
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePerformance and coke species of HZSM-5 in the isomerization of styrene oxide to phenylacetaldehyde
Ming-Lei Gou *,
Junqing Cai,
Wensheng Song,
Zhen Liu,
Yun-Lai Ren and
Qingshan Niu
*,
Junqing Cai,
Wensheng Song,
Zhen Liu,
Yun-Lai Ren and
Qingshan Niu
School of Chemical Engineering and Pharmaceutics, Henan University of Science and Technology, Luoyang, Henan 471023, PR China. E-mail: mingleigou@haust.edu.cn; Fax: +86 37964231914; Tel: +86 37964231914
First published on 14th September 2017
Abstract
The performance and coke species of HZSM-5 in the isomerization of styrene oxide to phenylacetaldehyde was investigated under gas phase, free of solvents. The reaction showed higher catalytic stability and phenylacetaldehyde selectivity at around 300 °C, lower feed rate (e.g., WHSV = 1.2–3 h−1) and higher flow rate of carrier gas (e.g., 120 mL min−1). Based on FT-IR spectra, the dimer formed via aldol condensation of phenylacetaldehyde was one of the precursors of coke species. TG results showed that there were two types of coke species. The soft coke, which can be removed via desorption between 200–400 °C, had less influence on the catalytic stability. The hard coke, which had a certain degree of crystallization (i.e., pregraphite-like carbon presenting in XRD patterns) and must be completely removed via burning with oxygen, causes major catalyst deactivation.
1. Introduction
Phenylacetaldehyde and its derivatives, which are valuable intermediates for the production of fragrances, pharmaceuticals, insecticides, fungicides and herbicides, can be obtained by isomerization of styrene oxide and its halogenated styrene oxides with acidic catalysts.1 A series of solid acid catalysts, such as mixed-metal oxides,2 silica-alumina gels,3,4 natural silicates,5 Nafion-H,6 heteropoly acids,7 zeolites,8–10 and so on, have been applied as active components for this process. However, the deactivation of catalysts is a main hindrance for their industrial application.
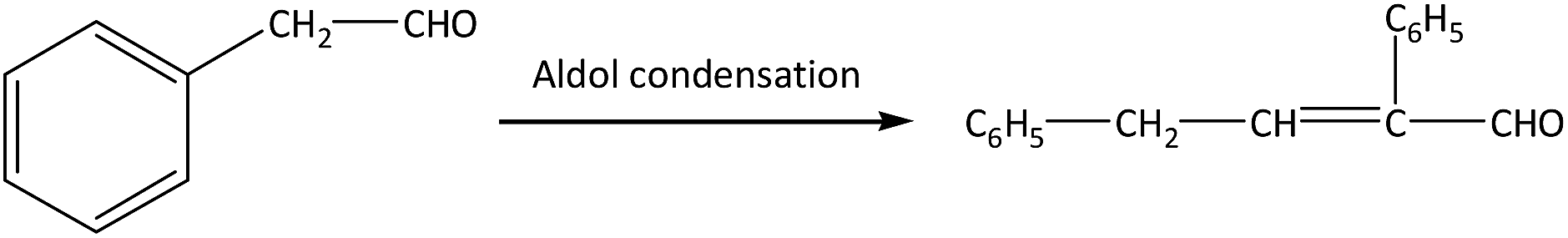 | (1) |
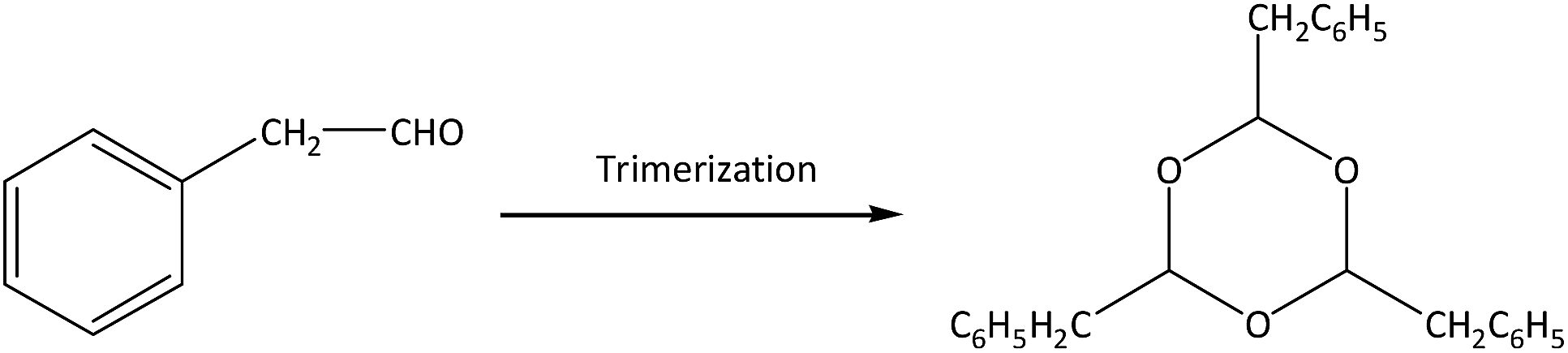 | (2) |
There are two main side reactions in this process: aldol condensation and trimerization of phenylacetaldehyde, as shown in eqn (1) and (2).11 Aldol condensation is not sensitive to the acidity of catalysts and may be a spontaneous process, while trimerization occurs at the external acid sites through an acid-catalyzed process.12 In order to control the side reactions and prevent catalyst deactivation, a large amount of solvents can be used to dilute reactants,7 but the subsequent separation is difficult and the energy consumption extensive. This process can also be operated under gas phase using inert gas (e.g., N2) as carrier gas, which facilitates the transport of reactants and products through the catalyst bed.13 On the premise of styrene oxide conversion, the shorter the contact time is between reactants and catalysts, the lower the probability of side reactions. Particularly, products can be separated in a simple manner without additional purification.
Zeolites are thought to be superior to other catalysts on account of the suppression of side reactions by shape selectivity, particularly the MFI-type zeolites.14 In this study, the catalytic performance of HZSM-5 under different conditions of temperature (200–400 °C), feed rate (weight hourly space velocity, WHSV = 1.2–6.0 h−1) and flow rate of carrier gas (30–120 mL min−1) were firstly examined, then the coke species and deactivation behavior of catalysts under different conditions were analyzed.
2. Experimental
2.1 Materials
Styrene oxide (>98 wt%) was purchased from TCI (Shanghai) Development Co., Ltd. and used without further purification. HZSM-5 (SiO2/Al2O3 = 135) was obtained from Catalyst Plant of Nankai University (Tianjin, People's Republic of China). Some physical and acidic properties of the catalyst are listed in Table 1.| Sample | SiO2/Al2O3 (mol mol−1) | BET surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore size Å | Weak acid (mmol g−1) | Strong acid (mmol g−1) | B/L (mol mol−1) | External acid (a.u.) |
|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: T = 200 °C, P = 1 atm, catalyst loading = 0.5 g, flow rate of N2 = 120 mL min−1, WHSV = 3.0 h−1, 7 h time on stream. | ||||||||
| Fresh HZSM-5 | 134.6 | 290.4 | 0.144 | 4.6 | 0.114 | 0.256 | 13.1 | Trace |
| Spent HZSM-5a | 135.1 | 15.7 | 0.004 | 1.5 | Trace | 0.120 | 8.9 | Trace |
2.2 Catalyst characterization
The bulk Si/Al ratios of catalysts were determined on a Bruker S4 Pioneer X-ray fluorescence (XRF) spectrometer. N2-adsorption was measured at 77 K on a Quantachrome Autosorb-1 instrument. The surface area was calculated according to the BET equation, and the pore volume and size were determined using the HK method.The powder X-ray diffraction (XRD) patterns were collected on a PANalytical X'Pert Pro diffractometer in the 2θ range of 5–50° with Co Kα radiation source (λ = 0.1789 nm).
The acidic properties of catalysts were measured by temperature-programmed desorption of NH3 (NH3–TPD) and FT-IR spectra of adsorbed pyridine (0.57 nm) and 2,4,6-trimethylpyridine (0.74 nm). NH3–TPD was performed on a conventional apparatus equipped with a thermal conductivity detector. The sample (100 mg, 20–30 mesh) was pretreated at 200 °C in nitrogen and then cooled down to ambient temperature. Sufficient NH3 was supplied into the system, followed by flushing with nitrogen at 150 °C for 1 h. The TPD profile was obtained by heating the sample from 150 to 500 °C at a rate of 15 °C min−1. Simultaneously, the desorbed NH3 was trapped in boric acid and then titrated with a standard H2SO4 solution.15
Brønsted (the band at 1545 cm−1) and Lewis acid sites (the band at 1445 cm−1) on the catalysts were characterized by FT-IR spectra of adsorbed pyridine, and FT-IR spectra with 2,4,6-trimethylpyridine adsorption was used to probe the external acid sites (the band at 1638 cm−1). FT-IR spectra were recorded on a Nicolet 380 spectrometer. The sample was pressed into a self-supporting wafer (diameter: 13 mm, weight: 15 mg) and pretreated at 500 °C for 2 h under vacuum. Probe molecule was adsorbed in situ at ambient temperature and then evacuated at 200 °C for 1 h. Finally, an IR spectrum was recorded, and a difference spectrum was obtained by subtracting the spectrum of the pretreated sample from the spectrum after probe adsorption.
FT-IR spectra of the fresh and spent catalysts were recorded on a Nicolet 380 spectrometer instrument at room temperature. The sample and KBr, with weight ratio of approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200, were pressed into a tablet and then measured between 400 and 4000 cm−1 with a resolution of 4 cm−1.
:200, were pressed into a tablet and then measured between 400 and 4000 cm−1 with a resolution of 4 cm−1.
The weight loss of the spent catalysts was measured by thermogravimetric analysis (TG-DTA) on a PerkinElmer Pyris 6 thermogravimetric analyzer at a heating rate of 10 °C from room temperature to 800 °C in air or nitrogen flow.
2.3 Isomerization of styrene oxide
The isomerization was carried out in a continuous flow fixed-bed reactor (stainless steel tube, i.d. = 9 mm) operated at different reaction conditions, including temperature (200–400 °C), feed rate (WHSV = 1.2–6.0 −1) and flow rate of N2 (30–120 mL min−1). The catalyst (0.5 g, 20–30 mesh) was pretreated at 500 °C for 2 h under nitrogen flow. Styrene oxide (>98 wt%, TCI Development Co., Ltd), free of any solvents, was introduced into the reactor using a HPLC pump (model Series II, LabAlliance, USA). Nitrogen was used as the carrier gas controlled by a mass flowmeter. After gas–liquid separation, the reactor effluent was collected in an ice trap and analyzed by GC (Agilent 6820, FID detector) equipped with VF-5ms capillary column (30 m, 0.25 mm, 0.25 μm). Identification of the products was achieved by GC-MS (Agilent 6890/5973, MSD detector).At the end of each experiment, the spent catalyst was unloaded after purging with nitrogen at 200 °C for 2 h to remove any possible residual reactants.
3. Results and discussion
3.1 Influence of temperature
Catalytic testing of HZSM-5 with time on stream (TOS) at different temperatures (200–400 °C) was carried out, as shown in Fig. 1. In blank experiments without the catalyst, the conversion of styrene oxide was between 1–5% under different temperatures (not shown here). The initial conversion of styrene oxide (TOS = 1.0 h) on HZSM-5 was above 99% at all temperatures, suggesting that the isomerization is a catalytic process. However, temperature had an obvious impact on the catalytic stability. To obtain quantitative measurement of catalytic stability, catalyst lifetime was defined as the time at which the initial conversion dropped by 2%, as denoted by the dashed line in Fig. 1. With increasing temperature from 200 °C to 300 °C, the catalyst lifetime increased from 7.2 h to 11.9 h. However, when the temperature rose to 350 °C and 400 °C, the lifetime instead decreased to 9.1 h and 8.4 h, respectively. Thus, the optimum temperature for this reaction over HZSM-5 is around 300 °C based on the catalytic stability.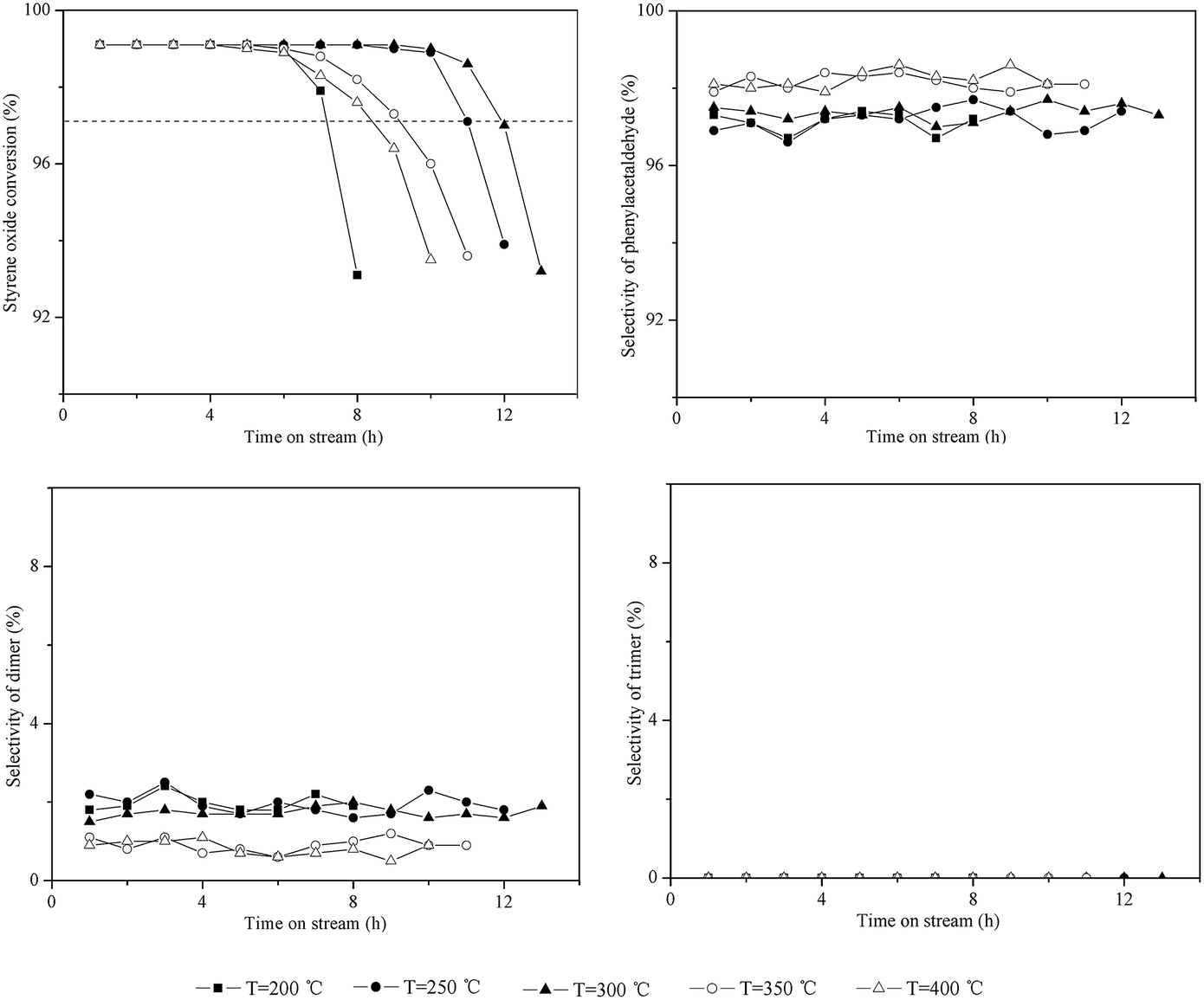 | ||
| Fig. 1 Influence of temperature on the isomerization of styrene oxide to phenylacetaldehyde. Reaction conditions: P = 1 atm, catalyst loading = 0.5 g, flow rate of N2 = 120 mL min−1, WHSV = 3.0 h−1. | ||
The main by-products in this reaction were the dimer (2,4-diphenyl-2-butenal) and trimer (2,4,6-tribenzyl-s-trioxane) of phenylacetaldehyde. The dimer was formed via aldol condensation of phenylacetaldehyde (as shown in eqn (1)), while the trimer was formed by trimerization of phenylacetaldehyde through an acid-catalyzed process (as shown in eqn (2)). Some other by-products, such as phenylethanol, phenylethanediol, styrene, and so on, were also detected by GC-MS, with total selectivity values less than 1%.
As shown in Fig. 1, the dimer selectivity remained lower at higher temperatures (such as 350 °C and 400 °C). However, the catalyst lifetime instead decreased as mentioned above. This may be due to the dimer being more readily converted into some high-boiling materials at higher temperature, which deposited on the surface of catalysts, leading to rapid deactivation of catalysts. This can be verified by TG analysis, as discussed in the following section.
For the trimer formation, three adsorbed precursor species should be in adjacent acid sites on the external surface of zeolites. HZSM-5 contained only a trace amount of external acid sites, thus leading to no formation of the trimer at 200–400 °C.
3.2 Influence of feed rate
Fig. 2 illustrates the catalytic performance of HZSM-5 with WHSV = 1.2–6 h−1. The initial conversion of styrene oxide (TOS = 1.0 h) was above 99% for all WHSV values. However, the catalytic stability and product distribution depended strongly on the WHSV. The catalyst lifetime was 24.4 h at WHSV = 1.2 h−1, while when WHSV = 6.0 h−1, the lifetime greatly decreased to 3.7 h. The dimer selectivity remained at 1–3% at WHSV = 1.2–3.0 h−1 from beginning to end of the reaction, and the corresponding phenylacetaldehyde selectivity remained greater than 96%. However, the dimer selectivity gradually increased to 6% with time on stream at WHSV = 6.0 h−1, and both the phenylacetaldehyde selectivity and catalyst lifetime obviously decreased. It can be assumed that the probability of side reactions (i.e., the aldol condensation) increases with increasing phenylacetaldehyde content on the catalyst surface at higher WHSV, which accelerates coke formation and deactivation of catalysts. This can be confirmed by FT-IR spectra and TG analysis of the spent catalysts as discussed in the following section.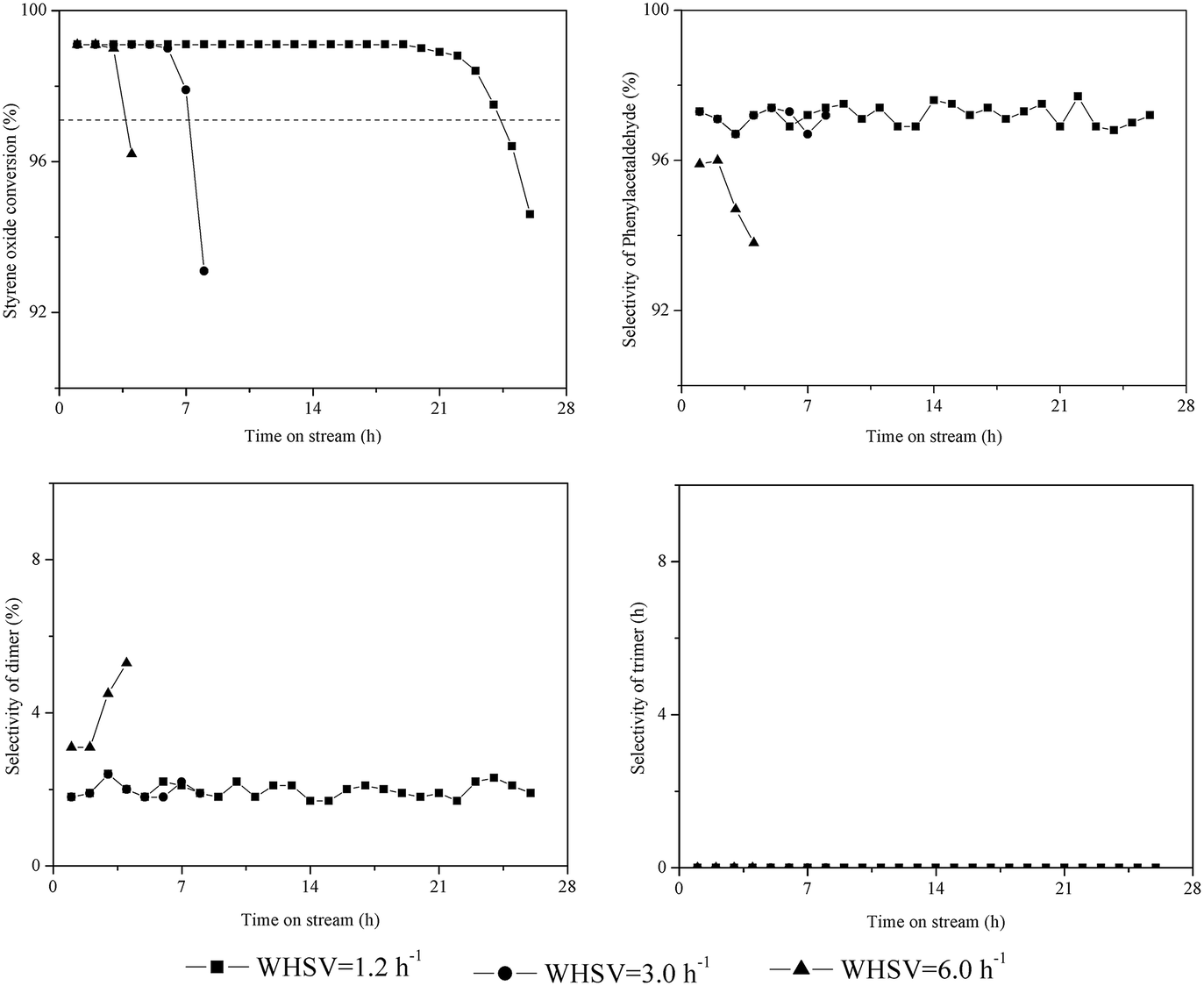 | ||
| Fig. 2 Influence of feed rate on the isomerization of styrene oxide to phenylacetaldehyde. Reaction conditions: T = 200 °C, P = 1 atm, catalyst loading = 0.5 g, flow rate of N2 = 120 mL min−1. | ||
The trimerization of phenylacetaldehyde could not happen on HZSM-5 at WHSV = 1.2–6.0 h−1, since the catalyst contained only a trace amount of external acid sites.
3.3 Influence of flow rate of carrier gas
As reported in the literature,3,13 a shorter contact time (3–5 s) between reactants and catalyst bed can efficiently decrease the probability of side reactions and prevent catalyst deactivation. Increasing the flow rate of carrier gas can facilitate the transport of reactants through the catalyst bed. Thus, the influence of flow rate of N2 (30–120 mL min−1) was investigated over HZSM-5. The results are shown in Fig. 3. The initial conversion of styrene oxide (TOS = 1 h) was greater than 99% at the flow rates of 30–120 mL min−1. However, the flow rate of N2 had an obvious impact on both the catalytic stability and product distribution. The dimer selectivity gradually increased from 3% to 7% as the flow rate of N2 decreased from 120 mL min−1 to 30 mL min−1; meanwhile, the catalyst lifetime decreased from 7.2 h to 5.3 h. It is evident that the probability of side reactions (i.e., the aldol condensation) increases as the flow rate of N2 decreases, thus resulting in more dimer formation and rapid deactivation of catalysts.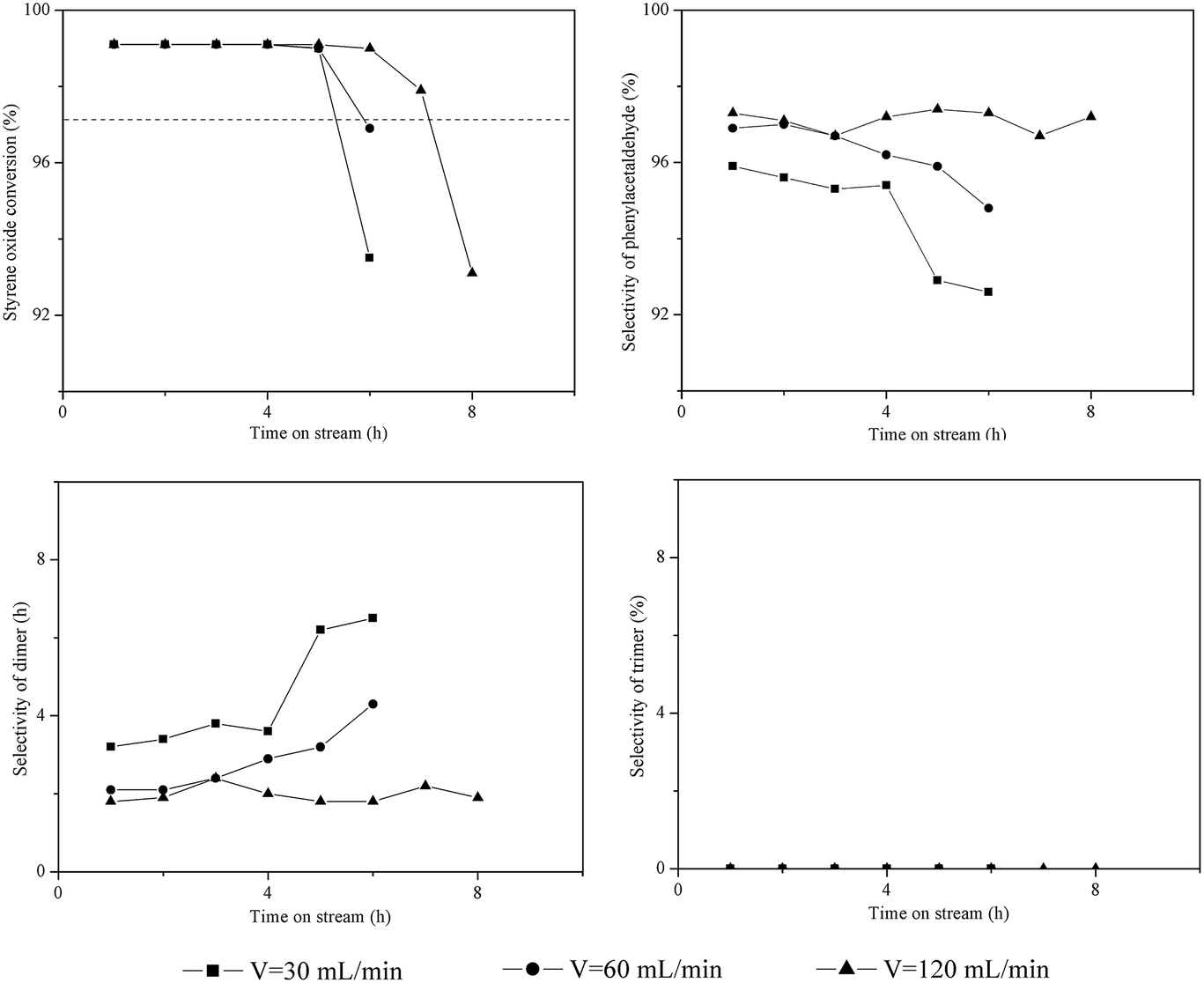 | ||
| Fig. 3 Influence of flow rate of carrier gas on the isomerization of styrene oxide to phenylacetaldehyde. Reaction conditions: T = 200 °C, P = 1 atm, catalyst loading = 0.5 g, WHSV = 3.0 h−1. | ||
The trimer was not generated on HZSM-5 at the N2 flow rate of 30–120 mL min−1, since it only had trace amount of external acid sites.
3.4 Coke species in the isomerization of styrene oxide
Table 1 shows the physical and acidic properties of the fresh and spent HZSM-5. When the reaction was continuously carried out for 7 h, the BET surface area, pore volume and pore size were greatly reduced to 15.7 m2 g−1, 0.004 cm3 g−1 and 1.5 Å. Meanwhile, the catalyst showed deactivation, as indicated by the decreasing styrene oxide conversion in Fig. 1. Styrene oxide is a highly reactive substance and can be completely catalyzed by the strong acid sites of HZSM-5 (0.048–0.606 mmol g−1).12 The spent HZSM-5 also contained some strong acid sites (0.120 mmol g−1), but gradually lost its activity. So it can be concluded that the coke deposited on the catalyst surface blocks the diffusion of reactants into the micropores and results in the deactivation of catalysts.The structure of HZSM-5 before and after reaction was examined by XRD, as shown in Fig. 4. The characteristic peaks at 2θ = 9–11° and 26–29° all matched well with typical MFI-type structure (JCPDS-PDF: 00-049-0657). However, a new peak at 2θ = 31.1° (labeled with arrow in Fig. 4) over the spent HZSM-5 was similar to the characteristic peak of graphite at 2θ = 30.7° (JCPDS-PDF: 00-001-0640). This kind of coke can be called pregraphite-like carbon.16 Therefore, the coke species on HZSM-5 from the isomerization of styrene oxide to phenylacetaldehyde has a certain degree of crystallization.
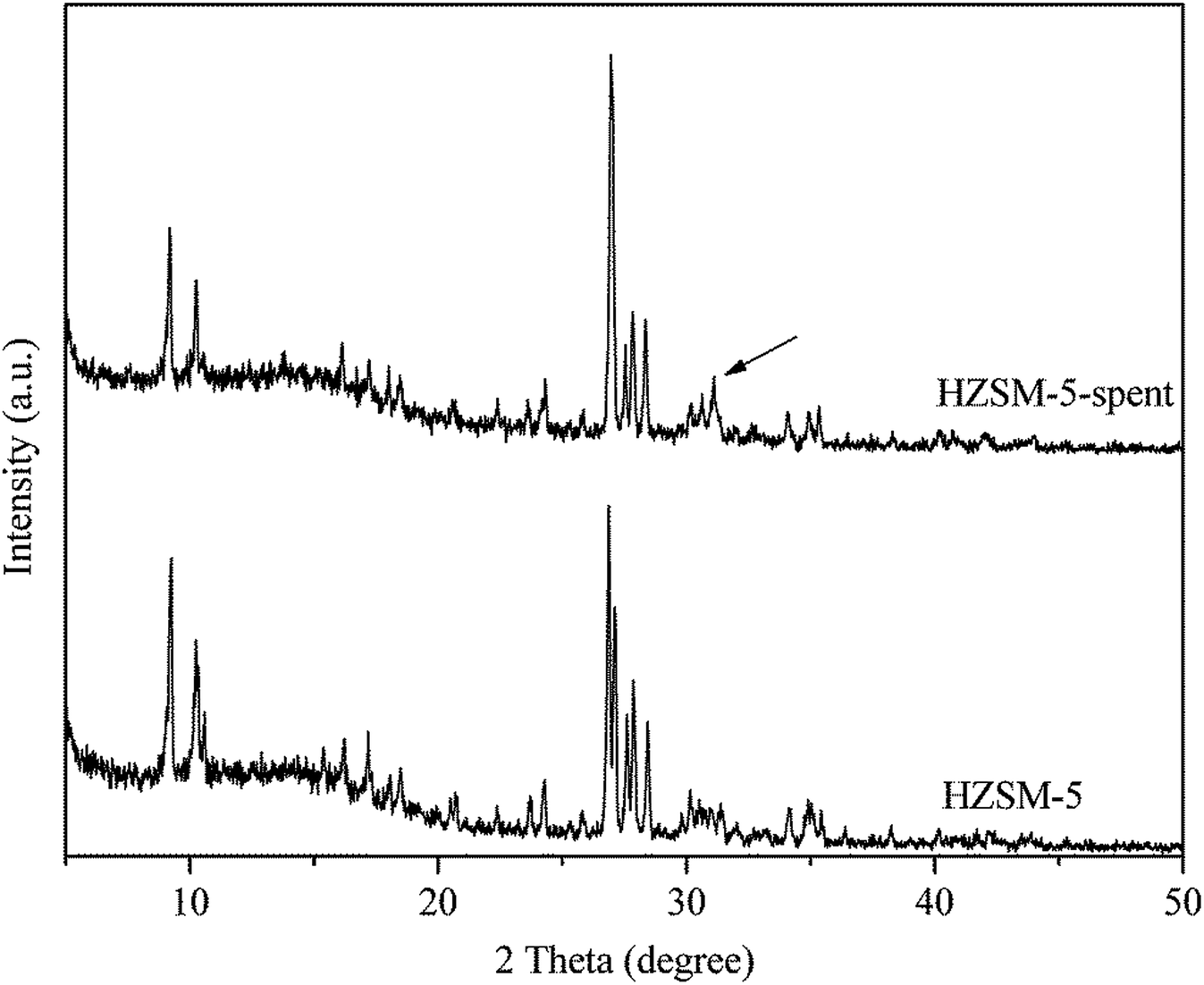 | ||
| Fig. 4 XRD patterns of the fresh and spent HZSM-5. Reaction conditions: T = 200 °C, P = 1 atm, catalyst loading = 0.5 g, flow rate of N2 = 120 mL min−1, WHSV = 3.0 h−1, 7 h time on stream. | ||
To obtain information on the nature of the coke species, FT-IR spectra of the fresh and spent HZSM-5 were investigated. There were two important regions against the characteristic bands of coke: (i) 2800–3100 cm−1 (as shown in Fig. 5(A)), and (ii) 1300–1700 cm−1 (as shown in Fig. 5(B)). In the range of 2800–3100 cm−1, the bands at 2870 cm−1 and 2925 cm−1 were assigned to the symmetric vibrations of –CH2 or –CH3 and the asymmetric vibration of –CH2.17 Taking into account the absence of the band at 2955 cm−1 (the asymmetric vibration of CH3), the band at 2870 cm−1 can be identified as the symmetric vibrations of –CH2. The bands beyond 3000 cm−1 usually belong to C–H stretching vibrations in unsaturated hydrocarbons. For instance, the bands at 3029 cm−1 and 3063 cm−1 were associated with the single-ring or alkyl aromatics,18 and the band at 3086 cm−1 was attributed to olefinic –CH groups.19 It is known that among the main products of this process, only the dimer (2,4-diphenyl-2-butenal) contained olefinic –CH groups. Thus, the aldol condensation of phenylacetaldehyde is one of the factors leading to coke formation in the isomerization of styrene oxide.
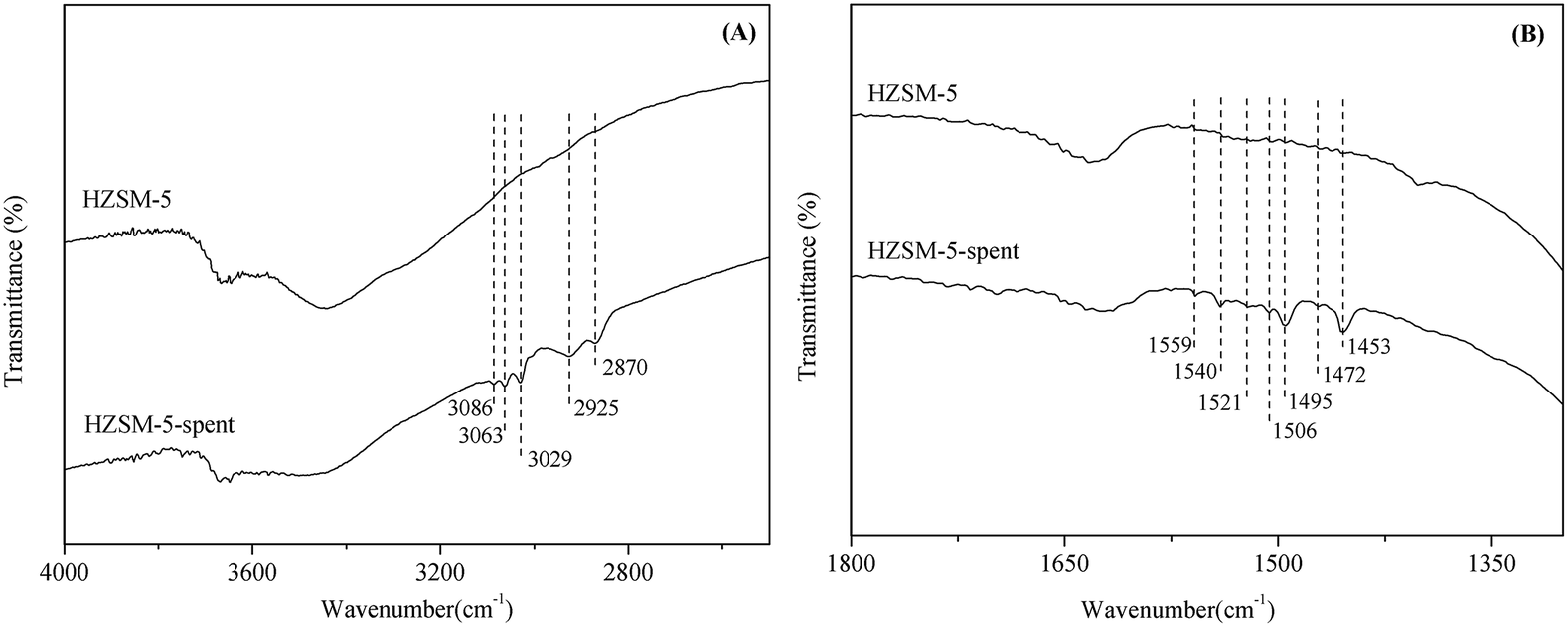 | ||
| Fig. 5 FT-IR spectra of the fresh and spent HZSM-5, (A) 2800–3100 cm−1, (B) 1300–1700 cm−1. Reaction conditions: T = 200 °C, P = 1 atm, catalyst loading = 0.5 g, flow rate of N2 = 120 mL min−1, WHSV = 3.0 h−1, 7 h time on stream. | ||
In the range of 1300–1700 cm−1, the distinct bands at 1453 cm−1 and 1495 cm−1 are due to benzene skeleton vibration,20 and the bands at 1506 cm−1 and 1540 cm−1 can be assigned to ring stretching and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations in alkyl or poly aromatics.21,22 It can be deduced that the aromatic-type of carbonaceous compounds were formed on the surface of zeolites. There were also three small bands at 1472 cm−1, 1521 cm−1 and 1559 cm−1, which were assigned to deformation vibrations of –CH or –CH2 groups and CC stretching vibrations of alkenes,23,24 thus again confirming that the dimer formed via aldol condensation of phenylacetaldehyde was one of the precursors for coke formation. However, the absence of the characteristic band of carbonyl group at around 1750 cm−1 implied that the dimer would then convert to polyaromatics and even the pregraphite-like carbon on the surface of HZSM-5.
C stretching vibrations in alkyl or poly aromatics.21,22 It can be deduced that the aromatic-type of carbonaceous compounds were formed on the surface of zeolites. There were also three small bands at 1472 cm−1, 1521 cm−1 and 1559 cm−1, which were assigned to deformation vibrations of –CH or –CH2 groups and CC stretching vibrations of alkenes,23,24 thus again confirming that the dimer formed via aldol condensation of phenylacetaldehyde was one of the precursors for coke formation. However, the absence of the characteristic band of carbonyl group at around 1750 cm−1 implied that the dimer would then convert to polyaromatics and even the pregraphite-like carbon on the surface of HZSM-5.
Weight loss of the spent catalysts at different reaction conditions was measured by TG, as listed in Table 2. The weight loss can be divided into three stages: stage I (<200 °C) was ascribed to the volatilization of adsorbed water and some volatile species, and stage II (200–400 °C) and stage III (400–700 °C) were ascribed to the desorption or combustion of coke species. The weight loss in stage II (so-called soft coke) varied slightly with different reaction conditions and was between 4.3–5.8%, while the weight loss in stage III (so-called hard coke) presented an obvious change as the temperature, feed rate and flow rate of N2 changed. From the above results, temperature, feed rate and flow rate of N2 all had obvious impacts on the catalytic stability and product distribution. Therefore, it is assumed that the soft coke has less influence on the catalytic stability, while the hard coke, which is the more polymerized coke species, would cause major catalyst deactivation.
| Spent catalyst | Coke (wt%) | ||
|---|---|---|---|
| Soft coke (200–400 °C) | Hard coke (400–700 °C) | Total | |
| a HZSM-5-T-200/250/300/350/400-7h means the reaction is carried out at 200, 250, 300, 350 or 400 °C with 7 h time on stream; other conditions are: P = 1 atm, catalyst loading = 0.5 g, flow rate of N2 = 120 mL min−1, WHSV = 3.0 h−1.b HZSM-5-W-1.2/6.0-7h means the reaction is carried out at WHSV = 1.2, 6.0 h−1 with 7 h time on stream; other conditions are: T = 200 °C, P = 1 atm, catalyst loading = 0.5 g, flow rate of N2 = 120 mL min−1.c HZSM-5-F-30/60-6h means the reaction is carried out at flow rate of N2 = 30, 60 mL min−1 with 7 h time on stream; other conditions are: T = 200 °C, P = 1 atm, catalyst loading = 0.5 g, WHSV = 3.0 h−1.d HZSM-5-T-200-7h-nitrogen means the TG analysis is operated in nitrogen atmosphere, reaction conditions are: T = 200 °C, P = 1 atm, catalyst loading = 0.5 g, flow rate of N2 = 120 mL min−1, WHSV = 3.0 h−1, 7 h time on stream. | |||
| HZSM-5-T-200-7ha | 5.6% | 5.9% | 11.5% |
| HZSM-5-T-250-7h | 5.1% | 4.1% | 9.2% |
| HZSM-5-T-300-7h | 4.8% | 3.6% | 8.4% |
| HZSM-5-T-350-7h | 4.9% | 4.5% | 9.4% |
| HZSM-5-T-400-7h | 5.0% | 4.9% | 9.9% |
| HZSM-5-W-1.2-7hb | 4.3% | 3.7% | 8.0% |
| HZSM-5-W-6.0-7h | 5.8% | 7.8% | 13.6% |
| HZSM-5-F-30-7hc | 5.7% | 6.6% | 12.3% |
| HZSM-5-F-60-7h | 5.7% | 6.3% | 12.0% |
| HZSM-5-T-200-7h-nitrogend | 5.8% | 2.7% | 8.5% |
As shown in Table 2, the amount of hard coke first decreased and then increased as the temperature increased from 200 °C to 400 °C, which was converse to the change in catalyst lifetime as shown in Fig. 1. Based on the FT-IR analysis, the dimer was one of the precursors for coke formation. The dimer selectivity remained lower at higher temperature (i.e., 350 °C and 400 °C); simultaneously, the amount of hard coke increased, and the catalyst lifetime decreased. So it can be verified from the above that the dimer more readily converts to some high-boiling materials at higher temperature, which deposit on the surface of the catalyst, resulting in a larger amount of hard coke and rapid deactivation of catalysts.
From Fig. 2 and 3, the dimer selectivity values increased and the catalyst lifetime decreased at higher feed rate (e.g., WHSV = 6.0 h−1) and lower N2 flow rate (e.g., 30 mL min−1). Based on the TG analysis in Table 2, the amount of hard coke increased as feed rate increased and N2 flow rate decreased. Therefore, it can be confirmed that the probability of side reactions (i.e., the aldol condensation) increases with longer contact time at higher feed rate and lower N2 flow rate, which accelerates the coke formation and deactivation of catalysts.
It is well known that most of the acid sites on HZSM-5 exist inside the micropores; thus, the catalytic stability is mainly determined by the reactions occurring at the internal acid sites. From FT-IR spectra and TG analysis, the dimer formed via aldol condensation of phenylacetaldehyde is one of the precursors of coke species. The dimer can then convert to some high-boiling materials (so-called hard coke), which would cause major catalyst deactivation, while the trimer is not responsible for the deactivation of HZSM-5.
As shown in Table 2, weight loss differences of the spent HZSM-5 in air and nitrogen atmosphere were also observed. The weight loss at stage II was similar whether in air (5.6%) or nitrogen (5.8%), showing that this soft coke can be completely removed via desorption from the catalysts between 200–400 °C. However, the weight loss at stage III in nitrogen atmosphere (2.7%) was lower than that in air (5.9%), indicating that this hard coke must be completely removed with oxygen. The weight loss above 700 °C in nitrogen was approximately 1.6%, which was due to part of the hard coke slowly decomposing to some volatile species. On the basis of XRD, the coke formation on HZSM-5 has a certain degree of crystallization, i.e., pregraphite-like carbon, which also has to be removed via burning with oxygen. By comparing the weight loss in air or nitrogen atmosphere, approximately 1.6% of the pregraphite-like carbon was formed on HZSM-5 in the isomerization of styrene oxide to phenylacetaldehyde at 200 °C, WHSV = 3.0 h−1 and N2 flow rate = 120 mL min−1.
4. Conclusions
Temperature, feed rate and flow rate of carrier gas all have an obvious impact on the catalytic stability and product distribution in the isomerization of styrene oxide to phenylacetaldehyde. The catalytic stability and phenylacetaldehyde selectivity are relatively higher at around 300 °C, at a lower feed rate (e.g., WHSV = 1.2–3 h−1) and higher flow rate of carrier gas (e.g., 120 mL min−1) due to lower probabilities of side reactions. By FT-IR spectra, the dimer formed via aldol condensation of phenylacetaldehyde is one of the precursors of coke species. TG results show that there are two types of coke species on the spent catalysts: soft coke and hard coke. The soft coke, which can be completely removed via desorption between 200–400 °C, has less influence on the catalytic stability, while the hard coke, which has a certain degree of crystallization (i.e., pregraphite-like carbon presenting in XRD patterns) and must be completely removed with oxygen, causes major catalyst deactivation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the Doctoral Scientific Research Foundation (No. 4008/13480049) and Youth Foundation (No. 2015QN009) of Henan University of Science and Technology.References
- W. F. Hölderich and U. Barsnick, in Fine chemicals through heterogeneous catalysis, ed. R. A. Sheldon and H. van Bekkum, Wiley-VCH, Weinheim, 1st edn, 2001, pp. 217–231 Search PubMed.
- H. Kochkar, J. M. Clacens and F. Figueras, Catal. Lett., 2002, 78, 91–94 CrossRef CAS.
- B. G. Pope, Production of arylacetaldehydes, US Pat., US4650908, 1987.
- F. Zaccheria, R. Psaro, N. Ravasio, L. Sordelli and F. Santoro, Catal. Lett., 2011, 141, 587–591 CrossRef CAS.
- E. Ruiz-Hitzky and B. Casal, J. Catal., 1985, 92, 291–295 CrossRef CAS.
- G. K. S. Prakash, T. Mathew, S. Krishnaraj, E. R. Marinez and G. A. Olah, Appl. Catal., A, 1999, 181, 283–288 CrossRef.
- V. V. Costa, K. A. da Silva Rocha, I. V. Kozhevnikov and E. V. Gusevskaya, Appl. Catal., A, 2010, 383, 217–220 CrossRef CAS.
- D. Brunel, M. Chamoumi, B. Chiche, A. Finiels, C. Gauthier, P. Geneste, P. Graffin, F. Marichez and P. Moreau, Stud. Surf. Sci. Catal., 1989, 52, 139–149 CrossRef.
- M. Chamoumi, D. Brunel, P. Geneste, P. Moreau and J. Solofo, Stud. Surf. Sci. Catal., 1991, 59, 573–579 CrossRef CAS.
- K. Smith, G. A. El-Hiti and M. Al-Shamali, Catal. Lett., 2006, 109, 77–82 CrossRef CAS.
- J. L. E. Brickson and G. N. Grammer, J. Am. Chem. Soc., 1958, 80, 5466–5469 CrossRef.
- M. L. Gou, R. Wang, Q. Qiao and X. Yang, Catal. Commun., 2014, 56, 143–147 CrossRef CAS.
- H. Smuda, W. Hoelderich, N. Goetz and H. G. Recker, Preparation of phenylacetaldehydes, US Pat., US4929765, 1990.
- W. Hoelderich, N. Goetz, L. Hupfer, R. Kropp, H. Theobald and B. Wolf, Phenylacetaldehydes and the preparation of phenylacetaldehydes, US Pat., US5225602, 1993.
- N. R. Meshram, S. G. Hegde, S. B. Kulkarni and P. Ratnasamy, Appl. Catal., A, 1983, 8, 359–367 CrossRef CAS.
- B. K. Vu, M. B. Song, I. Y. Ahn, Y. W. Suh, D. J. Suh, J. S. Kim and E. W. Shin, J. Ind. Eng. Chem., 2011, 17, 71–76 CrossRef CAS.
- M. Rozwadowski, M. Lezanska, J. Wloch, K. Erdmann, R. Golembiewski and J. Kornatowski, Chem. Mater., 2001, 13, 1609–1616 CrossRef CAS.
- L. Becker and H. Förster, Appl. Catal., A, 1997, 153, 31–41 CrossRef CAS.
- P. Castaño, G. Elordi, M. Olazar, A. T. Aguayo, B. Pawelec and J. Bilbao, Appl. Catal., B, 2011, 104, 91–100 CrossRef.
- P. Yin, Q. Xu, R. Qu and G. Zhao, J. Hazard. Mater., 2009, 169, 228–232 CrossRef CAS PubMed.
- T. C. Wen, J. B. Chen and A. Gopalan, Mater. Lett., 2002, 57, 280–290 CrossRef CAS.
- Y. Xiao, Z. Yao and D. Jin, Langmuir, 1994, 10, 1848–1850 CrossRef CAS.
- A. de-Lucas, P. Canizares, A. Duran and A. Carrero, Appl. Catal., A, 1997, 156, 299–317 CrossRef CAS.
- J. Yao, H. Wang, J. Liu, K. Y. Chan, L. Zhang and N. Xu, Carbon, 2005, 43, 1709–1715 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |