
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Five Co(II) coordination polymers with different counter anions based on [3,5-di(4H-1,2,4-triazol-4-yl)benzoato]− ligand: directed synthesis, structures and magnetic properties†
Wen-Hong Jianga,
Han-Zhong Zhanga,
Guang-Feng Houb,
Dong-Sheng Maa,
Bin Liua and
Ying-Hui Yu *a
*a
aSchool of Chemistry and Materials Science, Heilongjiang University, Harbin 150080, PR China. E-mail: yuyinghui@hlju.edu.cn; Fax: +86-451-86609151
bAgricultural College, Heilongjiang University, Harbin 150080, PR China
First published on 25th September 2017
Abstract
As a continuing challenge to obtain the desired coordination polymers in the synthesis stage, the counter ions, the metal–ligand ratio and the involvement of template have great impact on their structure construction and crystal growth. In this paper, three different crystals of coordination polymers 1–3 {{[CoL(μ2-OH)]}n (1), {[CoL2(H2O)]·H2O}n (2), {[Co3L3(H2O)]·3NO3·4H2O}n (3)} {L = [3,5-di(4H-1,2,4-triazol-4-yl)benzoate]−} were obtained in one pot with HL and Co(NO3)2. By tuning the reactants ratio and employing different templates, each complex could also be synthesized separately. Single-crystal X-ray analyses show that complex 1 exhibited a two-dimensional (2D) layered gek2 net structure. Complex 2 with deprotonated [L]− has two dimensional (2D) nets structure with 3,5L66 topology. Complex 3 displays a new 5,5,6,6-connected three-dimensional (3D) topological network with a point symbol of (410·65)(45·65)2(47·63)2(48·67). When the reactant Co(NO3)2 was changed to CoCl2 or CoSO4, 4 and 5 {{[CoL(Cl)]·H2O}n (4) and {[Co2L2(SO4)(H2O)]·2H2O}n (5)} was formed with a similar structure to that of 1. Complexes 4 and 5 are two-dimensional (2D) frameworks with 4,6L26 topology. Five complexes have been characterized by elemental analysis, infrared spectroscopy, powder X-ray diffraction and thermogravimetry measurements. The magnetic properties of 1–5 are also investigated. The magnetic property investigation reveals that complex 1 exhibits frequency-dependent behavior.
1. Introduction
As an emerging class of tunable hybrid materials, metal organic coordination polymers have been extensively explored not only for their aesthetics of crystalline structures1 but also for their promising functional purposes, such as storage,2 photoluminescence,3 separation,4 molecular magnetism,5 catalysis,6 sensor,7 ion exchange8 and optical device9 etc. Although much of initial interest and enthusiasm have been devoted to their design and synthesis, it remains to be a distant prospects to the rational construction of the expected architectures with desired properties, representing an ambitious goal for chemists. The difficulty in controlling the synthesis of complexes with well-defined architectures and associated functions owes to the influence of many factors, such as solvent system,10 ratio of the reactants,11 pH value of reaction mixture,12 anion,13 temperature,14 and reagent concentration15 etc. As an important role to maintain charge balance of the complex, the counter ion could greatly influence the topology and dimensionality of the complex, as the coordinated counter ion with a bridging mode could further extend the arrangement of metal node.16 Meanwhile, templates and molar ratio between the metal salt and the ligand also have an impact on the construction of final structures. Varied reactant ratios and the involvement of templates may perturb the equilibrium of the coordination reaction, thus affecting the crystal formation and eventually leading to a different structure.17 Further exploring the correlations between the counter anions, the ratio of reactants and template with the topological structures could be helpful for better understanding the self-assemble process in coordination polymers.Therefore, a judicious selection of functional ligands is important for deliberately achieving structural changes and the functional properties of these coordination polymers.18 Multi-carboxylic ligands with suitable spacers, especially benzoic acid-based ligands, the 1,2,4-triazole ligand and its derivatives are frequent choices for metal organic networks.19 As one of ongoing research in our laboratory, the coordination chemistry of a bifunctional ligand which combined with the triazole and carboxylate group have been continuously studied in our group.20 In this paper, [3,5-di(4H-1,2,4-triazol-4-yl)benzoate]− is adopted as a building block for the construction of coordination polymers based on cobalt salts with different counter ions. Five new coordination polymers {[CoL(μ2-OH)]}n (1), {[CoL2(H2O)]·H2O}n (2), {[Co3L3(H2O)]·3NO3·4H2O}n (3), {[CoL(Cl)]·H2O}n (4) and {[Co2L2(SO4)(H2O)]·2H2O}n (5) were successfully synthesized and characterized by single-crystal X-ray diffraction analysis, elemental analysis, IR analysis and thermal gravimetric analysis. The magnetic properties of 1–5 are also investigated. Magnetic investigation for complex 1 indicates that 1 demonstrates frequency-dependent magnetic behavior.
2. Experimental section
2.1 Materials and measurements
All chemicals purchased of reagent grade are used as received without further purification. The ligand 3,5-di(4H-1,2,4-triazol-4-yl)benzoic acid (HL) was synthesized according to the literature.20 Elemental analyses of C, H, N were processed on a Perkin-Elmer 2400 elemental analyzer. The IR spectra were obtained by using KBr disks on a Perkin-Elmer Spectrum 100 FT-IR spectrometer equipped with a DGTS detector (32 scans) in the range of 4000 to 500 cm−1. A Perkin-Elmer STA 6000 was used for thermal analyses under air atmosphere, with a heating rate of 10 °C min−1 in a temperature range from 50 to 800 °C. Bruker-D8 Advance X-ray diffractometer with Cu-Kα radiation (λ = 1.5406 Å) was employed for powder X-ray diffraction (PXRD) analysis. The magnetic susceptibility for complexes 1–5 were measured with a Quantum Design MPMS XL-7 SQUID-VSM magnetometer. The magnetic corrections were made by using Pascal's constants.2.2 Syntheses of 1–5
2.3 X-ray crystallography
Single-crystal X-ray diffraction data for complexes 1–5 were collected on a Rigaku R-AXIS RAPID imaging plate diffractometer with graphite-monochromated Mo Kα (λ = 0.71073 Å) at 291 K. Empirical absorption corrections based on equivalent reflections were applied. The structures of complexes 1–5 were solved by direct methods and refined by full-matrix least-squares methods on F2 with SHELXS-97 crystallographic software package.21 All the hydrogen atoms of ligands HL in complexes 1–5 were generated geometrically. The water molecules in 2, 3, 5 were located directly. The crystal parameters, data collection and refinement results for complexes 1–5 are summarized in Table 1. Selected bond lengths and angles of complexes 1–5 are listed in Table S1† and the H-bond lengths and angles are shown in Table S2† (see the ESI†). Crystallography data have been deposited to the Cambridge Crystallography Data Centre with deposition numbers CCDC: 1528099–1528103 for 1–5.†| Crystal parameters | 1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| a R1 = (Σ|Fo| − |Fc|)/Σ|Fo|. wR2 = [Σw(Fo2 − Fc2)2/Σw(Fo2)2]1/2. | |||||
| Empirical formula | C11H8N6O3Co | C22H18N12O6Co | C33H31N21O20Co3 | C11H9N6O3CoCl | C22H20N12O11Co2S |
| Formula weight | 331.16 | 605.41 | 1218.58 | 367.62 | 778.42 |
| Crystal system | Orthorhombic | Monoclinic | Monoclinic | Orthorhombic | Orthorhombic |
| Space group | Cmcm | P21/c | C2/c | Pmma | Pmm2 |
| a (Å) | 21.9686 | 16.8418(4) | 16.9247(4) | 7.1293(14) | 6.9023(2) |
| b (Å) | 6.8017 | 7.0186(2) | 17.5262(4) | 11.442(2) | 11.6321(4) |
| c (Å) | 20.0238 | 19.1189(4) | 15.1234(4) | 8.2387(16) | 8.0322(3) |
| α (°) | 90 | 90 | 90 | 90 | 90.00 |
| β (°) | 90 | 101.871(2) | 93.431(2) | 90 | 90.00 |
| γ (°) | 90 | 90 | 90 | 90 | 90.00 |
| V (Å3) | 2992.0(2) | 2211.63(9) | 4477.95(19) | 672.1(2) | 644.89(4) |
| Z | 8 | 4 | 4 | 2 | 1 |
| Dcaled (Mg cm−3) | 1.470 | 1.818 | 1.808 | 1.817 | 2.004 |
| μ (mm−1) | 1.165 | 0.851 | 1.205 | 1.498 | 1.459 |
| Collected/unique | 3119/1393 | 9165/3886 | 8411/3931 | 5072/680 | 1826/1114 |
| Rint | 0.0252 | 0.0270 | 0.0313 | 0.0346 | 0.0245 |
| GOF on F2 | 1.032 | 1.041 | 1.042 | 1.078 | 1.023 |
| R (I > 2σ(I)) | R1 = 0.0539 | R1 = 0.0373 | R1 = 0.0599 | R1 = 0.0297 | R1 = 0.0341 |
| wR2 = 0.1785 | wR2 = 0.0894 | wR2 = 0.1525 | wR2 = 0.0784 | wR2 = 0.0304 | |
| R (all data) | R1 = 0.0723 | R1 = 0.0515 | R1 = 0.0865 | R1 = 0.0325 | R1 = 0.0736 |
| wR2 = 0.1957 | wR2 = 0.0968 | wR2 = 0.1673 | wR2 = 0.0803 | wR2 = 0.0700 | |
3. Results and discussion
3.1 Crystal structure description of 1
Crystallographic analysis reveals that complex 1 crystallizes in the orthorhombic space group Cmcm. Each asymmetric unit contains one crystallographic independent Co(II), one [L]− and one μ2-bridging hydroxyl groups (Fig. 1a). Each Co(II) is coordinated by four triazole nitrogen atoms (N2, N2II, N3III, and N3IV) and two μ2-bridging hydroxyl groups (O2 and O2V). Thus the coordination environment of Co(II) atom could be described as a octahedral geometry with N4O2 donor set. The Co1–N bond lengths are from 2.189(5) Å to 2.196(1) Å, the Co1–O ones are 1.989(7) Å and 1.995(0) Å, respectively. The coordinated hydroxyl groups connect Co(II) cations to form chains along [010] direction. The central Co(II) cations further link each chain through N2, N3, N2I and N3I atoms of [L]− along [100] direction, eventually generating a 2D layered structure (Fig. 1b). The distance between two [Co]2+ cations is 3.400(9) Å.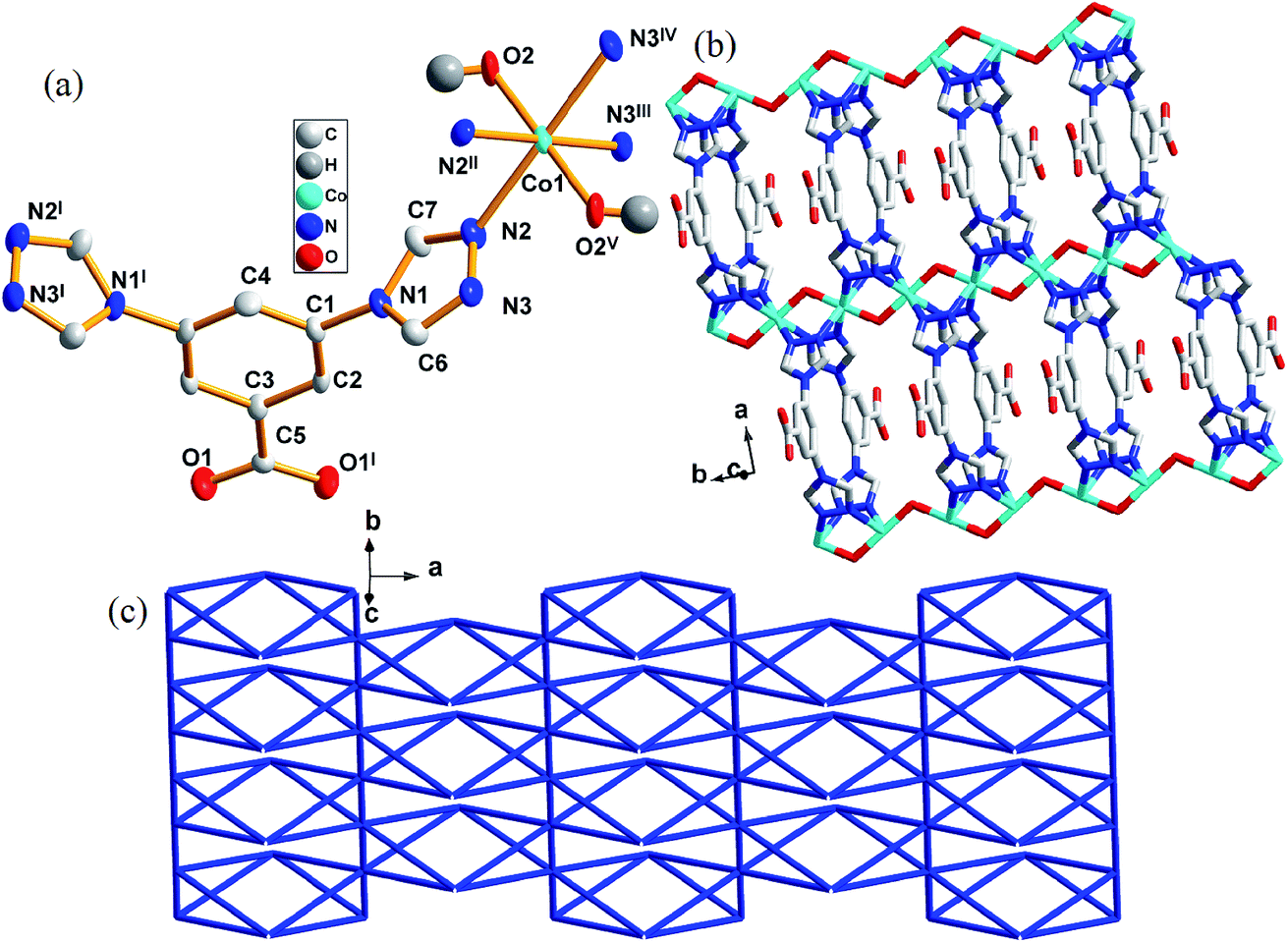 | ||
| Fig. 1 (a) Coordination environment of Co(II) atom in complex 1, the H-atoms were omitted for clarity. (b) 2D supramolecular structure of 1. (c) Schematic illustrating topology of the bimodal (4,6)-connected net of complex 1. Symmetry code: I = 2 − x, y, z; II = x, y, 1.5 − z; III = 1.5 − x, 0.5 + y, z; IV = 1.5 − x, 0.5 + y, 1.5 − z; V = 1.5 − x, −0.5 + y, 1.5 − z. | ||
A better insight into the structure 1 can be accessed by topological analysis by considering each ligand as a 4-connector and the central Co(II) atom as a 6-connector. Such connectivity repeats infinitely to give a 2D framework, as indicated in Fig. 1b. Analysis of the coordination sequence of 1 shows that its topology is related to gek2 by the TOPOS 4,22 both the ligand and metal nodes result in a symbol (32·44)(34·42·64·75) as shown in Fig. 1c.
3.2 Crystal structure description of 2
Complex 2 crystallizes in monoclinic space group P21/c. There is one Co(II) atom, two [L]−, one coordinated water molecules and one lattice water molecule in an asymmetric unit of 2 (lattice water molecule was omitted in Fig. 2a). Co1 is six coordinated by two nitrogen atoms from two different triazoles (N6II, N8III), three carboxylate oxygen atoms from three different [L]− anions (O1, O3, O4I) and one oxygen atom from one coordinated water molecule (O5), forming a distorted octagonal coordination geometry. The Co1–N bond lengths are 2.135(3) and 2.173(9) Å, the Co1–O ones are in the range of 2.073(7)–2.136(4) Å. O3 and O4 from one ligand linked two Co1 atoms with a distance of 4.990(0) Å between Co–Co.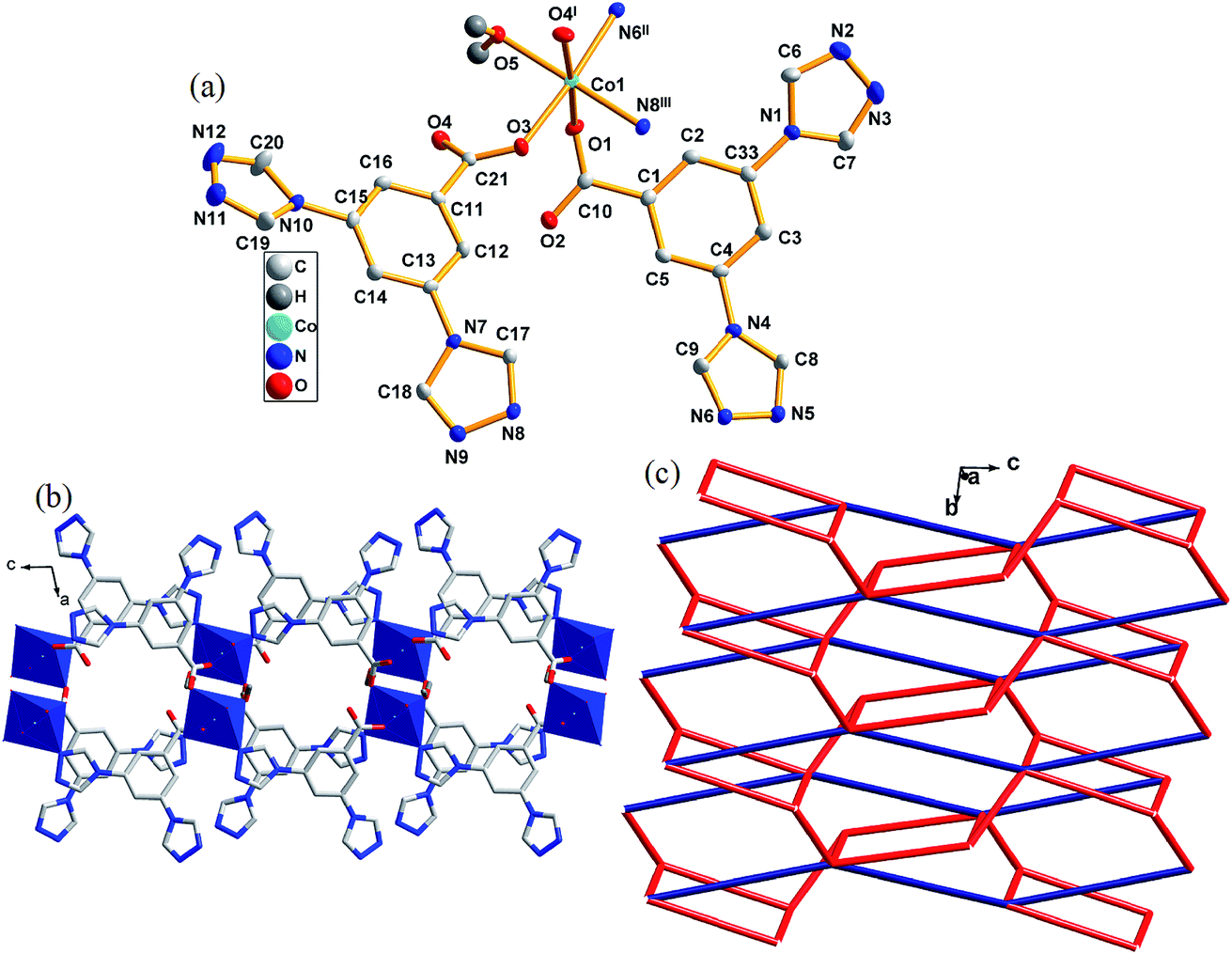 | ||
| Fig. 2 (a) The coordination environment of Co(II) atom in complex 2. The hydrogen atoms and the lattice water molecules were omitted for clarity. (b) 2D network in complex 2. (c) Schematic illustrating topology of 2D supramolecular structure in 2. Symmetry code: I = 1 − x, 0.5 + y, 0.5 − z; II = x, 1.5 − y, 0.5 + z; III = 1 − x, 2 − y, −z. | ||
In complex 2, N6 and O1 of [L]− link Co(II) to form a 1D chain along [001] direction. The central Co(II) atoms link O3 and O4 of the ligand to form a 1D chain along [010] direction, the 1D chain further links N8 of the ligand to form a 2D layered structure along [001] direction. The topological method is also employed to get a better structural insight of 2. As described above, each Co(II) atom is connected by five ligands, thus it can be considered as a five connector node. The two ligands connecting three or two Co(II) atoms can be regarded as three and two connectors, respectively. Such connectivity repeats infinitely to give a 2D network with a 3,5L66 topology, as schematically shown in Fig. 2c.
3.3 Crystal structures descriptions of 3
Complex 3 crystallizes in monoclinic space group C2/c. There is one-half Co atoms, one-half [L]− anions, a half coordinated water molecule, one-half [NO3]− anions and two lattice water molecules (solvent molecules were omitted) in an asymmetric unit of 3 (Fig. 3a). The Co1 atom with distorted octahedral coordination geometry is six coordinated by two carboxylate oxygen atoms (O1, O3III) from two different [L]−, one oxygen atom from coordination water molecule (O9) and three triazole nitrogen atoms (N1, N6I, N9II) from three different [L]−. Co2 is six-coordinated by six triazole nitrogen atom (N2II, N5IV, N8, N2VII, N5VI, N8V) from six different [L]− to complete the distorted octahedral coordination geometry.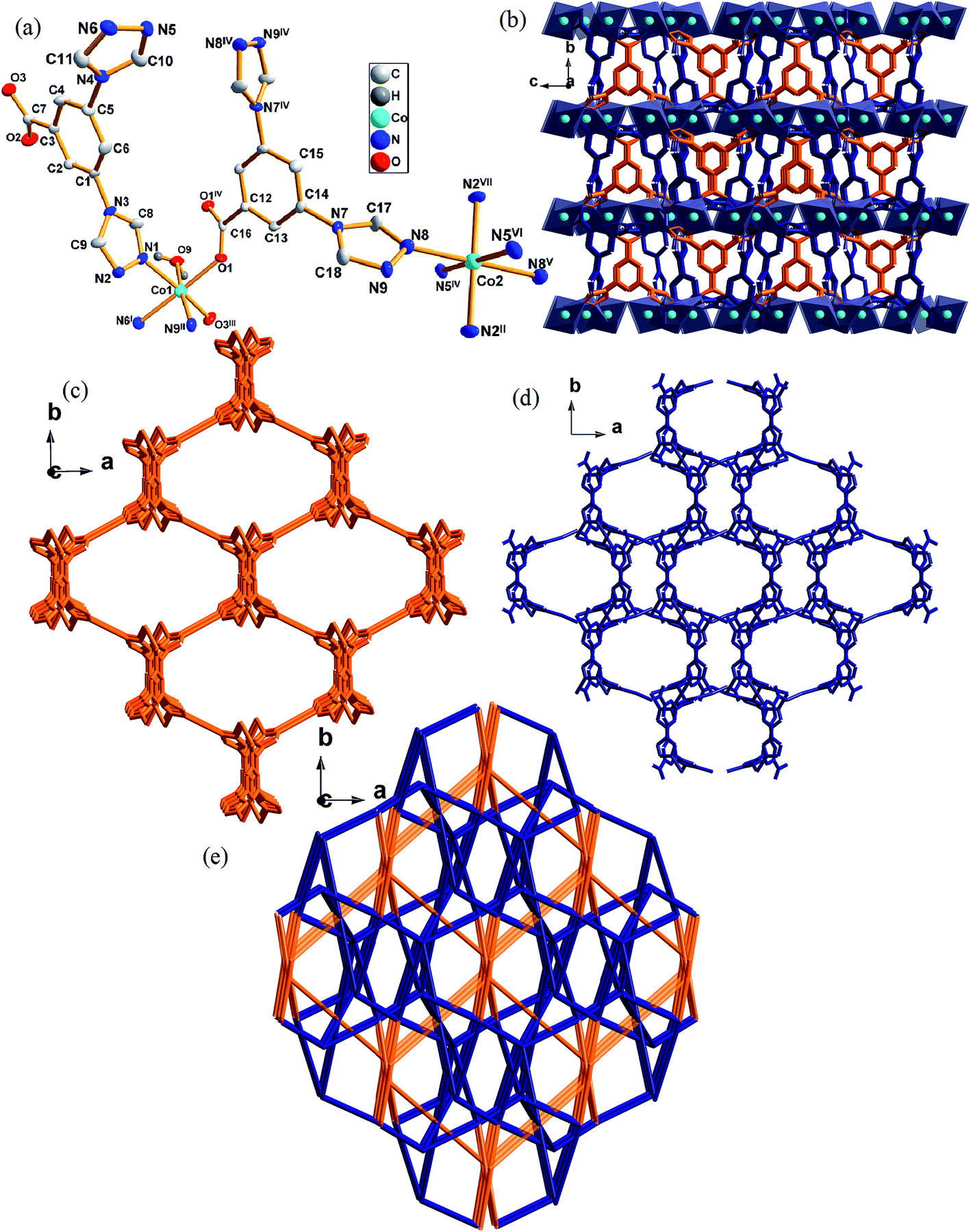 | ||
| Fig. 3 (a) Coordination environment of Co(II) atom in complex 3. The hydrogen atoms and solvent molecules were omitted for clarity. (b) 3D network in complex 3. (c) The six-connected coordination mode of [L]− in complex 3. (d) The five-connected coordination mode of [L]− in complex 3. (e) Schematic illustrating topology of complex 3. Symmetry code: I = 1.5 − x, 0.5 + y, 1.5 − z; II = 2 − x, −y, 1 − z; III = 0.5 + x, 0.5 − y, −0.5 + z; IV = 2 − x, y, 1.5 − z; V = 2.5 − x, −0.5 − y, 1 − z; VI = 0.5 + x, −0.5 − y, −0.5 + z; VII = 0.5 + x, −0.5 + y, z. | ||
CoN3O3 and CoN6 SBUs are linked by the ligands to form a 3D structure. The five-connected ligand links Co1 to form 1D chains along [001] direction, which further link Co2 giving rise to a 3D structure in Fig. 3c. The six-connected ligand links Co1 to a 2D layer structure, which further connects Co2 to generate a 3D structure in Fig. 3d. Two kinds of 3D structures are joint together through Co to eventually build the structure shown in Fig. 3e. The basic building block of this structure is the dinuclear [Co2(L)2]2+ entity, in which two [L]− link two metal nodes into a 3D structure with a Co–Co distance of 10.349(1) Å.
From a topological view of structure of 3, two kinds of Co(II) atoms can be considered as five and six connector nodes and two ligands can be regarded as five or six connectors, respectively. Such connectivity repeats infinitely to give a 3D framework with a point symbol (410·65)(45·65)2(47·63)2(48·67).
3.4 Crystal structures descriptions of 4 and 5
When the reactant Co(NO3)2 was changed to CoCl2 or CoSO4, a similar structure was formed to that of 1. Complex 4 crystallizes in the orthorhombic space group Pmma. There is one Co(II) atom, one [L]−, one [Cl]− and one lattice water molecule (lattice water were omitted) in an asymmetric unit of 4 (Fig. 4a). The Co1 atoms with a distorted octahedral coordination geometry is six coordinated by four nitrogen atoms (N2, N2IV, N2VI, N2VII) from four different [L]− and two [Cl]− (Cl1, Cl1V). However, complex 5 crystallizes in orthorhombic space group Pmm2 (Fig. 4b). The asymmetric unit of 5 contains one crystallographically independent [Co]2+ ion, one [L]−, a half [SO4]2−, a half coordinated water molecule and one lattice water molecule. Co1 is six-coordinated by four triazole nitrogen atoms (N1, N3, N1I, N3I) from four different [L]−, one oxygen atom (O1w) from terminal water molecule, one oxygen atom (O3) from [SO4]2− to complete the distorted octahedral coordination geometry.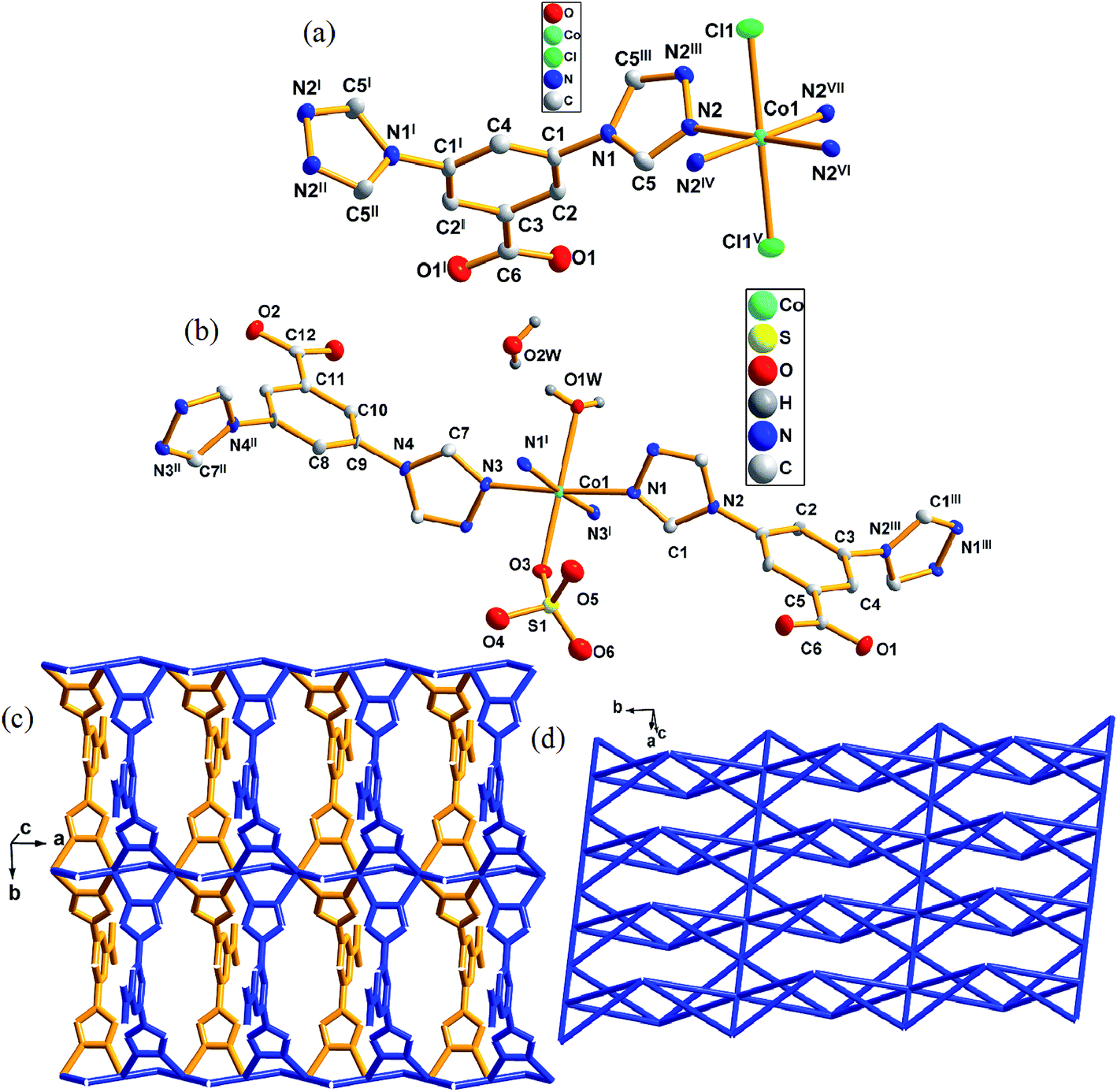 | ||
| Fig. 4 (a) Coordination environment of Co(II) atom in complex 4 (the hydrogen atoms and one lattice water molecules were omitted for clarity). (b) Single molecular structure of complex 5. The hydrogen atoms were omitted for clarity. (c) Two dimensional layers and its 4,6-connected net for 4 and 5. (d) Schematic illustration of 2D topological framework in complexes 4 and 5. Symmetry code for 4: I = 0.5 − x, 1 − y, z; II = x, 1 − y, z; III = 0.5 − x, y, z; IV = 1 − x, y, 1 − z; V = 0.5 + x, 2 − y, 1 − z; VI = 1 − x, 2 − y, 1 − z; VII = x, 2 − y, z. Symmetry code for 5: I = x, 1 − y, z; II = 1 − x, −y, z; III = 2 − x, 2 − y, z. | ||
In 4, the [Cl]− connect two Co(II) cations to form chains along [100] direction. The central Co(II) cations further link each chain through N2, N2I, N2II and N2III atoms of [L]− along [010] direction, eventually generating a 2D layered structure (Fig. 4c). The distance between two [Co]2+ cations is 3.564(7) Å. In complex 5, same coordination and connection modes as those in 4 generated same structures, however, the only difference between them was the [Cl]− anions in 4 changed to oxygen atoms from the coordination water molecule and [SO4]2−. In 5, the distance between two [Co]2+ ions are 3.375(9) and 3.526(4) Å.
On the other hand, each [L]− connects four Co(II) atoms with its two triazole groups. If the coordination linkage between [L]− was neglected, an infinite 1D chain was obtained via Co–Cl coordination. Each [L]− links Co atoms to form infinite 1D chains which were further linked by [Cl]− to give a 2D layer structure (Fig. 4c). According to the simplification principle, the central Co(II) atom could be viewed as a 6-connector and each [L]− ligand be considered as a 4-connector. The resulting structure of 4 and 5 are a 4,6-connected net with a schläfli symbol 4,6L26 (Fig. 4d).
In 1, 4 and 5, different counter anions generate different complexes based on the same central metal Co(II). In 1, the bond length of Co–O is 1.955(0) Å, that of Co–Cl in 4 is 2.485(3) Å and those of Co–O in 5 are 2.154(9) and 2.251(9) Å, respectively. Meanwhile, different anions also have an influence on the distances between Co–Co. Moreover, same connection modes are present in 1, 4 and 5.
3.5 Structures diversity
The selection of suitable ions is an important key factor in the design and synthesis of novel multidimensional coordination polymers with appealing functionality. In this work, we adopted [L]− and cobalt salts with different anions ([NO3]−, [Cl]− and [SO4]2−) for the construction of coordination polymers. Influenced by different counter anions, 1–5 demonstrate different frameworks based on Co(II) nodes. As illustrated in Fig. 5, HL ligands adopt a total of five types of coordination modes, acting as bi-(2), tri-(2), tetra-(1, 4 and 5), penta-(3) and hexa-dentate(3) ligands in 1–5. All [Co]2+ are six connected in complexes 1–5.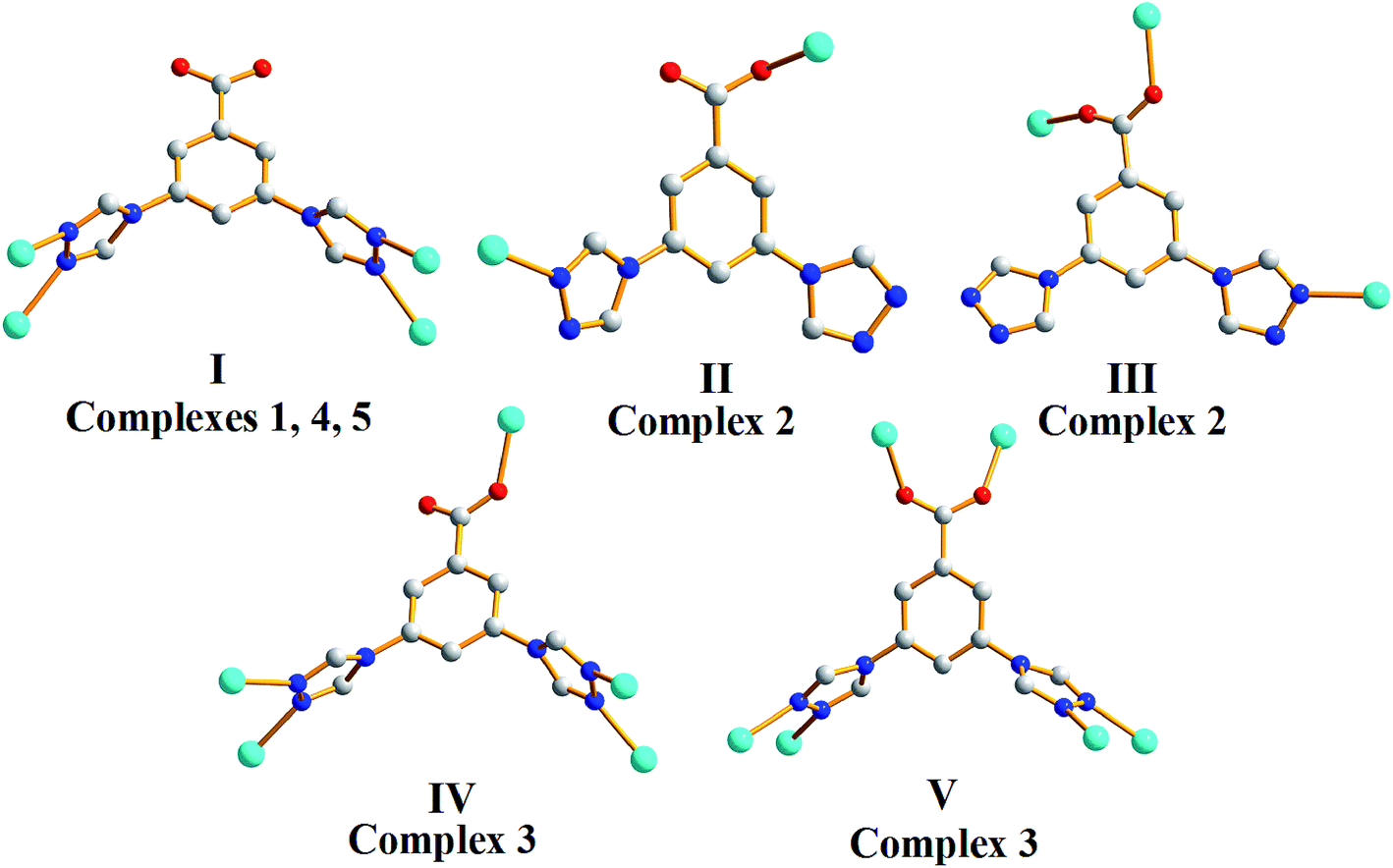 | ||
| Fig. 5 Five coordination modes of [L]− in complexes 1–5. | ||
As 1, 4 and 5 share the same coordination and connection mode, we will mainly discuss the structural differences of 1, 2 and 3. Generally, various coordination modes of the ligands provides the possibilities of the formation of versatile structures. In complex 1, the carboxyl group is not involved in the coordination, two triazoles of the ligand are four connected to form a 2D structure. In complexes 2 and 3, the carboxyl groups demonstrate both monodentate and bis-monodentate coordination modes. The ligands in 2 exhibit two kinds of coordination mode (II, III). One is two connected, the other one is three connected to form a 2D structure. In 3, the ligands also have two kinds of coordination mode (IV, V). One is five connected, the other one is six connected to give a 3D structure.
3.6 Directed synthesis of complexes 1, 2 and 3
Among these complexes, 1, 2 and 3 are originally obtained in one pot by the reaction of Co(NO3)2 and the ligand under solvothermal conditions (Fig. 6a). Afterwards, we successfully fulfilled the directed synthesis for each of them by tuning the ratio between reactants, employing different templates and counter anions. Complex 1 alone could be produced under the same solvothermal reaction conditions except supplying imidazole as template and employing CoCO3 instead of Co(NO3)2, the crystal appearance as shown in Fig. 6b. When tuning the molar ratio between Co(NO3)2 and the ligand to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2 was obtained separately (Fig. 6c). In the presence of 1,3,5-benzenetricarboxylic acid as template, only the crystals of 3 was achieved (Fig. 6d). From the hundreds of experiments, we found that different reaction conditions including metal salts and templates could not only lead to the production of different complexes mixtures but also have a strong influence on the crystal appearance. The reactant ratio, counter anion and the addition of different templates can impact the complex production with different structures. In our investigation, two different templates imidazole and 1,3,5-benzenetricarboxylic acid were found to play important role in the growth and formation of the crystals. Considering their different effect on the solution pH value, we were wondering if different pH value could have such big influence on the reaction. Accordingly, we employed another three polycarboxylic acids 1,4-benzenedicarboxylic, 1,2,3-benzenetricarboxylic acid or 1,2,4,5-benzenetetracarboxylic acid as templates, respectively, to conduct the reaction under the same conditions, however, we obtained mixtures of 1, 2, 3 with different quantities. Instead, only the crystals of 3 was achieved with 1,3,5-benzenetricarboxylic acid as template. The results revealed that this phenomenon might not be merely caused by the solution pH, which further confirms the template effect in the synthesis process. In the presence of templates, the synthetic process controls the structure of the complex and is therefore regarded as one way by which the structural characteristics of template molecules are transcribed into the resulting complex. Complexes 1, 2 and 3 demonstrate different coordination modes resulting in three different structures.
:1, 2 was obtained separately (Fig. 6c). In the presence of 1,3,5-benzenetricarboxylic acid as template, only the crystals of 3 was achieved (Fig. 6d). From the hundreds of experiments, we found that different reaction conditions including metal salts and templates could not only lead to the production of different complexes mixtures but also have a strong influence on the crystal appearance. The reactant ratio, counter anion and the addition of different templates can impact the complex production with different structures. In our investigation, two different templates imidazole and 1,3,5-benzenetricarboxylic acid were found to play important role in the growth and formation of the crystals. Considering their different effect on the solution pH value, we were wondering if different pH value could have such big influence on the reaction. Accordingly, we employed another three polycarboxylic acids 1,4-benzenedicarboxylic, 1,2,3-benzenetricarboxylic acid or 1,2,4,5-benzenetetracarboxylic acid as templates, respectively, to conduct the reaction under the same conditions, however, we obtained mixtures of 1, 2, 3 with different quantities. Instead, only the crystals of 3 was achieved with 1,3,5-benzenetricarboxylic acid as template. The results revealed that this phenomenon might not be merely caused by the solution pH, which further confirms the template effect in the synthesis process. In the presence of templates, the synthetic process controls the structure of the complex and is therefore regarded as one way by which the structural characteristics of template molecules are transcribed into the resulting complex. Complexes 1, 2 and 3 demonstrate different coordination modes resulting in three different structures.
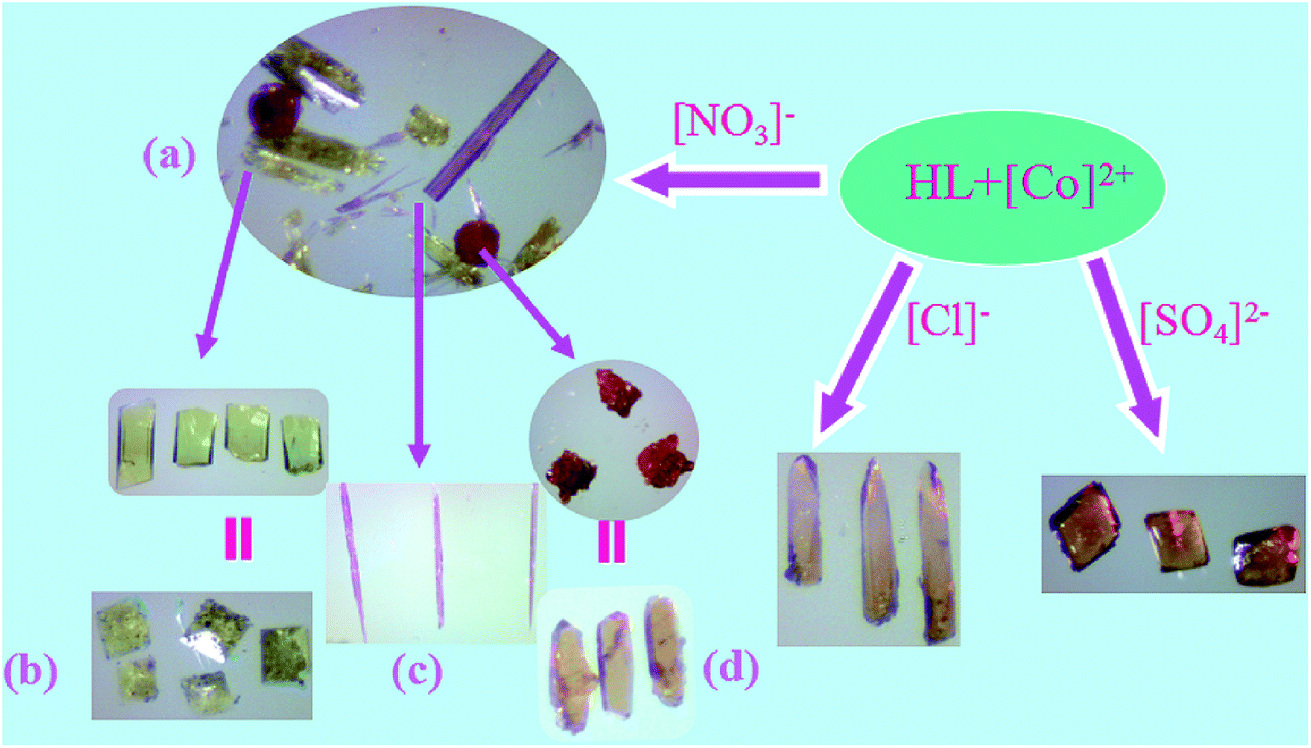 | ||
| Fig. 6 Crystal morphological comparison of complexes 1–5. | ||
3.7 Thermogravimetric analyses
The TGA measurements of complexes 1–5 were carried out in the temperature range 50–800 °C under atmosphere with a heating rate of 10 °C min−1 (Fig. S2†). Complex 1 shows the first weight loss of 5.75% corresponds to the departure of the hydroxyl groups in the range of 50–165 °C (calcd: 5.14%). Then the loss of organic ligands occurs in the range of 165–440 °C, and the remaining weight could be ascribed to CoO (obsd 22.21%, calcd 22.63%). In complex 2, the decomposition of the framework occurs at the range of 310–573 °C. With the loss of ligand molecule, the remaining weight can be attributed to the formation of CoO (obsd: 11.54%, calcd: 12.38%). For complex 3, TGA curve displays the first weight loss of 8.01% from 50 to 164 °C corresponding to the departure of lattice water molecules and coordinated water molecules (calcd: 7.39%). The second weight loss occurred from 164 to 440 °C, the remaining weight corresponds to the formation of CoO (obsd: 18.30%; calcd: 18.45%). Complex 4 exhibits the first weight loss of 4.88% at 246 °C which could be ascribed to the loss of lattice water molecule (calcd: 4.90%). Further decomposition from 246 to 675 °C is consistent with the loss of the ligand molecule (obsd: 75.68%; calcd: 74.32%). For complex 5, the first weight loss of 6.18% occurs at 356 °C corresponding to the release of water molecule (calcd: 6.94%). Further, the second weight loss was observed between 356–612 °C due to the release of ligand molecule. The remaining weight was ascribed to the formation of CoO (obsd: 19.15%; calcd: 19.25%).3.8 Magnetic property
Magnetic measurements were carried out on crystalline samples of 1–5, the phase purity of which was confirmed by XRPD (Fig. S3†). Solid-state magnetic susceptibility measurements for 1–5 were performed in the range 1.8–300 K under a field of 1000 Oe.In complex 1, a χMT value of 2.70 cm3 K mol−1 is higher than that for a spin-only case (1.87 cm3 K mol−1, S = 3/2) at 300 K, as expected for an orbital contribution.23 With the lowered temperature, the χMT value keeps decreasing until at ca. 35 K and rises to a maximum at 12 K before dropping. Such results indicate the presence of ferromagnetic interaction occurring in cobalt ions of Co–O–Co chains in 1. Spin–orbit coupling effects and the presence of possible antiferromagnetic interactions explained the decrease of the χMT plot upon cooling at high temperature. Fitting the data above 50 K by the Curie–Weiss law gives C = 3.60 cm3 K mol−1 and θ = −101.59 K (Fig. 7). Generally, the single-ion behavior of octahedral Co(II) could lead to a bigger negative θ value, however, such a large value could better be explained by the dominant antiferromagnetic coupling between Co(II) ions.
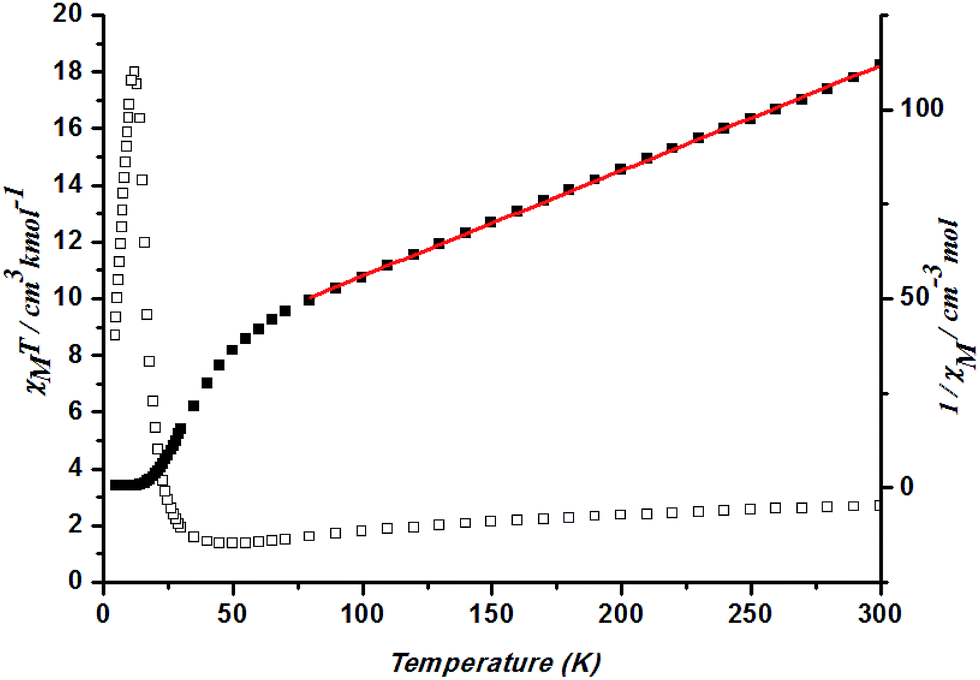 | ||
| Fig. 7 Plots of χMT vs. T for 1 and the temperature dependence of 1/χM, the best fit of the curve (red) to the Curie–Weiss law. | ||
In 1, the Co–O(H)–Co angles range between 117.19°, resulting in competing antiferromagnetic and ferromagnetic interactions. Considering the presence of the numerous magnetic exchange pathways in the chains and the adjacent coordinated octahedrons of the anisotropic Co(II) ions that tilt towards each other, it is possible for spin canting occurrence. As the shortest Co⋯Co distance across the bridge is 3.4009 Å, the magnetic exchange coupling through these bridges is expected to be very weak in 1. Fig. 8 shows an sudden increase to above 0.38 Nβ of magnetization below 2500 Oe and then increases linearly to 0.61 Nβ at 70 kOe without achieving saturation, which suggests the canted antiferromagnetism of 1. This could be further confirmed by the field dependence of magnetization at 2 K. The isonic anions between the Co(II) ions from neighboring chains could only lead to very weak magnetic interactions.
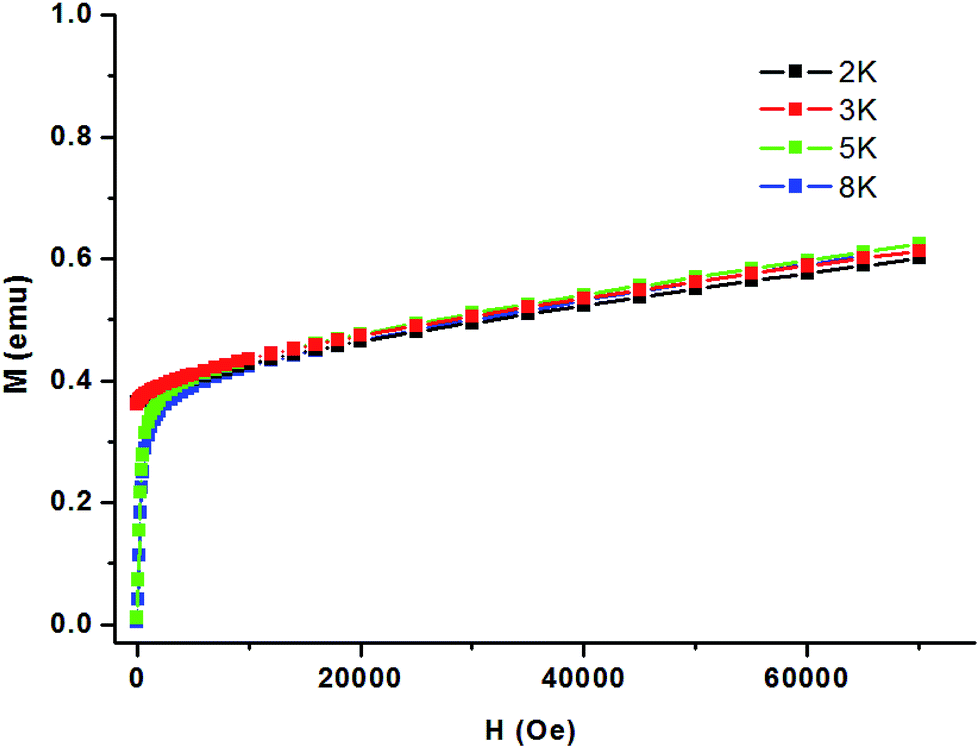 | ||
| Fig. 8 Magnetization M versus the magnetic field H for 1 at different low temperatures. | ||
Temperature dependent alternating-current (ac) magnetic susceptibilities of 1 is obtained at zero direct-current (dc) fields to further understand the low-temperature magnetic properties of 1. Slow magnetic relaxation was found to be present in 1, considering the frequency-dependent behaviors of in-phase (χ′M) and out-of-phase (χ′′M) components of the susceptibilities below ca. 12 K (ref. 24) (Fig. 9, left). Plotting lnτ against 1/T has been fitted following the Arrhenius equation τ = τ0exp(ΔE/kBT) (τ is the relaxation time). The calculated τ0 (1.05 × 10−8 s) and ΔE (89.5 K) suggests that the slow relaxation of magnetization in 1 is a thermally activated process (ΔE is the energy barrier to reverse the magnetization). The ΔE and τ0 values are in the normal range for superparamagnets including SCMs (10−6 to 10−13 s)25 (Fig. 9, right). The strong frequency dependence of the relaxation time and the physically reasonable value of τ0, without introducing any scaling factor, rule out the possibility that the system is a spin glass.
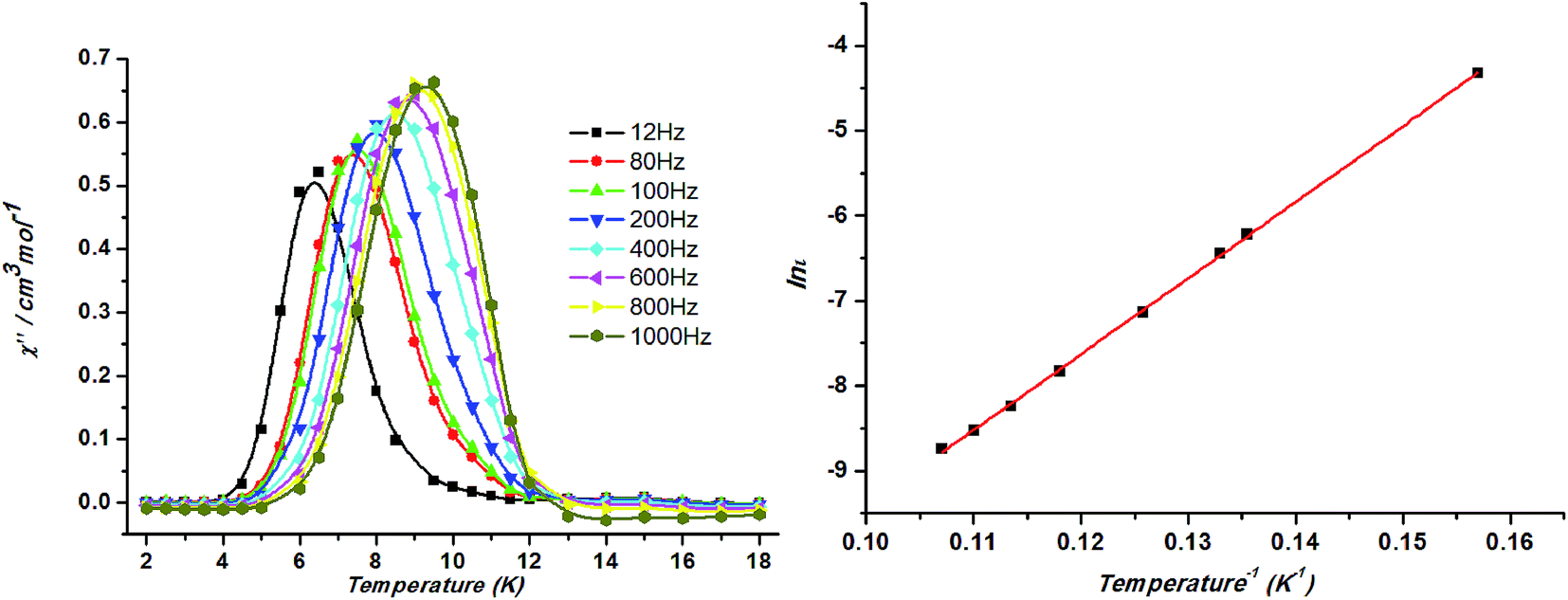 | ||
| Fig. 9 Frequency dependence of the out-of-phase ac susceptibility (χ′′) between 12 and 1000 Hz at Hdc = 0 Oe for 1 (left). Relaxation time of the magnetization ln(τ) vs. T−1 (Arrhenius plot using ac data) for 1. The line corresponds to the fit(right). | ||
A hysteresis loop is clearly observed at 1.8 K, a more distinct magnetization hysteresis loop is observed. It is also noteworthy that the hysteresis loops along the axis direction exhibit “steps” at the original region due to the effect of weak interchain interactions, indicating a hard-magnetic behavior of 1.
The magnetic susceptibility of 2 versus temperature was shown in Fig. 10. At 300 K, χMT value of 3.42 cm3 K mol−1 is higher than that for a spin-only case (1.87 cm3 K mol−1, S = 3/2) at 300 K, as expected for an orbital contribution. With the lowered temperature, the χMT value keeps decreasing. Such results indicate the presence of antiferromagnetic interaction occurring in cobalt ions in 2. Fitting the data above 50 K by the Curie–Weiss law gives C = 4.11 cm3 K mol−1 and θ = −58.80 K (Fig. 10).
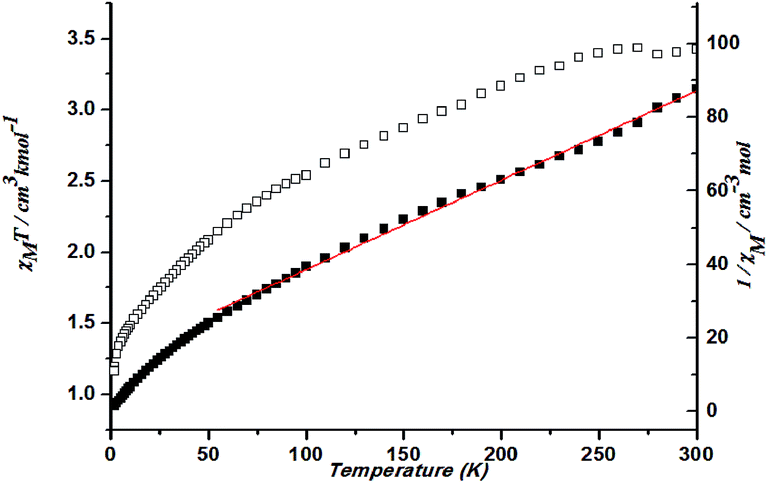 | ||
| Fig. 10 Plots of χMT vs. T for 2 and the temperature dependence of 1/χM, the best fit of the curve (red) to the Curie–Weiss law. | ||
In 3, the χMT value is 11.02 cm3 K mol−1 per Co3 unit is much larger than the spin value expected for three uncoupled high-spin Co(II) ion (5.65 cm3 K mol−1, S = 3/2),26 revealing a significant orbital contribution. The decrease of the χMT value or the negative θ value can be induced by antiferromagnetic coupling interactions within and between dimeric units as well as strong spin–orbit coupling. When the temperature is decreased, the χMT value increases slightly at ca. 5 K. Fitting the data above 50 K by the Curie–Weiss law gives C = 13.99 cm3 K mol−1 and θ = −82.02 K (Fig. 11). The large negative Weiss constant suggests the presence of spin–orbit coupling effect.
 | ||
| Fig. 11 Plots of χMT vs. T for 3 and the temperature dependence of 1/χM, the best fit of the curve (red) to the Curie–Weiss law. | ||
At 300 K, a χMT value of 6.67 cm3 K mol−1 is higher than that for a spin-only case (1.87 cm3 K mol−1, S = 3/2) in complex 4. This is also due to the large orbital contribution of Co(II) ion. With a decrease in temperature, the χMT keep decreasing. Spin–orbit coupling effects and the presence of possible antiferromagnetic interactions explained the decrease of the χMT plot upon cooling at high temperature. Fitting the data above 50 K by the Curie–Weiss law gives C = 11.93 cm3 K mol−1 and θ = −242.76 K (Fig. 12). Such a large value could better be explained by the spin–orbit coupling between Co ions.
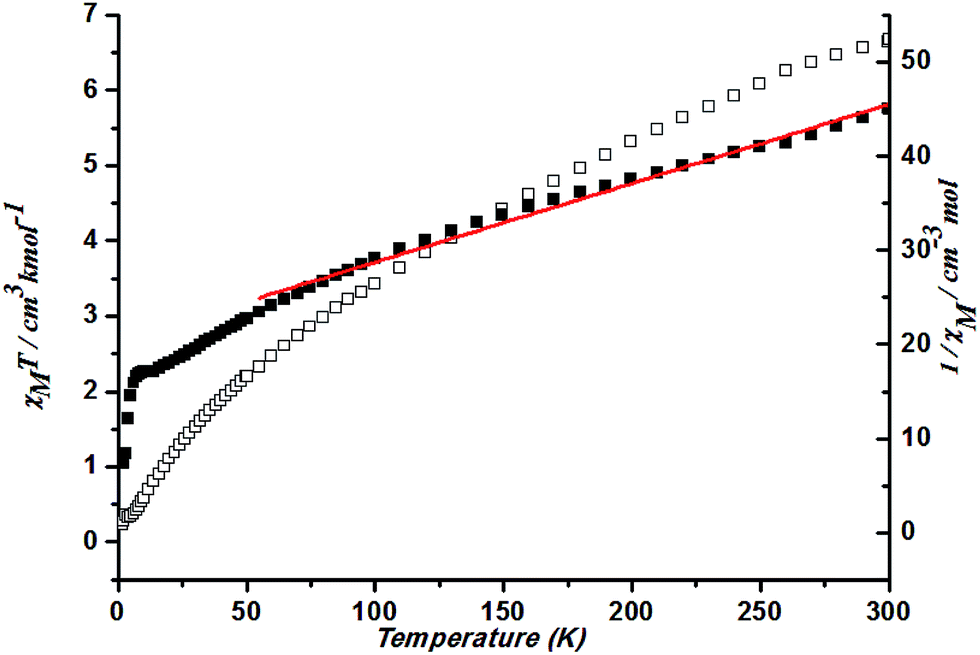 | ||
| Fig. 12 Plots of χMT vs. T for 4 and the temperature dependence of 1/χM, the best fit of the curve (red) to the Curie–Weiss law. | ||
At 300 K, a χMT value is 5.19 cm3 K mol−1 in complex 5. With a decrease in temperature, the χMT decreases gradually and then rapidly. Spin–orbit coupling effects and the presence of possible antiferromagnetic interactions explained the decrease of the χMT plot upon cooling at high temperature. The slow decrease is a typical manner of an orbitally degenerate 4T1g (d7 octahedral) system undergoing spin–orbit coupling. The rapid decrease suggests the possibility of antiferromagnetic coupling. Fitting the data above 50 K by the Curie–Weiss law gives C = 6.54 cm3 K mol−1 and θ = −80.29 K (Fig. 13).
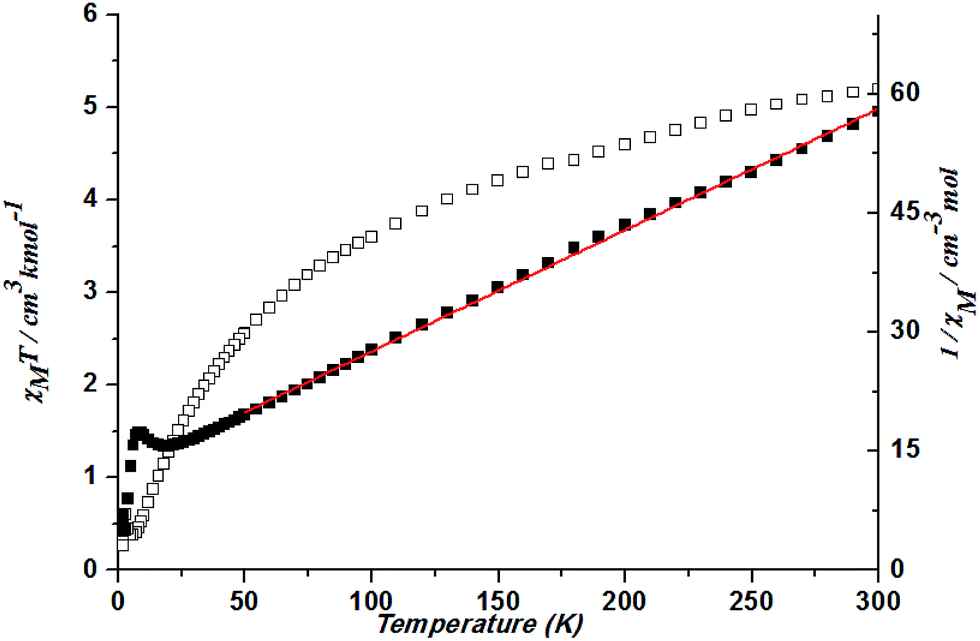 | ||
| Fig. 13 Plots of χMT vs. T for 5 and the temperature dependence of 1/χM, the best fit of the curve (red) to the Curie–Weiss law. | ||
4. Conclusion
In this paper, we have demonstrated the formation of five different coordination polymers 1–5 from the solvothermal reactions of [L]− with Co(NO3)2, CoCO3, CoCl2, CoSO4. It is noted that the counter anions play an important role in tuning the structural diversities of these frameworks. The crystals of 1–3 could be obtained in one pot and the directed synthesis for each of them was also fulfilled by tuning the ratio between reactants, employing different templates and counter anions. It also confirms the template effect in the synthesis process. The magnetic property investigation reveals that complex 1 exhibits frequency-dependent magnetic behavior.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (Nos. 21371052 & 21501050), Heilongjiang Postdoctoral Scientific Research Foundation (LBH-Q14138), and Natural Science Foundation of Heilongjiang University (QL201307).References
- (a) H. Li, B. Zhao, R. Ding, Y. Jia, H. Hou and Y. Fan, Cryst. Growth Des., 2012, 12, 4170 CrossRef CAS; (b) H. N. Miras, I. Chakraborty and R. G. Raptis, Chem. Commun., 2010, 46, 2569 RSC; (c) Z. F. Wu, B. Tan, Z. H. Deng, Z. L. Xie, J. J. Fu, N. N. Shen and X. Y. Huang, Eur. J. Inorg. Chem., 2016, 22, 1334 CAS; (d) J. F. Ma, J. Yang, G. L. Zheng, L. Li and J. F. Liu, Inorg. Chem., 2003, 42, 7531 CrossRef CAS PubMed.
- (a) R. J. Li, M. Li, X. P. Zhou, D. Li and M. O'Keeffe, Chem. Commun., 2014, 50, 4047 RSC; (b) D. M. Chen, N. Xu, X. H. Qiu and P. Cheng, Cryst. Growth Des., 2015, 15, 961 CrossRef CAS; (c) J. Duan, W. Jin and R. Krishna, Inorg. Chem., 2015, 54, 4279 CrossRef CAS PubMed.
- (a) S. Zhang, J. Ma, X. Zhang, E. Duan and P. Cheng, Inorg. Chem., 2015, 54, 586 CrossRef CAS PubMed; (b) W. Wang, J. Yang, R. Wang, L. Zhang, J. Yu and D. Sun, Cryst. Growth Des., 2015, 15, 2589 CrossRef CAS.
- A. Cingolani, S. Galli, N. Masciocchi, L. Pandolfo, C. Pettinari and A. Sironi, J. Am. Chem. Soc., 2005, 127, 6144 CrossRef CAS PubMed.
- (a) D. Shao, S. L. Zhang, L. Shi, Y. Q. Zhang and X. Y. Wang, Inorg. Chem., 2016, 55, 10859 CrossRef CAS PubMed; (b) C. G. Werncke, M. A. Bouammali, J. Baumard, N. Suaud, C. Martins, N. Guihery, L. Vendier, J. Zheng, J. B. Sortais, C. Darcel, S. Sabo-Etienne, J. P. Sutter, S. Bontemps and C. Pichon, Inorg. Chem., 2016, 55, 10968 CrossRef CAS PubMed; (c) Y. Kim, Y. S. Choi, H. J. Lee, H. Yoon, Y. K. Kim and M. Oh, Chem. Commun., 2014, 50, 7617 RSC; (d) J. Werner, M. Rams, Z. Tomkowicz and C. Näther, Dalton Trans., 2014, 43, 17333 RSC.
- (a) Y. H. Yu, H. T. Ye, G. F. Hou, C. Y. Ren, J. S. Gao and P. F. Yan, Cryst. Growth Des., 2016, 16, 5669 CrossRef CAS; (b) H. T. Ye, C. Y. Ren, G. F. Hou, Y. H. Yu, X. Xu, J. S. Gao, P. F. Yan and S. W. Ng, Cryst. Growth Des., 2014, 14, 3309 CrossRef CAS.
- B. Chen, Y. Yang, F. Zapata, G. Lin, G. Qian and E. B. Lobkovsky, Adv. Mater., 2007, 19, 1693 CrossRef CAS.
- (a) J. F. Ayme, J. Lux, J. P. Sauvage and A. Sour, Eur. J. Inorg. Chem., 2012, 18, 5565 CAS; (b) C. K. Brozek, L. Bellarosa, T. Soejima, T. V. Clark, N. Lopez and M. Dinca, Eur. J. Inorg. Chem., 2014, 20, 6871 CAS.
- G. W. Zhou, Y. Z. Lan, F. K. Zheng, X. Zhang, M. H. Lin, G. C. Guo and J. S. Huang, Chem. Phys. Lett., 2006, 426, 341 CrossRef CAS.
- (a) Q. Yang, X. Chen, Z. Chen, Y. Hao, Y. Li, Q. Lu and H. Zheng, Chem. Commun., 2012, 48, 10016 RSC; (b) X. Y. Chen, R. B. Huang, L. S. Zheng and J. Tao, Inorg. Chem., 2014, 53, 5246 CrossRef CAS PubMed.
- (a) F. Zhao, S. Jing, Y. Che and J. Zheng, CrystEngComm, 2012, 14, 4478 RSC; (b) L. Wang, Z. H. Yan, Z. Xiao, D. Guo, W. Wang and Y. Yang, CrystEngComm, 2013, 15, 5552 RSC.
- (a) D. Deng, L. Liu, B. M. Ji, G. Yin and C. Du, Cryst. Growth Des., 2012, 12, 5338 CrossRef CAS; (b) K. P. Rao, M. Higuchi, J. Duan and S. Kitagawa, Cryst. Growth Des., 2013, 13, 981 CrossRef CAS.
- (a) B. C. Tzeng, Y. C. Hung and G. H. Lee, Eur. J. Inorg. Chem., 2016, 22, 1522 CAS; (b) K. Y. Zou, Q. Zou, T. Han, Y. C. Liu, J. J. Wang, X. Zhang and Z. X. Li, J. Solid State Chem., 2016, 235, 85 CrossRef CAS.
- (a) W. G. Lu, L. Jiang and T. B. Lu, Cryst. Growth Des., 2010, 10, 4310 CrossRef CAS; (b) P. Cui, L. Ren, Z. Chen, H. Hu, B. Zhao, W. Shi and P. Cheng, Inorg. Chem., 2012, 51, 2303 CrossRef CAS PubMed.
- Z. M. Hao and X. M. Zhang, Cryst. Growth Des., 2007, 7, 64 CAS.
- (a) G. A. Farnum and R. L. LaDuca, Cryst. Growth Des., 2010, 10, 1897 CrossRef CAS; (b) P. Nielsen, H. Toftlund, A. D. Bond, J. F. Boas, J. R. Pilbrow, G. R. Hanson, C. Noble, M. J. Riley, S. M. Neville, B. Moubaraki and K. S. Murray, Inorg. Chem., 2009, 48, 7033 CrossRef CAS PubMed; (c) Z. Hulvey, E. Ayala, J. D. Furman, P. M. Forster and A. K. Cheetham, Cryst. Growth Des., 2009, 9, 4759 CrossRef CAS; (d) Y. Y. Liu, J. F. Ma, J. Yang and Z. M. Su, Inorg. Chem., 2007, 46, 3027 CrossRef CAS PubMed; (e) L. L. Liu, J. J. Huang, X. L. Wang, G. C. Liu, S. Yang and H. Y. Lin, Inorg. Chim. Acta, 2013, 394, 715 CrossRef CAS.
- (a) R. Seetharaj, P. V. Vandana, P. Arya and S. Mathew, Arabian J. Chem., 2016 DOI:10.1016/j.arabjc.2016.01.003; (b) N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933 CrossRef CAS PubMed; (c) T. Yu, S. Wang, X. Li, X. Gao, C. Zhou, J. Cheng, B. Li, J. Li, J. Chang, H. Hou and Z. Liu, CrystEngComm, 2016, 18, 1350 RSC.
- (a) B. Ding, P. Yang, Y. Y. Liu, Y. Wang and G. X. Du, CrystEngComm, 2013, 15, 2490 RSC; (b) B. Ding, Y. Y. Wang, S. X. Liu, X. X. Wu, Z. Z. Zhu, J. Z. Huo and Y. Y. Liu, CrystEngComm, 2015, 17, 5396 RSC; (c) N. Wang, Y. C. Feng, W. Shi, B. Zhao, P. Cheng, D.-Z. Liao and S.-P. Yan, CrystEngComm, 2012, 14, 2769 RSC; (d) H. A. Habib, A. Hoffmann, H. A. Hoppe, G. Steinfeld and C. Janiak, Inorg. Chem., 2009, 48, 2166 CrossRef CAS PubMed.
- (a) H. A. Habib, J. Sanchiz and C. Janiak, Dalton Trans., 2008, 1734 RSC; (b) H. A. Habib, A. Hoffmann, H. A. Hoppe and C. Janiak, Dalton Trans., 2009, 1742 RSC; (c) X. X. Lv, L. L. Shi, K. Li, B. L. Li and H. Y. Li, Chem. Commun., 2017, 53, 1860 RSC; (d) K. Li, X. X. Lv, L. L. Shi, L. Liu, B. L. Li and B. Wu, Dalton Trans., 2016, 15078 RSC.
- (a) B. Liu, Y. H. Yu, W. H. Jiang, G. F. Hou and J. S. Gao, Inorg. Chem. Commun., 2015, 54, 12 CrossRef CAS; (b) W. H. Jiang, J. L. Liu, L. Z. Niu, B. Liu, Y. H. Yu, G. F. Hou and D. S. Ma, Polyhedron, 2017, 124, 191 CrossRef CAS.
- G. M. Sheldrick, Programs for the Solution and Refinement of Crystal Structures, SHELXS-97, University of Götingen, Germany, 1997 Search PubMed.
- V. A. Blatov and A. P. Shevchenko, TOPOS-Version 4.0 professional (beta evaluation), Samara State University, Samara, Russia, 2006 Search PubMed.
- P. Mahata, S. Natarajan, P. Panissod and M. Drillon, J. Am. Chem. Soc., 2009, 131, 10140 CrossRef CAS PubMed.
- (a) N. F. Chilton, C. A. Goodwin, D. P. Mills and R. E. Winpenny, Chem. Commun., 2015, 51, 101 RSC; (b) D. N. Woodruff, R. E. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110 CrossRef CAS PubMed; (c) J. Zhang, K. Wang, X. Li and E. Gao, Inorg. Chem., 2014, 53, 9306 CrossRef CAS PubMed.
- (a) J. Y. Zhang, K. Wang, X. B. Li and E. Q. Gao, Inorg. Chem., 2014, 53, 9306 CrossRef CAS PubMed; (b) J. P. Zhao, Q. Yang, Z. Y. Liu, R. Zhao, B. W. Hu, M. Du, Z. Chang and X. H. Bu, Chem. Commun., 2012, 48, 6568 RSC.
- (a) S. J. Liu, L. Xue, T. L. Hu and X. H. Bu, Dalton Trans., 2012, 41, 6813 RSC; (b) X. Z. Song, S. Y. Song, M. Zhu, Z. M. Hao, X. Meng, S. N. Zhao and H. J. Zhang, Dalton Trans., 2013, 42, 13231 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data, IR data, powder X-ray diffraction patterns, and TGA curves for CPs 1–5. CCDC 1528099–1528103 for 1–5. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra07089f |
| This journal is © The Royal Society of Chemistry 2017 |