3D plot (cf. Fig. 4), an overview of all samples in the data can be observed and a clear grouping trend (R2X, 0.781; Q2, 0.706) between the control group, the OGD group, the DHI-treated group and the positive drug group could be observed. The OGD group vs. control group exhibited an improved separation. This observation indicates that OGD-processing may disturb the metabolism of the lipids compared to the normal state. DHI and the positive drug exhibited an effect on the OGD-induced damage, although the trajectory of the treated groups did not show a complete separation with the OGD group and was found not to return to a normal state.To further confirm the certain polar lipids used as selective and sensitive biomarkers for OGD-induced neuron injury, PLS-DA was applied to compare the lipid changes between the OGD model group and the control group. As demonstrated by the PLS-DA scores scatter plot (cf. Fig. 5A), a clear separation of the control group versus the OGD-model group could be observed. The cumulative R2X and Q2 were 0.876 and 0.869, respectively, in the PLS-DA model. No over-fitting could be observed according to the results of the chance permutation (cf. Fig. 5B). As shown here, the R2Y-intercept was 0.84 for the sham group compared to the model group. Furthermore, all green R2-values to the left were found to be lower than the original points to the right, indicating that the original model was valid. Afterwards, the significantly changed lipids of the OGD model group compared with control group were filtered out based on VIP values (VIP > 1) and t-test (P < 0.05).
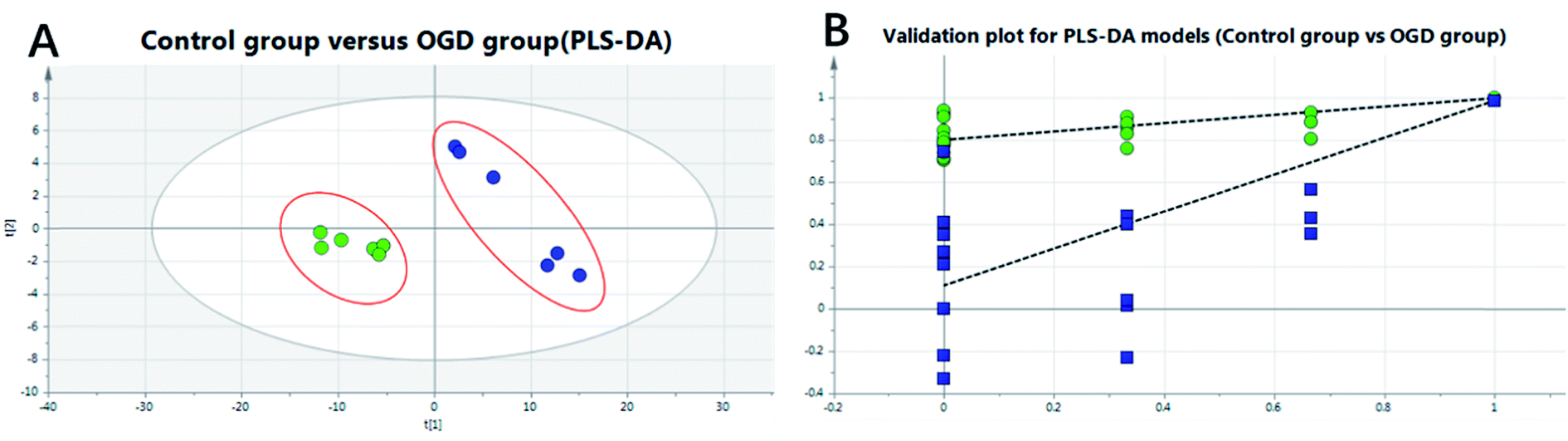 | ||
Fig. 5 The PLS-DA scores scatter plot obtained from control versus OGD model groups (A) and validation plot for PLS-DA models obtained from 20 permutation tests. (B) A:  : control group, : control group,  : OGD group; B: : OGD group; B:  , R2, , R2,  : Q2. : Q2. | ||
Subsequently, a total of 28 potential lipids biomarkers of OGD-induced cortical neuron damages, including 18 PCs, 9 SMs, and 1 Cer, were studied. The changed levels and names of the biomarkers are shown in Fig. 6. Specifically, the 12 lipids levels of PC (16:0/0:0), PC (0:0/17:0), PC (17:0/0:0), PC (0:0/18:0), PC (18:0/0:0), PC (O-16:0/0:0), PC (P-16:0/0:0), PC (O-18:0/0:0), PC (P-18:0/0:0), PC (P-18:0/18:2), PC (18:2/0:0), PC (20:2/0:0) were found to be significantly decreased in the OGD group. However, the levels of 6 lipids including PC (0:0/24:0), PC (24:0/0:0), PC (20:5/0:0), PC (22:6/14:0), PC (18:0/22:5), PC (14:0/0:0) were all found to be increased in the OGD group compared to the control group. With regards to SMs and Cers, 9 SMs, including SM (d16:1/16:0), SM (d16:1/17:0), SM (d17:1/24:1), SM (d18:0/16:0), SM (d18:1/16:0), SM (d18:1/17:0), SM (d18:1/24:1), SM (d18:2/23:0), SM (d18:2/24:0) and one Cer species, i.e. Cer (d14:1/18:0), were all found to be significantly decreased in the OGD group compared to the control group.
 | ||
| Fig. 6 Metabolic changes of lipids in the four groups. Blue, red, green and yellow box chart represent the normalized content in the control, OGD, DHI and positive groups, respectively. #P < 0.05, ##P < 0.01, ###P < 0.001 the OGD group versus the Control group; *P < 0.05, **P < 0.01, ***P < 0.001 the DHI group and positive group versus the OGD group OGD. (A) SMs; (B) PCs and Cers. | ||
After DHI treatment, 28 potential lipids biomarkers of OGD-induced cortical neuron damage were analyzed and 1 SMs level, i.e. SM (18:0,16:0), as well as 4 PCs levels, i.e. PC (17:0,0:0), PC (18:0,0:0), PC (16:0,0:0), and PC (P-16:0,0:0) in the DHI-treated group exhibited a normal-level tendency (P < 0.05). Moreover, after EDA (positive drug) treatment, 2 PCs levels, i.e. PC (0:0,18:0) and PC (20:5,0:0), could be found exhibiting a tendency of returning to normal levels (P < 0.05).
4. Discussion
The pathophysiological processes of ischemic stroke-induced brain injuries are complex and generally poorly understood, with many questions remaining unanswered. A variety of studies have been devoted towards explaining the mechanisms and providing better therapeutic approaches of ischemic stroke. DHI, a standardized commercial product derived from TCM, has long been used to treat and prevent ischemic stroke, although the mechanisms are still not fully understood. In this study, a MCAO-induced mouse injury model confirmed the protective effects of DHI by decreasing the infarct volume and improving neurological functions. Furthermore, an OGD-induced neuron injury model demonstrated the cell protective effect of DHI from various perspectives of cell viability, ROS, [Ca2+] levels and apoptosis. Based on the OGD-induced neuron injury model, a lipidomics profiling analysis with a targeted polar lipids extract was carried out to reveal the pathogenesis of acute ischemic stroke and the underlying mechanisms of DHI. For the first time, 28 biomarkers of PCs, SMs and Cers of neuron injury induced by OGD were identified according to the database generated by the standards and the perturbations could be partly reversed by DHI intervention. Monitoring the changes in these lipids may help shed light on the mechanism of OGD-induced neuron injury and the efficacy of DHI.4.1. Sphingomyelin metabolism
Sphingomyelin (SM), a sphingolipid species, is located in the membrane myelin sheath which surrounds some nerve cell axons.30 As an essential modulator, SM influences the membrane gathering of proteins involved in cellular proliferation, growth and apoptosis.31,32 Furthermore, SM represents an important source of ceramide.33 Previous studies have shown that the levels of SMs are crucial to ischemic stroke mostly based on tissue levels. In this study, lipidomics was conducted for the first time on the primary mouse cortical neurons subjected to OGD. Except for only one saturated acyl chains of SM (d18:0/16:0), the structures of the 9 identified SMs biomarkers mainly consisted of SMs with unsaturated acyl chains as sub-species of SM (d16:1/16:0), SM (d16:1/17:0), SM (d17:1/24:1), SM (d18:1/16:0), SM (d18:1/17:0), SM (d18:1/24:1), SM (d18:2/23:0), SM (d18:2/24:0).It has already been shown that the energy requirements of the brain are satisfied by the glucose metabolism and oxygen needed for the phosphorylation of ADP to ATP. ATP is vital to maintain intracellular homeostasis and transmembrane ion gradients of sodium, potassium, and calcium. Oxygen and glucose deprivation results in the rapid loss of ATP which further causes an uncontrolled calcium leakage at certain disease related events. As shown in Fig. 2B, the calcium levels are found to be significantly increased after OGD compared to the control group and the rapid increase of calcium leads to the activation of sphingomyelinase which plays an important role in the sphingomyelin cycle.34
In the sphingomyelin cycle, SMs are hydrolyzed to ceramides by the activation of sphingomyelinases.21 Ceramide, a hydrolyzed product of sphingomyelin, is reported to play an important role in cell death as well as cell cycle arrest and generally serves as a second messenger. Several signaling pathways can be regulated by ceramide, including a diverse range of protein kinases and phosphatases.35 Previous studies have shown that the further release of ceramide-1-phosphate (C1P), a hydrolyzed product of Cer, results in the activation of phospholipases.36 The activation of phospholipases further increases the hydration of PCs. Moreover, Cer has been shown to promote the release of arachidonic acid (ARAC) which acts as an inflammatory intermediate.36,37 The release of ARAC results in activation of cyclooxygenase (COX) and the generation of ROS which causes lipid peroxidation.38,39 Eventually, this process leads to apoptosis as highlighted in Fig. 7. In this study, compared to the control group, the levels of 9 significantly modified SM species found in the OGD model were shown to decrease by 10–20%, a finding that was in accordance with the hydrolyzation theory (cf. Fig. 6). As the levels of Cers were too low, only 3 Cers sub-species were found in the neuron samples and only the level of Cer (d14:1/18:0) changed significantly. However, the levels of ROS and apoptosis in the OGD group were confirmed and showed a remarkable increase compared to the control group.
 | ||
| Fig. 7 The injury mechanism of neurons subjected to OGD and the effects of DHI. Activation of phospholipases and sphingomyelinases following cerebral ischemia results in release of Cer and ArAc. LDH, lactic dehydrogenase; FFA, free fatty acid; COX, cyclooxygenase; ARAC, arachidonic acid. | ||
4.2. Phosphatidylcholines metabolism
Phosphatidylcholines (PCs) can not only be found in the composition of cell membranes, but also serve as nuclear receptor of peroxisome proliferator-activated receptors (PPARa) which describes a transcription factor regulating the expression of various genes that govern lipid metabolism.40 The metabolic products of PCs may act as secondary messengers for cellular regulation, e.g. fatty acids.41 Prior studies have provided evidence that degradation of membrane phospholipids may play a key role in ischemia injuries.14,42 As shown in Fig. 7, with uncontrolled leakage of calcium, phospholipases such as phospholipases A2 were also found to be activated, in addition to sphingomyelinase. Subsequently, free fatty acids (FFA), biologically active lipid mediators in the brain, were stimulated. The large polyunsaturated fatty acid (PUFA) pools lead to peroxidation of membrane lipids which further resulted in functional impairment.43In this study, of the 18 PCs biomarkers we observed that the levels of 12 PCs sub-species, i.e. PC (16:0/0:0), PC (0:0/17:0), PC (17:0/0:0), PC (0:0/18:0), PC (18:0/0:0), PC (O-16:0/0:0), PC (P-16:0/0:0), PC (O-18:0/0:0), PC (P-18:0/0:0), PC (P-18:0/18:2), PC (18:2/0:0), and PC (20:2/0:0) in the OGD group, were significantly decreased after ischemia compared to the control group which was consistent with levels reported previously.14,44 Among these suppressed biomarkers, more structures were found to contain saturated acyl chains. However, the levels of 6 PC sub-species, i.e. PC (0:0/24:0), PC (24:0/0:0), PC (20:5/0:0), PC (22:6/14:0), PC (18:0/22:5), and PC (14:0/0:0) were found to be increased which may account for a self-protective mechanism. The increased PC levels may also serve as endogenous neuroprotective cytokine, similarly to gangliosides (GM1).45 However, this notion as well as the underlying mechanisms requires further studies to be entirely confirmed.
4.3. Potential mechanism of DHI
DHI, a Chinese Materia Medica standardized product with multiple components, has been reported to exhibit various pharmacological activities. In this work, an OGD-induced primary mouse cortical neuron injury model was used for the first time to confirm the activities and neuroprotective effects of DHI in the reduction of neuronal damages and the excess generation of ROS, suppressing intracellular calcium influx and apoptosis. As shown in Fig. 6, we found that after DHI treatment, of the 28 potential lipids biomarkers of OGD-induced cortical neuron injury, only one SM level, i.e. SM (18:0,16:0) and 4 PCs levels, i.e. PC (17:0,0:0), PC (18:0,0:0), PC (16:0,0:0), and PC (P-16:0,0:0) in the DHI-treated group, exhibited a significant normal level tendency (P < 0.05) which suggests that the protective effects of DHI on cerebral ischemia are closely related to the regulation of the PCs metabolism. Furthermore, among the 6 significantly increased PCs biomarkers in the OGD model which may serve as endogenous neuroprotective cytokines, PC (18:0/22:5) was found to be increased in the DHI treated group (P < 0.05). However, upon EDA treatment, the results were not the same with DHI. It has already been shown that EDA as brain protecting agent, may act as a free-radical scavenger. Of the 28 potential lipids biomarkers of OGD-induced cortical neuron injury, 2 PC levels demonstrated a normal trend (P < 0.05) in the EDA treated group compared to the OGD model group. Between DHI and EDA, no common returned biomarkers were present, further indicating that the mechanism of DHI and EDA may not be equal.5. Conclusions
A UPLC-MS profiling analysis for neuronal polar lipids including PCs, SMs and Cers was developed and applied to explore the mechanisms of OGD-induced primary mouse cortical neuron injury and the therapeutic effects of DHI. Before lipidomics analysis, MCAO-induced mice were used to confirm the protective effects of DHI in vivo. After DHI treatment, we observed decreases in neurological deficits and cerebral infarct sizes, indicating that DHI exhibits therapeutic efficacy for the treatment of cerebral ischemia. Furthermore, primary mouse cortical neurons subjected to OGD were used to investigate the protective mechanisms of DHI in vitro. DHI was found to reduce neuronal damages and the excess generation of ROS. Furthermore, intracellular calcium surge and apoptosis were observed to be suppressed. Based on an OGD-induced neuron injury model, 28 biomarkers of PCs, SMs and Cers sub-species of neuron injury induced by OGD have been accurately identified. The perturbations could be partly reversed by DHI intervention such as PC (17:0,0:0), PC (18:0,0:0), PC (16:0,0:0), PC (P-16:0,0:0) and SM (18:0,16:0). We found that the therapeutic effects of DHI on cerebral ischemia are partially due to interferences with the PCs and SMs metabolisms. These results specifically shed light on the relationship between PCs, SMs, Cers sub-species and the mechanism of neuronal damages during ischemia stokes. Potentially, these modified lipids may be used as biomarkers of ischemic cerebral injury for clinical diagnosis and treatment.Conflicts of interest
All authors have approved of the manuscript and agree with submission to your esteemed journal. We declare no conflicts of interest.Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (No. 81330086, No.81403210) and the National Basic Research Program of China (No. 2015CB554406).Notes and references
- T. M. Woodruff, J. Thundyil, S. C. Tang, C. G. Sobey, S. M. Taylor and T. V. Arumugam, Mol. Neurodegener., 2011, 6, 11 CrossRef PubMed.
- S. N. Wang, T. Y. Xu, X. Wang, Y. F. Guan, S. L. Zhang, P. Wang and C. Y. Miao, CNS Neurosci. Ther., 2016, 22, 782–788 CrossRef CAS PubMed.
- J. S. Tauskela, A. Aylsworth, M. Hewitt, E. Brunette and G. A. Mealing, J. Neurochem., 2012, 122, 470–481 CrossRef CAS PubMed.
- Y. Fu, N. Zhang, L. Ren, Y. Yan, N. Sun, Y. J. Li, W. Han, R. Xue, Q. Liu, J. Hao, C. Yu and F. D. Shi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 18315–18320 CrossRef CAS PubMed.
- M. A. Moskowitz, E. H. Lo and C. Iadecola, Neuron, 2010, 68, 161 CrossRef CAS.
- X. Luo, X. Yu, X. Wu, Y. Cheng and H. Qu, Microchem. J., 2008, 90, 8–12 CrossRef CAS.
- X. Liu, Z. Wu, K. Yang, H. Ding and Y. Wu, J. Pharm. Biomed. Anal., 2013, 76, 70–74 CrossRef CAS PubMed.
- Y. He, H. Wan, Y. Du, X. Bie, T. Zhao, W. Fu and P. Xing, J. Ethnopharmacol., 2012, 144, 387–394 CrossRef CAS PubMed.
- J. Wei, Y. Zhang, Q. Jia, M. Liu, D. Li, Y. Zhang, L. Song, Y. Hu, M. Xian, H. Yang, C. Ding and L. Huang, Sci. Rep., 2016, 6, 29823 CrossRef CAS PubMed.
- H. Guo, M. J. Li, Q. Q. Liu, L. L. Guo, M. M. Ma, S. X. Wang, B. Yu and L. M. Hu, Neurochem. Res., 2014, 39, 1817–1824 CrossRef CAS PubMed.
- Y. Jiang and Y. J. Lian, Genet. Mol. Res., 2015, 14, 16929–16937 CrossRef CAS PubMed.
- M. Yang, J. Orgah, J. Zhu, G. Fan, J. Han, X. Wang, B. Zhang and Y. Zhu, Brain Res., 2016, 1642, 516–523 CrossRef CAS PubMed.
- S. Wang, H. Guo, X. Wang, L. Chai, L. Hu, T. Zhao, B. Zhao, X. Tan and F. Jia, Neural Regener. Res., 2014, 9, 1453–1459 CrossRef PubMed.
- S. R. Shanta, C. S. Choi, J. H. Lee, C. Y. Shin, Y. J. Kim, K. H. Kim and K. P. Kim, J. Lipid Res., 2012, 53, 1823–1831 CrossRef CAS PubMed.
- M. P. Mattson and R. G. Cutler, Adv. Cell Aging Gerontol., 2003, 12, 97–116 CAS.
- S. Furse and A. I. de Kroon, Mol. Membr. Biol., 2015, 32, 117–119 CrossRef CAS PubMed.
- P. C. Choy, M. Chan, G. Hatch and R. Y. Man, Mol. Cell. Biochem., 1992, 116, 53–58 CrossRef CAS PubMed.
- X. C. Jiang, F. Paultre, T. A. Pearson, R. G. Reed, C. K. Francis, M. Lin, L. Berglund and A. R. Tall, Arterioscler., Thromb., Vasc. Biol., 1996, 27, 527–535 Search PubMed.
- J. A. Clemens, D. T. Stephenson, E. B. Smalstig, E. F. Roberts, E. M. Johnstone, J. D. Sharp, S. P. Little and R. M. Kramer, Stroke, 1996, 27, 527–535 CrossRef CAS PubMed.
- R. Ohtani, H. Tomimoto, T. Kondo, H. Wakita, I. Akiguchi, H. Shibasaki and T. Okazaki, Brain Res., 2004, 1023, 31–40 CrossRef CAS PubMed.
- M. Nakane, M. Kubota, T. Nakagomi, A. Tamura, H. Hisaki, H. Shimasaki and N. Ueta, Neurosci. Lett., 2000, 296, 89–92 CrossRef CAS PubMed.
- A. A. Farooqui, W. Y. Ong and L. A. Horrocks, Neurochem. Res., 2004, 29, 1961–1977 CrossRef CAS PubMed.
- Y. Cui, X. Liu, X. Li and H. Yang, Int. J. Mol. Sci., 2017, 18, 1355 CrossRef PubMed.
- E. Z. Longa, P. R. Weinstein, S. Carlson and R. Cummins, Stroke, 1989, 20, 84–91 CrossRef CAS PubMed.
- S. Powell, A. Vinod and M. L. Lemons, J. Visualized Exp., 2014, 51991, DOI:10.3791/51991.
- H. Guo, S. Kong, W. Chen, Z. Dai, T. Lin, J. Su, S. Li, Q. Xie, Z. Su, Y. Xu and X. Lai, Neurochem. Res., 2014, 39, 2197–2210 CrossRef CAS PubMed.
- J. S. Tauskela, H. Fang, M. Hewitt, E. Brunette, T. Ahuja, J. P. Thivierge, T. Comas and G. A. Mealing, J. Biol. Chem., 2008, 283, 34667–34676 CrossRef CAS PubMed.
- R. Ye, N. Li, J. Han, X. Kong, R. Cao, Z. Rao and G. Zhao, Neurosci. Res., 2009, 64, 306–310 CrossRef CAS PubMed.
- G. Gong, L. Yuan, L. Cai, M. Ran, Y. Zhang, H. Gong, X. Dai, W. Wu and H. Dong, PLoS One, 2014, 9, 105944 CrossRef PubMed.
- R. A. Claus, M. J. Dorer, A. C. Bunck and H. P. Deigner, Curr. Med. Chem., 2009, 16, 1978–2000 CrossRef CAS PubMed.
- J. P. Slotte, Prog. Lipid Res., 2013, 52, 424–437 CrossRef CAS PubMed.
- Y. Barenholz and T. E. Thompson, Biochim. Biophys. Acta, Biomembr., 1980, 604, 129–158 CrossRef CAS.
- Y. A. Hannun and R. M. Bell, Science, 1989, 243, 500–507 CrossRef CAS PubMed.
- R. M. Adibhatla, R. Dempsey and J. F. Hatcher, Front. Biosci., 2008, 13, 1250–1270 CrossRef CAS.
- M. I. H. Bhuiyan, M. N. Islam, S. Y. Jung, H. H. Yoo, Y. S. Lee and C. Jin, Biol. Pharm. Bull., 2010, 33, 11–17 Search PubMed.
- G. F. Nixon, Br. J. Pharmacol., 2009, 158, 982–993 CrossRef CAS PubMed.
- B. J. Pettus, A. Bielawska, S. Spiegel, P. Roddy, Y. A. Hannun and C. E. Chalfant, J. Biol. Chem., 2003, 278, 38206–38213 CrossRef CAS PubMed.
- S. Schütze, K. Potthoff, T. Machleidt, D. Berkovic, K. Wiegmann and M. Krönke, Cell, 1992, 71, 765–776 CrossRef.
- A. H. Nave, K. S. Lange, C. O. Leonards, B. Siegerink, W. Doehner, U. Landmesser, E. Steinhagen-Thiessen, M. Endres and M. Ebinger, Atherosclerosis, 2015, 242, 496–503 CrossRef CAS PubMed.
- S. Kersten, Mol. Metab., 2014, 3, 354–371 CrossRef CAS PubMed.
- S. Koizumi, S. Yamamoto, T. Hayasaka, Y. Konishi, M. Yamaguchi-Okada, N. Goto-Inoue, Y. Sugiura, M. Setou and H. Namba, Neuroscience, 2010, 168, 219–225 CrossRef CAS PubMed.
- R. M. Adibhatla and J. F. Hatcher, Subcell. Biochem., 2008, 49, 241–268 Search PubMed.
- J. Kim, J. W. Lampe, T. Yin, K. Shinozaki and L. B. Becker, Mol. Cell. Biochem., 2015, 408, 273–281 CrossRef CAS PubMed.
- K. Hamazaki and H. Y. Kim, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2013, 88, 299–306 CrossRef CAS PubMed.
- L. Candelise and A. Ciccone, Stroke, 2002, 33, 2336 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09245h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |