
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Drowning in diversity? A systematic way of clustering and selecting a representative set of new psychoactive substances†
Mire Zloh *,
Eleftherios G. Samaras,
Jesus Calvo-Castro,
Amira Guirguis,
Jacqueline L. Stair and
Stewart B. Kirton*
*,
Eleftherios G. Samaras,
Jesus Calvo-Castro,
Amira Guirguis,
Jacqueline L. Stair and
Stewart B. Kirton*
Department of Pharmacy, Pharmacology and Postgraduate Medicine, School of Life and Medical Sciences, University of Hertfordshire, College Lane, AL10 9AB, UK. E-mail: m.zloh@herts.ac.uk; s.b.kirton3@herts.ac.uk
First published on 17th November 2017
Abstract
New psychoactive substances (NPS) can be generally described as a set of compounds that have been designed to mimic the effects of illegal recreational drugs, but are not subject to restriction or control with respect to existing regulations and legislation. In recent years, the number and chemical diversity of emergent NPS has increased substantially, and regulators have struggled to develop methods for accurate detection of NPS at the same rate. Existing approaches to NPS classification are pragmatic and/or semi-systematic and do not lend themselves to objective spectroscopic classification of emergent NPS. As such, this research discusses the identification of a systematic NPS classification based on chemical structures. A set of 478 NPS were grouped according to the similarity between their chemical structural features using hierarchical clustering and a maximum common substructure of 9 atoms, which included both hydrogen and heavy atoms. The rationale for including hydrogen atoms is that accurate spectroscopic identification of NPS will be dependent upon variations in substitution patterns in the molecules. This analysis generated 79 clusters, arising from 21 superclusters. The medoid substances of each cluster were used to form a dataset that was representative of the chemical space encompassed by known NPS. Subsequent categorisation of a test set of NPS showed that the test substances were assigned to an appropriate cluster when the Tanimoto similarity coefficient between the cluster medoid and the test substance was at least 0.5. This indicates that the cluster medoids could be used for assignment of emerging NPS to systematically-defined categories based on chemical structure. These medoids will also aid in the prediction of spectroscopic properties for emergent NPS, which will be invaluable for structure-based classifications and development of methods for detection of emerging NPS.
Introduction
New psychoactive substances (NPS), also inaccurately known as ‘legal highs’ or ‘designer drugs’, are defined by the United Nations Office on Drugs and Crime (UNDOC) as “substances of abuse, either in a pure form or a preparation, that are not controlled by the 1961 Single Convention on Narcotic Drugs or the 1971 Convention on Psychotropic Substances, but which may pose a public health threat”.1 NPS are designed with the intention to imitate the pharmacological effects of controlled substances such as cocaine, heroin and methamphetamine by slightly modifying the molecular structure of these existing controlled compounds, thus bypassing legislation to prevent their distribution, possession and consumption.2–4 According to the UNODC, the numbers, and rate, at which NPS are entering the market is increasing, and the chemical diversity of emergent NPS also continues to expand.4 As of 2016, more than 700 types of NPS had been reported by 109 countries.1Existing NPS are frequently rebranded i.e. the names and composition of the product are altered, and marketed as superior, but legal, alternatives to the banned substances they purport to replace or supplement,5–7 which results in added complexity to the NPS problem. The fact that NPS are being used for recreational purposes,8 are not fully risk assessed, and are not yet completely controlled by international drug conventions identifies them as a possible serious threat to public health.9 For this reason, a number of countries have recently introduced NPS legislation. For example, the UK recently enforced the Psychoactive Substance Act, a blanket ban on the supply, possession with the intention to supply, possession in custodial environments, production and importation of all substances that produce a psychoactive effect.10,11 As a result of this legislation, it is imperative that new tools and approaches are developed to more effectively tackle current NPS abuse, production and supply, given that these compounds will now be reaching users through more clandestine routes. The current state of the art for the detection of NPS includes “wet” laboratory-based techniques such as chromatography, mass spectrometry, nuclear magnetic resonance spectroscopy, gas chromatography-mass spectrometry and liquid chromatography mass spectrometry. Solid-state laboratory techniques including attenuated total reflectance Fourier transform infrared (ATRF-IR) and Raman spectroscopy have also gained popularity as techniques for identifying NPS, and recent studies have highlighted the importance and utility of handheld Raman devices for detection of NPS “in the field” (see e.g. (ref. 12) for a more in-depth discussion).
According to the International Narcotic Control Board, the growth in production and distribution of NPS is ‘escalating out of control’.13 The EMCDDA (European Monitoring Centre for Drugs and Drug Addiction) has stated that the number of NPS detected in Europe is rising, as demonstrated by a Europe-wide early warning system that detected 100 NPS in 2015.14 This explosion of NPS onto the market is causing a major challenge to drug control, as regulators struggle to monitor the compounds at the same pace as they appear, especially given the lack of information on chemistry, pharmacology and toxicology for new analogues. The number of known NPS, the rate of emergence and the often transient nature of some compounds are such that it is difficult to obtain the complete information on physicochemical and biological properties for all NPS to be able to inform relevant stakeholders.
Current classifications of NPS are pragmatic and non-systematic. They are either based on their chemical scaffold and/or pharmacological/clinical effect. NPS classified according to their chemical structure include phenethylamines, piperazines, synthetic cathinones and tryptamines. Conversely, classification of an NPS as a synthetic cannabinoid is based on its pharmacological action on the cannabinoid receptors, and therefore this class contains very structurally diverse molecules, as illustrated in Fig. 1. Whilst classification of NPS according to pharmacological action could be useful, it can be argued that it is not optimal from a systematic point of view. This is due to the relative promiscuity of a number of known NPS, and a relative dearth of knowledge around these substances with respect to their explicit pharmacological action (see e.g. (ref. 15)). For example, the cathinones exhibit a number of pharmacological responses including stimulant, empathogenic and antidepressant effects.16 This is thought to be related to the interaction of these compounds with a number of biological receptors including tyrosine and tryptophan hydroxylases.17,18 In addition, cathinones, like a number of other NPS classes, inhibit the re-uptake of the neurotransmitters dopamine, serotonin and norepinephrine by their respective monoamine transporter (MAT) proteins of the synaptic cleft, and induce the release of newly synthesised neurotransmitters to the synaptic cleft.19–21 Even for cases where the interaction between receptor and NPS appears less ambiguous, such as the interaction between synthetic cannabinoids and the CB1 receptor, the explicit pharmacological action can be difficult to determine as it is difficult to determine whether the NPS is acting as a full or partial agonist.22
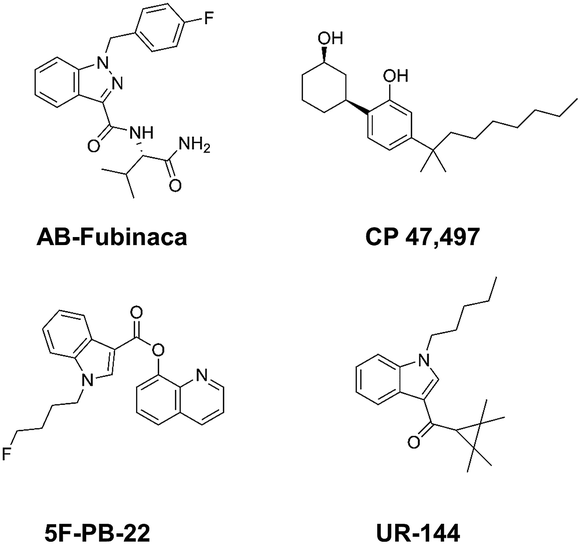 | ||
| Fig. 1 Exemplar chemical structures of some of the subcategories of known synthetic cannabinoids, demonstrating the structural diversity of this class of compounds. | ||
To add a further layer of complexity, clinical classifications aimed at the effective treatment of NPS intoxication also exist outside of the chemical scaffold/pharmacological effect classifications. Whilst the classification of NPS as hallucinogenics, stimulants, synthetic opioids, GABA A/B receptor agonists, dissociatives or depressants is useful for clinicians,23,24 these categories are not well defined as several NPS can have overlapping actions between more than one of these clinical categories.25
The approaches outlined above do not provide a consistent, systematic method for NPS classification. Consequently, only cursory assessments can be performed for emerging NPS, which may not provide enough information to assess their potential to cause harm for either clinicians or regulatory bodies. This means it is essential to explore new ways of efficiently and systematically identifying and classifying existing and emerging NPS.
One strategy to achieve this would be to group NPS according to their structural similarity, as structurally similar compounds are likely to have similar biological activities26 and exhibit similar spectroscopic behaviour. However, the increasing complexity and diversity of NPS prevents systematic classification with respect to their structural similarity by visual inspection alone. Hence, it is essential to process this information computationally in order to maximize the speed and accuracy at which results can be generated, and to provide an accessible mechanism by which the classification system can be iteratively updated as new chemical scaffolds emerge. In addition, systematic analyses could be used to identify mechanistic similarities and use them to accelerate screening and classification of NPS according to their physicochemical properties or mechanism of action.27
Although it is preferable to acquire a complete information set for every known NPS, this would be difficult to achieve because of limitations with respect to time constraints and the availability and costs of reference standards. Therefore, a credible alternative strategy would be the creation of a diverse subset comprising a number of molecules that serve as ‘representatives’ of the physicochemical properties of known NPS. Selecting structural representatives would reduce the number of molecules that need to be analysed, whilst retaining maximum information about the structural diversity of the whole dataset. Such a subset would have inherent variety due to the highly diverse chemical space that NPS cover, but be representative of known NPS.28 The results from the analysis of representative molecules would be used to infer the properties of structurally similar NPS29 and have the potential to identify and classify NPS emerging onto the market – a key point of interest for law enforcement agencies and associated scientific bodies worldwide.
Cluster analysis is an appropriate tool to help guide the identification of this diverse NPS dataset. Clustering techniques are generally employed as versatile data mining approaches to create groups of (structurally) similar molecules within a given set of compounds30 and to find molecules that hold central positions in the chemical space occupied by a cluster (i.e. medoids).31 Clustering followed by medoid identification provides a comprehensive and systematic way of grouping known NPS according to chemical structure and identifying those molecules that best represent the dataset as a whole. In this work, we demonstrate, for the first time, the use of hierarchical clustering and similarity calculation techniques to group NPS, with an aim to aid the development of novel tools for NPS detection and classification. This wealth of NPS structural data currently available provides an opportunity to explore how structural patterns might manifest and be used for the prediction of emergent NPS to help research scientists, legal authorities and healthcare professionals identify and classify them.
Experimental
Acquisition and preparation of NPS data
In January 2015, a list of 478 NPS molecules was obtained from the European Monitoring Centre for Drugs and Drug Addiction (EMCDDA).32 A list of emergent NPS that appeared on the market between February 2015 and March 2016 were subsequently supplied by the EMCDDA, and these 98 molecules became the test compounds used to interrogate the model generated. The majority of new analogues that appeared on the market belonged to three EMCDDA/EDND classes: synthetic cathinones (24), synthetic cannabinoids (24) and phenethylamines (10), whilst the rest were distributed almost evenly between the remaining classes. Both lists contained information including synonym names, systematic names and the NPS class for each molecule. The list was cross-checked with available information in the literature to ensure accuracy and completeness. For entries in the EMCDDA/EDND that record products comprising herbal extracts, such as NPS Kawa and Kratom, the active ingredients of these products were identified and added to the dataset. Such entries were labelled as “not present” in the initial list of NPS provided by EMCDDA, in order to highlight that they had arisen from manipulation of the dataset.The dataset was expanded by inclusion of the SMILES (Simplified Molecular Input Line Entry System) strings for each molecule, which were acquired from ChemSpider33 and the New Synthetic Drugs databases.34 A unique identifier was given for each NPS in the dataset to facilitate easier identification of molecules at later stages.
Hierarchical clustering (hydrogen atoms included)
A maximum common substructure (MCS) based approach, a well-established set of algorithms based on graph theory and used to identify structural overlap in chemical databases35 was used to identify structurally similar NPS. The NPS set represented by text file with SMILES strings for each molecule was used to carry out hierarchical clustering. The clustering was achieved by ChemAxon LibMCS [2] using normal mode and the ‘jcsearch’ algorithm, and the default settings (i.e. MCS mode = fast, minimal MCS size = 9, matching atom types = true, bond type = true, charge = true, keep rings = true, required cluster count = 1, maximal level count = 10). The effect of the common substructure composition on the number of clusters was evaluated by varying the MCS setting to include only heavy atoms, and to include all atoms. ChemAxon LibMCS uses chemical hashed fingerprints (a linear fingerprint that uses hashing to create the binary representation of the fingerprint) and the Tanimoto coefficient for the clustering of molecules. Although ChemAxon does have other similarity metrics that could have been exploited in these studies (e.g. Euclidean distances and variants of Tversky metrics), Tanimoto was chosen as it is a recognised industry standard, which has been shown to be amongst the best performing similarity metrics in a recent study.36 The recognition of a substructure that is common to a pair of molecules results in disjoint subsets, where one molecule becomes a member of a single cluster only. The outliers, molecules that do not share common scaffolds with the rest of the set, are classified as singletons and therefore do not impact on the appropriate clustering of the remainder of the set.Building representative molecules and classification of emerging NPS
The representative structures for each of the clusters found using hierarchical clustering were identified using dissimilarity calculations. A dissimilarity matrix for molecules was calculated for each cluster using the JKlustor tool from ChemAxon.37 The mean dissimilarity score was calculated for every molecule against all members of its cluster. The NPS with the lowest mean dissimilarity score in a cluster was selected as a representative molecule (medoid) for that cluster.The set of emerging NPS was then combined with the set of representative NPS and a second dissimilarity matrix calculated. The emerging NPS were assigned to the class to which the medoid with which it shared the lowest dissimilarity score belonged.
NMR spectroscopy
NMR spectra of the selected NPS in deuterated methanol (Sigma-Aldrich, as received) were acquired using JEOL EX-400 NMR spectrometer (1H operating frequency 400 MHz) at 298 K. NMR data were processed using JEOL Delta software and 1H chemical shifts were referenced against residual solvent peak (CD3OD at 3.31 ppm). 5F-PB-22, BB-22 and DOM reference standards materials were purchased from Chiron AS (Trondheim, Norway). In turn, 5-APB reference standard was purchased from LGC group (Teddington, UK). In all cases, reference standard materials were used as supplied without any further purification.Results and discussion
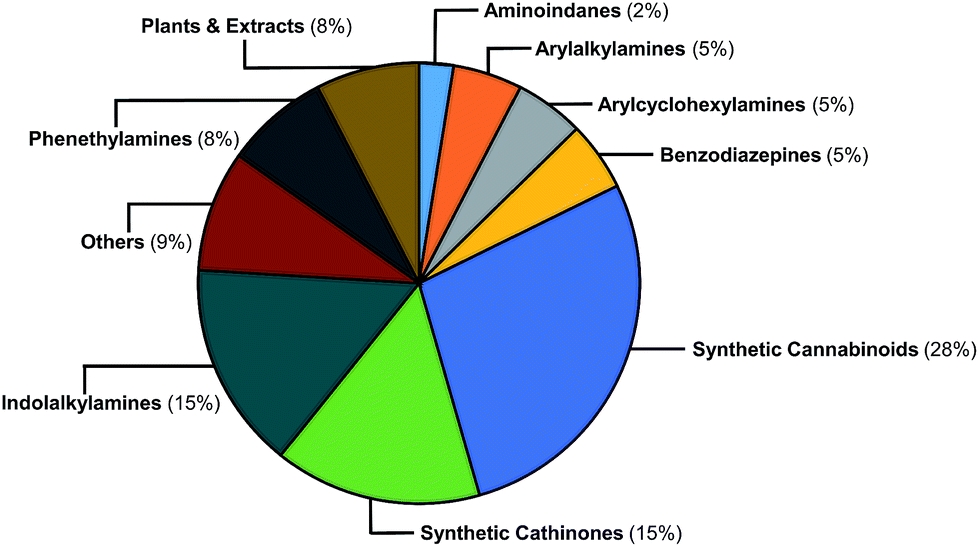
The EMCDDA's European Database on New Drugs (EMCDDA/EDND) classification32 of the 478 NPS compounds used in this study is based on chemical scaffold and/or pharmacological/clinical effect. Similar classifications are used by the United Nations Office on Drug and Crimes (UNODC).1 Fig. 2 shows the 478 NPS in the dataset grouped according to these existing classifications along with their relative abundance. The selection of representative NPS structures could have been based on identifying characteristic molecules from the EMCDDA/EDND classifications. However, more than 40% of NPS belong to the structurally diverse groups of ‘cannabinoids’, ‘opioids’, and ‘others’, that do not permit a straightforward classification by chemical structure. Furthermore, there is an overlap between the existing EMCDDA/EDND categories arising from similarities in chemical structures and scaffolds. For example, the phenylethylamine scaffold also exists as a substructure within the arylalkylamine and synthetic cathinones classes and in the derivatives of the psychedelic 2C-B series of compounds such as the phenylethylamine 25H-NBOMe analogues and ‘others’ class.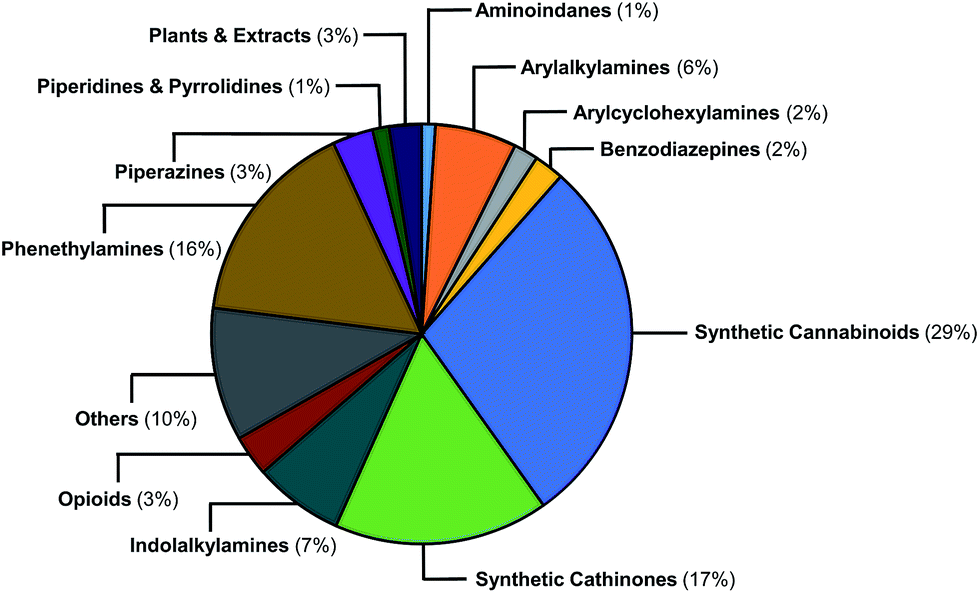 | ||
| Fig. 2 Graphical representation of the relative abundance of the 478 molecules in the initial dataset classified according to EMCDDA/EDND categories. | ||
In order to select a representative number of molecules from the 478 NPS dataset, specific criteria need to be considered. Exploring the diversity of large libraries and selection of representative structures for screening using in vitro assays are commonly based on the molecular properties38 and pharmacophoric features39 of the molecules. Also, structural and spectroscopic studies such as infrared,40 NMR, Raman or GC-MS, used in the classification of NPS would benefit from clustering of compounds based on their chemical fingerprints. The similarity of observed spectroscopic properties for compounds will most likely depend on the presence of functional groups and their relative chemical environments, including substitution patterns. Thus, hierarchical clustering and selection of representative NPS according to the chemical structural properties of the molecules in the dataset was carried out.
Application of hierarchical clustering techniques
Hierarchical clustering provides a compact representation of the NPS chemical space while preserving the relationships between members of the datasets in the form of dendrograms.41 It is often used in combination with the maximum common substructure (MCS) approach to group molecules with a common scaffold into one cluster.42 Hierarchical clustering was carried out through LibMCS,43 implemented in the JKlustor and JChem software.37 The minimal size of the maximum common substructure was set to an empirical threshold of 9 atoms, but crucially this included hydrogen atoms in addition to the heavy atoms normally considered when clustering compounds (referred to herein as the all-atom model). The rationale for including hydrogen atoms in the determination of clusters that may demonstrate similar spectroscopic patterns, was that substitution patterns in a molecule will influence the spectra generated, and the exclusion of the relative positions of H atoms when grouping molecules would have the potential to introduce error into these sets. The results of the clustering using an MCS with a minimal threshold of 9 atoms are shown in Table 1 with the identified MCSs for superclusters shown in Fig. 3.| MCS composition | Heavy atoms | All atoms |
|---|---|---|
| Number of superclusters | 48 | 21 |
| Number of clusters | 112 | 79 |
| Total number of clusters containing on compound (‘singletons’) | 19 | 13 |
| Number of clusters containing 2 compounds | 55 | 40 |
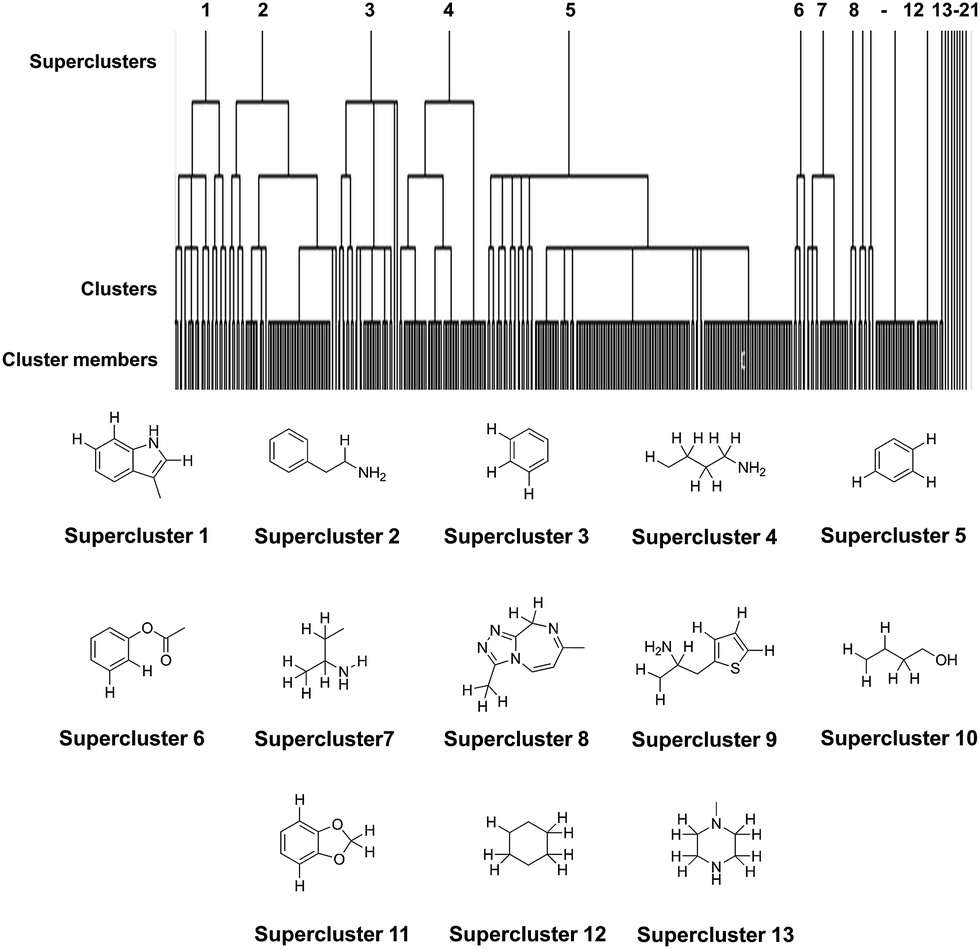 | ||
| Fig. 3 Dendrogram to illustrate the relationship between superclusters, clusters and cluster membership for the all atom analysis conducted on the NPS dataset (478 molecules). The fragments of structure containing the MCS that define the superclusters (excluding singletons) are given. Hydrogen atoms that can contribute to the MCS are shown explicitly. | ||
The all-atom clustering, which aims to group compounds together that are likely to have similar spectroscopic features, resulted in 21 superclusters, 79 clusters and 13 singletons. When compared to the heavy atom clustering alone (48 superclusters, 112 clusters and 19 singletons), it is clear that the all-atom approach provides a reasonable balance between the number of possible representative NPS and the diversity of structures in the clusters. The lower number of representatives and singletons arising from the all-atom approach is also better suited for future experimental studies when taking into consideration practical constraints such as availability of NPS reference standards and their costs. Therefore, the result of the all-atom NPS dataset clustering was used to identify the representative NPS subset.
Inspection of the distribution of NPS shows that superclusters 2, 4 and 5 have the largest number of members (67, 57 and 197, compounds respectively: Fig. 4 and Table S1†). This is not surprising as their MCSs are fragments that are commonly observed in NPS (Fig. 3). The EMCDDA/EDND classification was not well conserved after the all-atom hierarchical clustering based on chemical structure was completed. Molecules belonging to one particular EMCDDA/EDND class are often observed grouping into two or more superclusters. For example, molecules belonging to the aminoindanes, benzodiazepines, piperazine derivatives and opioids are split between two or more different superclusters. The exceptions to this observation are NPS from the ‘piperidines & pyrrolidines’ and ‘arylalkylamines’ class, which group together into one supercluster (supercluster 5).
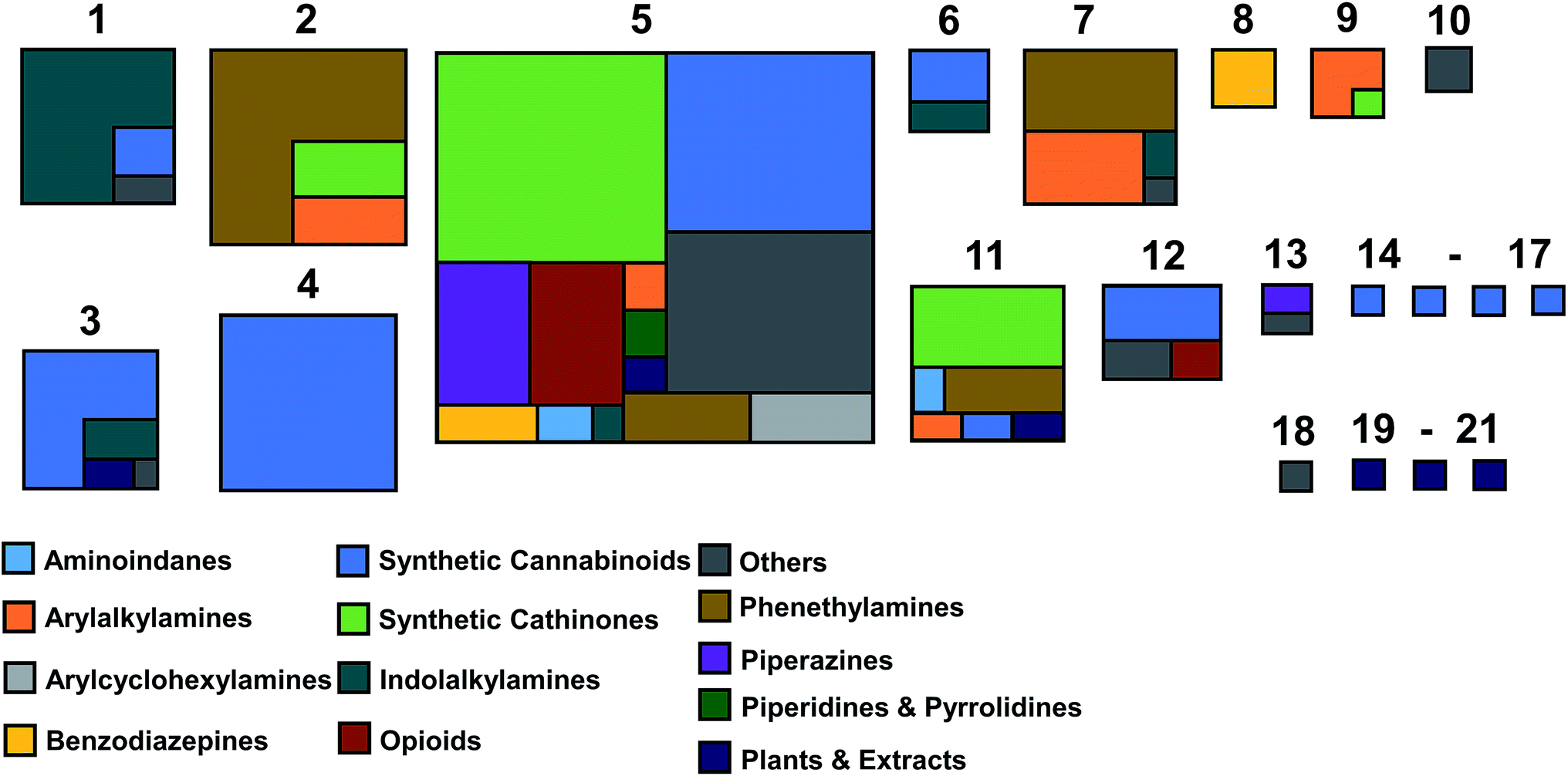 | ||
| Fig. 4 Illustration of the proportion of compounds according to the EMCDDA/EDND NPS categories found in each of the dendrogram's superclusters. The relative size of each box is proportional to the number of compounds contained in the supercluster. | ||
Cannabinoids, the largest and most structurally diverse group in the initial dataset are distributed across 11 superclusters, with the majority found in superclusters 3, 4 and 5. Supercluster 5 contains the greatest spread of molecules with respect to EMCDDA/EDND classification (with all classes represented). This is attributable to the supercluster 5 MCS (a tri-substituted benzene ring) which is commonly observed in known NPS.
Detailed analysis of cluster membership was carried out using calculated pairwise dissimilarity values between cluster members using ChemAxon's JChem software suite.37 The maximum pairwise dissimilarity coefficient observed between individual members of a supercluster varied between 0.19 and 0.84. Unsurprisingly, higher pairwise dissimilarity values were often observed for superclusters that had the largest number of members and/or a greater range of EMCDDA/EDND classes represented within them (e.g. 0.82 was the maximum pairwise dissimilarity coefficient observed in supercluster 5, a supercluster containing 197 NPSs from all EMCDDA/EDND classes). However, it was not anticipated that superclusters containing a small number of members would also exhibit high pairwise dissimilarity values (e.g. a dissimilarity value of 0.84 was observed for supercluster 13, where the three members all contain a variant of the piperazine ring defined the supercluster parent fragment but then diversify to become considerably different with respect to molecular size and extended chemical structure).
It is notable that a maximum pairwise dissimilarity value of <0.5 was observed for 87% of all clusters. This suggests that for the majority of the clusters there is a genuine structural relationship between cluster members. For the superclusters containing the greatest number of NPS (superclusters 2 and 5), the maximum pairwise dissimilarity coefficient observed was higher than 0.5 in three cases only (clusters 2.8, 5.14 and 5.18). However, these three clusters account for 65% and 75% of the members for superclusters 2 and 5, respectively. As this is a possible limitation of our approach, future work could be carried out on further decomposition of clusters with large membership into smaller groups, in order to establish a finer-grained representation of NPS chemical space.
Selection of a representative subset of molecules from the NPS dataset of 478 molecules
The medoid is the member of a set whose dissimilarity to other members in the set is, on average, the lowest. As such, the medoid is normally chosen as a representative for that set. For each of the 79 all-atom clusters, a dissimilarity matrix was generated using JKlustor by calculating pairwise dissimilarity scores for each of the compounds, and the compound with the lowest overall mean dissimilarity score was identified as the medoid. The medoids for each cluster were selected to form a subset of NPS representative of known NPS chemical space.Of the 79 clusters identified, 13 were orphan clusters or ‘singletons’ (clusters that contained only one molecule). At the time of writing, this indicated that, there were no other known NPS with similar chemical structures, it was deemed reasonable to exclude them from the representative dataset. The medoids for the remaining clusters were selected to represent the diversity of NPS chemical space. In the cases where a cluster had only two molecules, both molecules could be considered as equally representative, and in these cases the “medoid” molecule was selected based on criteria including its perceived availability, the current level of interest in the NPS research community for that molecule, and cost. For clusters having two or more molecules with identical mean dissimilarity scores, the same criteria were applied. Examples of selected representative NPS are shown in Fig. 5. All representative NPS, including singletons, are illustrated in S1–S21.†
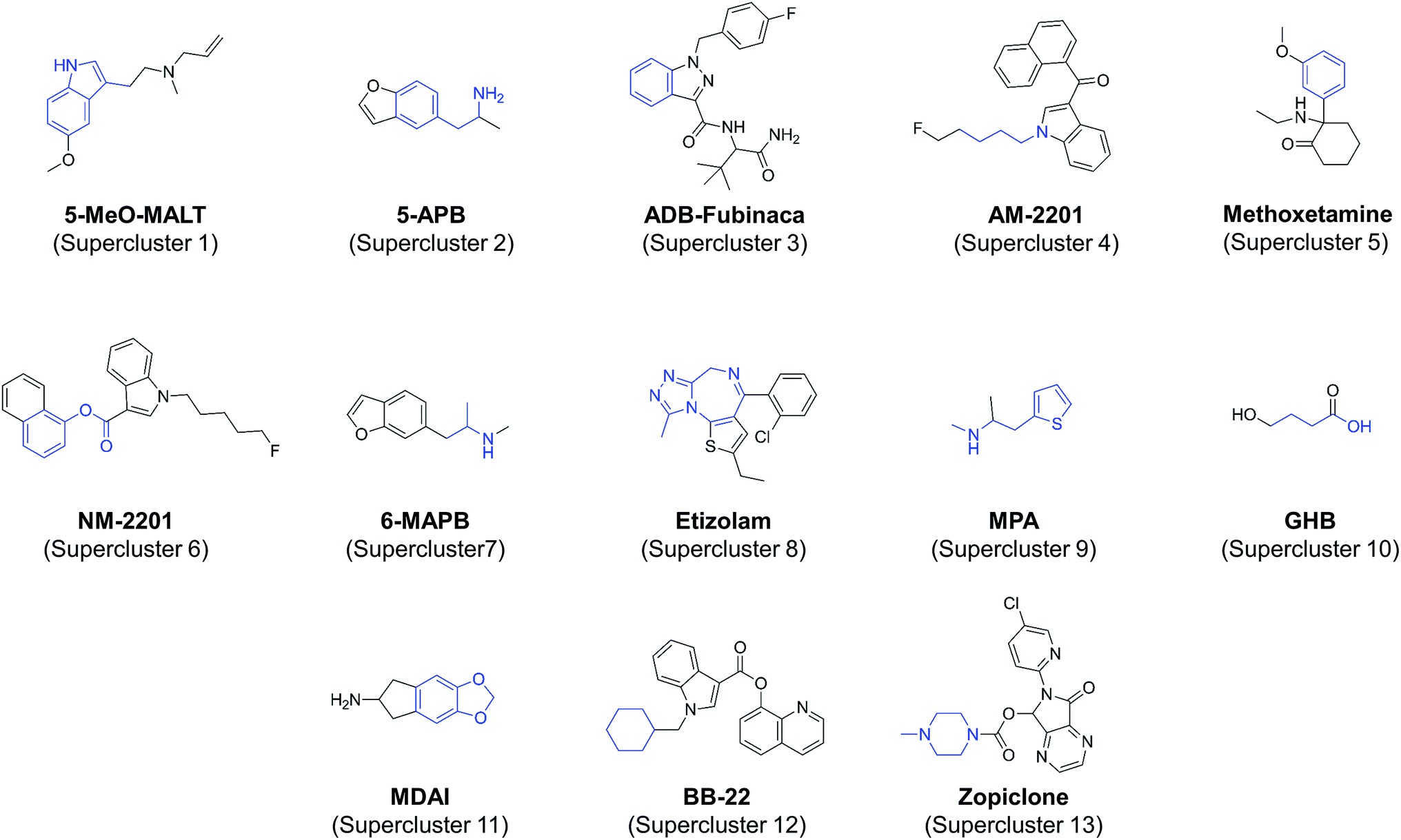 | ||
| Fig. 5 Selected representative NPS for each supercluster containing at least two compounds. The supercluster fragment is shown in blue. Hydrogen atoms present in the supercluster fragment are not shown explicitly. | ||
The majority of the EMCDDA/EDND classes are exemplified in the set of representative structures (Fig. 6) although it should be noted that classes with smaller number of members (opioids, piperidines & pyrrolidines, and piperazine derivatives) do not have representatives in the selected set. This is not unexpected, as most of their members (89%) were assigned to supercluster 5, specifically to clusters 5.14 and 5.18, while the remainder were assigned to clusters 12 and 13. As clusters 5.14 and 5.18 are two of the largest clusters in the set (with 80 and 62 members, respectively), it may be desirable to expand the set of representative molecules by inclusion of the most representative piperidines & pyrollidines, piperazines and opioids especially if an increase in their popularity amongst users of the latter is considered.44 However, at this stage, our systematic approach based on chemical structure alone indicates that explicit inclusion of compounds from these categories is not strictly necessary.
 | ||
| Fig. 6 Graphical representation showing the relative abundance of molecules grouped according to the EMCDDA/EDND classification system that are present in the set that contains the 79 NPS identified by the all atom clustering method. | ||
In contrast, it is interesting that the aminoindanes appear relatively overrepresented in the medoid dataset. 1-Indanamine and 2-indanamine were both selected to be part of the dataset. However, this is because 1 and 2-indanamine are sorted into different clusters, as a consequence of the hierarchical clustering strategy employed. This indicates that a single change of the position of substitution can result in significant dissimilarity between two molecules (dissimilarity coefficient of 0.35), which impacts on the clustering results and the objective selection of representative structures. This lends credence to the all-atom approach to clustering, which specifically considers substitution patterns.
In order to further interrogate the outcome of the all atom clustering methodology, the largest pairwise dissimilarity value between the medoid and the other cluster members was identified. In 28 clusters (42% of the total number of clusters that were not singletons), the largest dissimilarity found for any cluster member with respect to the medoid was less than 10%. This number increased to 37 (56% of clusters that were not singletons) with a threshold for the largest dissimilarity between the medoid and individual cluster members was increased to 20%.
Clusters characterised by greater pairwise dissimilarities between cluster members and the medoid were also examined. 6 clusters (9%) contain a compound with greater than a 70% pairwise dissimilarity value with respect to the medoid. The largest dissimilarity was found in cluster 5.18, where an 87% dissimilarity was calculated between 4-MEC (medoid) and 1-harmine. The level of dissimilarity within clusters can be reduced by increasing the minimal number of atoms in the maximum common substructure from 9. However, this would skew the balance between identifying enough structures to be representative of the NPS chemical space, whilst maintaining a sufficiently low number so that these structures could be obtained and analysed. Consequently, it was concluded that hierarchical clustering and chemical similarity can be used for the identification of representative compounds, one from each cluster, which will represent the diversity of the chemical structural space of known NPS. In addition, the striking similarity observed between the members of each cluster in most cases (vide supra) despite the large complexity and diversity of the initial dataset, indicates that the choice of representatives can be extended to cluster members other than medoids.
After the selection of the cluster representatives, the dissimilarity matrix between the 79 molecules identified was calculated. These molecules are, as expected, very structurally diverse, which is reflected in the range of pairwise dissimilarity scores (0.654 to 0.942). This suggests that the structural diversity of the initial NPS dataset was maintained in the representative subset. These studies suggest that a structure-based hierarchical clustering method using an MCS approach has identified molecules that could rationalize structural and molecular properties of known NPS chemical space. For example, the structural features that are present in the MCS of a supercluster can lead to characteristic signals in spectra that can be replicated in the spectra of the cluster members.
It has been shown that similarity between complex proteins can be established using their NMR fingerprints.45 Such studies can be extended into identification of substances by confirming the presence of peaks and specific multiplicity patterns found in the NMR spectra of MCSs and compared to those found in the NMR spectra of other NPS. The expansion of 1D 1H NMR spectra of selected representative NPS and members of their clusters are shown in Fig. 7 to support this statement. Although the number of peaks in the NMR spectrum of 5F-PB-22 differs from the number of peaks present in BB-22 (a representative of supercluster 12), associated to their distinct number of inequivalent hydrogen atoms, the specific pattern of quinolin-8-yl 1H-indole-3-carboxylate substructure can be observed in both spectra. The comparison of the NMR spectra of DOM (di-2,5-dimethoxy-4-methylamphetamine, a representative of the cluster 2.8) and 5-APB indicates that the alkylamine moiety of these two molecules have similar positions and splitting patterns. Such information can be utilised to develop pattern recognition algorithms to compare the spectra of NPSs and aid their classification.
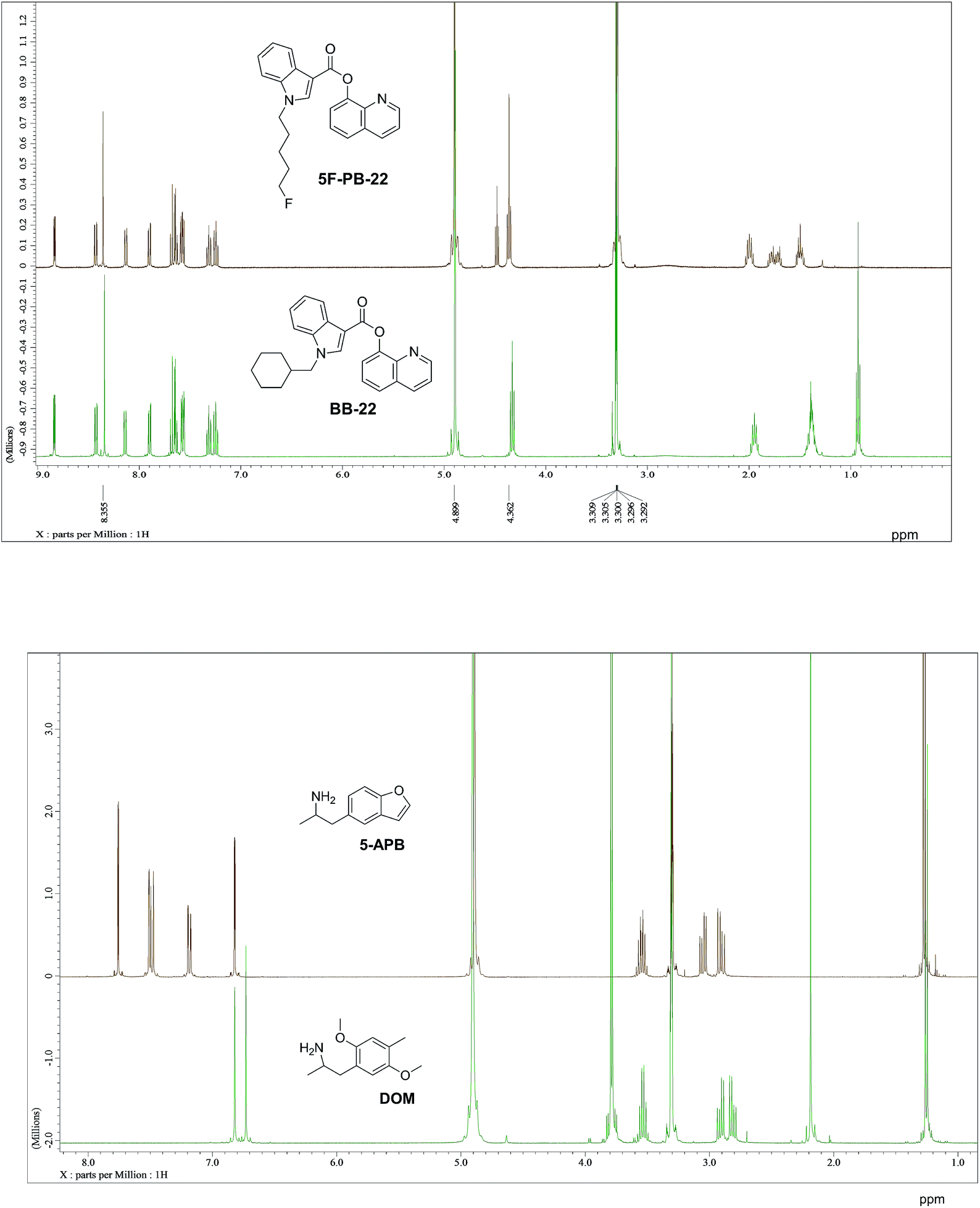 | ||
| Fig. 7 1D 1H NMR spectra of representative NPS (green) and the members of their respective clusters (brown). Full spectral range for 5-APB and DOM available in ESI.† | ||
Assignment of emerging NPS to clusters
The number of NPS recorded by the EMCDDA/EDND increased by 98 during the period from February 2015 until March 2016. In order to test the proposed all atom structural classification, the structural diversity of these new analogues (which we have termed “test compounds”) have been explored. This was achieved by calculating pairwise dissimilarity scores (a) for all molecules in the test set, compared to all other molecules in the test set and (b) between each of the test molecules and the 79 representative molecules of the NPS subset. The analysis indicated that some of the test compounds were similar to one another. For example the pairwise dissimilarity score for two cannabinoid analogues in the test set (5F-EMB-PINACA and AMB-CHMINACA) was 0.05. Conversely, others in the test set were unique in comparison to the rest of the set (e.g. the mean dissimilarity score of DMBA with respect to the remainder of the test set was 0.82).This chemical diversity and rate of emergence of NPS may present difficulty when developing tools for monitoring and identifying new analogues. Therefore, the test compounds were compared to the 79 representative molecules from the initial NPS dataset. The range of pairwise dissimilarity values for the compounds in the test set resulted in a minimum dissimilarity of 65% and a maximum dissimilarity of 92%. This demonstrates the chemical diversity in emergent NPS, which helps to contextualise the challenge in developing tools to quickly and accurately identify these compounds. This spread in diversity is close to that for the representative NPS subset of 79 molecules (pairwise dissimilarity ranges between 65% and 94%) which was specifically selected to be as diverse as possible.
The lowest pairwise dissimilarities between the 79 representative NPS and the test compounds was observed for molecules that belong to the EMCDDA/EDND synthetic cathinone and cannabinoid classes, whilst the maximum pairwise dissimilarity was observed for test compounds that were classified as synthetic cannabinoids and ‘others’ (dissimilarity coefficients between the test and representative molecules ranging from 0.65 to 0.76). This observation is not surprising as the synthetic cannabinoids and ‘others’ NPS classes are highly variable in their origin and chemical scaffolds (vide supra).
The calculated dissimilarity coefficients between the test set compounds and the cluster representatives were used to predict the supercluster/cluster classification the test compounds would belong to. The cluster to which a test compound was assigned was based on the NPS from the representative set with which it had the lowest pairwise dissimilarity coefficient (highest similarity). Any discrepancies between the classifications of the test compounds according to the all atom model, as compared to that classification given by the EMCDDA/EDND were noted. In order to be considered as correctly classified test compounds had to demonstrate pairwise dissimilarity coefficients lower than 0.5 when compared to a medoid in the representative sample from the same EMCDDA/EDND class. Test compounds that showed a dissimilarity value below the threshold for a substance from a different EMCDDA/EDND class was deemed to be misclassified. 67 out of the 98 (66%) test compounds were grouped in agreement with their ECMDDA/EDND classification i.e. the test compounds were most similar to an NPS from the representative dataset that belonged to the same class as that assigned to the emergent molecules by the EMCDDA/EDND.
The anticipated classification was achieved for most of the test compounds, including the diverse cannabinoid structures (selected examples are shown in Table 2) e.g. an emergent cannabinoid, CBL-018 was assigned to cluster 6.2, as its calculated dissimilarity coefficient to cluster medoid, PB22, was 0.1. Similarly, the test compound, AB-CHMFUPPYCA was correctly assigned as a synthetic cannabinoid. Although its dissimilarity coefficient to AB-Fubinaca, the medoid of cluster 3.1, was 0.49, this was the lowest pairwise dissimilarity recorded by the test compound with respect to the 79 molecules in the representative subset, and was considered a successful classification. Other examples of successful classifications of test set compounds include molecules classified as synthetic cathinones, indolalkylamines (Table 2), phenethylamines, arylcyclohexylamines and benzodiazepines (data not shown). The only “misclassified” test set compound was 4-fluoromethylphenidate, which was classified into piperidines & pyrrolidines by the EMCDDA/EDND, compared to a synthetic cathinone (dissimilarity score of 0.48) using the all atom clustering approach presented in this paper. This could be due to the fact that there is no molecule from the “piperidine & pyrrolidine” class in the set of objectively identified representative NPS, which may indicate a limitation of the first iteration of this classification system.
| Cluster representative | Emergent NPS | Dissimilarity score | Cluster allocation | Predicted EMCDDA/EDND classification | Agreement between classification systems |
|---|---|---|---|---|---|
 |
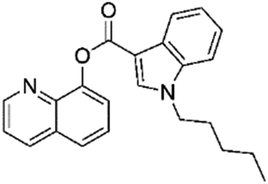 |
0.10 | 6.2 | Cannabinoids | Y |
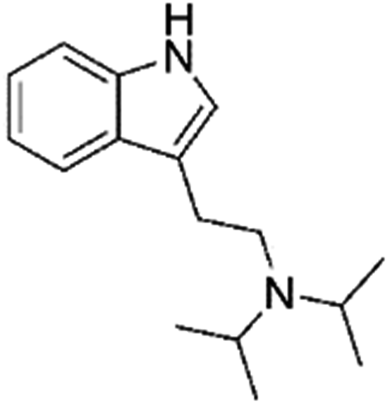 |
 |
0.11 | 3.9 | Indolalkylamine | Y |
 |
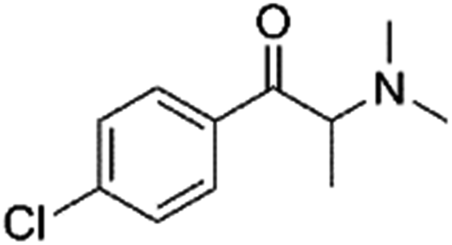 |
0.29 | 5.18 | Synthetic cathinone | Y |
 |
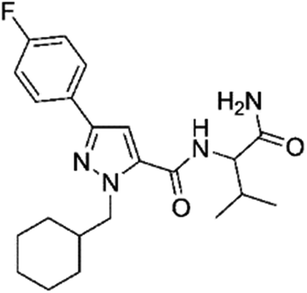 |
0.49 | 3.1 | Cannabinoids | Y |
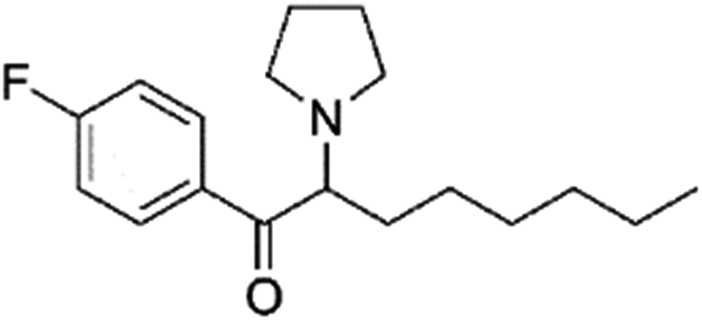 |
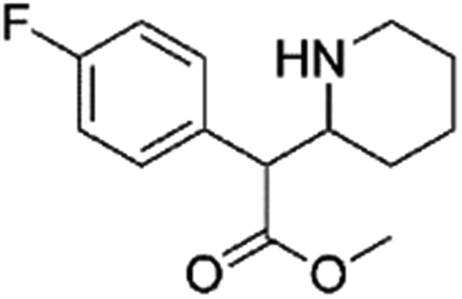 |
0.48 | 2.3 | Synthetic cathinone | N |
The remainder of the test compounds (30 out of 108 molecules) were not definitively assigned to a cluster as a result of this experiment, and were deemed to be unclassified. These molecules had pairwise dissimilarity coefficients greater than 0.5 when compared to the set of 79 representative NPS. 15 (50%) of these unclassified molecules were from the EMCDDA/EDND class “others”, which by its nature is a catch-all class used to pragmatically assign a label to emergent NPS that otherwise defy labelling. As such the expectation that these compounds could be classified correctly using the clustering approach outlined here is ambitious, and it is unsurprising that there is such a high failure rate for these molecules.
It is acknowledged that the clustering approach has some limitations, which arise mainly due to the small size of the subset identified to represent the complex chemical space of different and diverse NPS classes. These limitations can be overcome by an incremental increase of the number of compounds in the set of representative structures. These can be identified using the all atom clustering approach on molecules, which have emerged onto the market since January 2015 and which are currently unclassified by the model.
However, it is also noted that the molecular similarity calculated can be used to correctly classify NPS whose structural features are present in the set of representative molecules. The robustness of the approach used in the selection of representative molecules ensured that the majority of the chemical features of the diverse NPS chemical scaffolds in the initial set are successfully mapped to the representative subset. Based on this, successful classification, it can be postulated that a representative subset can be used to represent structural and molecular properties of the larger NPS chemical scaffold and predict some of the properties of the emerging NPS.
Conclusions
The aim of this work was to identify a ‘representative’ subset of NPS that could be used in future experimental studies to exemplify the entire NPS chemical space known to date. This aims at reducing the number of NPS needed to be studied for purposes of accurate and efficient identification, whilst retaining maximum physicochemical diversity between the members of the subset. The all-atom hierarchical clustering method proved to be a suitable approach to group the whole dataset of NPS into clusters with distinct maximum common substructures. Clustering of the dataset showed that NPS from different EMCDDA/EDND classes were grouped, such that none of the clusters formed exclusively from a single EMCDDA/EDND-defined class of NPS. This is most likely due to their similar molecular properties, activity against similar targets in the central nervous system (CNS) and presence of common structural features. This experiment resulted in the selection of 79 compounds that can be used to represent the NPS dataset as a whole. Although there are clusters that display higher degrees of structural diversity between individual cluster members, it was demonstrated that for 73% of the clusters identified the maximum pairwise dissimilarity score between any two cluster members was below 0.5. This would allow the development of new approaches for classification and identification of emergent NPS e.g. it was demonstrated that common patterns exist in the NMR spectra of selected representative NPS and cluster members. Furthermore, the ab initio classification of 98 test compounds that were not present in the initial set was explored by calculating dissimilarity scores between test compounds and the 79 representative molecules. It was observed that structural dissimilarity between the test molecules and a representative NPS t of less than 0.5 can be used as a criterion for accurately classifying emergent NPS to the anticipated EMCDDA/EDND class, as long as the structural features of that class are contained in the set of representative molecules. In addition, it can be postulated that the representative subset can be used to illustrate structural and molecular properties of the larger NPS set and predict some of the properties of emerging NPSs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the European Commission for funding (Drug Prevention and Information Programme 2014–16, con-tract no. JUST/2013/DPIP/AG/4823, EU-MADNESS project and JUST/ISEC/DRUGS/AG/6428, EPSNPS project). EMCDDA and Dr Michael Evans-Brown (EMCDDA) are acknowledged for provision of the NPS data. Mr John Corkery is greatly acknowledged for his helpful discussions.Notes and references
- UNODC, What are NPS?.
- F. Measham, in Novel Psychoactive Substances, ed. P. I. D. M. Wood, Academic Press, Boston, 2013, pp. 105–127 Search PubMed.
- E. Underwood, Science, 2015, 347, 469–473 CrossRef CAS PubMed.
- Executive Summary: Conclusions and Policy Implications, in World Drug Report 2017, ed. J. Gibbons, UNODC, Vienna, 2017, pp. 13–21 Search PubMed.
- M. Baron, M. Elie and L. Elie, Drug Test. Anal., 2011, 3, 576–581 CrossRef CAS PubMed.
- S. D. Brandt, H. R. Sumnall, F. Measham and J. Cole, Drug Test. Anal., 2010, 2, 377–382 CrossRef CAS PubMed.
- J. Ramsey, P. I. Dargan, M. Smyllie, S. Davies, J. Button, D. W. Holt and D. M. Wood, QJM, 2010, 103, 777–783 CrossRef CAS PubMed.
- C. O'Neill, J. Community Pract., 2014, 87, 45–47 Search PubMed.
- UNODC, The Challenge of New Psychoactive Substances, UNODC, Vienna, 2013 Search PubMed.
- Psychoactive Substances Act 2016, The National Archives, The Stationery Office Limited, Norwich, UK, 2016 Search PubMed.
- M. Rychert and C. Wilkins, Drug Test. Anal., 2016, 8, 768–778 CrossRef CAS PubMed.
- S. Assi, A. Guirguis, S. Halsey, S. Fergus and J. L. Stair, Anal. Methods, 2015, 7, 736–746 RSC.
- International Narcotic Control Board, Report of the International Narcotics Control Board for 2011, New York, 2011 Search PubMed.
- EMCDDA-Europol, European drug market reports. In-depth analysis, Publications Office of the European Union, Luxembourg, 2016 Search PubMed.
- A. Guirguis, J. M. Corkery, J. L. Stair, S. B. Kirton, M. Zloh and F. Schifano, Hum. Psychopharmacol. Clin. Exp., 2017, 32, e2598 CrossRef PubMed.
- The Vaults of Erowid, https://erowid.org/psychoactives/psychoactives.shtml, accessed October 30, 2017.
- M. Coppola and R. Mondola, Toxicol. Lett., 2012, 211, 144–149 CrossRef CAS PubMed.
- N. V. Cozzi, M. K. Sievert, A. T. Shulgin, P. Jacob and A. E. Ruoho, Eur. J. Pharmacol., 1999, 381, 63–69 CrossRef CAS PubMed.
- M. P. Gygi, J. W. Gibb and G. R. Hanson, J. Pharmacol. Exp. Ther., 1996, 276, 1066–1072 CAS.
- M. P. Gygi, A. E. Fleckenstein, J. W. Gibb and G. R. Hanson, J. Pharmacol. Exp. Ther., 1997, 283, 1350–1355 CAS.
- M. Sparago, J. Wlos, J. Yuan, G. Hatzidimitriou, J. Tolliver, T. A. Dal Cason, J. Katz and G. Ricaurte, J. Pharmacol. Exp. Ther., 1996, 279, 1043–1052 CAS.
- S. Tai and W. E. Fantegrossi, Curr. Addict. Rep., 2014, 1, 129–136 CrossRef PubMed.
- F. Schifano, L. Orsolini, G. Duccio Papanti and J. M. Corkery, World Psychiatr., 2015, 14, 15–26 CrossRef PubMed.
- D. Abdulrahim and O. Bowden-Jones, Novel Psychoactive Treatment UK Network NEPTUNE: Guidance on the Clinical Management of Acute and Chronic Harms of Club Drugs and Novel Psychoactive Substances, London, 2015 Search PubMed.
- S. L. Hill and S. H. L. Thomas, Medicine, 2009, 37, 621–626 CrossRef.
- M. A. Johnson and G. M. Maggiora, Concepts and applications of molecular similarity, John Wiley & Sons, New York, 1990 Search PubMed.
- D. Kokel, J. Bryan, C. Laggner, R. White, C. Y. J. Cheung, R. Mateus, D. Healey, S. Kim, A. A. Werdich, S. J. Haggarty, C. A. MacRae, B. Shoichet and R. T. Peterson, Nat. Chem. Biol., 2010, 6, 231–237 CrossRef CAS PubMed.
- R. D. Clark and W. J. Langton, J. Chem. Inf. Comput. Sci., 1998, 38, 1079–1086 CrossRef CAS.
- A. Yosipof and H. Senderowitz, J. Chem. Inf. Model., 2014, 54, 1567–1577 CrossRef CAS PubMed.
- A. R. Leach and V. J. Gillet, in An Introduction To Chemoinformatics, Springer, Netherlands, Dordrecht, 2007, pp. 119–139 Search PubMed.
- Z. Lepp, C. Huang and T. Okada, J. Chem. Inf. Model., 2009, 49, 2429–2443 CrossRef CAS PubMed.
- EMCDDA.
- H. E. Pence and A. Williams, J. Chem. Educ., 2010, 87, 1123–1124 CrossRef CAS.
- Home Office, Annual Report on the Home Office Forensic Early Warning System (FEWS). A system to identify New Psychoactive Substances (NPS) in the UK, Home Office, 2015 Search PubMed.
- E. Duesbury, J. D. Holliday and P. Willett, MATCH Commun. Math. Comput. Chem., 2017, 77, 213–232 Search PubMed.
- D. Bajusz, A. Rácz and K. Héberger, J. Cheminf., 2015, 7, 20 Search PubMed.
- Chemaxon, JChem, ChemAxon, 2015 Search PubMed.
- J. W. Godden, L. Xue, D. B. Kitchen, F. L. Stahura, E. J. Schermerhorn and J. Bajorath, J. Chem. Inf. Comput. Sci., 2002, 42, 885–893 CrossRef CAS PubMed.
- P. J. Therese, D. Manvar, S. Kondepudi, M. B. Battu, D. Sriram, A. Basu, P. Yogeeswari and N. Kaushik-Basu, J. Chem. Inf. Model., 2014, 54, 539–552 CrossRef CAS PubMed.
- K. Varmuza, P. N. Penchev and H. Scsibrany, J. Chem. Inf. Comput. Sci., 1998, 38, 420–427 CrossRef CAS.
- G. M. Downs and J. M. Barnard, Rev. Comput. Chem., 2002, 18, 1–40 CAS.
- M. Stahl and H. Mauser, J. Chem. Inf. Model., 2005, 45, 542–548 CrossRef CAS PubMed.
- Chemaxon, Library MCS, 2008 Search PubMed.
- S. Elliott and J. Evans, Forensic Sci. Int., 2014, 243, 55–60 CrossRef CAS PubMed.
- B. Japelj, G. Ilc, J. Marušič, J. Senčar, D. Kuzman and J. Plavec, Sci. Rep., 2016, 6, 32201 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full list of NPS representative molecules, distribution of these in the dendrogram's superclusters and full details for the application of non-hierarchical clustering techniques. See DOI: 10.1039/c7ra09066h |
| This journal is © The Royal Society of Chemistry 2017 |