
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBeneficial effects of Trametes versicolor pretreatment on saccharification and lignin enrichment of organosolv-pretreated pinewood
Gurshagan Kandholaa,
Kalavathy Rajan bc,
Nicole Labbéc,
Stephen Chmelyc,
Nelson Heringera,
Jin-Woo Kima,
Elizabeth E. Hoodd and
Danielle Julie Carrier*ab
bc,
Nicole Labbéc,
Stephen Chmelyc,
Nelson Heringera,
Jin-Woo Kima,
Elizabeth E. Hoodd and
Danielle Julie Carrier*ab
aDepartment of Biological and Agricultural Engineering, Institute for Nanoscience and Engineering, University of Arkansas, 101 Biosystems Engineering Soil Science 2506 E J. Chapman Dr., Knoxville, TN 37996, Fayetteville, AR 72701, USA. E-mail: dcarrie1@utk.edu; Tel: +1 865 974 7305
bDepartment of Biosystems Engineering and Soil Science, The University of Tennessee, Knoxville, TN 37996, USA
cCenter for Renewable Carbon, The University of Tennessee Institute of Agriculture, Knoxville, TN 37996, USA
dArkansas Biosciences Institute, Arkansas State University, Jonesboro, AR 72401, USA
First published on 25th September 2017
Abstract
While previous studies have shown that white-rot fungal pretreatment reduces the severity of chemical pretreatments and improves enzymatic saccharification yields, very few have investigated the synergistic effects of fungal pretreatment combined with organosolv pretreatment on lignin recovery and quality. In this study, loblolly pine chips were incubated with Trametes versicolor for 15, 30 and 45 days, prior to organosolv pretreatment and enzymatic saccharification. Fungal pretreatment for 15 days improved the saccharification yield by 23%, and higher amounts (56%) of lignin-enriched fractions were obtained. Fungal pretreatment for 45 days led to extensive depolymerization, structural modification and enrichment of lignin in the organosolv precipitates (OP). Characterization of the OP fraction showed that samples pretreated for 30 and 45 days contained higher amounts of hydroxyl groups and p-hydroxyphenyl (H) subunits, and showed increases in depolymerization of carbohydrates as well as decreases in G/H lignin ratio. It became apparent that further investigations are needed to explore the benefits of combining fungal and organosolv pretreatments for improving lignin yields from softwoods.
1. Introduction
Loblolly pine (Pinus taeda) is a widely cultivated and the most commercially important forest species in the southern United States; it is predominant in about 11.7 million hectares (29 million acres) and accounts for over one-half of the standing pine volume.1 Loblolly pine is an extremely fast-growing softwood species and is extensively used in the pulp and paper industry. Several countries have well-established systems in place for sustainable management of coniferous forests, where the main product is high-quality sawn timber. A considerable fraction of harvested biomass is still left unutilized, making available an abundant resource of lignocellulosic biomass throughout the year that could be used as biorefinery feedstock.2 On the other hand, deconstruction of softwoods is a major challenge due to its high lignin content, high cellulose crystallinity, and strong binding of lignin with the cellulose and hemicellulose fractions at the cellular level. It is essential to investigate the efficacy of various physical, chemical, physico-chemical, and biological pretreatment strategies for reducing the recalcitrance of this valuable softwood resource.Biological pretreatment of woody biomass with lignin-degrading fungi, is a low cost and environment-friendly strategy that can result in significant energy savings when coupled with conventional mechanical and chemical pulping operations.3,4 White-rot and brown-rot fungi are commonly used for biodegradation of lignocellulosic biomass; they act by depolymerizing and/or mineralizing lignin and in some cases, hemicellulose. Previously published studies explore the mechanistic actions of white-rot fungi such as Ceriporiopsis subvermispora and Trametes versicolor in reducing the recalcitrance of lignocellulosic biomass and consequently, improving its enzymatic digestibility;5,6 however, only a few studies present its potential in terms of lignin isolation.7 Biological pretreatment is often combined with chemical pretreatments, for example, the organosolv process, which uses organic solvents such as, ethanol, methanol, butanol and acetone, at high temperatures ranging between 150 and 200 °C, to fractionate lignocellulosic biomass.8 Originally developed as an alternative to Kraft pulping, the ethanol organosolv process has demonstrated great potential in enhancing the enzymatic digestibility of the cellulosic fraction as well as recovering high-quality lignin as a relatively pure co-product.9–11 Studies have suggested that combining fungal pretreatment and organosolv fractionation was effective in improving the enzymatic saccharification of pine cellulose,12 increasing ethanol production efficiency13 and reducing the severity of chemical pretreatments.14 There are, however, no reports demonstrating the combined effects of fungal and organosolv pretreatments on recovery and properties of lignin from pine.
It is essential for a biorefinery to maximize resource utilization in order to achieve economic feasibility and this can be realized by careful isolation of each lignocellulosic component and converting them into valuable products.15 While fungal pretreatment studies have focused on optimizing the treatment conditions for delignification and enhancement of enzymatic conversion of cellulose into sugars, about 40–60% of lignin is left underutilized in the biodegraded residual biomass.7 Also there has been little research on characterization of lignin isolated from biodegraded biomass, which is partly due to the lack of fractionation technology that can efficiently isolate lignin. Cellulolytic enzymes were reported to be used to isolate lignin (CEL) from white-rot fungi pretreated bamboo, which was characterized using thermo-chemical measurements, but the yields were not reported.7 Overall, there is a dearth of research aimed at examining the combination of fungal and organosolv pretreatments on lignin recovery and elucidation of its structural and chemical properties. With that in mind, we pretreated loblolly pinewood with white-rot fungus, T. versicolor, for 15, 30, and 45 days and evaluated the resulting changes in saccharification efficiency. Fungal pretreatment was followed by ethanol organosolv pretreatment, where the liquid fraction precipitates were extensively characterized using spectroscopic and chromatographic techniques. Understanding fungal-altered lignin properties is essential for lignin-based product development in bio-refineries.
2. Materials & methods
2.1. Biomass and chemicals
Loblolly pine (Pinus taeda), grown in the School of Forestry & Natural Resources POW Camp, in Monticello, AR and harvested in September 2015, was used in this study. The stem wood had a diameter at breast height (diameter measured at 1.37 m above the ground) of 0.31–0.36 m. The particle size of debarked wood was reduced using a laboratory wood chipper and chips of size 2.0–3.5 mm (that passed through and were retained between US 6 and 10 mesh sieves, respectively) were separated and stored at −20 °C until further use. For compositional analysis, the pine chips were air-dried to a moisture content of less than 10%, ground using a Thomas Wiley® Mini-Mill (Swedesboro, NJ), and passed through a 20 mesh screen to obtain particles of 0.263 mm in size. The structural carbohydrates and lignin content of the pine biomass were determined as described in the NREL (National Renewable Energy Laboratory, Golden, CO) laboratory analytical protocols (LAPs).16Commercial standards of glucose and hydroxymethylfurfural (HMF) were purchased from Alfa-Aesar (Haverhill, MA); xylose, galactose, arabinose, mannose, and furfural were purchased from Sigma-Aldrich (St. Louis, MO). Sulfuric acid (98%) and ethanol (99.5%) were purchased from VWR International (Westchester, PA). Calcium carbonate was obtained from Thermo Fisher Scientific (Fair Lawn, NJ). Distilled water was prepared using a Direct-Q® system (Millipore, Billerica, MA) that had 0.182 MΩ m resistivity at 25 °C. Potato Dextrose Agar (PDA) and Potato Dextrose Broth (PDB) were obtained from EMD (Gibbstown, NJ) and HiMedia (Mumbai, India), respectively.
2.2. Preparation of fungal inoculum
The strain of T. versicolor used in this study (HBB-2740) was isolated from Nantahala National Forest, NC and supplied by the United States Department of Agriculture, Madison, WI. The strain was maintained at 4 °C on agar slants containing 20 g L−1 PDA and 20 g L−1 agar. Fungal inoculum for pine biomass fermentation was obtained by growing a mycelial mat on sterile, pre-poured PDA plates and by transferring 5 mycelial discs, each of 8 mm in diameter, into a 1 L flask containing 200 mL of sterile, PDB media prepared at a concentration of 25 g L−1. The inocula were maintained unshaken at 28 °C, for 10 days, in a forced-air incubator (VWR, Chicago, IL).2.3. Pretreatment of wood chips with T. versicolor
Pine wood chips (10 g dry weight) were soaked in 200 mL water in 1 L flasks. The excess water was drained after 15 h and the wood chips were autoclaved at 121 °C for 15 min to eliminate any possibility of prior microbial contamination. Mass loss resulting from sterilization (1.0 ± 0.1%) was accounted for, while calculating the mass and component losses during fungal pretreatment. The fungal inoculum, along with the liquid medium, was carefully transferred to each flask containing pre-soaked and autoclaved pine chips under sterile conditions. The fungal mycelia was allowed to grow on the pine biomass for 15, 30, and 45 days at 28 °C, inside a forced-air incubator. Each treatment was done in duplicate. At the end of each incubation period, the fungal mat that had formed on pine chips was manually removed. The pine chips were subsequently washed with 1 L of 0.1 N NaOH, rinsed twice with 1 L distilled water, and stored at 4 °C until further analysis. An uninoculated sample, consisting of only pine chips incubated with 200 mL of PDB, was prepared as control.2.4. Organosolv pretreatment
The organosolv process was conducted in a 1 L stainless steel, Parr reactor (Moline, IL) at a total solids loading of 4%, where 6 g of dry ground pine chips were reacted with 150 mL of ethanol and water mixture (65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35, v/v) that contained 1% H2SO4 (v/v), for 1 h at 170 °C; these conditions were chosen based on a previous study by Heringer (2016).17 After the reaction, the solid pre-hydrolyzate was separated from the liquid fraction using vacuum filtration. The solid fractions were washed with two consequent portions of 150 mL distilled water, prior to enzymatic digestibility studies. The organosolv liquid fraction was used for anti-solvent precipitation and characterization. The control was uninoculated pine chips that were also subjected to organosolv pretreatment.
:35, v/v) that contained 1% H2SO4 (v/v), for 1 h at 170 °C; these conditions were chosen based on a previous study by Heringer (2016).17 After the reaction, the solid pre-hydrolyzate was separated from the liquid fraction using vacuum filtration. The solid fractions were washed with two consequent portions of 150 mL distilled water, prior to enzymatic digestibility studies. The organosolv liquid fraction was used for anti-solvent precipitation and characterization. The control was uninoculated pine chips that were also subjected to organosolv pretreatment.
2.5. Anti-solvent precipitation and characterization
Organosolv precipitates (OP) were recovered via anti-solvent extraction from the liquid fraction using distilled water (5 times the volume of liquor); the precipitate was filtered with a Whatman® nylon membrane filter (0.45 μm) and air-dried to a constant weight. The overall scheme for isolation of the OP fraction is given in Fig. 1. The molecular weight of OP was determined using size exclusion chromatography (SEC) (Tosoh EcoSec, Griesheim, Germany), where all samples were derivatized using an aceto-bromination reagent, as described by Guerra et al. (2008),18 and dissolved in THF prior to SEC analysis. Polystyrene calibration standards at a range of 600 to 7500000 g mol−1 were used for molecular weight computation.
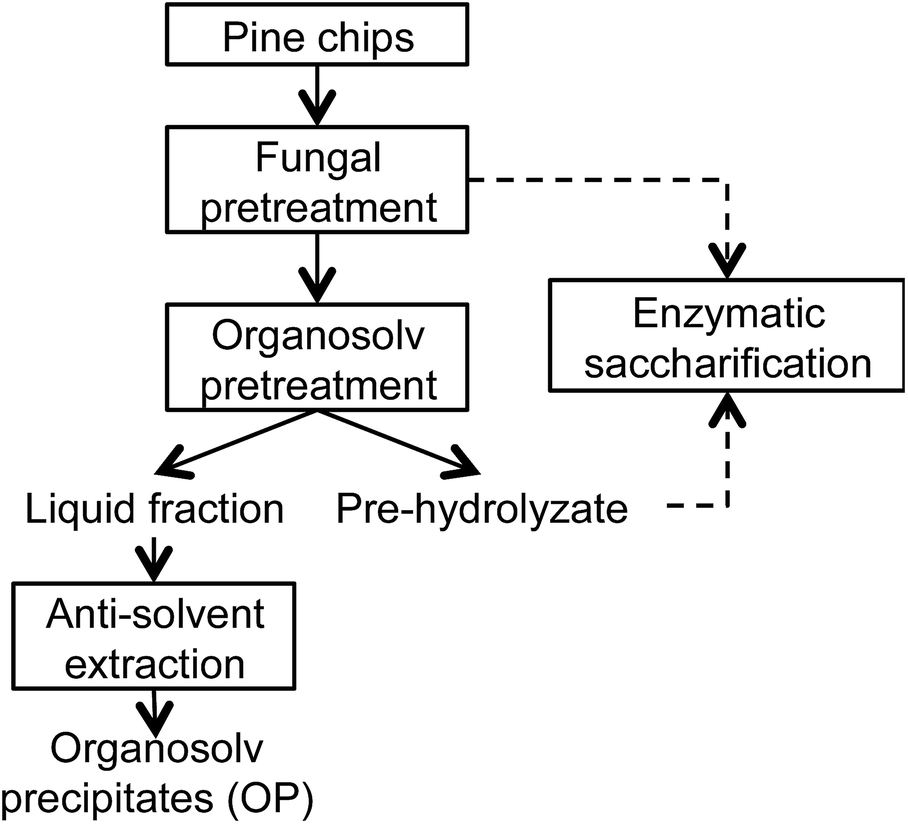 | ||
| Fig. 1 Scheme for Trametes versicolor and ethanol-organosolv pretreatments of loblolly pine chips. | ||
A Fourier transform infrared (FTIR) spectrophotometer (Perkin-Elmer, Waltham, MA), fitted with a diamond ATR (Attenuated Total Reflectance) cell was used to collect the infrared absorption spectrum of OP from 4000 to 600 cm−1, at 32 scans per spectrum and 4 cm−1 resolution. Five FTIR spectra were collected per OP sample and FTIR bands were assigned based on previous publications.19–21
For 31P NMR (nuclear magnetic resonance) spectroscopy, 20 to 40 mg of OP were dissolved in 400 μL of 1.6:1 (v/v) deuterated pyridine and chloroform. Then 50 μL of relaxation reagent, chromium(III) acetylacetonate at 11.4 g L−1, and 100 μL of internal standard (IS) solution, endo-N-hydroxy-5-norbornene-2, 3-dicarboximide (21.5 g L−1), were added. Finally, 100 μL of phosphitylating agent, 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane, were added; the samples were thoroughly mixed before being subjected to NMR acquisition. The phosphorous acquisition parameters were: 1 s acquisition time, 5 s relaxation delay and a total of 512 scans. The spectra were calibrated to the signal of water derivatized product of IS i.e., 132.2 ppm. Chemical shifts corresponding to aliphatic, guaiacyl (G), syringyl (S) and p-hydroxyphenyl (H) lignin OH groups were assigned based on Balakshin and Capanema (2015).22
For pyrolysis-gas chromatography mass spectrometry (py-GC/MS) analysis, 200–230 μg of OP were loaded in small stainless steel cylinders (4 mm OD × 8 mm ht) and subjected to pyrolysis at 500 °C. The pyrolysis products were separated using a Perkin-Elmer Clarus 680 gas chromatography system fitted with an Elite 1701 MS capillary column (60 m × 0.25 mm ID by 0.25 μm film thickness). Ultra high pure helium was used as the carrier gas, set at a flow rate of 1 mL per minute and 6.9 kPa pressure. The temperature ramp program was as follows: 4 min at 50 °C, ramp 4.65 °C per second to 290 °C, hold at 290 °C for 5 min. The pyrolysis products were analyzed using a Clarus SQ 8C mass spectrometer (source temperature 270 °C, 70 eV electron ionization), and compared to the NIST library for identification based on their mass fragmentation patterns. About 200 chromatographic peaks with S/N ≥ 2000 were extracted from the pyrograms using TurboMass software and the individual peak area was normalized to the total peak area. Each treatment group was analyzed in triplicate.
2.6. Enzymatic hydrolysis and sugar analysis
All fungal pretreated biomass and organosolv pre-hydrolyzates were air-dried to a moisture content of 28–30% and milled to particle size below 850 μm. Weight of pretreated material equivalent to 0.1 g of glucan was mixed with 10 mL of buffer solution (0.05 M sodium citrate) at pH 4.8 and was hydrolyzed for up to 72 h, at 50 °C, using Accellerase® 1500 (Genencor-DuPont, Palo Alto, CA) enzyme, which was loaded at 15 FPU g−1 glucan. The liquid hydrolyzate was collected and stored at 4 °C, prior to glucose content measurement. Sample preparation for High Performance Liquid Chromatography (HPLC) analysis was done by filtering all the liquid process samples using a 0.2 μm syringe filter assembly (Thermo Fisher Scientific, Nashville, TN), in order to remove any particulates. Composition of sugars and byproducts in the liquid fraction were analyzed using HPLC protocol as reported by Sluiter et al. (2008).16 The quantification of monomers, organic acids, and furans was done using in-house calibration curves.2.7. Statistical analysis
Statistical software JMP Pro (version 12.1.0) was used to perform Student's t-test and one-way ANOVA for comparison of treatment means. All differences are reported at a significance level of p ≤ 0.05. Principal component analysis (PCA) of the infrared spectra, in the range of 4000 to 600 cm−1, was performed using the Unscrambler X v. 10.4 software (CAMO software, Oslo, Norway). The IR spectra were grouped into four classes based on the duration of fungal pretreatment i.e., 0 day, 15 days, 30 days and 45 days and PCA was performed to determine any significant variations in chemical bonding structure of OP as a result of biological pretreatment. Prior to PCA, the FTIR data were pre-processed as follows; initially the data were normalized to the mean absorption value and then fully corrected for multiplicative scatter in order to compensate for scatter-effects and baseline shifts in the spectra. During PCA, the first five factor loadings were plotted to gather information about major components responsible for variability in the IR spectra. After PCA, the scores plot was used to classify the sample groups according to their similarity (sample groups in neighboring coordinates are more alike than those farther away) and the component loadings plot was used indicate the extent of variation in IR spectra corresponding to that component.3. Results
The chemical composition of pine wood chips on an oven dry basis was determined to be: glucan 38.6 ± 0.5%, lignin 37.4 ± 0.1%, mannan 7.2 ± 0.2%, xylan 6.7 ± 0.1%, galactan 5.3 ± 0.2%, arabinan 1.5 ± 0.1%, ethanol extractives 5.4 ± 0.3% and negligible amounts of ash (0.1%). Glucan and lignin accounted for the majority (∼74%) of the total dry solids. Total hemicellulose (xylan, mannan, and galactan) content was 22.5%, with arabinan as a relatively minor component.3.1. Effect of fungal pretreatment on pine deconstruction
The effect of T. versicolor pretreatment on glucan, xylan, galactan, mannan and lignin as a function of incubation time is presented in Fig. 2A. The average lignin content of fungal pretreated biomass decreased from 37.4% to 29.2% and the xylan content was reduced by 16% when compared to the uninoculated control. Correspondingly, fungal pretreated wood chips were enriched in glucan and mannan; the average glucan content increased from 38.6% to 43.7% and average mannan content increased from 7.2% to 10.0% (Fig. 2A). However, no significant differences in the overall chemical composition were observed among the tested incubation periods of 15, 30, and 45 days. The overall mass loss was 15% and it occurred within the first 15 days of fungal growth; it remained constant thereafter. Losses in glucan content were minimal, reaching only 3.7% after 30 days. Compared to previous reports,6,23 T. versicolor pretreatment of loblolly pine resulted in significantly higher reduction of xylan and lignin content.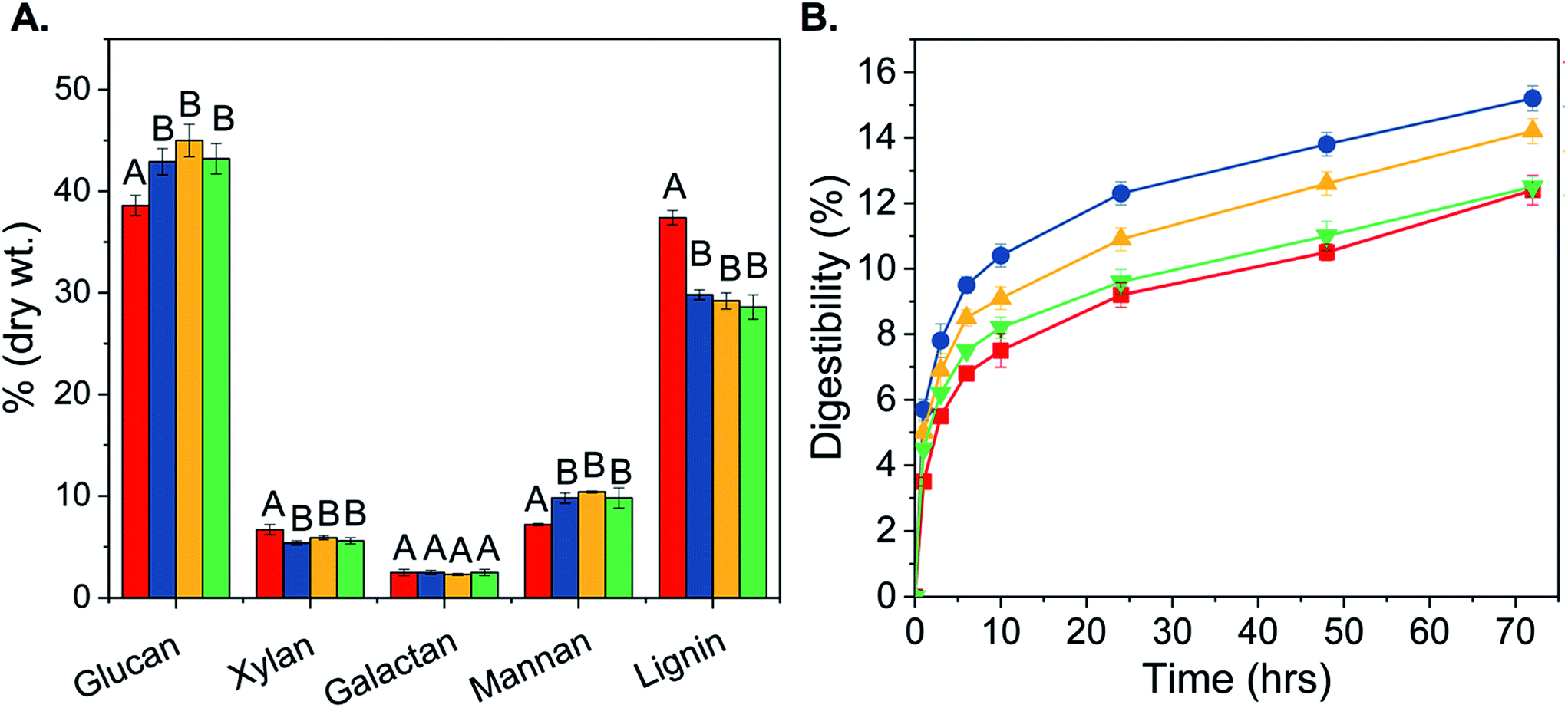 | ||
| Fig. 2 (A) Chemical composition changes in biologically pretreated loblolly pine wood chips with Tramestes versicolor, incubated for 0 (red), 15 (blue), 30 (yellow) and 45 (green) days. All values represent means of 2 measurements and error bars represent standard deviation. Bars not connected by the same letter are statistically significant at p ≤ 0.05 as per the Student's t-test. (B) Average digestibility of fungal pretreated pine chips after 0 (red square), 15 (blue circle), 30 (yellow triangle) and 45 (green inverted triangle) days of incubation. Error bars represent standard deviation for N = 3. | ||
The T. versicolor strain used in this study (HBB-2740) was previously selected for its potential to secrete extracellular enzymes, especially laccases.24 HBB-2740 was reported to produce 12 units per mL of laccase activity after 14 days of growth. And these T. versicolor laccases were shown to reduce lignin-derived phenols and xylo-oligosaccharides in biomass prehydrolyzates by 76% and 64%, respectively.24 Other potential extracellular enzymes include xylanases and peroxidases that are involved in degradation of hemicellulose and lignin, respectively, and also play a role in the initial colonization, growth, and metabolism of fungi.25,26 White rot fungi can also produce a metabolite called 3-hydroxyanthranilate,27 which laccases can use to depolymerize lignin even in the absence of lignin peroxidase and manganese peroxidase enzymes, as well as externally added radical mediators.28 A similar mechanism could be present in HBB-2740, thus explaining its high delignification potential.
3.2. Effects of fungal and organosolv pretreatments on pine digestibility
Digestibility of 15, 30 and 45 days fungal pretreated pine was calculated and expressed as the percentage of glucose released in solution per g of glucan present in the biomass (Fig. 2B). Maximum digestibility of 15.2% was obtained for 15 days pretreated material, followed by 30 (14.2%) and 45 (12.5%) days pretreated materials. Glucose yields from 15 and 30 days fungal pretreated pine were significantly higher than the control, indicating better susceptibility of fungal pretreated pine to enzymatic hydrolysis. The observed decrease in digestibility upon prolonged incubation with T. versicolor, i.e. for 30 and 45 days, could be due to an increase in cellulose crystallinity as a result of selective fungal degradation of amorphous components.29 Overall, 15 days of fungal incubation was sufficient to improve the final hydrolysis yield of untreated pine by 22.6%.Fungal pretreatment of pine was followed by organosolv pretreatment; Table 1 presents the overall mass balance for 0, 15, 30, and 45 days fungal pretreated pine chips, before and after organosolv pretreatment. Structural carbohydrate analysis of the solid fraction showed that it was completely devoid of hemicellulose fraction namely xylan, arabinan, galactan and mannan (Table 1), indicating that hemicellulose was completely solubilized in the liquid fraction, similar to previous studies.30,31 The glucan fraction was completely digestible, as 100% digestibility was achieved within 24 h of enzymatic hydrolysis for the fungal-organosolv treated pine solids (data not shown). This conversion efficiency could be attributed to the decrease in degree of polymerization of cellulose and increase in accessible surface area.10,30 Lignin removal and redistribution could also be a contributing factor.30,32 Recent studies have shown that the ease of cellulose hydrolysis is not only dependent on the total lignin content of the pretreated material,9 but also on the structural and chemical properties of residual lignin.32,33
| Treatment | Fungal pre-treated pine composition | FOa prehydrolyzate | FOa liquid fraction | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Glucan (%) | Lignin (%) | Total solids yield (%) | Glucan content (%) | OPb (%) | HMF (%) | Furfural (%) | Formic acid (%) | Acetic acid (%) | |
| a FO – sequential fungal and organosolv pretreatments.b OP – organosolv precipitates. OP yields not connected by the same letter are significantly different at p ≤ 0.05 as per Student's t-test. All values represent means and standard deviations for N = 2. | |||||||||
| 0 day | 37.9 ± 1.0 | 36.1 ± 0.7 | 11.4 ± 0.3a | 29.3 ± 0.1 | 45.5 ± 0.1a | 0.1 ± 0.0 | 0.2 ± 0.0 | 0.6 ± 0.0 | 0.2 ± 0.0 |
| 15 days | 42.9 ± 1.3 | 29.8 ± 0.5 | 18.8 ± 2.5b | 32.0 ± 0.9 | 55.2 ± 1.8b | 0.1 ± 0.0 | 0.2 ± 0.0 | 0.5 ± 0.0 | 0.1 ± 0.0 |
| 30 days | 45.0 ± 1.6 | 29.2 ± 0.8 | 16.3 ± 0.1b | 15.5 ± 0.4 | 55.5 ± 1.0b | 0.1 ± 0.0 | 0.2 ± 0.0 | 0.5 ± 0.0 | 0.1 ± 0.0 |
| 45 days | 43.2 ± 1.5 | 28.6 ± 1.2 | 12.8 ± 0.1a | 6.8 ± 0.0 | 56.0 ± 1.1b | 0.1 ± 0.0 | 0.2 ± 0.0 | 0.5 ± 0.0 | 0.0 ± 0.0 |
3.3. Characterization of organosolv precipitates (OP)
Mass balance of the organosolv process (Table 1) showed that, as a result of combining fungal pretreatment with organosolv fractionation, 55 to 56% of OP were recovered from pine. For non-pretreated pine, the OP yield was only 46% of dry biomass. Fungal pretreatment thus resulted in higher OP yields, which could potentially generate additional revenue and justify the use of fungal pretreatment on a commercial scale.Overall, pretreatment with fungi had a significant impact on the molecular weight distribution of OP (Fig. 3); OP from all the fungal pretreated material displayed a bimodal distribution as opposed to the non-pretreated material. There was a decrease in large molecular weight compounds in the differential distribution curve (Fig. 3) and a subsequent increase in small molecular weight compounds as a function of fungal pretreatment time. It is possible that solubilization of hemicellulose happening at 30 and 45 days of fungal pretreatment may have led to the generation of furan-like byproducts, thereby increasing the small molecular weight components and OP's dispersity. This observation is supported by results obtained from FTIR as well as pyrolysis GC/MS, discussed later, which clearly indicated that fungal-pretreated OP had more carbohydrate derivatives.
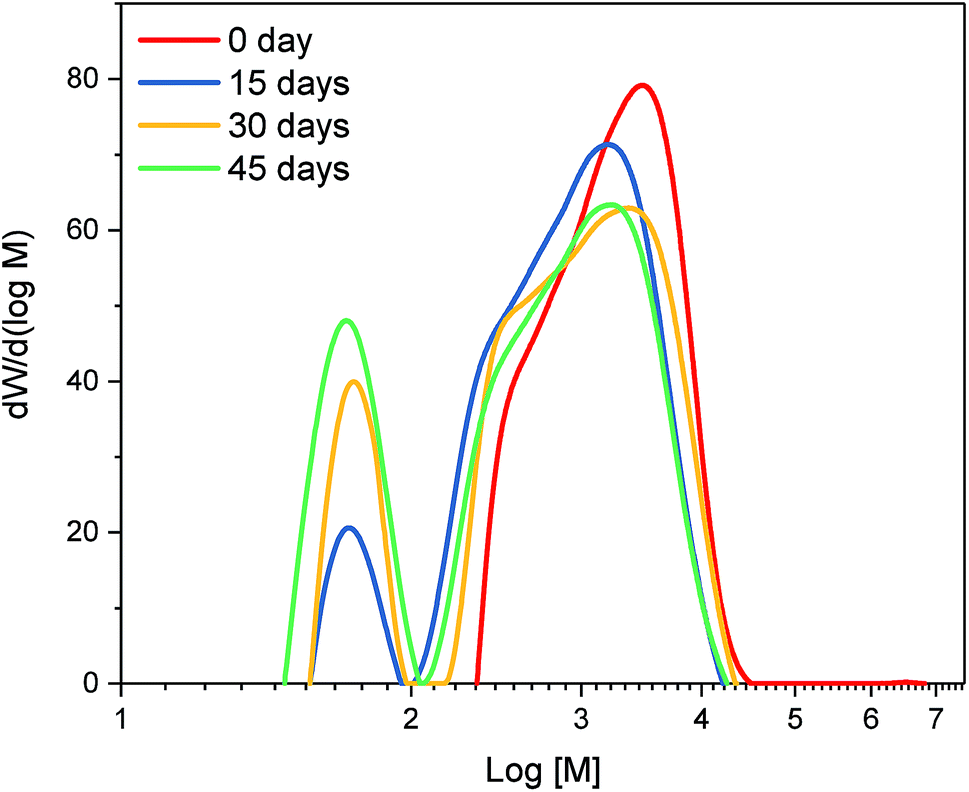 | ||
| Fig. 3 Differential molecular weight distribution of organosolv precipitates (OP). Different OP was obtained from loblolly pine chips pre-treated with T. versicolor for variable durations. | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch, aromatic skeletal vibrations and C–C, C–O and CO stretches, respectively (Fig. 4A). The OP infrared spectra also showed other absorption bands characteristic to lignin, such as 2999 and 2931 cm−1, corresponding to C–H stretch in methyl and methylene groups and asymmetric CH2 valence vibrations, respectively. The PCA-scores plot of the FTIR spectra (Fig. 4B) showed that OP from the control and 15 days pretreated pine have similar chemical features, but differ from 30 and 45 days pretreated pine samples. Principal component-1 (PC-1) accounted for 71% of the variations observed in the FTIR spectra, specifically explaining how the 45 day OP differed from the control and 15 day OP. The loadings plot of PC-1 showed that 45 days of fungal incubation led to lignin with higher CC skeletal vibrations of condensed G rings, corresponding to 1575 cm−1 (Fig. 4C). The 45 day OP also displayed higher asymmetric bending of C–H from methoxy groups and out of plane C–OH bending, corresponding to 1443 cm−1 and 643 cm−1, respectively. Oxidative lignolysis by fungal peroxidases followed by organosolv fractionation have been reported to induce cleavage of α–β, β–1 and β–O–4 linkages, giving rise to condensed lignin and methoxy groups.25
O stretch, aromatic skeletal vibrations and C–C, C–O and CO stretches, respectively (Fig. 4A). The OP infrared spectra also showed other absorption bands characteristic to lignin, such as 2999 and 2931 cm−1, corresponding to C–H stretch in methyl and methylene groups and asymmetric CH2 valence vibrations, respectively. The PCA-scores plot of the FTIR spectra (Fig. 4B) showed that OP from the control and 15 days pretreated pine have similar chemical features, but differ from 30 and 45 days pretreated pine samples. Principal component-1 (PC-1) accounted for 71% of the variations observed in the FTIR spectra, specifically explaining how the 45 day OP differed from the control and 15 day OP. The loadings plot of PC-1 showed that 45 days of fungal incubation led to lignin with higher CC skeletal vibrations of condensed G rings, corresponding to 1575 cm−1 (Fig. 4C). The 45 day OP also displayed higher asymmetric bending of C–H from methoxy groups and out of plane C–OH bending, corresponding to 1443 cm−1 and 643 cm−1, respectively. Oxidative lignolysis by fungal peroxidases followed by organosolv fractionation have been reported to induce cleavage of α–β, β–1 and β–O–4 linkages, giving rise to condensed lignin and methoxy groups.25
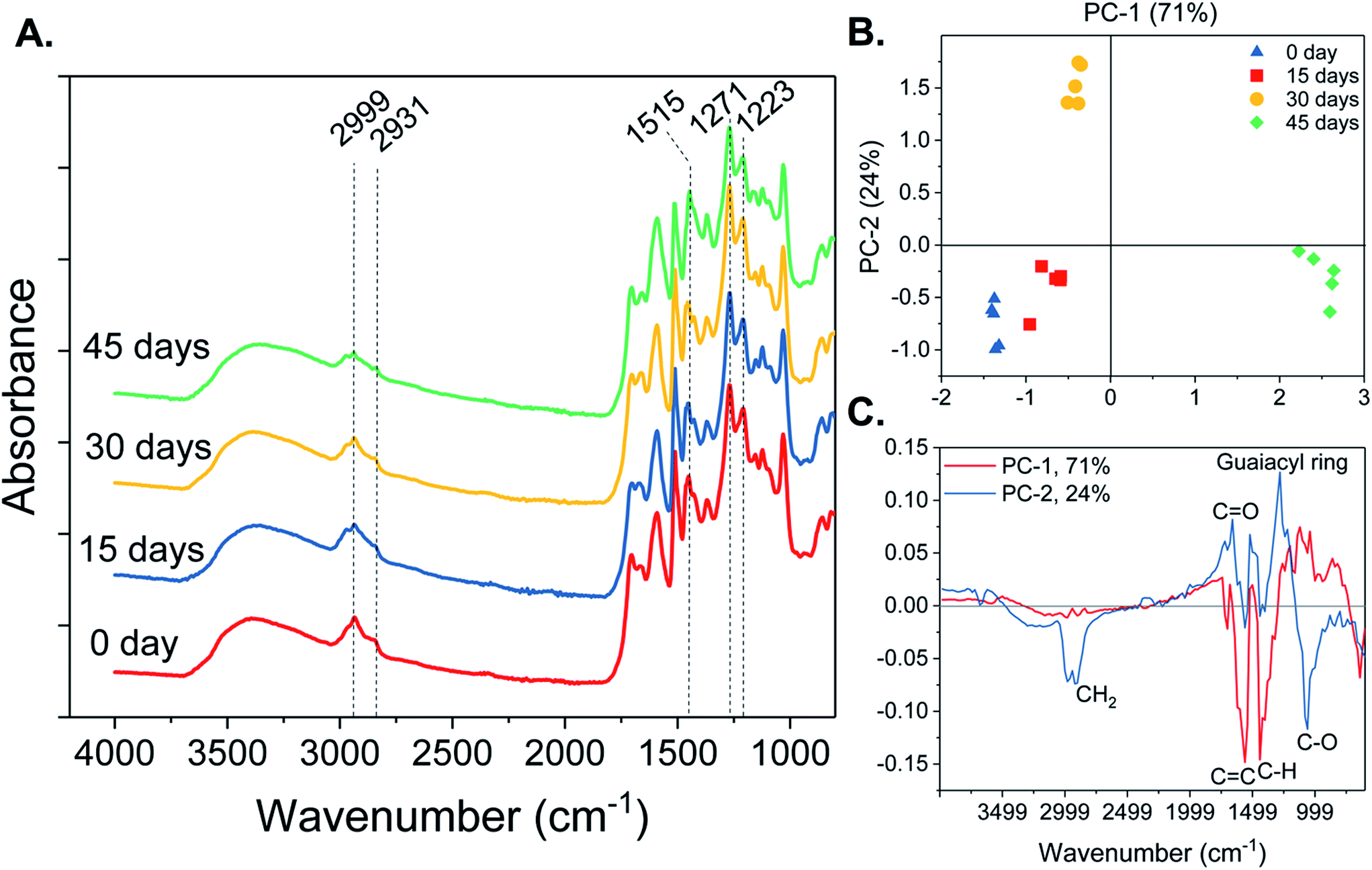 | ||
| Fig. 4 (A) Fourier transform infrared (FTIR) absorption spectra of organosolv precipitates (OP) obtained from fungal pretreated pine chips, incubated for 0, 15, 30 and 45 days. (B) Principal component analysis (PCA)-scores scatter plot and (C) PCA-loadings plot for OP FTIR spectra in the range of 4000 to 600 cm−1. | ||
The PC-2 explained 24% of the variations observed in 30 day FTIR spectra from that of the control and 15 day OP (Fig. 4B). The loadings plot for PC–2 (Fig. 4C) showed that, 30 day OP exhibited higher CH2 vibration and C–O vibration mainly from C3/HO3, corresponding to 2923 and 1067 cm−1, respectively. This adds credence to the conclusion that prolonged fungal pretreatment led to increased removal of carbohydrates in the OP. The 30 day OP also displayed lower CO stretch corresponding to 1663 cm−1 and lower C–O stretch in guaiacyl ring corresponding to 1283 cm−1. This shows that fungal demethoxylation could cause reduction in G-lignin, which is also supported by subsequent findings in py-GC/MS analysis.
| OH groups | Assignment, δ (ppm) | OH groups in OPa (mol kg−1) | ||
|---|---|---|---|---|
| 15 days | 30 days | 45 days | ||
| a Internal standard was endo-N-hydroxy-5-norbornene-2, 3-dicarboximide, organosolv precipitates (OP) were fractionated from T. versicolor pre-treated pinewood. | ||||
| Aliphatic | 150.0–145.0 | 0.71 | 0.80 | 0.97 |
| 5-Substituted phenolic | 143.8–140.3 | 0.69 | 1.32 | 1.73 |
| Free guaiacyl | 140.1–138.4 | 1.20 | 1.94 | 2.15 |
| p-Hydroxyphenyl | 138.2–137.0 | 0.27 | 0.40 | 0.55 |
| COOH | 135.8–133.7 | 0.08 | 0.14 | 0.21 |
| Grand total | — | 2.95 | 4.60 | 5.61 |
| Peak ID | Originb | Fungal incubation time (days) | |||
|---|---|---|---|---|---|
| 0 | 15 | 30 | 45 | ||
| a All peak areas were normalized to the total peak area. Means and standard deviations were calculated for N = 3.b C – carbohydrate, G – guaiacyl lignin, H – p-hydroxyphenyl lignin, FA – ferulic acid.c Values in parenthesis indicate organosolv lignin yields per unit of biomass, after adjusted for purity. | |||||
| 1-Methyl-1-cyclopenten-3-one | C | 0.3 ± 0.0 | |||
| 2-Ethyl-5-methylfuran | C | 0.1 ± 0.0 | 0.1 ± 0.0 | ||
| 2-Ethylfuran | C | 0.9 ± 0.0 | 0.1 ± 0.0 | ||
| 2-Methyl-2-cyclopentenone | C | 0.2 ± 0.0 | 0.2 ± 0.0 | ||
| 2,5-Dimethylfuran | C | 0.3 ± 0.0 | 0.4 ± 0.0 | 0.3 ± 0.0 | |
| 2(3H)-Furanone, 5-methyl– | C | 0.6 ± 0.0 | 0.3 ± 0.0 | 0.5 ± 0.1 | |
| 5-Methyl furfural | C | 0.3 ± 0.0 | 0.3 ± 0.0 | ||
| Acetic acid | C | 0.8 ± 0.0 | 0.6 ± 0.1 | ||
| Acetic aldehyde | C | 1.3 ± 0.2 | 0.7 ± 0.0 | 0.8 ± 0.1 | |
| Acetylfuran | C | 0.2 ± 0.0 | 0.3 ± 0.0 | ||
| Cyclopentenone | C | 0.3 ± 0.0 | 0.6 ± 0.1 | ||
| Levulinic acid, ethyl ester | C | 2.0 ± 0.5 | 1.1 ± 0.1 | 0.5 ± 0.0 | |
| Levulinic acid, methyl ester | C | 0.3 ± 0.0 | 1.1 ± 0.1 | ||
| Vinyl methyl ketone | C | 1.0 ± 0.0 | 1.5 ± 0.1 | 1.0 ± 0.1 | |
| Acetophenone, 2,4′-dihydroxy-3′-methoxy– | G | 2.6 ± 0.3 | 0.9 ± 0.0 | 3.0 ± 0.3 | 1.4 ± 0.1 |
| Acetovanillone | G | 1.0 ± 0.0 | 0.8 ± 0.0 | 1.5 ± 0.1 | 1.2 ± 0.1 |
| Ethyl homovanillate | G | 1.3 ± 0.0 | 0.6 ± 0.0 | 1.0 ± 0.0 | |
| Guaiacylacetone | G | 3.6 ± 0.6 | 1.3 ± 0.0 | 1.6 ± 0.1 | |
| o-Allylguaiacol | G | 0.2 ± 0.0 | 0.5 ± 0.0 | 0.5 ± 0.0 | |
| o-Guaiacol | G | 2.9 ± 0.4 | 1.8 ± 0.1 | 3.9 ± 0.7 | 3.3 ± 0.2 |
| o-Isoeugenol | G | 0.5 ± 0.0 | 0.7 ± 0.0 | 0.9 ± 0.0 | 1.0 ± 0.0 |
| p-Ethylguaiacol | G | 0.5 ± 0.0 | 0.5 ± 0.0 | 0.9 ± 0.0 | 0.8 ± 0.0 |
| p-Methylguaiacol | G | 3.2 ± 0.1 | 1.6 ± 0.3 | 1.9 ± 0.1 | |
| Vanillic acid | G | 2.5 ± 0.4 | 2.0 ± 0.5 | 1.9 ± 0.1 | 3.0 ± 0.2 |
| Vanillin | G | 1.7 ± 0.1 | 0.6 ± 0.0 | 2.1 ± 0.1 | 2.2 ± 0.1 |
| 4-Vinylguaiacol | G/FA | 0.9 ± 0.0 | 0.9 ± 0.0 | 1.1 ± 0.2 | 1.0 ± 0.0 |
| 3-Methoxy-2-methylphenol | H | 0.9 ± 0.0 | |||
| 3,4-Dimethylphenol | H | 0.1 ± 0.0 | |||
| 3,5-Dihydroxyacetophenone | H | 0.7 ± 0.0 | |||
| m-Cresol | H | 3.1 ± 0.5 | 0.2 ± 0.0 | ||
| m-Cresol methyl ether | H | 0.2 ± 0.0 | 0.2 ± 0.0 | ||
| o-Cresol | H | 2.2 ± 0.2 | 0.6 ± 0.0 | 1.4 ± 0.1 | 1.5 ± 0.0 |
| p-Creosol | H | 1.0 ± 0.0 | 0.9 ± 0.0 | 2.7 ± 0.3 | 0.2 ± 0.0 |
| Phenol | H | 1.8 ± 0.1 | 2.4 ± 0.6 | 2.4 ± 0.3 | 2.6 ± 0.1 |
| Phenol, 4-methoxy-3-(methoxymethyl) | H | 0.3 ± 0.0 | 0.4 ± 0.0 | 0.2 ± 0.0 | |
| % ligninc | 60.0 ± 2.6 (27.3 ± 0.6) | 49.7 ± 2.9 (27.4 ± 0.4) | 53.9 ± 1.7 (%) (29.9 ± 0.2) | 57.9 ± 2.2 (32.4 ± 0.5) | |
| G/H | 3.4 | 1.7 | 2.2 | 1.8 | |
| % carbohydrate | 7.9 ± 1.1 | 13.0 ± 1.5 | 14.8 ± 1.7 | 20.0 ± 1.7 | |
Similar to the FTIR spectroscopic studies, the py-GC/MS results also showed that the proportion of carbohydrate derivatives in OP increased from 0 to 45 days (Table 3). The py-GC/MS results also showed that the total G lignin-type pyrolysis products increased from 15 to 45 days. On the other hand, the G/H ratio of OP lignin decreased from 3.4 to 1.8 in 0 to 45 days, indicating an overall increase in H lignin content. This could possibly be attributed to demethoxylation of G lignin units by fungal laccases.36
4. Discussion
There is an increasing interest in the extraction of lignin as a co-product in bio-refinery operations.9,11 By altering its chemistry through oxidative functionalization, lignin could be used for different applications in polymer, fine chemicals and pharmaceutical industries.34 Organosolv fractionation results in solubilization of large lignin fragments into the pretreatment liquor in a pure and sulfur-free form, which can easily be precipitated and recovered by changing the pH of the liquor and/or by anti-solvent extraction.37 While the structure and chemical composition of organosolv lignin has been previously characterized,37 the effect of combining organosolv process with fungal pretreatment on the chemical composition of lignocellulosic biomass and the structure and properties of organosolv lignin is not well elucidated. There is only one prior study that explored this concept and they reported a 25% increase in G/H ratio in cellulolytic enzyme lignin (CEL), which was recovered from bamboo pretreated with Echinodontium taxodii.7 This study showed that the G/H ratio of T. versicolor pre-treated organosolv pine lignin actually decreased by 53%, which implies that different combination of biological and chemical pretreatments have the potential to customize the resultant lignin composition.Despite the fact that fungal pretreatment resulted in significant delignification within 15 days, only small increases were noted in enzymatic hydrolysis yields, corroborating the previous reports that fungal pretreatment cannot be used as a stand-alone pretreatment for maximal conversion of the cellulose fraction into glucose.6,12 Organosolv pretreatment results in higher yields of pure cellulose with reduced crystallinity and degree of polymerization that are desirable outcomes in pretreatment choices as applied to conversion of biomass in bio-refinery operations.8,9 Our results demonstrated that the organosolv pretreatment resulted in 100% saccharification within 24 h of enzymatic hydrolysis, with or without fungal pretreatment. On the other hand, the combination of fungal and organosolv pretreatments was highly effective in improving the organosolv lignin yield, despite lower purity. Lodgepole pine infested with mountain pine beetle, Dendroctonus ponderosae, and insignis pine bio-treated with brown-rot fungus, Gloeophyllum trabeum, displayed higher organosolv lignin yields in the liquid fraction, when compared to those obtained with healthy lodgepole pine and non-pretreated insignis pine, respectively.9,11 Higher delignification rates were also observed during organosolv pulping of hardwood Eucalyptus grandis that was earlier subjected to pretreatment with T. versicolor for 75 days, as compared to the uninoculated control.38 Additionally, our study demonstrated that there is a synergy between fungal and organosolv pretreatments that resulted in higher organosolv lignin yields. With better organosolv pretreatment conditions it will be possible to improve the organosolv lignin purity.
Physico-chemical analysis of OP showed that lignin components from 30 and 45 day pretreated samples were extensively depolymerized and structurally different from the control and 15 day samples. During initial colonization of woody biomass, T. versicolor is known to oxidize lignin and use it as an energy source. Previous studies determined that T. versicolor exhibited its peak laccase activity at 10–14 days of growth.24 Studies have also shown that, during later stages of fungal growth, non-enzymatic degradation of lignocellulose takes place, mostly targeting cellulose and hemicellulose components.27 This was substantiated by GPC, FTIR and py-GC/MS results, all of which showed an increase in degraded carbohydrate components in 30 and 45 day-OP. Prolonged incubation of pinewood chips with T. versicolor led to an observed increase in aliphatic, phenolic and carboxylic OH groups, which could be due to the combined enzymatic and non-enzymatic degradation of both phenolic and non-phenolic components of lignin. Also, preferential degradation of G-lignin by T. versicolor coupled with possible demethoxylation could have led to a higher H-lignin content in 45 day pretreated OP and consequently, a lower G/H ratio. The properties of higher phenol content and lower MW of fungal-pretreated OP make it suitable for products such as antioxidants, resins and chemicals.37,39 In particular, the presence of high amounts of free phenolic OH groups has been associated with higher reactivity of lignin towards chemical modification and potential applications in phenol-formaldehyde resin production.39 The fact that fungal pretreatment produces lower MW organosolv lignin components with higher OH content opens an avenue for future research in bio-degraded lignin-based applications.
5. Conclusions
Through the results of our study, it can be concluded that there is a synergy between fungal and organosolv pretreatments, and that possibly fungal pretreatment could be included in biomass storage steps.40 Using this combination, higher saccharification yields and larger amounts of lignin-enriched fractions were obtained. Fungal pretreatment also resulted in compositional changes of OP and associated variations in organosolv lignin properties. Incubation with T. versicolor for 30 and 45 days resulted in increased depolymerization of carbohydrates, as well as increase in H-type lignin and a corresponding decrease in G/H lignin ratio. Future work should be aimed at examining the integration of fungal pretreatment with lignin fractionation processes, especially in shortening T. versicolor treatment time in an industrial setting. It would also be critical to further understand the structural and chemical changes caused by fungal pretreatment, as these changes could improve the chances of lignin to be incorporated in value-added products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by the USDA National Institute of Food and Agriculture, capacity grant (S15-723-15-1) and the Center for Advanced Surface Engineering under the National Science Foundation grant (OIA-1457888) and the Arkansas EPSCoR Program, ASSET III.References
- D. N. Wear, J. Prestemon, R. Huggett and D. Carter, in The Southern Forest Futures Project Technical Report, ed. D. N. Wear and J. G. Greis, U.S. Department of Agriculture Forest Service, Southern Research Station, Ashville, NC, 2013, ch. 9, pp. 183–212 Search PubMed.
- M. Normark, S. Winestrand, T. A. Lestander and L. J. Jonsson, BMC Biotechnol., 2014, 14, 1–12 CrossRef PubMed.
- K. Messner and E. Srebotnik, FEMS Microbiol. Rev., 1994, 13, 351–364 CrossRef CAS.
- S. K. Gulsoy and H. Eroglu, Int. Biodeterior. Biodegrad., 2011, 65, 644–648 CrossRef CAS.
- A. Guerra, R. Mendonca and A. Ferraz, Enzyme Microb. Technol., 2003, 33, 12–18 CrossRef CAS.
- A. Aguiar, D. Gavioli and A. Ferraz, Fungal Biol., 2014, 118, 935–942 CrossRef CAS PubMed.
- K. Yan, F. Liu, Q. Chen, M. Ke, X. Huang, W. Hu, B. Zhou, X. Zhang and H. Yu, Biotechnol. Biofuels, 2016, 9, 1–12 CrossRef PubMed.
- X. Zhao, K. Cheng and D. Liu, Appl. Microbiol. Biotechnol., 2009, 82, 815–827 CrossRef CAS PubMed.
- X. Pan, D. Xie, R. W. Yu and J. N. Saddler, Biotechnol. Bioeng., 2008, 101, 39–48 CrossRef CAS PubMed.
- L. F. D. Rio, R. P. Chandra and J. N. Saddler, Appl. Biochem. Biotechnol., 2010, 161, 1–21 CrossRef PubMed.
- P. Sannigrahi, A. J. Ragauskas and S. J. Miller, Energy Fuels, 2009, 24, 683–689 CrossRef.
- M. J. Ray, D. J. Leak, P. D. Spanu and R. J. Murphy, Biomass Bioenergy, 2010, 34, 1257–1262 CrossRef CAS.
- A. Fissore, L. Carrasco, P. Reyes, J. Rodriguez, J. Freer and R. T. Mendonca, J. Ind. Microbiol. Biotechnol., 2010, 37, 893–900 CrossRef CAS PubMed.
- M. Monrroy, J. Ibanez, V. Melin, J. Baeza, R. T. Mendonca, D. Contreras and J. Freer, Enzyme Microb. Technol., 2010, 47, 11–16 CrossRef CAS.
- D. M. Alonso, S. H. Hakim, S. Zhou, W. Won, O. Hosseinaei, J. Tao, V. Garcia-Negron, A. H. Motagamwala, M. A. Mellmer, K. Huang, C. J. Houtman, N. Labbé, D. P. Harper, C. Maravelias, T. Runge and J. A. Dumesic, Sci. Adv., 2017, 3, 1–7 Search PubMed.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Determination of structural carbohydrates and lignin in biomass, NREL Laboratory Analytical Procedure, National Renewable Energy Laboratory, Golden, CO, 2008 Search PubMed.
- N. B. Heringer, Undergraduate Thesis, University of Arkansas, 2016 Search PubMed.
- A. Guerra, L. A. Lucia and D. S. Argyropoulos, Holzforschung, 2008, 62, 24–30 CrossRef CAS.
- M. Fan, D. Dai and B. Huang, in Fourier Transform - Materials Analysis, ed. M. S. Salih, InTech, Rijeka, Croatia, 2012, ch. 3, pp. 45–68 Search PubMed.
- S. Mahajan, D. Jeremic, R. E. Goacher and E. R. Master, Appl. Microbiol. Biotechnol., 2012, 94, 1303–1311 CrossRef CAS PubMed.
- M. Schwanninger, J. C. Rodrigues, H. Pereira and B. Hinterstoisser, Vib. Spectrosc., 2004, 36, 23–40 CrossRef CAS.
- M. Balakshin and E. Capanema, J. Wood Chem. Technol., 2015, 35, 220–237 CrossRef CAS.
- A. Ferraz, J. Rodriguez, J. Freer and J. Baeza, Microbiol. Biotechnol., 2001, 17, 31–34 CrossRef CAS.
- R. K. Kapoor, K. Rajan and D. J. Carrier, Bioresour. Technol., 2015, 189, 99–106 CrossRef CAS PubMed.
- R. ten Have and P. J. M. Teunissen, Chem. Rev., 2001, 101, 3397–3414 CrossRef CAS PubMed.
- M. Dashtban, H. Schraft, T. A. Syed and W. Qin, Int. J. Biochem. Mol. Biol., 2010, 1, 36–50 CAS.
- C. Eggert, U. Temp, J. F. D. Dean and K. E. L. Eriksson, FEBS Lett., 1996, 391, 144–148 CrossRef CAS PubMed.
- C. Eggert, U. Temp and K. E. L. Eriksson, FEBS Lett., 1997, 407, 89–92 CrossRef CAS PubMed.
- F. Nazarpour, D. K. Abdullah, N. Abdullah and R. Zamiri, Materials, 2013, 6, 2059–2073 CrossRef CAS PubMed.
- H. Amiri and K. Karimi, Bioprocess Biosyst. Eng., 2015, 38, 1959–1972 CrossRef CAS PubMed.
- E. Araque, C. Parra, J. Freer, D. Contreras, J. Rodriguez, R. Mendonca and J. Baeza, Enzyme Microb. Technol., 2008, 43, 214–219 CrossRef CAS.
- S. Singh, G. Cheng, N. Sathitsuksanoh, D. Wu, P. Varanasi, A. George, V. Balan, X. Gao, R. Kumar, B. E. Dale, C. E. Wyman and B. A. Simmons, Frontiers in Energy Research, 2015, 2, 1–12 CrossRef.
- S. Nakagame, R. P. Chandra and J. N. Saddler, Biotechnol. Bioeng., 2010, 105(5), 871–879 CAS.
- H. Lange, S. Decina and C. Crestini, Eur. Polym. J., 2013, 49, 1151–1173 CrossRef CAS.
- X. Geng and K. Li, Appl. Microbiol. Biotechnol., 2002, 60, 342–346 CrossRef CAS PubMed.
- O. Skyba, C. J. Douglas and S. D. Mansfield, Appl. Environ. Microbiol., 2013, 79, 2560–2571 CrossRef CAS PubMed.
- P. Sannigrahi and A. J. Ragauskas, in Aqueous pretreatment of plant biomass for biological and chemical conversion to fuels and chemicals, ed. C. E. Wyman, John Wiley & Sons, Ltd, Chichester, UK, 2013, ch. 10, pp. 201–222 Search PubMed.
- A. Ferraz, R. Mendonça and F. T. da Silva, J. Chem. Technol. Biotechnol., 2000, 75, 18–24 CrossRef CAS.
- N. E. El Mansouri and J. Salvado, Ind. Crops Prod., 2006, 24, 8–16 CrossRef CAS.
- N. Frederick, M. Li, D. J. Carrier, M. Buser and M. Wilkins, AIMS Bioeng., 2016, 3, 389–399 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |