
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An Fe3O4@P4VP@FeCl3 core–shell heterogeneous catalyst for aerobic oxidation of alcohols and benzylic oxidation reaction†
Ruilian Lia,
Jian Zhaob,
Fengxia Yangb,
Yingchao Zhangb,
Daniele Ramellac,
Yu Peng *a and
Yi Luan*b
*a and
Yi Luan*b
aHunan Agricultural University, Hunan 410128, P. R. China. E-mail: pengy7505@hunau.net; Tel: +86-731-84617022
bSchool of Materials Science and Engineering, University of Science and Technology Beijing, 30 Xueyuan Road, Haidian District, Beijing 100083, P. R. China. E-mail: yiluan@ustb.edu.cn
cTemple University-Beury Hall, 1901, N. 13th Street, Philadelphia, PA 19122, USA
First published on 3rd November 2017
Abstract
A novel magnetic Fe3O4@P4VP(poly(4-vinylpyridine))@FeCl3 core–shell structure was successfully synthesized. Its Fe3O4@P4VP core was initially prepared via polymerization of 4-vinyl pyridine on the surface of Fe3O4 microspheres. The successful introduction of the FeCl3 moiety as a catalytic active site was achieved through coordination interaction between P4VP and FeCl3. The obtained Fe3O4@P4VP@FeCl3 catalyst was applied in the selective oxidation of alcohols using molecular oxygen as the oxidant. It was shown that a variety of alcohol substrates is tolerated under optimized reaction conditions. Additionally, benzylic oxidation of hydrocarbon compounds was also evaluated using Fe3O4@P4VP@FeCl3 as the catalyst and tBuOOH as the oxidant, achieving high yields and very good selectivities. The heterogeneity of the Fe3O4@P4VP@FeCl3 core–shell catalyst was tested and the initial activity was maintained after five reuses.
1. Introduction
The catalytic selective oxidation of alcohols to the corresponding carbonyl compounds is one of the most industrially significant reactions in fine and industrial organic chemistry.1 The use of transition metal-based homogeneous2 and heterogeneous3 catalysts is a good strategy as the use of stoichiometric oxidants such as chromate or permanganate is replaced by atom-economical clean oxidants. However, heterogeneous processes are preferable for oxidation, as they display several advantages during the purification and recovery steps of products and catalysts.4 Oxidizing alcohols heterogeneously using molecular oxygen is an ideal reaction process, since it produces H2O as the sole by-product.5 Recently, aerobic oxidations received much attention because of their use of inexpensive oxidants and controllable selectivities. Studies about homogenous catalysts for aerobic oxidation reactions are abundant in the literature.6 Several successful noble metal catalytic systems, such as gold,7 palladium,8 platinum,9 rhodium,10 ruthenium,11 etc. were reported for having achieved efficient oxidation of alcohols.Over the years, the utilization of relatively inexpensive transitional metals like copper or iron have been considered an approach towards the replacement of such expensive precious metal catalysts. Although copper derived catalysts have been studied thoroughly over the years as an alternative to platinum-based ones in the oxidation of alcohols,12 iron derived ones remain relatively underexplored. Iron is less expensive and more naturally abundant than copper and other transitional metals. Since the first iron-promoted oxidation of alcohols was reported in 2002,13 this field witnessed an increase in iron use for the promotion of oxidation reactions. Wang and coworkers reported a FeCl3-TEMPO-NaNO2 system,14 while Ma and colleagues have published an Fe(NO3)3-TEMPO-NaCl system15 for oxidation reactions. However, homogeneous iron catalytic systems make the recovery of the catalyst extremely difficult, which might pose limitations to industrial scale applications. Thus, in recent years, numerous heterogeneous supports, such as molecular sieves,16 aluminosilicates,17 graphene-oxide18 polymer19 and inorganic microspheres, have been developed and applied to the alcohol oxidation reaction.20
Although heterogeneous iron catalysts were achieved in a stable and reusable manner, they commonly showed lower efficiency and metal leaching compared to their homogeneous counterparts. Among these heterogeneous catalysts, core–shell structures have been considered highly practical as they take advantage of their unique physicochemical and multi-functional properties.21 Magnetic core-shell structures are of particular interest as an effective separation method since they could easily and quickly respond to an external magnetic field.22 Many magnetic core–shell composites have been developed utilizing different coatings, such as silica, carbon, polymeric and porous materials on a Fe3O4 core.23 Several hybrid magnetic core–shell nanocatalysts have been utilized in reductions, oxidations, epoxidations, coupling reactions and photo-catalytic reactions.24 To the best of our knowledge, the combination of magnetic functionality and iron catalytic sites have not been reported in the literature yet. In this work, a core–shell Fe3O4@P4VP@FeCl3 catalyst was synthesized and it was characterized by transmission electron microscopy (TEM), powder X-ray diffraction (PXRD), FTIR spectroscopy, thermogravimetric analysis (TGA) and vibrating sample magnetometry (VSM). Numerous pyridine functional groups provided multi-dentate coordination to the iron(III) ion, which helps stabilizing the metal center. The aerobic oxidation of alcohols and benzylic carbons were both investigated using our synthesized Fe3O4@P4VP@FeCl3 catalyst. High chemical stability and low metal leaching were observed for the oxidation catalysis using our core–shell catalyst.
2. Experimental section
2.1 General information
All reaction substrates and solvents were purchased from Sigma–Aldrich, Alfa Aesar or Aladdin. Ferric chloride hexahydrate (FeCl3·6H2O), polyvinyl pyrrolidone (PVP; Mr = 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), divinylbenzene (DVB, 80%), potassium persulfate (KPS, 99%) were purchased from Alfa Aesar and were used as received. Poly(acrylic acid) (PAA; Mr = 1800) and 2,2′-azobis (2-methylpropionamidine) dihydrochloride (97%) were purchased from Sigma–Aldrich. Anhydrous sodium acetate (99%) was supplied by Aladdin. 4-Vinylpyridine monomer (96%, 4VP; Aldrich) was stabilized with 100 ppm hydroquinone and used after distillation.
000), divinylbenzene (DVB, 80%), potassium persulfate (KPS, 99%) were purchased from Alfa Aesar and were used as received. Poly(acrylic acid) (PAA; Mr = 1800) and 2,2′-azobis (2-methylpropionamidine) dihydrochloride (97%) were purchased from Sigma–Aldrich. Anhydrous sodium acetate (99%) was supplied by Aladdin. 4-Vinylpyridine monomer (96%, 4VP; Aldrich) was stabilized with 100 ppm hydroquinone and used after distillation.
2.2 Characterization
The morphology and size of the as-obtained samples were characterized on a ZEISS SUPRA55 scanning electron microscopy (FESEM). High-resolution transmission electron microscopy (HRTEM) studies were carried out on a JEOL JEM 2010 transmission electron microscope with an accelerating voltage of 200 kV. The phase structure was determined by powder X-ray diffraction (p-XRD) experiments on a DSADVANCE diffractometer with Cu Kα radiation scanning from 10° to 80°. Fourier-transformed infrared spectra (FTIR) were recorded using a NICOLET 6700 infrared spectrophotometer using KBr pellet samples. The magnetization curves of samples were measured by a MPMS-XL superconducting quantum interference device (SQUID) at room temperature. The thermogravimetric analysis (TGA) was carried out on a NETZSCH STAf409 at a heating rate of 10 °C min−1 under nitrogen. X-ray photoelectron spectroscopy data were obtained with a PHI Quantera SXM. Gas chromatography-mass spectrum was recorded using Agilent 7890A/5975C. Products were determined by GC-MS using internal standard technique, and nitrobenzene was used as an internal standard.2.3 Synthesis of the Fe3O4@P4VP materials
The PAA-modified Fe3O4 nanoparticles were prepared through an improved one-step solvothermal method reported in the literature.25 In a typical procedure, FeCl3·6H2O (1.08 g), PAA (0.108 g), and sodium acetate (9 g) were all dissolved in ethylene glycol (40 mL) to form a homogeneous solution. Then, the solution was transferred to a 50 mL Teflon-lined stainless-steel autoclave and maintained at 200 °C for 12 h. After being cooled to room temperature, the obtained black products were washed with deionized water and ethanol several times, and then dried in a vacuum for 12 h. The core–shell Fe3O4@P4VP microspheres were prepared as follows: PAA-modified Fe3O4 (0.1 g) was dispersed with PVP (0.15 g) in 100 mL deionized water under ultrasonication. After being transferred into a 250 mL four-necked flask, an emulsion contained PVP (0.05 g), 4-VP (0.125 g), DVB (0.125 g), and deionized water (20 mL) were added. All of the solution was bubbled with nitrogen for 12 h under stirring. When the temperature was raised to 70 °C, a KPS solution (0.25 mL, 0.04 g mL−1) was added into the reaction system. After 4 h of polymerization, the obtained products were washed with ethanol several times and dried under vacuum for 12 h.2.4 Synthesis of the Fe3O4@P4VP@FeCl3
5.0 g Fe3O4@P4VP nanoparticles were dispersed in 100 mL of FeCl3·6H2O ethanol solution (0.2 M). The solution was stirred for 12 h at 70 °C in an oil bath. The sample was recovered using a permanent magnet and washed with ethanol, and dried under vacuum at 80 °C. The amount of iron was analyzed by inductively coupled plasma-atomic emission spectrometry (ICP-AES); the contents of iron in Fe3O4@P4VP@FeCl3 was determined to be 4.36 wt%.2.5 General procedure for the aerobic oxidation of alcohol
In general, the catalytic reaction was carried out under the following conditions: 2 mol% Fe3O4@P4VP@FeCl3 core–shell catalyst (mole percent was based on iron), 0.1 mmol NaNO2, 0.2 mmol of 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO) and 1.0 mmol alcohol were mixed in 2.5 mL of CH3CN. The air in the catalytic reaction system was evacuated and oxygen gas was supplied through an O2 balloon. After each catalytic cycle, the solution was magnetically separated and the filtered liquid solution was analysed via gas chromatography-mass spectrometry using nitrobenzene as the internal standard.2.6 General procedure for the oxidation of hydrocarbon compounds
In general, a mixture of 2 mol% Fe3O4@P4VP@FeCl3 core–shell catalyst (mole percent was based on iron), 1.0 mmol of diphenylmethane, tert-butyl hydroperoxide (TBHP, 5.0–6.0 M in decane, 545.0 μL, 3.0 mmol) and 1.0 mL acetonitrile was mixed in a 25 mL single-necked flask fitted with a reflux condenser. The mixture was heated at 80 °C for 24 h under air atmosphere in an oil bath. After each catalytic cycle, the solution was magnetically separated and the filtered liquid solution was analysed via gas chromatography-mass spectrometry using nitrobenzene as the internal standard.2.7 Leaching test
The Fe3O4@P4VP@FeCl3 core–shell catalyst was magnetically separated after 4 h reaction time, the conversion of benzyl alcohol and selectivity of benzaldehyde was tested by GC/MS using nitrobenzene as the internal standard. The mixture was further stirred for an additional 8 hours. After the reaction, products were again analysed by GC/MS using nitrobenzene as the internal standard.3. Results and discussion
The Fe3O4 microspheres modified with PAA were synthesized by a solvothermal method reported in literature.25 These microspheres were composed of tiny Fe3O4 nanocrystals within 10 nm (Fig. 2a). The P4VP shell was successfully grafted on Fe3O4 to form core–shell composite microspheres held together by hydrogen bonds between carboxylic groups of PAA chains and pyridine. The diameters of the magnetic core and polymer shell were about 200 and 38 nm, respectively (Fig. 2b). Furthermore, the SEM images show that Fe3O4 and Fe3O4@P4VP microspheres are monodispersed with a narrow size distribution of about 200 nm and 280 nm, respectively (Fig. 2c and d).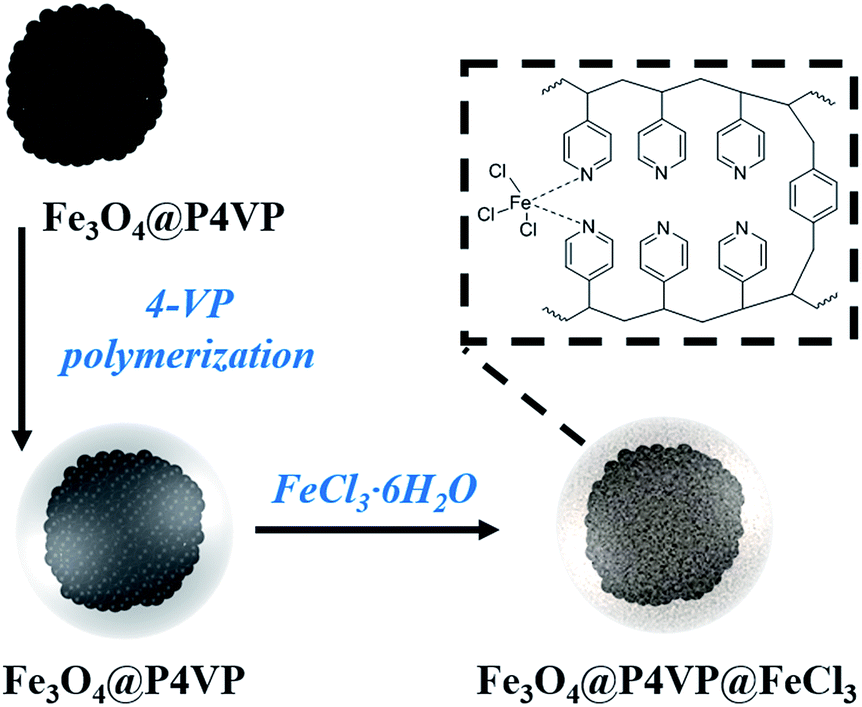 | ||
| Fig. 1 Schematic illustration of the synthesis of Fe3O4@P4VP@FeCl3. | ||
 | ||
| Fig. 2 TEM images of (a) Fe3O4, (b) Fe3O4@P4VP; SEM images of (c) Fe3O4, (d) Fe3O4@P4VP. | ||
Fe3O4@P4VP@FeCl3 core–shell catalysts were successfully prepared taking advantage of the multi-dentate coordination between FeCl3 and poly(4-vinylpyridine) (Fig. 1). Several nitrogen sites on poly(4-vinylpyridine) ensures the stability of Fe3+ ions on the surface of the core–shell structure. The EDX elemental maps further provides the firm evidence of the synthesis of Fe3O4@P4VP@FeCl3 (Fig. 3). Fe3O4 nanoparticles were encapsulated into polymer shells, and the iron(III) was well grafted on the surface of Fe3O4@P4VP. The iron elemental maps of the Fe3O4@P4VP@FeCl3 also demonstrated an excellent distribution of iron content on the surface. The existence of iron content in the Fe3O4 core structure was also observed, as shown in Fig. 3.
 | ||
| Fig. 3 HRTEM images of the Fe3O4@P4VP@FeCl3 and EDX elemental maps of Fe, Cl and N, respectively. | ||
Wide-angle p-XRD spectra of Fe3O4 nanoparticles, Fe3O4@P4VP core/shell nanospheres, and Fe3O4@P4VP@FeCl3 core–shell structure were collected and shown in Fig. 4. The diffraction peaks of Fe3O4 (Fig. 4a) agree with standard JCPDS 75-1609.39 indicating a face-centered cubic lattice. After coating with P4VP, the peaks of the core–shell Fe3O4@P4VP microspheres are highly similar as those for Fe3O4 due to the low-crystalline nature of the polymeric shell (Fig. 4b). As expected, the introduction of the FeCl3 to the Fe3O4@P4VP nanostructure did not affect the crystalline structures of Fe3O4@P4VP significantly. The high chemical stability of Fe3O4 and P4VP ensures the structure integration in presence of acidic FeCl3·6H2O.
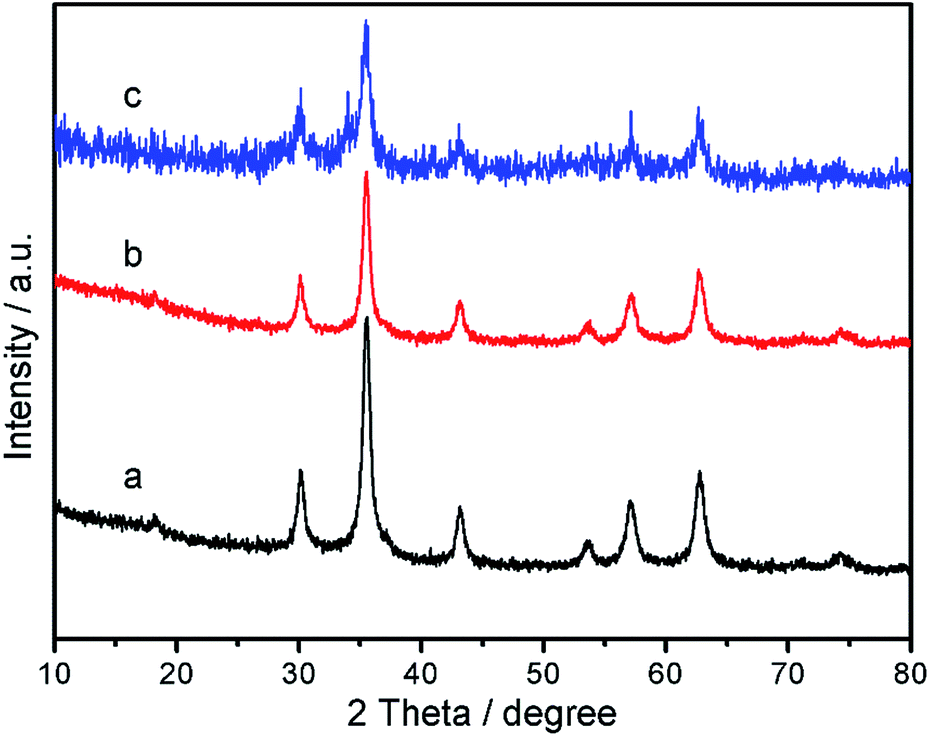 | ||
| Fig. 4 PXRD patterns of (a) Fe3O4, (b) Fe3O4@P4VP and (c) Fe3O4@P4VP@FeCl3. | ||
The FTIR spectrum of the product is shown in Fig. 5. The relatively high intensity of the band at 592 cm−1 is characteristic of the Fe–O vibrations. The characteristic absorption of the band at 1707 and 1165 cm−1 corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the carboxylic acid group and in-plane deformation of C–O–H stretching, which proves that –COOH functional groups are located on the surface of Fe3O4 nanoparticles (Fig. 5a). In the FTIR spectrum of Fe3O4@P4VP microspheres, the characteristic absorptions at 1603, 1562, and 1417 cm−1 are attributed to the vibration of the pyridine ring. The band at 1603 cm−1 corresponds to the stretching vibration absorption of the C–N bond, and the bands at around 1562 and 1417 cm−1 are attributed to the stretching vibration absorption of the CC bond, which further prove the encapsulation of P4VP on the surface of the Fe3O4 particles (Fig. 5b). Furthermore, a new peak was observed at 1627 cm−1 after the introduction of the FeCl3 moiety, which shows chemical interaction between the iron and the polymer (Fig. 5c).
O stretching of the carboxylic acid group and in-plane deformation of C–O–H stretching, which proves that –COOH functional groups are located on the surface of Fe3O4 nanoparticles (Fig. 5a). In the FTIR spectrum of Fe3O4@P4VP microspheres, the characteristic absorptions at 1603, 1562, and 1417 cm−1 are attributed to the vibration of the pyridine ring. The band at 1603 cm−1 corresponds to the stretching vibration absorption of the C–N bond, and the bands at around 1562 and 1417 cm−1 are attributed to the stretching vibration absorption of the CC bond, which further prove the encapsulation of P4VP on the surface of the Fe3O4 particles (Fig. 5b). Furthermore, a new peak was observed at 1627 cm−1 after the introduction of the FeCl3 moiety, which shows chemical interaction between the iron and the polymer (Fig. 5c).
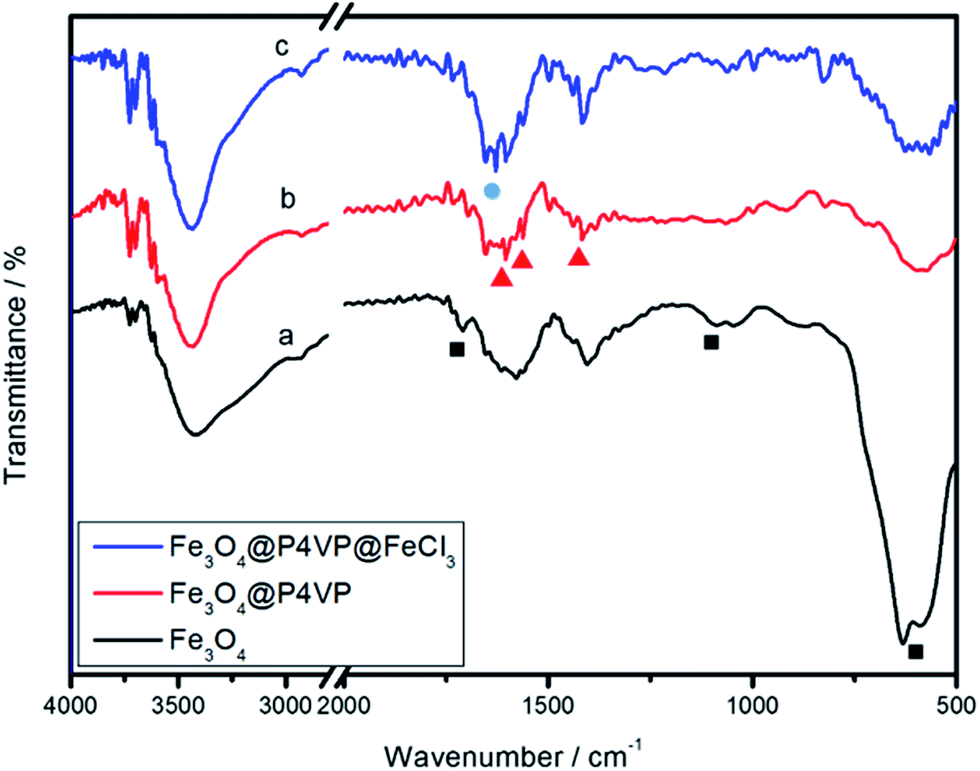 | ||
| Fig. 5 FTIR spectra of (a) Fe3O4, (b) Fe3O4@P4VP and (c) Fe3O4@P4VP@FeCl3. | ||
The thermal stability of the Fe3O4, Fe3O4@P4VP and Fe3O4@P4VP@FeCl3 nanostructures was also examined by TGA in the temperature range of 50–800 °C (Fig. 6). The mass loss of Fe3O4 was due to the decomposition of organic PAA layer on the surface of Fe3O4 microspheres (Fig. 6a). Fe3O4@P4VP microspheres show two steps of mass loss. The first one, from room temperature to 440 °C, is related to the desorption of adsorbed water and the degradation of secondary nucleation polymer chains on the surface of microsphere. The second loss, from 440 to 800 °C, is instead correlated to the polymer shell and the PAA modification on the Fe3O4 (Fig. 6b). For the Fe3O4@P4VP@FeCl3 catalyst, the main decomposition stage was similar to Fe3O4@P4VP microspheres. The results demonstrated that the Fe3O4@P4VP@FeCl3 catalyst is thermally stable under the conditions of the aerobic oxidation reaction.
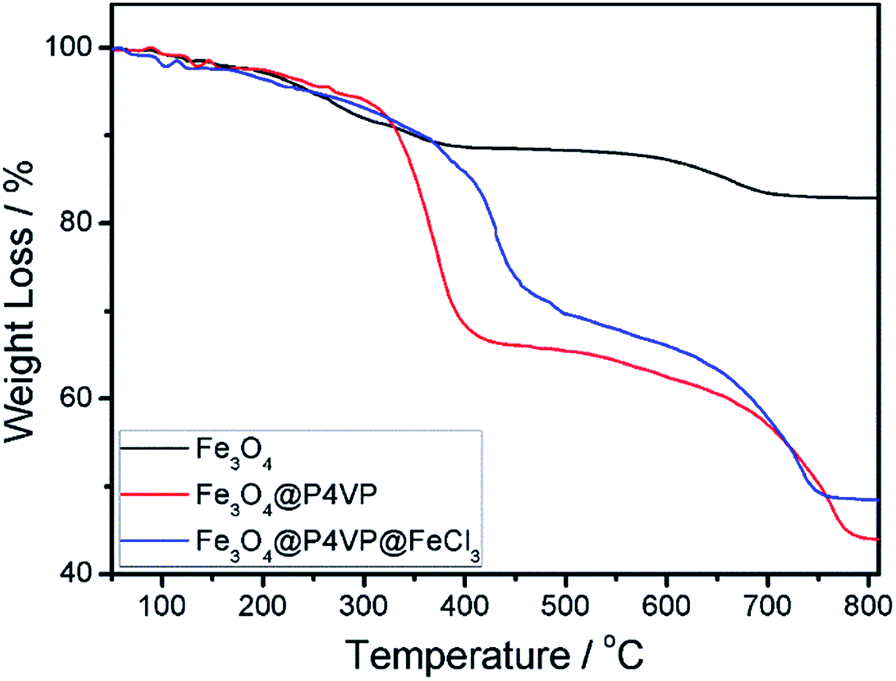 | ||
| Fig. 6 TGA of (a) Fe3O4, (b) Fe3O4@P4VP and (c) Fe3O4@P4VP@FeCl3. | ||
The magnetic performance of the Fe3O4@P4VP@FeCl3 core–shell nanostructure was investigated by SQUID at room temperature. The magnetization curves of synthesized samples are shown in Fig. 7. The magnetization curves of composite nanospheres exhibit no remanence at room temperature. Again, the results indicate their superparamagnetism, which is crucial for controllable flocculation and dispersion in solution by an external magnetic field. A saturation magnetization value of the Fe3O4@P4VP nanospheres of 47.2 emu g−1 ensures its rapid recoverability as catalyst support under an external magnetic field. The value is lower than that of Fe3O4 nanoparticles (70.8 emu g−1) due to the capsulation of magnetic components into the P4VP. In addition, Fe3O4@P4VP@FeCl3 catalyst could be easily recovered by an external magnetic field as shown in Fig. 7; its saturation magnetization value is 39.8 emu g−1.
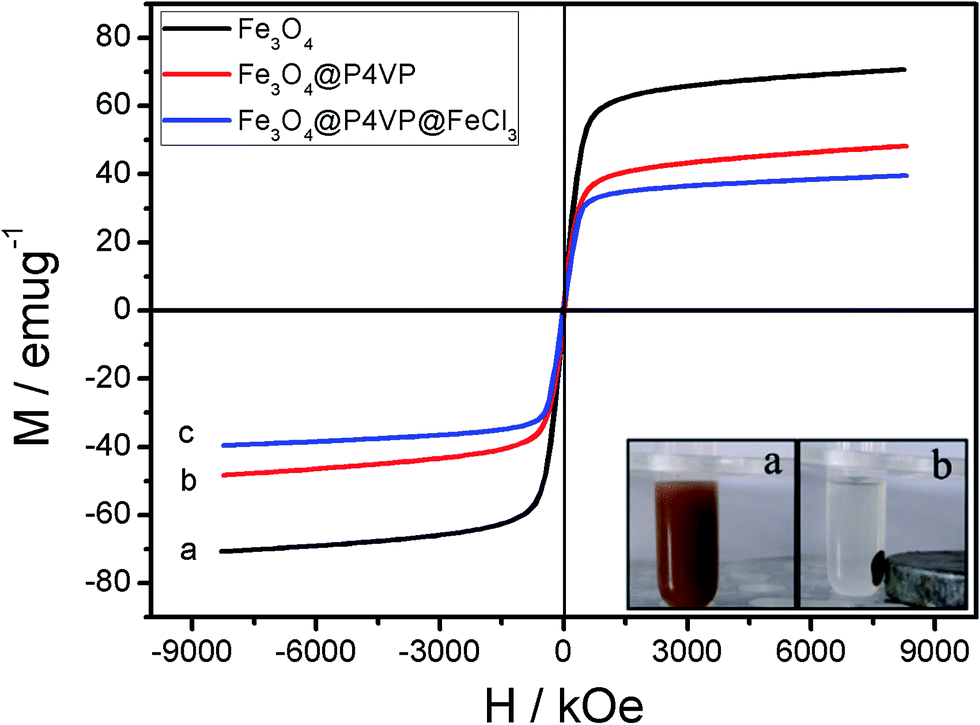 | ||
| Fig. 7 Room-temperature magnetic hysteresis loops of (a) Fe3O4, (b) Fe3O4@P4VP and (c) Fe3O4@P4VP@FeCl3. Photos of the insets depict magnetic recycling of Fe3O4@P4VP@FeCl3 catalyst. | ||
The high resolution XPS spectra is obtained and the results are shown in Fig. 8, which shows the XPS spectra of the Fe3O4@P4VP@FeCl3 core–shell structure. The strong peaks in the Fe3O4@P4VP@FeCl3 signal are attributed to the N 1s and C 1s binding energies, respectively. The binding energies at 711 and 55 eV belong to the Fe 3p and Fe 2p2/3, respectively. The peaks at 268 and 198 eV are due to the existence of Cl 2p and Cl 2s binding, respectively. The results strongly indicate the existence of iron(III) moieties on the surface of the Fe3O4@P4VP@FeCl3 catalyst.
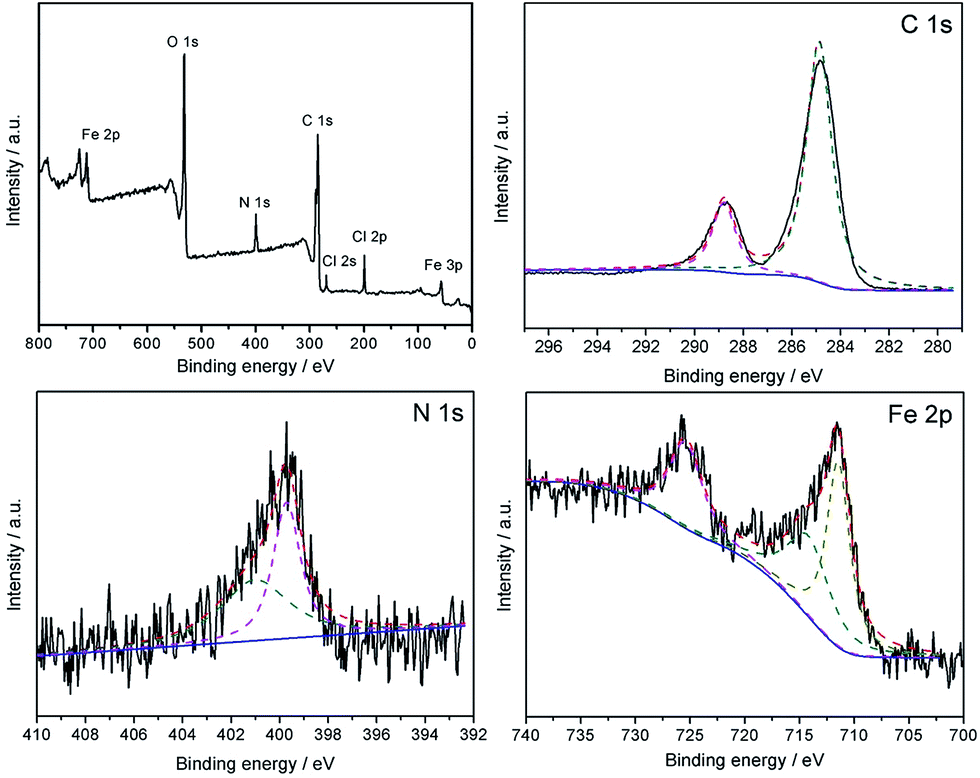 | ||
| Fig. 8 XPS Spectra of Fe3O4@P4VP@FeCl3. | ||
The catalytic activity of the synthesized Fe3O4@P4VP@FeCl3 toward the aerobic oxidation reaction was evaluated employing alcohol substrates and using TEMPO as the radical initiator. The control experiment showed that no benzaldehyde was detected in absence of catalyst (Table 1, entry 1). Also, the employment of Fe3O4 and Fe3O4@P4VP as catalysts failed to oxidize any benzyl alcohol to its corresponding aldehyde (Table 1, entries 2 and 3). Homogeneous iron catalysts were evaluated and almost quantitative conversion was observed. However, slightly lowered selectivity was observed due to overoxidation to benzoic acid (Table 1, entries 4–5). The amount of Fe3O4@P4VP@FeCl3 added in this reaction was 2 mol%, based on the same iron loading; quantitative yields were achieved using KNO2 additive (Table 1, entry 6). Several additives were tested under the same reaction conditions and KNO2 was the most optimal one. Basic inorganic additives were tested and only low to medium yields were achieved (Table 1, entries 7–8). A basic organic additive was evaluated but a poor yield was observed (Table 1, entry 9). Further solvent screening showed that CH3CN is the most suitable solvent for the aerobic oxidation of benzyl alcohol in the presence of Fe3O4@P4VP@FeCl3 (Table 1, entries 6, 10–12). Aromatic solvents such as toluene gave lower yields (Table 1, entry 10). Moreover, oxygen containing solvents, such as THF and ethanol, provided much lower yield, presumably due to their ability to excessively coordinate the metal (Table 1, entries 11 and 12).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Solvent | Additive | Selec.b | Yieldb |
| a Reaction conditions: 1.0 mmol of benzyl alcohol, 2 mol% of iron catalyst, 5.0 mL of solvent, 0.2 mmol of TEMPO and 0.2 mmol of additive; 60 °C, 12 h under 1 atm O2.b Selectivities and yields were calculated by GC-MS using nitrobenzene as the internal standard. | |||||
| 1 | — | CH3CN | — | — | <5% |
| 2 | Fe3O4 | CH3CN | KNO2 | — | <5% |
| 3 | Fe3O4@P4VP | CH3CN | KNO2 | — | <5% |
| 4 | FeCl3·6H2O | CH3CN | KNO2 | 91% | 99% |
| 5 | Fe(NO3)3 | CH3CN | KNO2 | 78% | 99% |
| 6 | Fe3O4@P4VP@FeCl3 | CH3CN | KNO2 | 99% | 99% |
| 7 | Fe3O4@P4VP@FeCl3 | CH3CN | NaHCO3 | 99% | 76% |
| 8 | Fe3O4@P4VP@FeCl3 | CH3CN | Na2CO3 | 99% | 68% |
| 9 | Fe3O4@P4VP@FeCl3 | CH3CN | Pyridine | 99% | 43% |
| 10 | Fe3O4@P4VP@FeCl3 | PhCH3 | KNO2 | 95% | 71% |
| 11 | Fe3O4@P4VP@FeCl3 | THF | KNO2 | 99% | 55% |
| 12 | Fe3O4@P4VP@FeCl3 | EtOH | KNO2 | 99% | 21% |
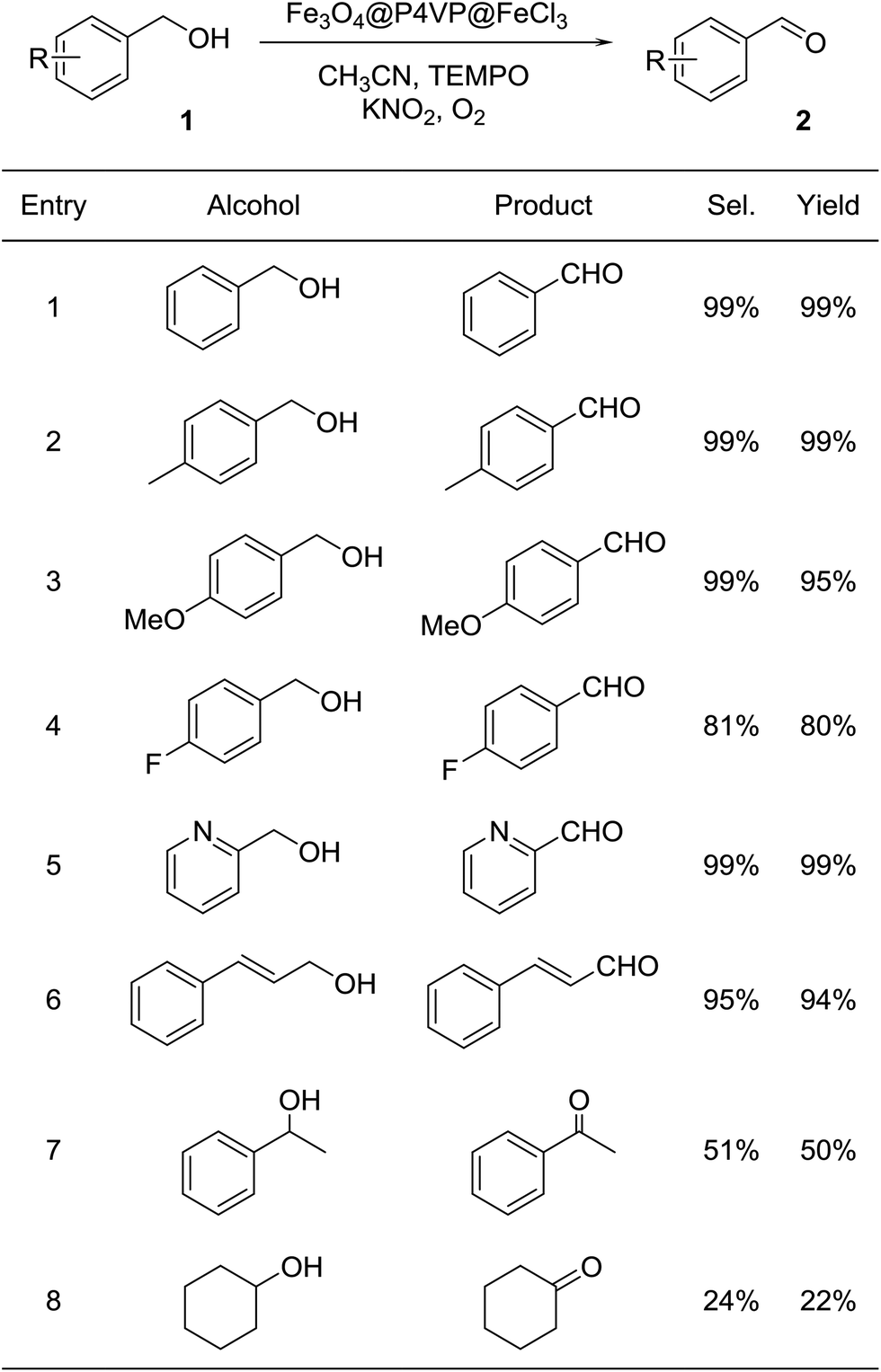
With the optimal reaction conditions in hand, a variety of alcohol substrates were tested using Fe3O4@P4VP@FeCl3 core–shell catalyst. Benzyl alcohol was transformed to the corresponding benzaldehyde in 99% yield after 12 h (Table 2, entry 1). For the aerobic oxidation of benzyl alcohol in presence of Fe3O4@P4VP@FeCl3 as the catalyst, the turnover number (TON) was calculated to be 50. p-Methyl benzyl alcohol and p-methoxy benzyl alcohol were reactive as electron-rich benzyl alcohols under the optimal reaction conditions, affording 99% and 95% yield, respectively (Table 2, entries 2 and 3). Alcohol substrates bearing electron-withdrawing functional groups, such as p-fluorobenzyl alcohol, were also tolerated. A slightly lowered yield was observed due to the reduction in electron efficiency (Table 2, entry 4). The heterocyclic alcohol pyridin-2-ylmethanol was selectively converted to its corresponding aldehyde, in the presence of the Fe derived core–shell catalyst (Table 2, entry 5). Moreover, cinnamaldehyde was obtained in good yield starting from the corresponding allylic alcohol (Table 2, entry 6). The transformation of secondary alcohols to ketones is also highly important in synthetic chemistry; 1-phenylethanol and cyclohexanol were both tested as secondary alcohols (Table 2, entries 7 and 8). Unfortunately, only 50% and 22% of acetophenone and cyclohexanone respectively were obtained.
| a Reaction conditions: 1.0 mmol of benzyl alcohol, 2 mol% of Fe3O4@P4VP@FeCl3 catalyst, 5.0 mL of CH3CN, 0.2 mmol of TEMPO and 0.2 mmol of additive; 60 °C, 12 h under 1 atm O2. |
|---|
 |
The oxidation of diphenylmethane 3a for the generation of benzophenone 4a was carried out in 2.0 mL acetonitrile at 80 °C for 24 h (Table 3). The activation of C–H bond is difficult and tert-butyl hydroperoxide (TBHP) was employed as the oxidant under extended reaction time. The catalytic system has been re-optimized to adapt the benzylic oxidation reaction of hydrocarbon derivatives. In the absence of any additive, FeCl3·6H2O and Fe(NO3)3·9H2O failed to promote any conversion (Table 3, entries 1 and 3). The employment of basic pyridine additive strongly increased the yield of the desired benzophenone product (Table 3, entries 2 and 4). Initially, 91% conversion and 99% selectivity were obtained for Fe3O4@P4VP@FeCl3 catalyst in the absence of additive (Table 1, entry 5). The high efficiency of Fe3O4@P4VP@FeCl3 catalyst was due to the pyridine sites on the surface of the heterogeneous iron catalyst. The addition of a small amount of homogeneous pyridine can further boost the conversion to over 99% (Table 1, entry 6). Furthermore, the amount of pyridine can be reduced to 2 mol% without affecting the yield and selectivity of the diphenylmethane oxidation (Table 1, entry 7). In addition, other basic additives were examined. However, almost no improvement was observed when triethylamine (Et3N) was employed (Table 3, entry 8). Hexamethylenetetramine (HMTA) failed to increase the yield as well (Table 3, entry 9). These results indicate that pyridine was the best base additive among the organic bases screened. Our Fe3O4@P4VP@FeCl3 catalyst was designed so that the heterogeneous P4VP layer would provide large amounts of pyridine sites. These heterogeneous pyridine sites were highly helpful for the oxidation reaction process, while homogeneous FeCl3 failed to convert diphenylmethane to the desired product. Several organic solvents were screened and acetonitrile demonstrated good compatibility in the catalytic system, which resulted in 99% isolated yield (Table 3, entry 2). Other solvents showed a trend similar to the results in Table 1. Therefore the optimal catalytic system was set to be 2.0 mol% Fe3O4@P4VP@FeCl3 catalyst, 0.02 equivalent of pyridine additive and 3.0 equivalents of TBHP in acetonitrile solvent.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Additive | Selec. | Yieldb |
| a Reaction conditions: 1.0 mmol 3d, 2.0 mol% Fe3O4@P4VP@FeCl3 catalyst, 5 mol% additive, 3.0 mmol TBHP, 2.0 mL acetonitrile, 80 °C for 24 h.b Isolated yield.c 2 mol% pyridine. | ||||
| 1 | FeCl3·6H2O | None | — | <5% |
| 2 | FeCl3·6H2O | Pyridine | 99% | 91% |
| 3 | Fe(NO3)3·9H2O | None | — | <5% |
| 4 | Fe(NO3)3·9H2O | Pyridine | 99% | 75% |
| 5 | Fe3O4@P4VP@FeCl3 | None | 99% | 84% |
| 6 | Fe3O4@P4VP@FeCl3 | Pyridine | 99% | 99% |
| 7c | Fe3O4@P4VP@FeCl3 | Pyridine | 99% | 99% |
| 8 | Fe3O4@P4VP@FeCl3 | Et3N | 99% | 85% |
| 9 | Fe3O4@P4VP@FeCl3 | HMTA | 99% | 87% |
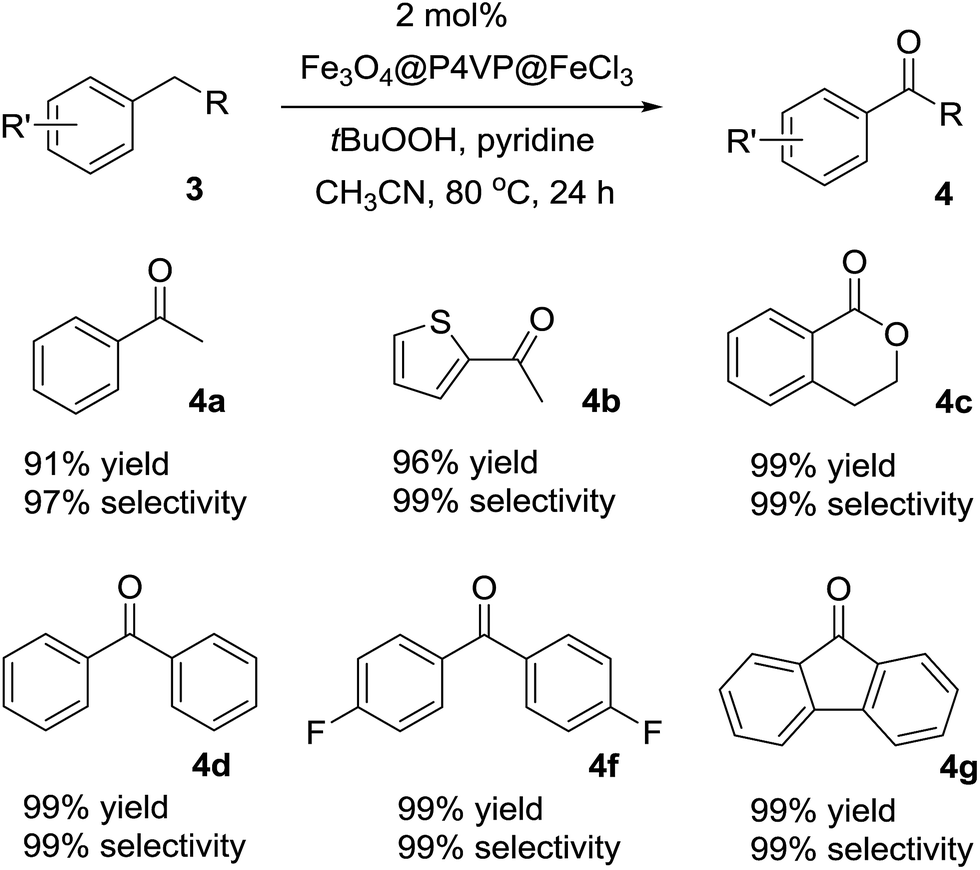
The optimized reaction conditions were utilized for the oxidation of other substrates in the presence of tBuOOH (Table 4). C–H activation/oxidation was further extended to other hydrocarbon compounds. Ethylbenzene 3a was oxidized to 1-phenylethanone 4a in 91% conversion and 97% selectivity (Table 4, compound 4a). Ethylthiophene 3b containing a heterocyclic moiety was also employed as the substrate and over 96% yield and up to 99% selectivity were achieved (Table 3, compound 4b). Isochromane 3c underwent oxidation to give 4-isochromanone 4c in high yield (Table 4, compound 4c). The oxidation of diphenylmethane 3d and bis(4-fluorophenyl)methanone 3f for the generation of corresponding ketones 4d and 4f were smooth (Table 4, compounds 4d and 4f). Moreover, by oxidation of 9H-fluorene 3g, the reaction proceeded smoothly to form the corresponding ketones in high yield and selectivity (Table 4, compound 4g). In summary, the Fe3O4@P4VP@FeCl3 was a versatile catalyst for benzylic methylene compounds (Table 4).
 |
The recyclability of the catalyst was studied under the optimized conditions and utilizing 2 mol% and 0.5 mol% of Fe3O4@P4VP@FeCl3 catalyst in acetonitrile (Fig. 9). The same batch of the Fe3O4@P4VP@FeCl3 catalyst was reused for five catalytic cycles. Conversion and selectivity were retained at 99% and the yield of benzaldehyde was compromised only slightly after five cycles (Fig. 9). At a lower catalyst loading of 0.5 mol%, 44% yield was achieved in the initial cycle. Only slight yield decrease (98%) was observed after 5 cycles of reaction (Fig. 9). The ability to retain conversion rates and selectivities after five cycles indicates that the core–shell heterogeneous catalyst is highly stable. TEM and p-XRD spectra showed no significant difference between fresh Fe3O4@P4VP@FeCl3 and five-time reused samples (Fig. S1 and S2†).
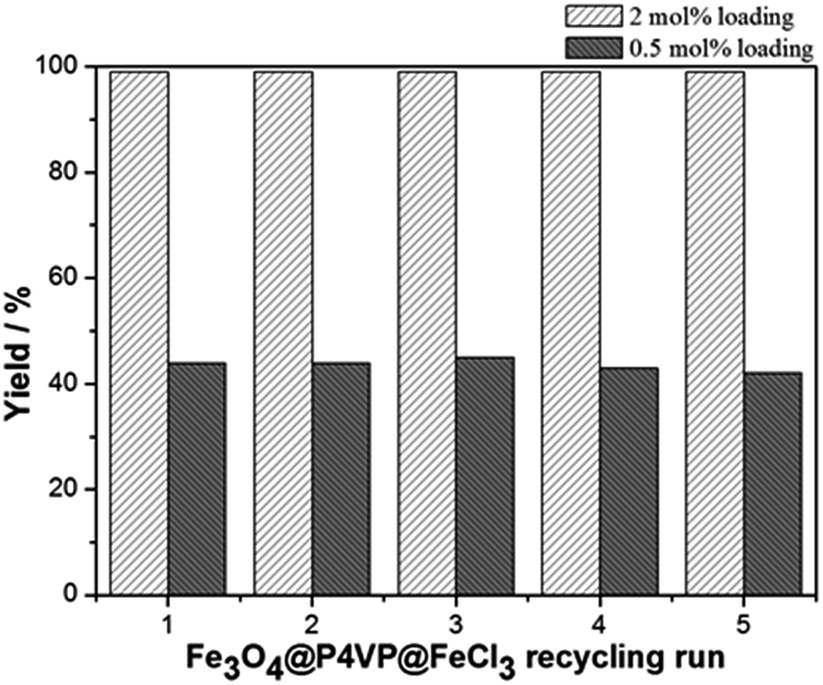 | ||
| Fig. 9 Core-shell iron catalyst recyclability test at 2 mol% and 0.5 mol% catalyst loading for aerobic oxidation of benzyl alcohol. | ||
The hot filtration test was conducted to confirm the heterogeneous nature of the catalytic aerobic oxidation and low reaction leaching (Fig. 10). The Fe3O4@P4VP@FeCl3 catalyst was isolated using an external magnet after 4 hours of reaction and the mixture was stirred for another 8 h. Conversion of benzyl alcohol stopped after the catalyst was removed at 4 h reaction time. This result indicates that the multi-dentate Fe⋯N bonds ensure the stability of the Fe3O4@P4VP@FeCl3 catalyst during the liquid oxidation process. Furthermore, the reaction solution after solid catalyst removal was tested by ICP-AES, which suggested 2.3 ppm of iron content. These results demonstrated the high stability of Fe3O4@P4VP@FeCl3 core–shell catalyst during the oxidation reaction condition.
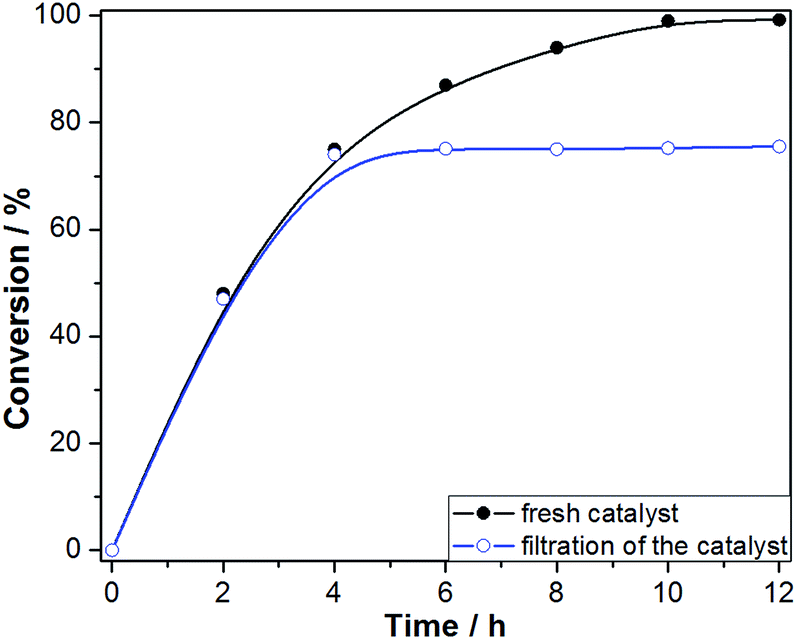 | ||
| Fig. 10 Hot filtration test of Fe3O4@P4VP@FeCl3 in aerobic oxidation of benzyl alcohol. | ||
Several control experiments were designed and conducted in order to reveal the reaction mechanism. First of all, the oxidation reaction was carried out without TEMPO (Scheme 1, reaction 1). In the absence of radical initiator, reaction was not able to proceed. This observation suggested radical reaction pathway. Furthermore, reaction was performed under open air condition, decent yield of the desired aldehyde product was obtained (Scheme 1, reaction 2). Oxygen was crucial in the oxidation process and the certain density of oxygen ensures the high conversion of the alcohol product.
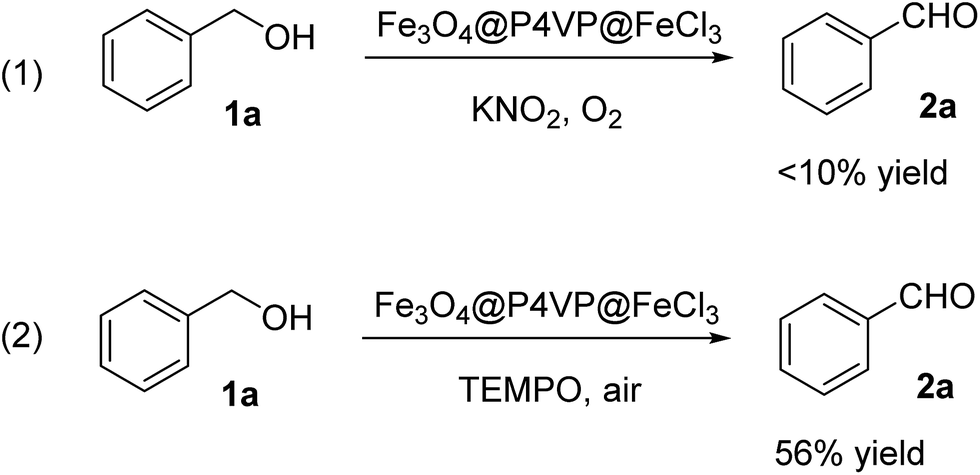 | ||
| Scheme 1 Controlled experimental reaction. | ||
A overall mechanism of Fe(III) promoted catalytic oxidation can be described as a redox reaction involving two separated cycles in Fig. 11. KNO2 was acting as a source of NO2. Therefore, TEMPO is envisioned to carry out the main oxidation reaction of alcohols with the help of Fe(III) that initiates a series of electron and proton transfer steps in the right cycle where TEMPO-Fe(III) is reduced to generate TEMPOH-Fe(II). The overall oxidation process involves the oxidation of Fe(II) to Fe(III) by NO2 and the oxidation of TEMPOH to TEMPO by Fe(III). The conversion of NO to NO2 was performed in the presence of molecular oxygen.
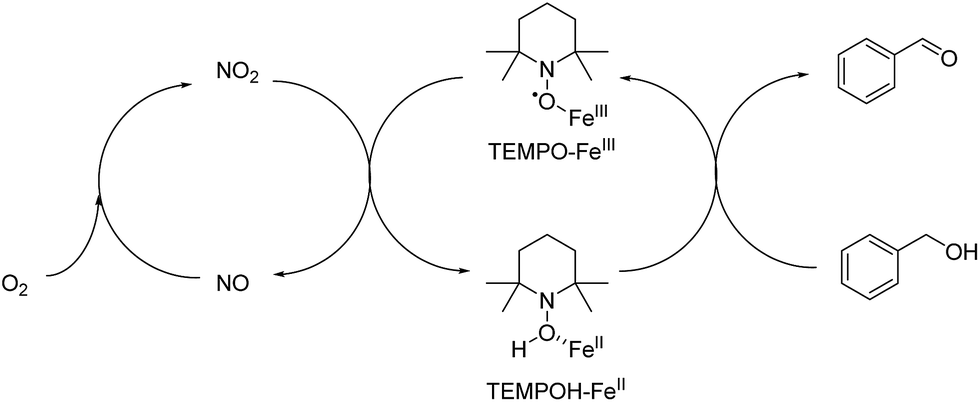 | ||
| Fig. 11 Reaction mechanism of Fe(III) promoted oxidation. | ||
4. Conclusions
In summary, a novel magnetic core–shell Fe3O4@P4VP@FeCl3 structure was designed, prepared and fully characterized. The iron(III) immobilized core–shell microspheres were composed by a magnetic Fe3O4 core and a P4VP middle layer. Taking advantage of active iron(III) catalytic site, the magnetic composite microspheres were utilized as an efficient catalyst for the selective aerobic oxidation of alcohols. A variety of alcohol substrates were tolerated under our optimized conditions at only 2 mol% catalyst loading. In addition, benzylic oxidation of hydrocarbon compounds was also carried out using the Fe3O4@P4VP@FeCl3 catalyst and tBuOOH oxidant. The initial catalytic activity of the Fe3O4@P4VP@FeCl3 catalyst was retained after at least five consecutive reaction cycles. A hot filtration test suggested low leaching of iron into the solution. Further applications of the various heterogeneous core–shell catalysts are currently under investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by Beijing Natural Science Foundation (2172037) and National Natural Science Foundation of China (No. 51503016). P. Y. thanks the State Key Laboratory of Chemical Resource Engineering and Y. L. thanks the Fundamental Research Funds for the Central Universities (Grant No. FRF-TP-16-004A3) for funding support.Notes and references
- Q. Cao, L. M. Dornan, L. Rogan, N. L. Hughes and M. J. Muldoon, Chem. Commun., 2014, 50, 4524–4543 RSC.
- (a) R. A. Sheldon, Catal. Today, 2015, 247, 4 CrossRef CAS; (b) C. Parmeggiani and F. Cardona, Green Chem., 2012, 14, 547 RSC; (c) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC; (d) Q. Cao, L. M. Dornan, L. Rogan, N. L. Hughes and M. J. Muldoon, Chem. Commun., 2014, 50, 4524 RSC; (e) M. J. Schultz and M. S. Sigman, Tetrahedron, 2006, 62, 8227 CrossRef CAS.
- (a) Z. Guo, B. Liu, Q. Zhang, W. Deng, Y. Wang and Y. Yang, Chem. Soc. Rev., 2014, 43, 3480 RSC; (b) S. E. Devis, M. S. Lde and R. J. Davis, Green Chem., 2013, 15, 17 RSC.
- Z. Guo, B. Liu, Q. Zhang, W. Deng, Y. Wang and Y. Yang, Chem. Soc. Rev., 2014, 43, 3480–3524 RSC.
- M. Hajimohammadi, N. Safari, H. Mofakham and F. Deyhimi, Green Chem., 2011, 13, 991 RSC.
- B. Chen, L. Wang and S. Gao, ACS Catal., 2015, 5, 5851–5876 CrossRef CAS.
- (a) P. Miedziak, M. Sankar, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, D. W. Knight, S. H. Taylor, C. J. Kiely and G. J. Hutchings, Catal. Today, 2011, 164, 315 CrossRef CAS; (b) Y. Li, Y. Gao and C. Yang, Chem. Commun., 2015, 51, 7721 RSC; (c) X. Yu, Y. Huo, J. Yang, S. Chang, Y. Ma and W. Huang, Appl. Surf. Sci., 2013, 280, 450 CrossRef CAS; (d) R. H. Adnan, G. G. Andersson, M. I. J. Polson, G. F. Metha and V. B. Golovko, Catal. Sci. Technol., 2015, 5, 1323 RSC; (e) T. Wang, X. Yuan, S. Li, L. Zeng and J. Gong, Nanoscale, 2015, 7, 7593 RSC; (f) C. D. Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077 RSC.
- (a) K. M. Gligorich and M. S. Sigman, Chem. Commun., 2009, 3854 RSC; (b) Y. Ye, M. D. Johnson, T. Diao, M. H. Yates and S. S. Stahl, Green Chem., 2010, 12, 1180 RSC; (c) B. Karimi, S. Abedi, J. H. Clark and V. Budarin, Angew. Chem., Int. Ed., 2006, 45, 4776 CrossRef CAS PubMed; (d) B. Karimi, A. Zamani, S. Abedi and J. H. Clark, Green Chem., 2009, 11, 109 RSC; (e) D. Sahu, C. Sarmah and P. Das, Tetrahedron Lett., 2014, 55, 3422 CrossRef CAS; (f) S. F. J. Hackett, R. M. Brydson, M. H. Gass, I. Harvey, A. D. Newman, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2007, 46, 8593 CrossRef CAS PubMed; (g) H. Wu, Q. Zhang and Y. Wang, Adv. Synth. Catal., 2005, 347, 1536 Search PubMed; (h) X. Ye, M. D. Johnson, T. Diao, M. H. Yates and S. S. Stahl, Green Chem., 2010, 12, 1180 RSC; (i) B. Karimi, D. Elhamifar, J. H. Clark and A. J. Hunt, Org. Biomol. Chem., 2011, 9, 7420 RSC; (j) H. Yang, X. Han, Z. Ma, R. Wang, J. Liu and X. Ji, Green Chem., 2010, 12, 441 RSC; (k) C. M. A. Parlett, D. W. Bruce, N. S. Hondow, A. F. Lee and K. Wilson, ACS Catal., 2011, 1, 636 CrossRef CAS; (l) J. Chen, Q. Zhang, Y. Wang and H. Wan, Adv. Synth. Catal., 2008, 350, 453 CrossRef CAS; (m) B. Karimi, A. Zamani and J. H. Clark, Organometallics, 2005, 24, 4695 CrossRef CAS; (n) M. J. Schultz, C. C. Park and M. S. Sigman, Chem. Commun., 2002, 3034 RSC.
- (a) Y. Hong, X. Yan, X. Liao, R. Li, X. Xu, L. Xiao and J. Fan, Chem. Commun., 2014, 50, 9679 RSC; (b) T. Lu, Z. Du, J. Liu, H. Ma and J. Xu, Green Chem., 2013, 15, 2215 RSC; (c) T. Wang, H. Shou, Y. Kou and H. Liu, Green Chem., 2009, 11, 562 RSC; (d) K. Kon, S. M. A. H. Siddiki and K.-i. Shimizu, J. Catal., 2013, 304, 63 CrossRef CAS; (e) T. Wang, C.-X. Xiao, L. Yan, L. Xu, J. Luo, H. Shou, Y. Kou and H. Liu, Chem. Commun., 2007, 4375 RSC.
- (a) A. Wusiman and C.-D. Lu, Appl. Organomet. Chem., 2015, 29, 254 CrossRef CAS; (b) J. Olguin, H. Muller-Bunz and M. Albrecht, Chem. Commun., 2014, 50, 3488 RSC; (c) L. Liu, M. Yu, B. B. Wayland and X. Fu, Chem. Commun., 2010, 46, 6353 RSC.
- (a) K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2002, 41, 4538 CrossRef CAS; (b) Z. W. Yang, X. Zhao, T. J. Li, W. L. Chen, Q. X. Kang, X. Q. Xu, X. X. Liang, Y. Feng, H. H. Duan and Z. Q. Li, Catal. Commun., 2015, 65, 34 CrossRef CAS; (c) D. Canseco-Gonzalez and M. Albrecht, Dalton Trans., 2013, 42, 7424 RSC.
- (a) B. Xu, J. P. Lumb and B. A. Arndtsen, Angew. Chem., Int. Ed., 2015, 54, 4208 CrossRef CAS PubMed; (b) L. Vanoye, M. Pablos, C. D. Bellefon and A. Favre-Reguillon, Adv. Synth. Catal., 2015, 357, 739 CrossRef CAS; (c) B.-T. Chen, K. V. Bukhryakov, R. Sougrat and V. O. Rodionov, ACS Catal., 2015, 5, 1313 CrossRef CAS; (d) G. Zhang, C. Yang, E. Liu, L. Li, J. A. Golen and A. L. Rheingold, RSC Adv., 2014, 4, 61907 RSC; (e) P. Gamez, I. W. C. E. Arends, J. Reedijk and R. A. Sheldon, Chem. Commun., 2003, 2414–2415 RSC; (f) G. Zhang, X. Han, Y. Luan, Y. Wang, X. Wen, L. Xu, C. Ding and J. Gao, RSC Adv., 2013, 3, 9255 Search PubMed; (g) S. Samanta, S. Das, P. K. Samanta, S. Dutta and P. Biswas, RSC Adv., 2013, 3, 19455 RSC.
- S. E. Martin and D. F. Suarez, Tetrahedron Lett., 2002, 43, 4475 CrossRef CAS.
- N. Wang, R. Liu, J. Chen and X. Liang, Chem. Commun., 2005, 5322 RSC.
- J. Liu and S. Ma, Org. Lett., 2013, 15, 5150 CrossRef CAS PubMed.
- (a) M. A. Chari and K. Syamasundar, Synthesis, 2005, 5, 0708–0710 CrossRef; (b) M. R. Farsani and B. Yadollahi, J. Mol. Catal. A: Chem., 2014, 392, 8–15 CrossRef CAS; (c) Y. Kuang, Y. Nabae, T. Hayakawa and M. Kakimoto, Appl. Catal., A, 2012, 423–424, 52–58 CrossRef CAS; (d) P. Das, N. Aggarwal and N. R. Guha, Tetrahedron Lett., 2013, 54, 2924–2928 CrossRef CAS.
- F. Rajabi, A. Pineda, S. Naserian, A. M. Balu, R. Luque and A. A. Romero, Green Chem., 2013, 15, 1232 RSC.
- M. Mahyari and A. Shaabani, Appl. Catal., A, 2014, 469, 524 CrossRef CAS.
- P. R. Likhar, R. Arundhathi, S. Ghosh and M. L. Kantam, J. Mol. Catal. A: Chem., 2009, 302, 142–149 CrossRef CAS.
- J. Mao, X. Hu, H. Li, Y. Sun, C. Wang and Z. Chen, Green Chem., 2008, 10, 827–831 RSC.
- R. G. Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef PubMed.
- M. B. Gawande, A. Goswami, T. Asefa, H. Guo, A. V. Biradar, D. L. Peng, R. Zboril and R. S. Varma, Chem. Soc. Rev., 2015, 44, 7540–7590 RSC.
- (a) C. Hui, C. Shen, J. Tian, L. Bao, H. Ding, C. Li, Y. Tian, X. Shi and H. J. Gao, Nanoscale, 2011, 3, 701–705 RSC; (b) Y. D. Chiang, S. Dutta, C. T. Chen, Y. T. Huang, K. S. Lin, J. C. S. Wu, N. Suzuki, Y. Yamauchi and K. C. W. Wu, ChemSusChem, 2015, 8, 789–794 CrossRef CAS PubMed.
- (a) Q. Zhang, I. Lee, J. B. Joo, F. Zaera and Y. Yin, Acc. Chem. Res., 46, 1816–1824 Search PubMed; (b) P. Hu, J. V. Morabito and C. K. Tsung, ACS Catal., 2014, 4, 4409–4419 CrossRef CAS; (c) Y. Long, M. Xie, J. Niu, P. Wang and J. Ma, Appl. Surf. Sci., 2013, 277, 288–292 CrossRef CAS; (d) X. Zhang, J. Wu, G. Meng, X. Guo, C. Liu and Z. Liu, Appl. Surf. Sci., 2016, 366, 486–493 CrossRef CAS; (e) Z. Sun, H. Li, G. Cui, Y. Tian and S. Yan, Appl. Surf. Sci., 2016, 360, 252–262 CrossRef CAS.
- H. Deng, X. Li, Q. Peng, X. Wang, J. Chen and Y. Li, Angew. Chem., Int. Ed., 2005, 44, 2782–2785 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09005f |
| This journal is © The Royal Society of Chemistry 2017 |