
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Monte Carlo study of chemical reaction equilibria in pores of activated carbons†
Sylwester Furmaniak *a,
Piotr A. Gaudenb,
Piotr Kowalczykc and
Andrzej Patrykiejewd
*a,
Piotr A. Gaudenb,
Piotr Kowalczykc and
Andrzej Patrykiejewd
aStanisław Staszic University of Applied Sciences in Piła, Podchorążych Street 10, 64-620 Piła, Poland. E-mail: sfurmaniak@pwsz.pila.pl
bPhysicochemistry of Carbon Materials Research Group, Faculty of Chemistry, Nicolaus Copernicus University in Toruń, Gagarin Street 7, 87-100 Toruń, Poland
cSchool of Engineering and Information Technology, Murdoch University, Murdoch 6150, WA, Australia
dDepartment for the Modelling of Physico-Chemical Processes, Faculty of Chemistry, Maria Curie Skłodowska University in Lublin, Gliniana Street 33, 20-031 Lublin, Poland
First published on 22nd November 2017
Abstract
This work has presented the results of a rather extensive Monte Carlo study concerning the effects of confinement on the reactions taking place in the pores of activated carbons. We have considered here three simple model reactions: isomerisation, dimerisation and synthesis, and investigated how the changes in the carbon porosity, the values of the equilibrium constant, and the energetic parameters of the reacting molecules influence the chemical equilibria. The obtained results show that the main factors affecting the reaction equilibria in pores are the latest ones. When the adsorption energy of the product molecules is higher than that of the reactants, the confinement causes a rise in the reaction yield. In the opposite situation (preferential adsorption of the reactants), the product mole fraction inside the pores is lower than in the bulk phase. It has been shown that the porous structure of activated carbons plays a very important role. The reduction of pore diameters may either increase or decrease the reaction yield, depending on the relative adsorption energy of the reactants and the products. If the product molecules are bigger than the reactant molecules, the presence of pores accessible for the reactant molecules, but inaccessible for the product, causes additional reduction of the reaction yield regardless of the magnitudes of the energetic parameters of the reacting species.
1. Introduction
In 1994, Johnson et al.1 and, independently, Smith and Triska2 published a description of methodology allowing the modelling of chemical reactions with the help of the Monte Carlo simulation method. This technique (called the reaction ensemble Monte Carlo, RxMC) predicts the locations of equilibrium states according to the laws of statistical physics. In order to use the technique, one needs to know the ideal-gas free energies (including the intramolecular contributions due to vibrational, rotational and electronic degrees of freedom) for the reacting species and the potentials describing the interactions in the system (adequately defined force field). Since the chemical reactions are modelled by some additional “reaction” steps (a simultaneous deletion of the reactant molecules and insertion of the product molecules according to the reaction stoichiometry), the RxMC simulations can be performed under different conditions, like isobaric or isochoric, and in different statistical ensembles.3 The RxMC simulation method and its various applications have been very well reviewed in the papers by Turner et al.4 and by Dubbeldam et al.3One should note that the RxMC simulation technique has been successfully used to study reactions at high temperatures and/or under high pressure, reactions in solutions, at the interfaces between different phases and, what is of particular interest here, the equilibrium states of reactions under non-ideal conditions occurring in porous solids or near solid surfaces. The studies of chemical reactions taking place in the pores of different adsorbents are important from the practical point of view, and allow to show that the confinement may substantially affect the reaction equilibria.5–8 One should note that in the case of reactions occurring in pores, the accurate experimental determinations of equilibrium constants may be extremely challenging (if possible at all). This is caused by, among other things, the difficulties in accurate measurements of concentrations of the reacting species in pores.4,9,10
Borówko et al.5 used the RxMC method to simulate the reaction process on the model surface (being either a non-interacting hard wall and an attractive wall). The molecules of the reagents were modelled as non-interacting hard-spheres. Despite the fact that this pioneering work used some rather artificial systems (lacking the attractive interactions between the reacting molecules), it confirmed the usefulness of the method for modelling the influence of external fields (the adsorbing wall) on the chemical equilibrium. In the next paper, Borówko and Zagórski6 extended their considerations to ideal slit-like pores. They studied the equilibrium states of chemical reactions, taking into account both the interactions between reactant or product and the pore walls, as well as the interactions between the reacting molecules. Their results gave clear evidence of the significant impact of an external field generated by the pore walls on chemical equilibria, i.e., the changes (an increase or decrease) of the yield of reaction in the pore in comparison with the bulk system. The studies conducted over the last two decades have involved some more complex porous systems. For example, Turner et al.11,12 studied the ammonia synthesis in the pores being replicas of some real carbons obtained from the reverse Monte Carlo method. The comparison between the ideal slit-like pores and more realistic models of carbon demonstrated that the corrugation of pore walls and the pore connectivity did not affect the yield of ammonia synthesis much. This conclusion was the consequence of a relatively high simulation temperature, because a lower reaction temperature could possibly magnify the effects of the geometric pore characteristics. One should note that despite the fact that the RxMC method has been known for more than 20 years, a relatively small number of reports concerned the modelling of reaction equilibria in the pores.4–8,11–23 The results of RxMC simulations of reactions in porous systems published hitherto may be grouped into the following classes involving different pore models: (a) ideal pores having a well-defined geometry (infinite slit-like and cylindrical pores/nanotubes),4–8,12–17,22,23 (b) inorganic ordered adsorbents, i.e. silica,17,22 silicalite-1,18,19 pillared clays,20 MCM-41,20 and zeolites (MFI, TON, LTL and FER)19 or (c) replicas of real carbons obtained from the Reverse Monte Carlo method (RMC).8,11,21 On the other hand, different processes were simulated: (a) simple model reactions involving LJ-type interaction potentials: isomerisation reaction A ↔ B,15 dimerisation/association reaction A + A ↔ B (with the formation of a spherical product)5,16,24 or A + A ↔ A2 (with the formation of a non-spherical linear product)5 and (b) real reactions: ammonia synthesis,8,11–13,20 nitric oxide dimerisation,8,13,14,21 the hydrogen iodide decomposition,7,8,12 the propene metathesis,18,19 and CO2 methanation,17,22 and xylene isomerisation.23 It should be pointed out that the pore chemistry and the pore morphology can also be investigated.22
To summarize, the results of the above-described selected works (on the modelling of chemical reactions using RxMC method) allow a better understanding of the factors affecting the reaction equilibrium in pores. However, many issues remain unsolved. In part, this is connected to the method of modelling the pore structure. On one hand, the isolated infinite ideal slit-like pores are only a rough approximation of the real carbons. The pores of real materials are finite and interconnected, have different widths and their walls are always heterogeneous to some extent.25–27 On the other hand, the replicas of real carbons (although they are more realistic and reflect the properties of such materials rather well) do not allow the modifications to their properties, e.g. porosity. Hence, a systematic study aiming at the evaluation of the influence of porosity changes on the reaction equilibrium may be impossible to conduct with the use of the “inflexible” replicas. In order to perform such a study, other virtual porous carbons (VPCs) models seem to be more useful.28,29 The use of VPCs makes it possible to generate a series of materials with a gradually changing structure. However, according to our knowledge, there are no reports based on the series of realistic model carbons with systematically modified porosities. Typically, such studies are based only on one or few replicas of particular real carbons.8,11,21 The study described in this paper is an attempt to fill the gap.
We have considered here different simple model reactions, such as the isomerisation (A ↔ B), the dimerisation (A + A ↔ B) and the synthesis (A + B ↔ C), on the series of VPCs of systematically changing porosity. We have not strictly defined the chemical nature of the reacting molecules (they have been modelled as the single Lennard-Jones centres). This facilitates a systematic study allowing to determine qualitative relationships between the porous structure of carbons and the reaction equilibria, for various combinations of the energetic parameters of the reacting molecules and/or the different values of the equilibrium constant.
2. Simulation details
2.1 Simulation boxes
We have used the series of nine VPCs described in detail in the earlier work.30 The model carbons (shown schematically in Fig. 1a) have been generated using the simple Metropolis Monte Carlo method, and one of the most sophisticated carbon force fields, i.e. the environment-dependent interaction potential for carbon (EDIP) proposed by Marks.31,32 All the structures have been placed in the cubic simulation box (4.5 × 4.5 × 4.5 nm) with periodic boundary conditions in all three directions. The subsequent VPCs in the series differ by the density of carbon atoms. The following values of the density have been considered 0.5, 0.6, 0.7, 0.8, 0.9, 1.0, 1.1, 1.2, and 1.3 g cm−3. Each structure is denoted as dx.x where x.x is the carbon density.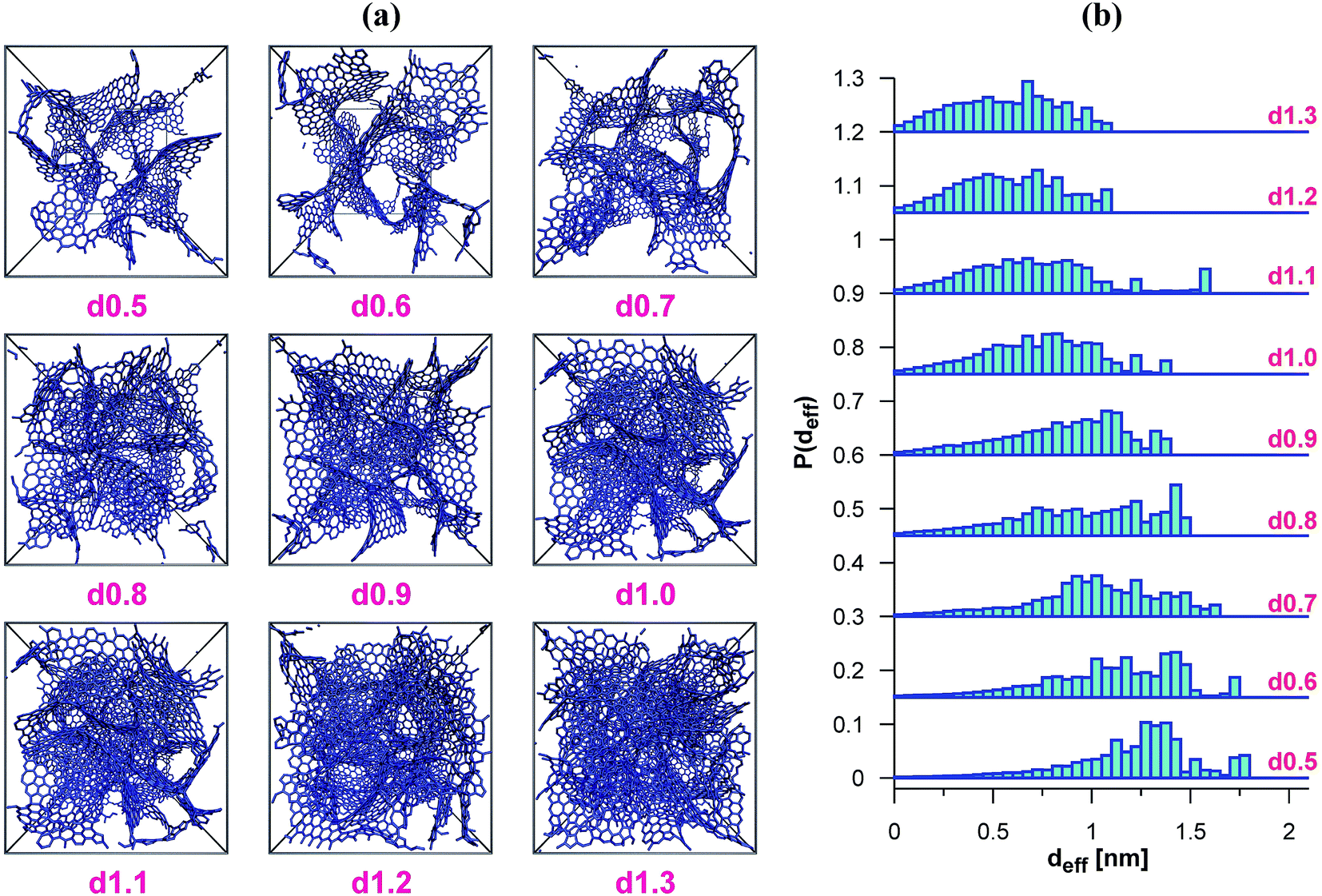 | ||
| Fig. 1 (a) The schematic representation of the considered VPCs (the frames reflect the size of the simulation box). (b) The Histograms of the pores diameters obtained with the BG method (subsequent histograms are shifted by 0.15 in comparison with the previous ones). | ||
The porosity of the carbonaceous adsorbents have been characterized by a geometrical method proposed by Bhattacharya and Gubbins (BG).33 The implementation of the method was described in details elsewhere.30,34 The BG method makes it possible to determine the histograms of effective pore diameters (deff). The data may be also used to calculate the average diameter of pores accessible for the molecules having a given diameter (i.e. the pores wider than the assumed minimum diameter (dmin)):
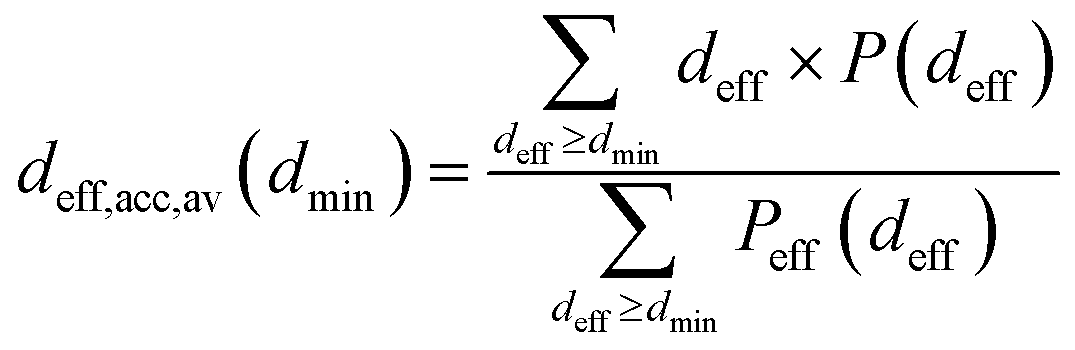 | (1) |
2.2 Chemical equilibria modelling
The majority of the studies5–8,11–16,18–21 using the RxMC method to model reaction equilibria in pores described in literature may be divided into two groups. The first group (ref. 5–7 and 15) utilizes a single simulation box containing the adsorbent structure. The initial numbers of the reacting molecules are placed into the box of constant volume (or under a constant pressure) kept during the simulation. The second group (ref. 8, 11–14, 16 and 18–21) is based on the formalism of the constant pressure Gibbs ensemble Monte Carlo.4 Here, the simulation system is composed of two subsystems. One of them is the box containing the adsorbent, while the other box contains only the bulk gaseous phase. Since the transfer of molecules between the subsystems is allowed and the first box has constant volume, the size of the bulk system is modified during the simulations in order to keep the constant pressure. This method is closer to the real experimental situation, in which the reactants are usually adsorbed from the gaseous phase. The use of Grand Canonical Monte Carlo technique (GCMC) may be an alternative for the direct modelling of the bulk reservoir. Hansen et al.18 found that the GCMC simulations are superior to the Gibbs ensemble simulations for reactions where the bulk-phase equilibrium can be calculated in advance, and does not have to be simulated simultaneously with the molecules inside the pore. In the case of such simulations, the assumption of chemical equilibrium in the bulk phase determines the values of chemical potential of the reacting species. In the GCMC simulations these chemical potentials are the same also in the pores. So, the conditions of chemical equilibrium are fulfilled also in the adsorbed phase. This state may be achieved only with the use of GCMC trial moves (creation and annihilation of the molecules of all the reacting species). The additionally performed attempts of reaction steps (especially when they are occurring with a low probability) may not give an important contribution to the determination of equilibrium composition. The same conclusions may be derived also from thermodynamic consideration. Chemical potentials of the mixtures components in two different phases are equal if the phases are in equilibrium. One can assume that there is chemical equilibrium in one of the phases (the sum of the products of the stoichiometric coefficients of all the reacting species and their chemical potentials are equal to zero, i.e. Σμiνi = 0).1,2,4 In such a situation (equality of μi in both phases) the condition of chemical equilibrium is fulfilled also in the other phase even if the reaction in this phase does not occur. The fact that reaction steps are not necessary in such simulations is indirectly also confirmed by the results obtained by Lísal et al.21 They showed that the composition of the mixture reacting in pores may be predicted with the use of single component adsorption isotherms and ideal adsorbed solution model.Since our aim is to directly simulate reactions in pores, including adsorption equilibrium with bulk phase, we have modified the scheme of GCMC simulations with the chemical reaction proposed by Hansen et al.18 In order to make the reaction steps responsible for determination of the adsorbed mixture composition (mainly the amount of the product molecules), we have assumed that the reaction can occur only in the pores (i.e. the adsorbent may be formally treated as a catalyst). Thus, the bulk phase contains only the reactants (the desorption of the product molecules has not been directly considered). In this case, the modelling of gaseous reservoir is not necessary. The adsorption of the reactants may be modelled with the use of the trial moves utilized during the Grand Canonical Monte Carlo simulations (GCMC), i.e. the creation of molecules at randomly selected positions and the annihilation of randomly chosen molecules in the simulation box.36 The other trial moves that have been used to modify the state of the system involved the random displacements of the also randomly chosen molecules and the (forward and backward) reactions steps, which are responsible for the creation and destruction of the product molecules in the pores.
The equation representing the chemical reaction may be written in the general form as:
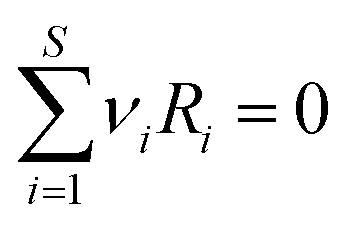 | (2) |
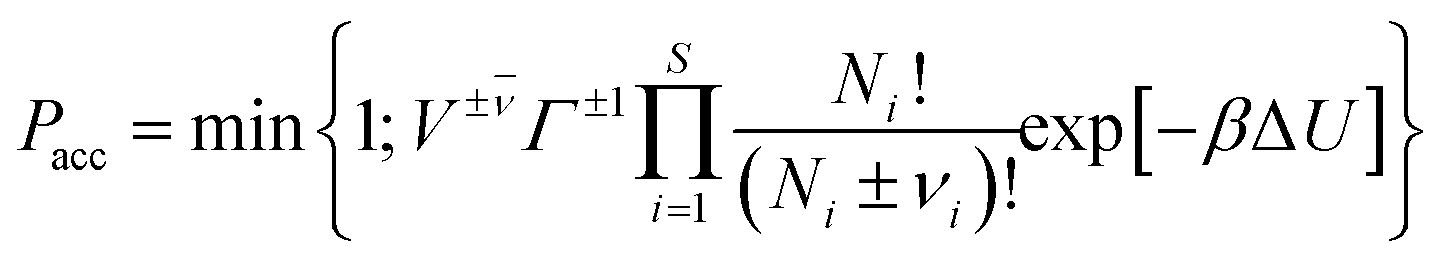 | (3) |
![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) is the sum of stoichiometric coefficients:
is the sum of stoichiometric coefficients:
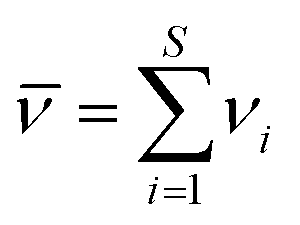 | (4) |
The parameter Γ (the reaction quotient) is related to the bulk equilibrium constant, and is defined as:
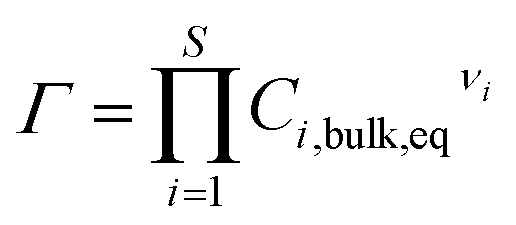 | (5) |
≠ 0). The optional sign (i.e. ‘±’) in the eqn (3) is positive (‘+’) for the forward reaction step and negative (‘−’) for the backward one.
We have used the above-described approach to model chemical equilibria in the pores of all the VPCs studied (cf. Fig. 1 and Table 1). Three model reactions have been considered:
| VPC | NSa | deff ≥ 0.3500b nm | deff ≥ 0.4410c nm |
|
||||
|---|---|---|---|---|---|---|---|---|
| deff,acc,av [nm] | Vacc | deff,acc,av [nm] | Vacc | |||||
| [nm3 per box] | [cm3 g−1] | [nm3 per box] | [cm3 g−1] | |||||
| a The number of carbon atoms in the simulation box.b The pores accessible to the reactants and also to the products of isomerisation reaction.c The pores accessible to the products of dimerisation and synthesis reactions. | ||||||||
| d0.5 | 2262 | 1.225 | 69.84 | 1.548 | 1.233 | 69.11 | 1.532 | 0.0105 |
| d0.6 | 2737 | 1.126 | 65.33 | 1.197 | 1.138 | 64.26 | 1.177 | 0.0167 |
| d0.7 | 3192 | 1.055 | 60.73 | 0.954 | 1.070 | 59.30 | 0.931 | 0.0241 |
| d0.8 | 3658 | 1.004 | 56.17 | 0.770 | 1.025 | 54.20 | 0.743 | 0.0365 |
| d0.9 | 4119 | 0.909 | 51.80 | 0.631 | 0.933 | 49.50 | 0.603 | 0.0465 |
| d1.0 | 4573 | 0.792 | 46.87 | 0.514 | 0.820 | 43.71 | 0.479 | 0.0723 |
| d1.1 | 5035 | 0.792 | 42.23 | 0.421 | 0.832 | 38.20 | 0.380 | 0.1055 |
| d1.2 | 5492 | 0.676 | 36.39 | 0.332 | 0.722 | 31.17 | 0.285 | 0.1678 |
| d1.3 | 5949 | 0.674 | 32.08 | 0.270 | 0.716 | 27.84 | 0.235 | 0.1520 |
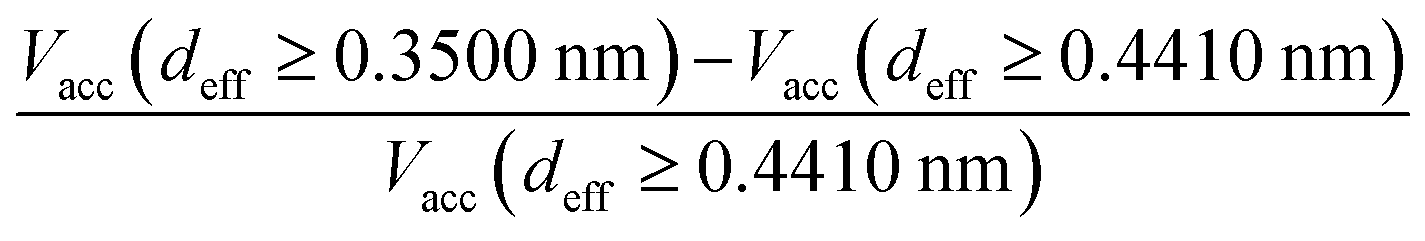
(a) isomerisation:
| A ↔ B | (6) |
(b) dimerisation:
| A + A ↔ B | (7) |
| A + B ↔ C | (8) |
In order to simplify the calculations, we have assumed spherical symmetry for all the reacting molecules (A, B and C). The physical interaction between all the species involved has been assumed as described by the Lennard-Jones (LJ) potential. The cross-interaction parameters have been calculated using the usual Lorentz–Berthelot mixing rules.36 It should be noted that for simplicity, during the discussion in paragraph 3, we have given only the values of the potential well depth of interactions between the molecules of the same type (i.e. εii). However, the analysed changes are also the consequence of differences in energy of interactions with other species including adsorbent atoms and here the assumed mixing rules are also crucial. For the carbon atoms, we have used the following values of LJ parameters: σSS = 0.34 nm and εSS/kB = 28 K.37 The interactions between all the pairs have been cut at the distance equal to 5 × σij. We have not applied the long range tail correction since the assumed conditions are far from any points of phase transitions. Our study has had qualitative character and this numerically costly procedure was not necessary. We have also fixed the LJ parameters for the A molecules by taking σAA = 0.35 nm and εAA/kB = 100 K (in all the cases). Besides, in the case of synthesis reaction, we have assumed that the collision diameter for molecules of reactant B is the same as for A, i.e. σBB = σAA = 0.35 nm. The collision parameters of the product molecules have been calculated assuming that the volume of the product molecule is equal to the sum of the volumes of reactant molecules. Only in the isomerisation reaction the volumes of the reactants and products are the same. This arbitrary assumption reduces the number of parameters whose influence should be tested. Analogical simplifications were also applied by others.6 Thus, in the case of the isomerisation reaction, σBB = σAA, for the dimerisation and the synthesis reactions, we have taken σBB = 21/3 × σAA and σCC = (σAA3 + σBB3)1/3, respectively. In consequence, the product collision diameter for the isomerisation reaction was equal to 0.3500 nm and for the dimerisation and the synthesis reactions it was equal to 0.4410 nm. We are aware, of course, that in the case of dimerisation and synthesis reactions in the narrow pores, the assumption of spherical symmetry for the products of reactions is a rather crude approximation. The non-sphericity of the products may affect the results considerably, and this problem will be addressed in the future papers.
All the reaction equilibria have been studied at the fixed temperature of T = 298 K. Each simulation (for the chosen set of parameters: the equilibrium constant (Γ), the LJ parameters, and the pressure of the reactant(s) in the gaseous phase) has consisted of 1 × 107 cycles. During one cycle, 5000 iterations have been preformed. Each iteration has been a single attempt to change the system state via a randomly chosen trial move (the creation or the annihilation of the reactant molecule and displacement of randomly chosen molecule (both the reactant as well as the product)). In addition, during randomly chosen iterations the (forward or backward) reaction step has been performed (with probability equal to 1/500). During the reaction steps, the position of created molecule (one of the molecules) in half of the randomly chosen cases was the same as the position of removed molecule (one of the molecules). In other cases, new molecules have been created in the randomly chosen position in the box. Table S1 in the ESI† collects the acceptation criteria of different trial moves. The first 4 × 106 cycles (2 × 1010 GCMC trial moves and ca. 4 × 107 attempts of the reaction step) have been treated as equilibration and the data collected during the next 6 × 106 cycles (3 × 1010 GCMC trial moves and ca. 6 × 107 attempts of the reaction step) have been used to calculate averages. In the case of isomerisation and dimerisation reactions, we have assumed the pressure of the reactant (A) in the gaseous phase to be equal to 0.1 MPa. In the case of the synthesis reaction, we have assumed the total pressure of the reactants in the gaseous phase (i.e. pA + pB) to be equal to 0.1 MPa. For all the considered reactions and the combination of parameters, we have also simulated the reaction equilibria in the bulk phase. The methodology of these computations has been exactly the same as described above (i.e. the reactant(s) molecules have been created and destructed according to the GCMC trial moves and the product molecules have been introduced/removed into/from the simulation box via the reaction steps in the absence of adsorbent), but we have used the box of the size 20 × 20 × 20 nm without the adsorbent. The results for the bulk phase have been treated as reference.
3. Results and discussion
3.1 Characteristics of VPCs
In Table 1 we have summarized the parameters characterizing the VPCs considered, and the histograms of their pore diameters have been shown in Fig. 1b. It is evident that all the structures are strictly microporous, i.e. the pore sizes do not exceed 2 nm. The lack of wider pores results from the relatively small sizes of the model structures. Real carbons usually contain wider pores, but the micropores are predominantly responsible for their adsorption properties.25–27,38 One should note that the synthesis methods leading to highly microporous carbons have also been reported, and these materials seem to be very promising adsorbents.25,27From the results given in Fig. 1b it follows that the porosity of the VPCs considered here changes systematically, i.e. passing from the d0.5 carbon to the d1.3 one, the contributions vanish gradually due to the wider pores. The last two VPCs (i.e. d1.2 and d1.3) have only quite small pores, about 1 nm in diameter. In consequence, the average pore diameter decreases as well (Table 1). The only exception is the d1.1 structure, which contains a small amount of wider pores (ca. 1.6 nm) and its average pore diameter deviates slightly from the general trend.
3.2 Isomerisation reaction
We have begun the discussion of the chemical reactions in VPCs by considering the isomerisation reaction. Fig. S1 in the ESI† compares the densities of the reacting molecules in pores and the mole fractions of the product in different VPCs, obtained for different values of the equilibrium constant (Γ). The assumed values of Γ correspond to the following ratios of average numbers of A and B molecules for reaction in the bulk phase: 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 2:1, 1:1, 1:2 and 1:4. The simulations have been carried out assuming that the potential well depth for the product molecules (εBB/kB = 120 K) is slightly higher than for the reactant molecules (εAA/kB = 100 K). The concentrations of the reactant and product molecules increase with passing from the carbon d0.5 to d1.3, regardless of the value of equilibrium constant, since a gradual decrease of the pore diameters causes the rise in adsorption. In general, an increase of the equilibrium constant is expected to cause a rise in the amount of the product in the system, and this is illustrated by the results given in Fig. S1 in the ESI.† The carbon porosity affects the densities of reacting molecules, but it does not affect appreciably the reaction yield. The analysis of the product mole fractions reveals that in all the systems the reacting mixture in pores is richer in B molecules, than for the same reaction taking place in the bulk phase. Moreover, the effect of confinement on the reaction yield is gradually enhanced when the average pore size decreases. The observed behaviour results from the differences in the energetic parameters of the reacting molecules (higher ε value for the product). Thus, the rise in the carbon density and/or the reduction in the pore diameters enhances the adsorption of the product molecules (B). Besides, the confinement and the resulting adsorption of the species result in their higher concentration, as compared with the bulk.
:1, 2:1, 1:1, 1:2 and 1:4. The simulations have been carried out assuming that the potential well depth for the product molecules (εBB/kB = 120 K) is slightly higher than for the reactant molecules (εAA/kB = 100 K). The concentrations of the reactant and product molecules increase with passing from the carbon d0.5 to d1.3, regardless of the value of equilibrium constant, since a gradual decrease of the pore diameters causes the rise in adsorption. In general, an increase of the equilibrium constant is expected to cause a rise in the amount of the product in the system, and this is illustrated by the results given in Fig. S1 in the ESI.† The carbon porosity affects the densities of reacting molecules, but it does not affect appreciably the reaction yield. The analysis of the product mole fractions reveals that in all the systems the reacting mixture in pores is richer in B molecules, than for the same reaction taking place in the bulk phase. Moreover, the effect of confinement on the reaction yield is gradually enhanced when the average pore size decreases. The observed behaviour results from the differences in the energetic parameters of the reacting molecules (higher ε value for the product). Thus, the rise in the carbon density and/or the reduction in the pore diameters enhances the adsorption of the product molecules (B). Besides, the confinement and the resulting adsorption of the species result in their higher concentration, as compared with the bulk.
In order to check the effects of the differences in the adsorption energetics of reacting molecules, we have performed the simulation for different values of the potential well depth of the product, assuming that Γ = 1, i.e. for the A to B ratio equal to 1:1, when the reaction takes place in the bulk phase. The obtained results are shown in Fig. S2 in the ESI.† As expected, when εBB = εAA (εAA/kB = 100 K) the average concentrations of the reactant and the product molecules are the same. The differences in adsorption energetics of the reacting species cause that the preferentially adsorbed compound (A or B) dominates (it should be noted that the number of (A) molecules is mainly determined by its pressure in the bulk phase which is the same for all the systems). Of course, these energetic parameters hardly affect the composition of the bulk reacting mixture. Since the densities of the reacting species are low, intermolecular interactions have a negligible impact. When the product molecules (B) are preferentially adsorbed, the reaction yield in pores is higher than in the bulk phase. On the other hand, when εBB < εAA,, an opposite effect appears, and the reaction yield in pores is lower than in the bulk. A comparison of the data for different VPCs clearly reveals some regularities. The reduction in the pore sizes and/or the rise in the carbon density cause the product mole fraction to increase (decrease), when the product adsorption energy becomes higher (lower).
3.3 Dimerisation reaction
Before we start the discussion of results for the dimerisation reaction, one important fact should be noted. In the case of interactions with complex structures (for example carbonaceous), the energy of adsorbed molecules depends not only on the potential well depth but also on the collision diameter as, for example, in the analytical Steele potential (interactions with ideal flat walls).37 However, this dependence is not clearly defined in the case of disordered structure. For the dimerisation (and also synthesis reactions) we have assumed that the size (collision diameter) of the product molecules (B) is bigger than of the reactant molecules (A). Therefore, it is very hard to find the value of ε for (B) that would result in the same energetics of adsorption of the A and B species. However, we have found the base value (εBB/kB = 60 K) which results in the similar energetics of solid–fluid interactions.Fig. S3 in the ESI† presents the composition of the reacting phase in the dimerisation reaction occurring in our porous materials for different values of the equilibrium constant (in this case we have assumed that εBB is 20% higher than the base value). The assumed values of Γ correspond to the following ratios of average numbers of A and B molecules for reaction in the bulk phase: 4:1, 2:1, 1:1, 1:2 and 1:4. The observed regularities are very similar to those found for the isomerisation reaction (cf. Fig. S2 in the ESI†). However, the average densities of the product molecules (B) are lower, since the volume occupied by a single (B) molecule is now higher. The assumed value of the potential well depth for the product molecules results in their higher adsorption, and hence in the higher reaction yield than in the gaseous phase. Regardless of the value of the equilibrium constant, the mole fractions of the product increases up to the d1.0 carbon, while it is nearly constant, or decreases slightly when Γ is sufficiently high, for the VPCs of high concentration of micropores. A careful analysis of the results reveals also a small anomaly observed for the carbon d1.2, which points to a slightly lower reaction yield than the adjacent structures (d1.1 and d1.3). Fig. 2 presents the product mole fraction in different porous carbons obtained for the systems characterized by different values of potential well depth for B molecules. The middle value of εBB/kB = 60 K has been chosen is such a way that the reaction yields for all the carbons have been similar to the values corresponding to the reaction in the bulk. This value of εBB corresponds to a similar adsorption energetics of (A and B) molecules. Similarly to the case of the isomerisation reaction, the values of εBB higher (lower) than 60 K, lead to the higher (lower) product mole fraction than in the bulk phase. The reduction of pore sizes and/or the rise of the carbon density significantly changes the product mole fraction. This effect is strong for the carbons up to d1.0, and becomes rather small for the carbons d1.1, d1.2 and 1.3. A small negative anomaly for the carbon d1.2 is observed again (with the exception of the case with the lowest value of εBB/kB = 36 K).
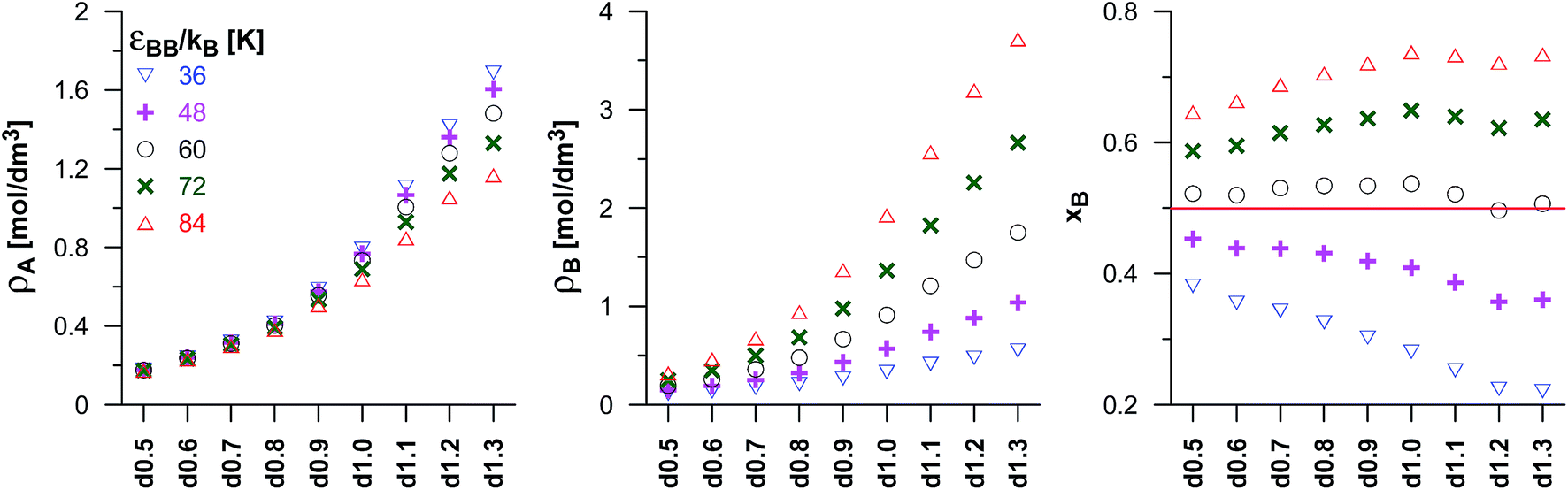 | ||
| Fig. 2 The comparison of the composition of the reacting phase (T = 298 K) in pores of all the considered VPCs for the dimerisation reaction (A + A ↔ B). The data corresponding to different values of the potential well depth for product molecules (εBB) and the same values of εAA and equilibrium constant (Γ = 41.1 nm3). The subsequent panels present the average densities of the reactant (ρA) and the product (ρB) in pores (the densities have been calculated per the volume of accessible pores) and the mole fraction of the product in the mixture (xB). The xB mole fractions for the reactions in the bulk phase are shown as horizontal solid lines. It should be noted that these lines are overlapped. | ||
3.4 Synthesis reaction
In the case of synthesis reaction, the situation becomes more complex. The system contains two kinds of reactants (A and B) molecules in the bulk phase, and three kinds of particles (the reactants A and B, and the product C), and there are more parameters that have to be taken into account. The first series of the calculations performed has concerned the effects due to the value of equilibrium constant on the reaction yield. Therefore, we have kept all other parameters fixed. We have also assumed equimolar composition of the bulk phase, i.e. pA = pB. However, the reactants A and B have been assumed to be characterized by different values of the potential well depth (εAA/kB = 100 K and εBB/kB = 110 K). In consequence, the component B adsorbs more strongly and its density in the system is higher. For the assumed value of εCC/kB = 72 K for the product C, its concentrations in porous materials are larger than in the gaseous phase (the used values of Γ correspond to the following ratios of the numbers of molecules A:B:C: 4:4:1, 2:2:1, 1:1:1, 1:1:2 and 1:1:4 in the bulk). The results obtained have been shown in Fig. S4 in the ESI.† Of course, the rise in the value of Γ increases the reaction yield. The higher concentration of C molecules hardly affects the concentrations of the reactants (A and B) for the carbons from d0.5 up to the d0.8, but leads to a decrease of their concentrations in highly microporous VPCs. The differences between the product mole fractions in pores and in the gaseous phase for all the values of Γ are always positive, and again a small anomaly for the carbon d1.2 is observed, i.e. the product (the reactants) mole fraction of product is slightly lower (are slightly higher) than in the case of adjacent VPCs (d1.1 and d1.3).
We have also studied the effects due to changes in the potential well depths of the product molecules (εCC) on the reaction equilibrium, and the results have been presented in Fig. 3. The assumed value of Γ corresponds to the A:B:C ratio in the bulk phase equal to 1:1:1 and the values of the other parameters have been kept the same as before. The observed changes are qualitatively consistent with the previously discussed effects of the changes of the energetic parameter of the product molecules for the dimerisation reaction (Fig. 2). Thus, the middle value of εCC (60 K) results in similar mole fractions of the product in pores and in the bulk phase. The higher values of εCC lead to higher reaction yields than those obtained for the reaction in the gas phase, and the lower values of εCC result in the lower values of xC. The increase as well as the decrease of the reaction yield becomes higher when average pore size decreases up to the carbon d1.0. The last three VPCs in the series (d1.1–d1.3) have been found to have similar composition of the reacting mixture. One should note, however, that the anomaly for the d1.2 structure appears again (it is well seen for the higher values of εCC).
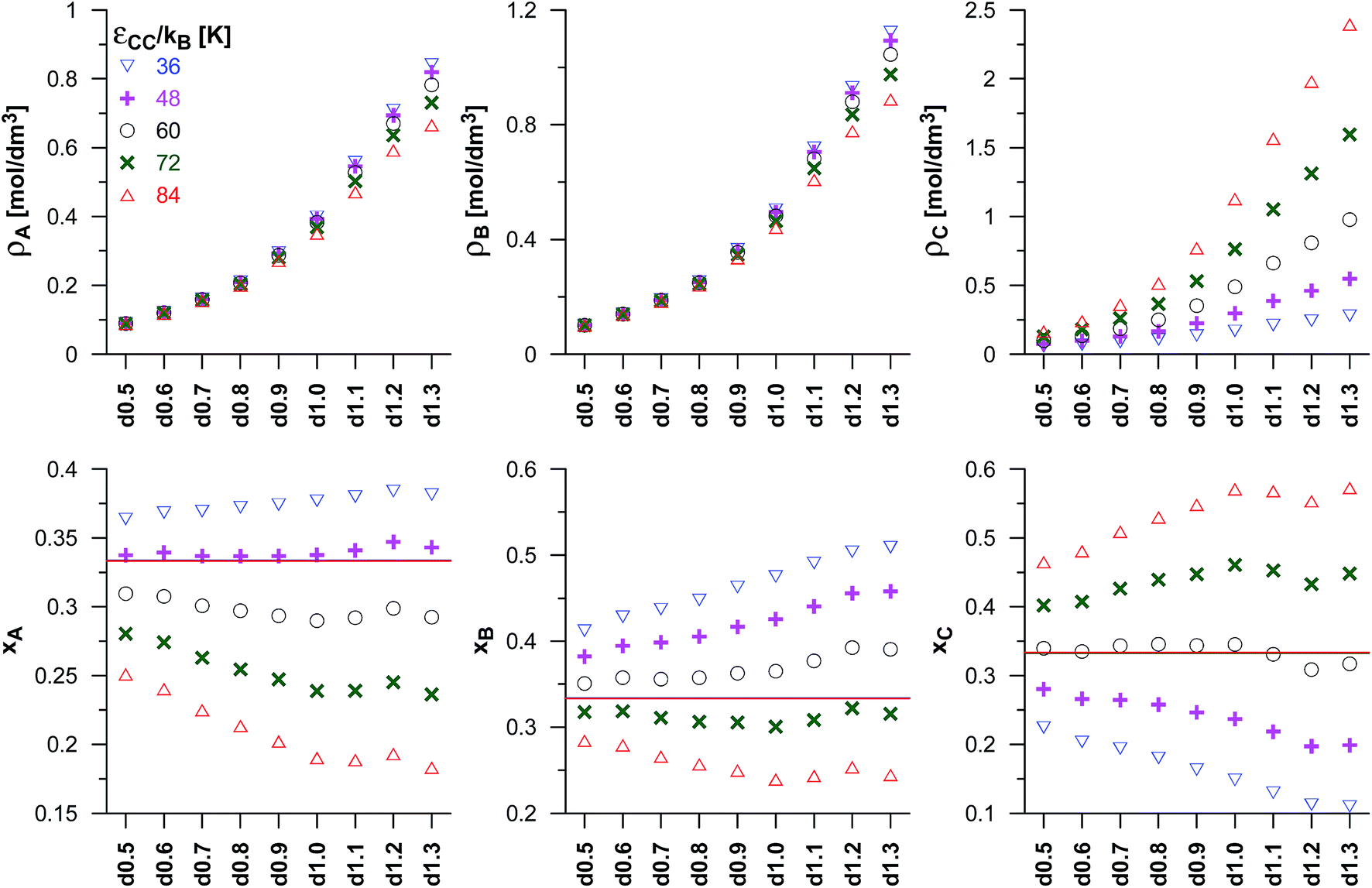 | ||
| Fig. 3 As in Fig. 2 but for the synthesis reaction (A + B ↔ C). The effects of the changes in potential well depth for product molecules (εCC) for the fixed values of the equilibrium constant (Γ = 82.3 nm3) and the potential well depth of the reactant B molecules (εBB/kB = 110 K). The upper panels present the average densities of the reacting species (ρA, ρB and ρC). The lower panels show their mole fractions in the mixture (xA, xB and xC). The mole fractions for the reaction taking place in the bulk phase are marked by horizontal lines. | ||
The reaction yield depends not only on the energy adsorption of the product molecules (εCC), but it is also affected by the adsorption of the reactant molecules (A and B). We have considered the changes in εBB only, while εAA is fixed and equal to 100 K. Fig. 4 shows the changes of the reacting phase composition in different carbons obtained for various choices of the potential well depth for the reactant B. As expected, the rise in εBB causes a gradual increase of the density and mole fraction of the adsorbed B molecules. One should note that the increase of εBB does not affect the adsorption of the other reactant A and of the product C in the carbons from d0.5 up to d0.9. In the case of highly microporous VPCs (d1.0–d1.3), a slight decrease of the numbers of A and C molecules has been found. It may be interpreted as the effect of blocking the high-energetic adsorption centres by the adsorbed B molecules. The effect of the rise of the number of B molecules in pores (connected with the increase of εBB) on the number of product molecules is rather surprising. According to the Le Chatelier's principle, an increase of the reactant concentration should lead to the increase of the product concentration. This has not been observed. However, as already mentioned, the changes of the concentration of B in the carbons d0.5 to d0.9 do not affect the concentration of C for the carbons d0.5 d0.9, and for the most microporous VPCs (d1.0–d1.3) even cause a small decrease of the C density. In order to explain this observation, a simple thermodynamic argument can be used. One can assume that the reaction occurs in the pores as well as in the bulk phase, but the transport of the product molecules between gaseous phase and pores is forbidden. The partial pressure of C in the bulk for given values of the reactants partial pressures is determined by the equilibrium state. Under given conditions, the composition of the reacting gaseous phase is almost independent on the energetic parameters of the B molecules (see the horizontal lines in the lower panels of Fig. 4), the partial pressure of C is the same in all the systems. There are the following equilibria in the system: Abulk + Bbulk ↔ Cbulk, Abulk ↔ Apores, Bbulk ↔ Bpores, and Apores + Bpores ↔ Cpores. Of course, the chemical potential of every component in all the phases must be the same, since the molecules in pores and in the bulk phase are in equilibrium. Despite the fact that C molecules are not transferred from the bulk phase to the pores (and vice versa), the chemical potential of this compound in both phases is determined by the reaction equilibrium and it is the same. Therefore, the amount of the product in pores is mainly determined by its hypothetical partial pressure in the bulk (almost the same for all the systems), and hence similar product densities may be expected for the given carbon. The observed small decrease of the product density in the most microporous VPCs may be interpreted as the effect of blocking the adsorption places by preferentially adsorbed B molecules.
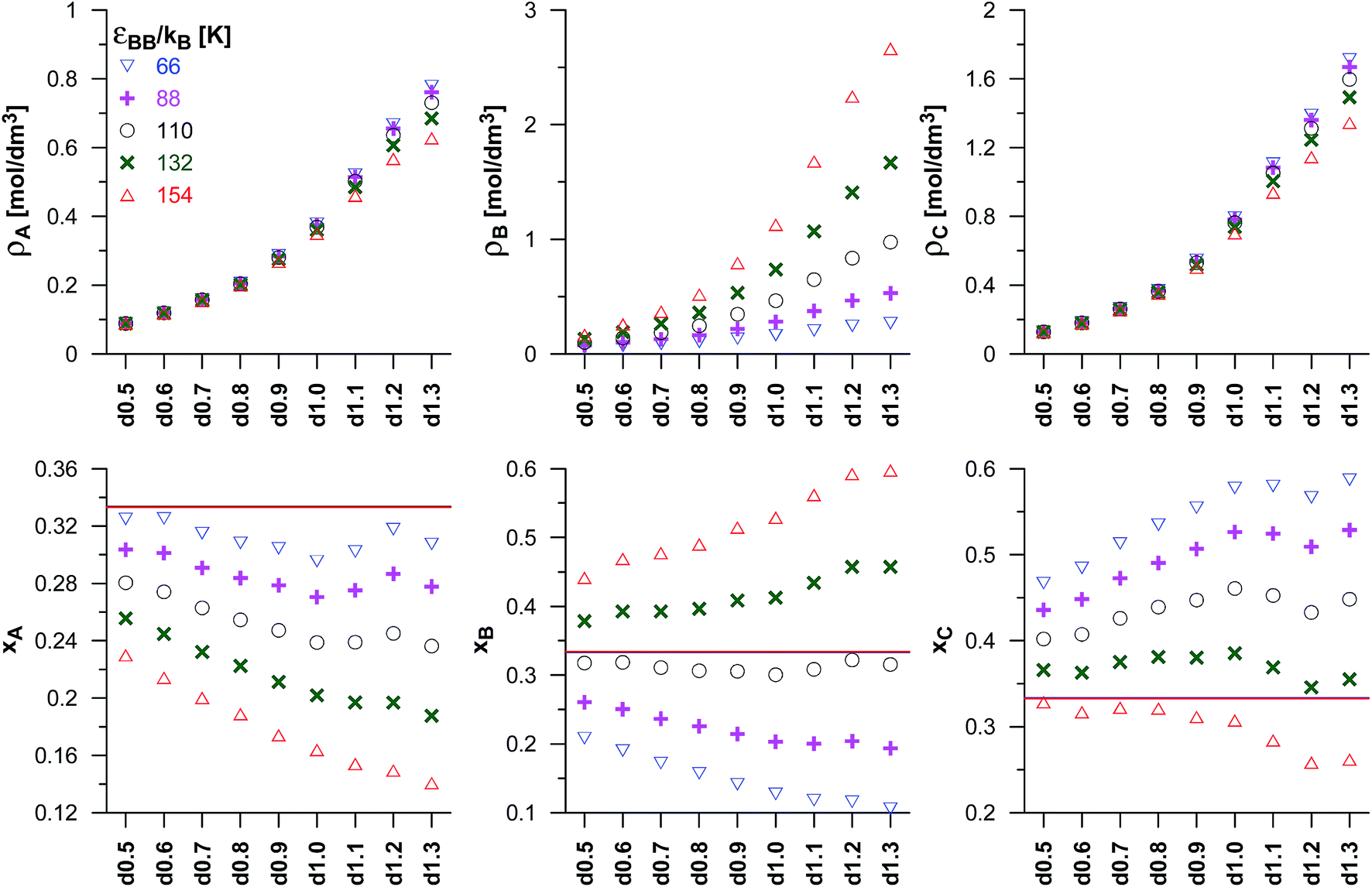 | ||
| Fig. 4 As in Fig. 3, but for the fixed values of the equilibrium constant (Γ = 82.3 nm3) and of the potential well depth of the product (εCC/kB = 72 K), and for different values of the potential well depth for reactant B (εBB). The values of the reactants pressure in the bulk phase have been fixed and equal to pA = pB = 0.05 MPa. The horizontal lines in lower panels mark the mole fractions for the reaction taking place in the bulk phase (it should be noted that these lines are overlapped). | ||
Finally, we have also studied the effects of the bulk phase composition on reaction equilibria in pores of the selected carbons. We have fixed the energetic parameters of all the components, the equilibrium constant, and assumed a constant value of the total pressure of both reactants in the gaseous phase (i.e. pA + pB = 0.1 MPa). Then, we have changed the partial pressures of the components. The simulation results have been shown in Fig. 5. Apart from the obvious changes of the reactants mole fractions, we have observed a clear maximum of the product mole fraction when the partial pressures of A and B are (the same in the bulk) nearly the same in porous systems. This, of course, results from the reaction stoichiometry, and the small asymmetry of xC in porous systems results from different values of εAA/kB = 100 K and εBB/kB = 110 K. It is noteworthy that the product mole fraction in porous systems is considerably higher than in the bulk.
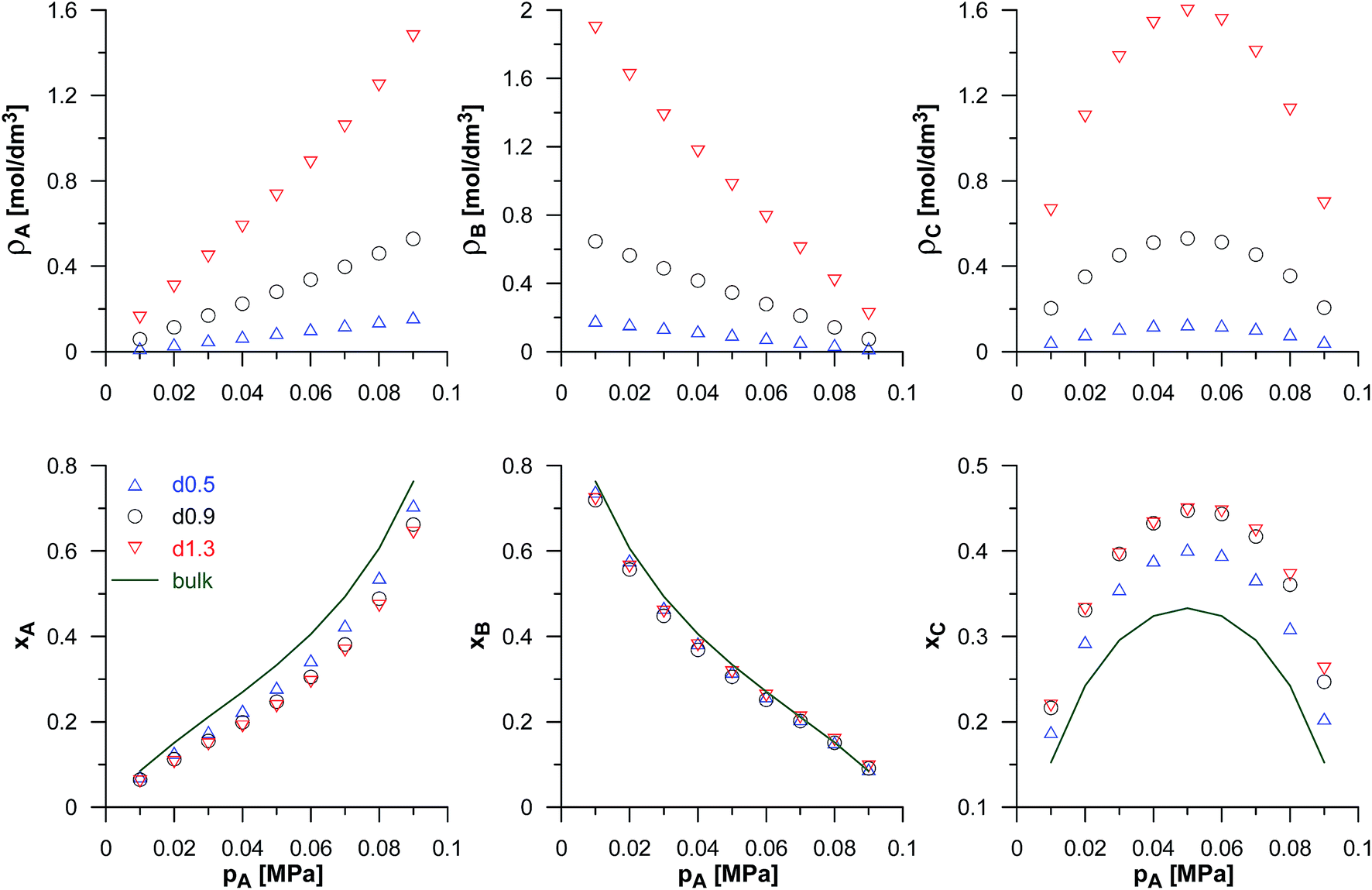 | ||
| Fig. 5 Effects of the bulk phase composition (total pressure of the reactants in the bulk phase pA + pB = 0.1 MPa) on the composition of reacting phase in pores of selected VPCs (d0.5, d0.9 and d1.3) for the synthesis reaction (A + B ↔ C). The applied values of parameters: Γ = 82.3 nm3, εBB/kB = 110 K, and εCC/kB = 72 K. The average densities and mole fractions of the reacting species in pores have been shown in upper and lower panel, respectively. The mole fractions for the reaction in the bulk phase are presented as lines. | ||
3.5 General relationships
The above discussed qualitative relationships between the reaction equilibria in pores of activated carbons with systematic changes in their porosity have shown that the adsorption energetics plays an important role. Here, we try to determine some quantitative dependencies. One of the most obvious parameters characterizing porous structures is the average pore diameter. This parameter is also closely related to the energetics of adsorption. Fig. 6–8 show the correlations between the product mole faction and the reciprocal of the average diameter of pores accessible for the product molecules (see Table 1) for different reactions. The lowest value of 1/deff,acc,av is related to the initial VPC in the series (i.e. d0.5) and the highest values are related to the final VPCs (i.e. d1.2 and d1.3). In the case of isomerisation reaction (Fig. 6), we have found practically linear changes of xB with 1/deff,acc,av. In this case, the reactant (A) and the product molecules (B) are of the same size, so that they demonstrate the same geometrical constrains within the porous structure. By increasing the equilibrium constant, in the systems with all the energetic parameters fixed, the reaction yield increases when the average diameter of accessible pores becomes lower. On the other hand, for a given value of the equilibrium constant, the relative energy of adsorption of the reactant and of the product determine whether the product mole fraction increases (εAA < εBB) or decreases (εAA > εBB). This reflects the fact that when the reactant is more strongly adsorbed than the product, the reaction is hindered, while in the case of stronger product adsorption, the reaction yield becomes higher.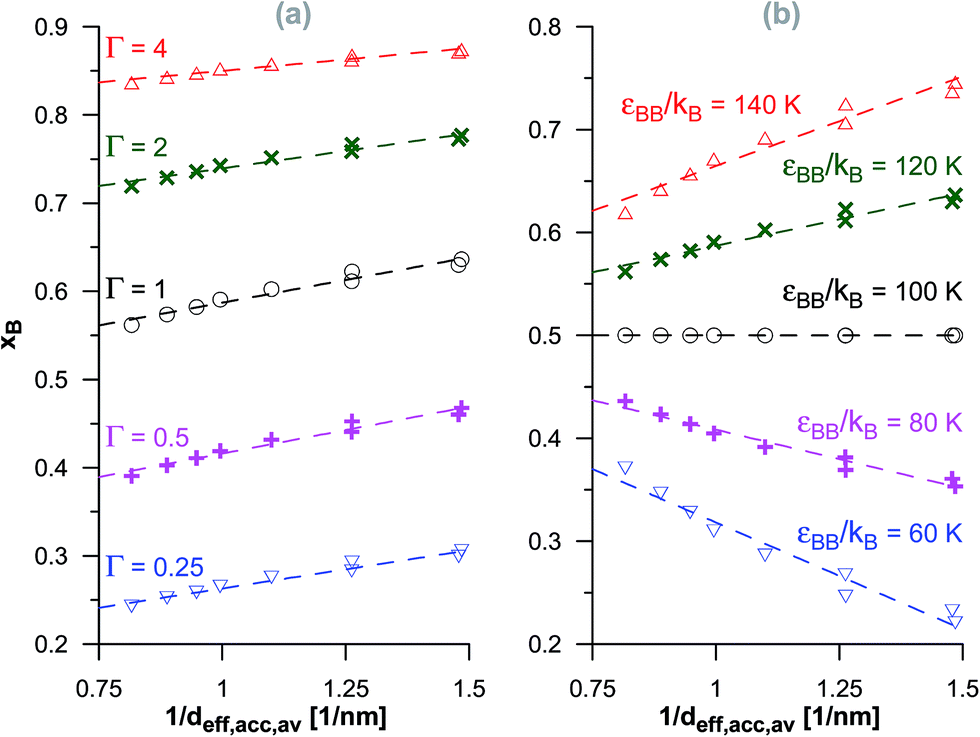 | ||
| Fig. 6 The changes of the product mole fraction (xB) for the isomerisation reaction versus the reciprocal of the average diameter of the pores accessible for the product molecules (1/deff,acc,av), obtained for the systems presented in Fig. S1† (a) and in Fig. S2† (b). The meaning of symbols as in Fig. S1 and S2 in the ESI.† The dashed lines have been drawn to guide the eye. | ||
In the case of the dimerisation (Fig. 7) and synthesis (Fig. 8) reactions, the situation is different, since the product molecules are larger than the reactant molecules. In the case of systems with large contribution of wider pores, the confinement effects are rather limited, which causes the linear changes of the product mole fraction versus 1/deff,acc,av as well as versus the adsorption energy of the product molecules to appear again. However, in the case of highly microporous structures, the deviations from linearity are observed, since the accessibility of very narrow pores to the product molecules is considerably hindered. This causes that the reaction yields are rather similar for those VPCs. Fig. 9 shows the ratio of the volume accessible only to the reactant molecules (0.3500 nm ≤ deff < 0.4410 nm) to the volume accessible to all the molecules (deff ≥ 0.4410 nm). This ratio increases when the contribution due to narrow pores becomes higher. This fact (the presence of pores inaccessible to the product) explains why the yield of dimerisation and synthesis reactions does not increase proportionally to the reduction in the pore diameters for highly microporous carbons. The highest value of the analyzed parameter is observed for the structure d1.2, and it is responsible for the already mentioned anomalies in the behaviour of this carbon.
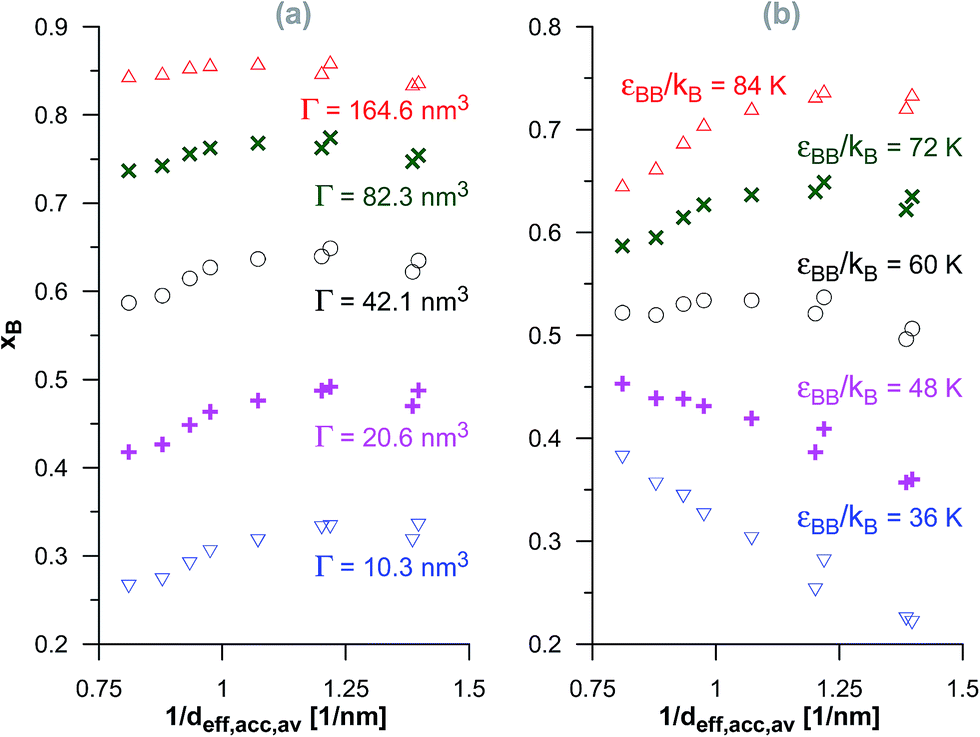 | ||
| Fig. 7 The changes of the product mole fraction (xB) for the dimerisation reaction versus the reciprocal of the average diameter of the pores accessible for the product molecules (1/deff,acc,av) for all the systems presented in Fig. S3 in the ESI† (a) and in Fig. 2 (b). The meaning of symbols as in Fig. S3† and 2. | ||
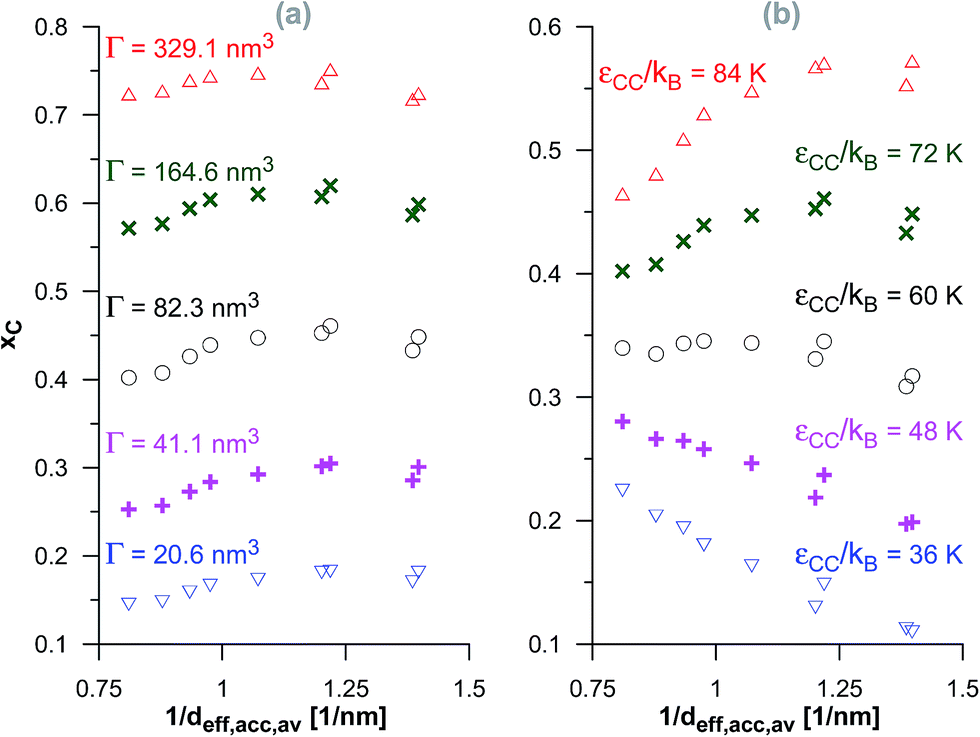 | ||
| Fig. 8 The changes of the product mole fraction (xC) for the synthesis reaction versus the reciprocal of the average diameter of the pores accessible for the product molecules (1/deff,acc,av) for all the systems presented in Fig. S4 in the ESI† (a) and in Fig. 3 (b). The meaning of symbols as in Fig. S4† and 3. | ||
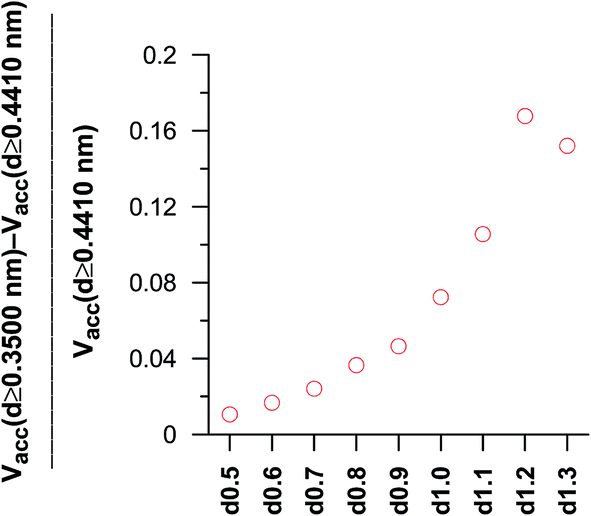 | ||
| Fig. 9 The comparison of the ratio of the volume accessible only for the reactants molecules to the volume accessible for both the reactants and the product molecules in the dimerisation and synthesis reactions. | ||
To summarize, the chemical equilibria in activated porous carbons have been found to be influenced by the energetic parameters of the reacting molecules as well as by the geometrical confinement. When the adsorption energy of the product is higher than that of the reactant or the reactants, the reaction yield is higher than in the bulk. In this case, the reduction of pore sizes leads to higher reaction yields. In the opposite situation (the preferential adsorption of the reactant or reactants), the yield of reaction under confinement is lower than in the bulk phase. In such cases, the adsorbents with wider pores are a better medium for the reactions. Besides, when the product molecules are larger than the reactant, the presence of pores inaccessible to the product causes a reduction of the reaction yield, regardless of the energetic parameters of the reacting species.
Finally, one may raise a question about the relation between the systems with chemical reaction in pores in equilibrium with the bulk containing non-reacting reactant/reactants (such systems have been considered in this paper) and the systems with chemical equilibria in both the pores and the gaseous phase. We have performed some additional simulations, taking the isomerisation and dimerisation reactions as examples, and using the selected carbons of different microporosity (d0.5, d0.9 and d1.3). The concentration of the reactant in the bulk phase has been controlled by its pressure (we have assumed a fixed value). Assuming that the reaction occurs also in the gaseous phase (cf. the discussion of the results given in Fig. 4), one can expect the concentration and the partial pressure of the product to be determined by the value of the reaction constant. These values for the bulk can be calculated easily. We have considered only a low pressure, implying that the bulk density is also low. Under such conditions, the intermolecular interactions hardly affect the equilibrium state. We have considered the systems characterized by different values of Γ, and performed the simulations using two different schemes. In the first scheme, the reactant molecules (A) have been created and annihilated in the pores according to the GCMC trial moves (adsorption equilibria with the bulk phase), and the product molecules (B) have been formed or destructed only via the reaction step. In the other scheme, adsorption equilibria concerned B molecules, and A molecules were created and destructed only during the reaction. The comparison of the results obtained with the help of both schemes has been shown in Fig. S5 and S6 in the ESI.† As can be observed, the densities of the reacting molecules in pores are the same in both cases. These results suggest that the methodology of simulations used here (assuming that the reaction takes place in the pores only) is equivalent to the systems in which the reaction occurs in the pores and in the bulk phase.
3.6 Comparison with the results of other studies
As it was mentioned in the Introduction, experimental studies of chemical equilibria are rather difficult to carry out. Hence, the reports on the measurements of compositions of mixtures reacting in carbonaceous pores are also rare. Typically, such studies are restricted to examination of the particular materials.39–43 There is lack of reports based on the series of materials significantly differing in porosity. If the series of materials were considered, the main idea was to study the effects of surface functionalities or other chemical modifications.10,44,45 However, in the current study we have neglected the influence of chemical heterogeneity (the raw carbons have been considered) and we plan to study the effects due to the presence of different surface groups on chemical equilibria in the future. So, the influence of changing in the porous structure of activated carbons has not been investigated sufficiently. According to our knowledge, there is also a lack of theoretical studies designed to evaluate directly such effects in realistic models of heterogeneous carbon pores. The only reports concerning this problem are based on the model of ideal pores (slit-like or cylindrical ones).4–8,12–17,22,23The results of theoretical studies are usually affected by the choice of the model reaction (for example the NO dimerisation or the NH3 synthesis).8,11–14,20,21 Several reports confirmed the positive effect of confinement on the reaction yield.6,8,11,12,21 However, such conclusions are the consequence of energetics of the reacting molecules adsorption (higher energy for product molecules). In our investigation we have tested systematically different combinations of energetic parameters of the species and we have also shown that the opposite situation (i.e. lowering of the reaction yield under confinement) may occur. As we have discussed above, such behaviour has been observed if the energy of the product molecules adsorption is lower than for the reactant(s). Similar observations were reported by Domínguez.15 However, his study was restricted only to the isomerisation reaction in ideal slit-like pores. It should be also noted that, despite the lower reaction yield in the discussed case, the confined reacting mixture has higher density than he corresponding bulk phase.
When it comes to the effects of the pore diameters, qualitative regularities reported by us are compatible with the result of simulations in ideal pores.4–8,12–17,22,23 In the case of energetic preferences of the product adsorption, the reduction of pore diameters causes the rise in the product mole fraction. Such behaviour was also observed for the NO dimerisation in slit-like and/or cylindrical pores,8,13,14 the NH3 synthesis8,12,13 and the model dimerisation reaction (A + A ↔ B) in slit-like pores.6 In the latest case, the potential well depth for the reactant and the product molecules was assumed to be the same. However, the collision diameter for the B molecules was higher and, as we have discussed in the Section 3.3, it resulted in the higher interaction energy of the B molecules with the walls. Clearly, the model of ideal pores is only the rough approximation. It does not reflect the complex nature of the porosity of real adsorbents. Ideal models neglect many features of the pores as, for example, the distribution of their diameters, their finite sizes, heterogeneity or connectivity.26–29 Such simplifications can also affect the results of the modelled reactions under confinement. This was demonstrated, among others, by Lísal et al.21 They compared the results of RxMC simulation of the NO dimerisation in ideal slit-like pores and in three models/replicas of disordered nanoporous carbons obtained from the reverse Monte Carlo method. The use of such realistic replicas led to significantly different behaviour of reacting mixtures in comparison to the ideal pores.
In the context of the above-mentioned experimental difficulties and insufficiency of other results of theoretical research, the current study is the first systematic report on the relationships between the chemical equilibria and the porous structure of carbons and the energetic parameters of reacting molecules. Despite the fact that qualitative regularities described by us are based on the simple model reactions, we believe that our conclusions are universal and they can be adapted also to the real reactions.
4. Conclusions
We have presented a systematic Monte Carlo study concerning the effects of confinement in pores of activated carbons of different distribution of pore diameter on the reaction equilibria. We have examined how the porosity and the values of the equilibrium constant and the energetic parameters of the reacting molecules affect the reaction yield. The performed simulations have confirmed that the proposed modification of the scheme of reactive Monte Carlo simulations in the grand canonical ensemble is useful to model equilibria of reactions occurring under confinement, in porous materials. This has resulted in the determination of general qualitative relationships between the porous structure and chemical equilibria, despite the fact that our simulations have been based on simplified assumptions (the simplest models of the reacting molecules).The obtained results have shown that the reaction equilibria in pores are considerably affected by the energetic parameters of the reacting species. However, when the product molecules are larger than the reactant molecules, the geometrical confinement becomes an important factor as well.
We plan to study more complex systems, involving explicitly non-sphericity of the products of dimerisation and synthesis reactions. In such cases, the geometrical confinement may lead to the phenomena and orderings that can not be treated using the simplest spherical models of molecules.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. F. gratefully acknowledges the financial support of the National Science Centre (Poland) grant no. 2014/15/D/ST4/03760. The authors acknowledge the use of the computer cluster at Poznań Supercomputing and Networking Centre (Poznań, Poland) and the Information and Communication Technology Centre of the Nicolaus Copernicus University (Toruń, Poland). The authors also thank Grzegorz Szymański (Nicolaus Copernicus University in Toruń, Poland) for fruitful discussion on experimental measurements of chemical equilibria and real reactions occurring in the porous carbonaceous materials.References
- J. K. Johnson, A. Z. Panagiotopoulos and K. E. Gubbins, Mol. Phys., 1994, 81, 717–733 CrossRef CAS.
- W. R. Smith and B. Triska, J. Chem. Phys., 1994, 100, 3019–3027 CrossRef CAS.
- D. Dubbeldam, A. Torres-Knoop and K. S. Walton, Mol. Simul., 2013, 39, 1253–1292 CrossRef CAS.
- C. H. Turner, J. K. Brennan, M. Lísal, W. R. Smith, J. K. Johnson and K. E. Gubbins, Mol. Simul., 2008, 34, 119–146 CrossRef CAS.
- M. Borówko, A. Patrykiejew, S. Sokołowski, R. Zagórski and O. Pizio, Czech J. Phys., 1998, 48, 371–388 CrossRef.
- M. Borówko and R. Zagórski, J. Chem. Phys., 2001, 114, 5397–5403 CrossRef.
- C. H. Turner, J. K. Brennan, J. K. Johnson and K. E. Gubbins, J. Chem. Phys., 2002, 116, 2138–2148 CrossRef CAS.
- E. E. Santiso, A. M. George, C. H. Turner, M. K. Kostov, K. E. Gubbins, M. Buongiorno-Nardelli and M. Śliwinska-Bartkowiak, Appl. Surf. Sci., 2005, 252, 766–777 CrossRef CAS.
- K. Kaneko, N. Fukuzaki, K. Kakei, T. Suzuki and S. Ozeki, Langmuir, 1989, 5, 960–965 CrossRef CAS.
- Y. Nishi, T. Suzuki and K. Kaneko, J. Phys. Chem. B, 1997, 101, 1938–1939 CrossRef CAS.
- C. H. Turner, J. Pikunic and K. E. Gubbins, Mol. Phys., 2001, 99, 1991–2001 CrossRef CAS.
- C. H. Turner, J. K. Brennan, J. Pikunic and K. E. Gubbins, Appl. Surf. Sci., 2002, 196, 366–374 CrossRef CAS.
- C. H. Turner, J. K. Johnson and K. E. Gubbins, J. Chem. Phys., 2001, 114, 1851–1859 CrossRef CAS.
- M. Lísal, J. K. Brennan and W. R. Smith, J. Chem. Phys., 2006, 124, 064712 CrossRef PubMed.
- H. Domínguez, Rev. Mex. Fis., 2012, 58, 378–383 Search PubMed.
- M. Lísal, M. Predota and J. K. Brennan, Mol. Simul., 2013, 39, 1103–1120 CrossRef.
- T. N. Le, Molecular Transport and Reactivity in Confinement, PhD thesis, University College London, 2016.
- N. Hansen, S. Jakobtorweihen and F. J. Keil, J. Chem. Phys., 2005, 122, 164705 CrossRef PubMed.
- S. Jakobtorweihen, N. Hansen and F. J. Keil, J. Chem. Phys., 2006, 125, 224709 CrossRef CAS PubMed.
- X. Peng, W. Wang and S. Huang, Fluid Phase Equilib., 2005, 231, 138–149 CrossRef CAS.
- M. Lísal, P. Cosoli, W. R. Smith, S. K. Jain and K. E. Gubbins, Fluid Phase Equilib., 2008, 272, 18–31 CrossRef.
- T. Le, A. Striolo, C. H. Turner and D. R. Cole, Sci. Rep., 2017, 7, 9021 CrossRef PubMed.
- R. G. Mullen and E. J. Maginn, J. Chem. Theory Comput., 2017, 13, 4054–4062 CrossRef CAS PubMed.
- E. O. Fetisov, I.-F. W. Kuo, C. Knight, J. VandeVondele, T. Van Voorhis and J. I. Siepmann, ACS Cent. Sci., 2016, 2, 409–415 CrossRef CAS PubMed.
- H. Marsh and F. Rodriguez-Reinoso, Activated Carbon, Elsevier, Amsterdam, 2006 Search PubMed.
- Activated Carbon Surfaces in Environmental Remediation, ed. T. J. Bandosz, Academic Press, New York, 2006 Search PubMed.
- R. C. Bansal and M. Goyal, Activated Carbon Adsorption, CRC Press, Boca Raton, 2005 Search PubMed.
- M. J. Biggs and A. Buts, Mol. Simul., 2006, 32, 579–593 CrossRef CAS.
- A. P. Terzyk, S. Furmaniak, P. A. Gauden, P. J. F. Harris and P. Kowalczyk, in Novel Carbon Adsorbents, ed. J. M. D. Tascón, Elsevier, Amsterdam, 2012, pp. 61–104 Search PubMed.
- S. Furmaniak, Comput. Methods Sci. Technol., 2013, 19, 47–57 CrossRef.
- N. A. Marks, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 63, 035401 CrossRef.
- N. Marks, J. Phys.: Condens. Matter, 2002, 14, 2901–2927 CrossRef CAS.
- S. Bhattacharya and K. E. Gubbins, Langmuir, 2006, 22, 7726–7731 CrossRef CAS PubMed.
- S. Furmaniak, A. P. Terzyk, P. A. Gauden, N. A. Marks, R. C. Powles and P. Kowalczyk, J. Colloid Interface Sci., 2011, 360, 211–219 CrossRef CAS PubMed.
- S. Furmaniak, P. A. Gauden, A. P. Terzyk and P. Kowalczyk, Condens. Matter Phys., 2016, 19, 13003 CrossRef.
- D. Frenkel and B. Smit, Understanding Molecular Simulation, Academic Press, San Diego, 1996 Search PubMed.
- W. A. Steele, The Interaction of Gases with Solid Surfaces, Pergamon Press, Oxford, 1974 Search PubMed.
- J. Rouquerol, F. Rouquerol and K. S. W. Sing, Adsorption by Powders and Porous Solids, Academic Press, London, 1999 Search PubMed.
- Carbon Materials for Catalysis, ed. P. Serp and J. L. Figueiredo, Wiley, New Jersey, 2009 Search PubMed.
- P. Serp and B. Machado, Nanostructured Carbon Materials for Catalysis, Royal Society of Chemistry, Cambridge, 2015, vol. 23 Search PubMed.
- F. Rodriguez-Reinoso, Carbon, 1998, 36, 159–175 CrossRef CAS.
- G. Szymański, in Carbon Materials - Theory and Practice, ed. A. P. Terzyk, P. A. Gauden and P. Kowalczyk, Research Signpost, Kerala, India, 2008, pp. 517–570 Search PubMed.
- R. W. Coughlin, Ind. Eng. Chem. Prod. Res. Dev., 1969, 8, 12–23 CAS.
- G. S. Szymański, T. Grzybek and H. Papp, Catal. Today, 2004, 90, 51–59 CrossRef.
- B. J. Ku, J. K. Lee, D. Park and H.-K. Rhee, Ind. Eng. Chem. Res., 1994, 33, 2868–2874 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08992a |
| This journal is © The Royal Society of Chemistry 2017 |