
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Elastomers built up through the π–π stacking association of polycyclic planar aromatic diimides†
Yuji Higaki abc,
Yudai Kiyoshimac,
Ken Suzukic,
Hirofumi Kabayamac,
Noboru Ohtae,
Yongsok Seoad and
Atsushi Takahara*abc
abc,
Yudai Kiyoshimac,
Ken Suzukic,
Hirofumi Kabayamac,
Noboru Ohtae,
Yongsok Seoad and
Atsushi Takahara*abc
aInstitute for Materials Chemistry and Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: takahara@cstf.kyushu-u.ac.jp
bInternational Institute for Carbon-Neutral Energy Research (WPI-I2CNER), Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
cGraduate School of Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
dIntellectual Textile Research Center, RIAM School of Materials Science and Engineering, Seoul National University, Shillim9dong 56-1, Kwanakku, Seoul, Republic of Korea
eJapan Synchrotron Radiation Research Institute/SPring-8, 1-1-1 Kouto, Sayo-cho, Sayo-gun, Hyogo 679-5198, Japan
First published on 28th September 2017
Abstract
Polymers composed of polycyclic aromatic planar diimide (PCAD) units and flexible rubbery poly(propylene glycol) (PPG) chains were synthesized, and the morphology and viscoelasticity were investigated. The polymers were synthesized by polycondensation of naphthalene or perylene tetracarboxylic dianhydrides with PPG-diamines and subsequent thermal imidization. The thermal and viscoelastic properties were measured by differential scanning calorimetry and dynamic mechanical analysis, respectively. The molecular aggregation structure was explored by infrared spectroscopy, wide-angle X-ray diffraction and small-angle X-ray scattering measurements. The PCAD units associate in the PPG continuum by the electromagnetic interaction of π electrons on the planar aromatic rings to build a stacked structure. The morphology and viscoelastic characteristics depend on the PCAD structure and molecular weight of the PPG segments, and the polymers composed of perylene diimide units and high molecular weight PPG chains show rubber elasticity because of the well-segregated morphology. The thermal stability was enhanced by PPG chain end capping by thermally stable PCAD units.
Introduction
Polycyclic aromatic diimides (PCAD), such as perylene-3,4,9,10-diimide (PDI) and 1,4,5,8-naphthalene diimide (NDI), have attracted interest due to their conductivity as well as thermal and photo-stabilities.1,2 The strong π–π stacking interaction of the large conjugated planar aromatic polycyclics triggers supramolecular assembly.3 It produces a well-aligned anisotropic fibrous structure that can be applied to a variety of functional supramolecular assemblies, and optoelectronic devices including organic n-type semi-conducting polymers.1,2,4,5 Aromatic polyimides that consist of aromatic diimide repeating units are known as one of the best thermally stable polymers.6,7 The aromatic diimide structure produces large cohesive energy and small entropic gain via melting because of the polar, planar, symmetric, and rigid structure. The chemical structure is also stable because of the high bonding energy of the aromatics and heterocycles due to the resonance effect. The supramolecular assembly and the outstanding thermal stability of aromatic polyimides demonstrate the superior cohesive binding of PCAD.Thermoplastic elastomers are a class of polymeric material that combine rubber elasticity and thermal plasticity. The thermally activated reversible aggregation of the cohesive components in the polymers causes the thermoplastic elasticity. Block copolymers composed of rubbery segments (soft segments) with major component and glassy or crystalline segments (hard segments) produce phase-separated mesoscopic morphology, and the domains of cohesive hard segments behave as physical cross-linking points to give a network structure.8–10 Segmented polyurethane(urea)s also show rubber elasticity by constructing a network structure of hard segment domains via complementary hydrogen bonding of urethane(urea) groups.11–14 Various reversible network polymeric materials including thermoplastics and gels that are built up via supramolecular cohesive interaction have been proposed so far, and the toughening effect and self-healing capability have been demonstrated.15–25
In this paper, a new type of elastomers composed of PCAD moieties and rubbery flexible polyether chains is proposed, and the molecular aggregation state and viscoelasticity are investigated. A special focus of this study is to shed light on the effect of the two structural parameters; number of polycyclic structure (i.e. size of conjugated planar structure) and the rubbery chain length (i.e. volume fraction of the rigid and flexible components), on the thermal stability, morphology, and viscoelastic properties. We successfully developed a thermoplastic elastomer that consist of PDI units and poly(propylene glycol) (PPG) segments (Fig. 1). The elastomers with PCAD assembly network are expected to bring impacts in both fundamental understanding of self-assembly of PCAD units in rubbery polymer matrix along with the thermal and mechanical stabilities, and future applications to the functional elastomeric materials such as anisotropic conductive elastomers.
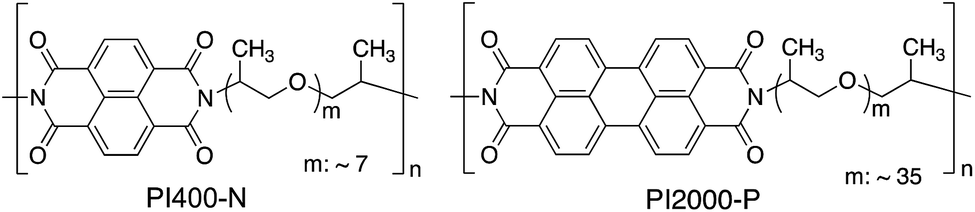 | ||
| Fig. 1 Chemical structures of polymers alternating polycyclic aromatic diimide units (naphthalene diimide or perylene diimide) and poly(propylene glycol)s. | ||
Experimental
Materials
1,4,5,8-Naphthalenetetracarboxylic dianhydride (NTCDA, Tokyo Chemical Industry) was purified by sublimation. 3,4,9,10-Perylenetetracarboxylic dianhydride (PTCDA, Tokyo Chemical Industry) was used as received. Poly(propylene glycol) bis(2-aminopropyl ether)s (PPG-diamine, Mn: 400, 2000, Sigma Aldrich) were dried under reduced pressure at room temperature for over 24 hours prior to use. Dehydrated tetrahydrofuran (THF, Wako Pure Chemical Industries, 99.5+%) and imidazole (Tokyo Chemical Industry, 98.0+%) were used without further purification. Sample codes of the polymers consist of the number averaged molecular weight of PPG-diamine (400 or 2000) and type of PCAD units (N: naphthalene diimide, P: perylene diimide).Synthesis of the polymers with naphthalene diimide units (PI400-N)
A two-necked flask was dried by thermal baking under vacuum and purged with dry argon. PPG-diamine (Mn: 400, 0.63 g, 1.6 mmol), NTCDA (0.40 g, 1.5 mmol), and THF (2.9 mL) were introduced in the two-necked flask, and stirred by magnetic stirrer at room temperature for 12 hours. The solution was transferred to a polytetrafluoroethylene Petri dish. The solvent was dried under ambient condition for 12 hours, and the film was heated at 473 K under vacuum for 3 hours to obtain the polymer film.Mn: 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Mw/Mn: 1.74. 1H NMR (400 MHz, CDCl3): δ/ppm 0.75–1.43 (br m, CH3), 3.12–3.66 (br m, –CHx–O–) where x = 1 or 2, 3.90 and 4.16 (br m, –N–CH2–), 5.44 (br m, –N–CH–), 8.70 (s, naphthalene protons). FT-IR (KBr, cm−1): 3000–2800 (C–H), 1708 (asymmetric C
000, Mw/Mn: 1.74. 1H NMR (400 MHz, CDCl3): δ/ppm 0.75–1.43 (br m, CH3), 3.12–3.66 (br m, –CHx–O–) where x = 1 or 2, 3.90 and 4.16 (br m, –N–CH2–), 5.44 (br m, –N–CH–), 8.70 (s, naphthalene protons). FT-IR (KBr, cm−1): 3000–2800 (C–H), 1708 (asymmetric C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1667 (symmetric CO), 1334 (C–N).
O), 1667 (symmetric CO), 1334 (C–N).
Synthesis of the polymers with perylene diimide units (PI2000-P)
A two-necked flask was dried by thermal baking under vacuum and purged with dry argon. PPG-diamine (Mn: 2000, 2.52 g, 1.3 mmol), PTCDA (0.50 g, 1.3 mmol), and imidazole (9.0 g) were introduced in the two-necked flask, and stirred by mechanical stirrer at 423 K for 12 hours. The product was washed with pure water to remove imidazole, and then dried under reduced pressure. The precursor polymer, poly(amic acid), was dissolved in chloroform. The solution was introduced in a polytetrafluoroethylene Petri dish. The solvent was dried under ambient condition for 12 hours, and the film was heated at 473 K under vacuum for 12 hours to obtain the polymer film.Mn: 18500, Mw/Mn: 1.46. 1H NMR (400 MHz, CDCl3): δ/ppm 1.06 (br s, CH3), 3.20–3.68 (br m, –CHx–O–) where x = 1 or 2, 3.92 and 4.13 (br m, –N–CH2–), 5.40 (br m, –N–CH–), 8.36–8.58 (br m, perylene protons). FT-IR (KBr, cm−1): 3020–2790 (C–H), 1697 (asymmetric CO), 1655 (symmetric CO), 1344 (C–N).
Measurement
Proton nuclear magnetic resonance spectroscopic measurements were performed at 298 K with an AVANCE-III400 spectrometer (Bruker, Billerica, MA; 400 MHz). Chloroform-d (CDCl3) was used as a deuterated solvent, and tetramethylsilane (TMS) was used as an internal standard.Infrared spectroscopic measurements were conducted with a Spectrum100 infrared spectrometer (PerkinElmer Inc., Waltham, MA) by KBr tablet method under nitrogen. IR spectra were recorded by integrating 128 scans with resolution of 4 cm−1.
Number average molecular weight (Mn) and polydispersity index (Mw/Mn) were determined by size exclusion chromatography (SEC) system HLC-8220GPC (TOSOH Bioscience, Tokyo, Japan) equipped with two polystyrene gel columns TSKgel SuperH4000 and TSKgel SuperH6000 (TOSOH Bioscience, Tokyo, Japan) and a refractive index (RI) detector. THF was used as an eluent at a flow rate of 0.5 mL min−1 at 313 K. The columns were calibrated with six polystyrene standards (Mn = 4920–3000000).
Differential scanning calorimetry (DSC) was performed using a DSC6220 calorimeter (Hitachi High-Tech Science Corporation, Tokyo, Japan) in a temperature range of 173–423 K at a scanning rate of 10 K min−1 under dry nitrogen gas.
Wide angle X-ray diffraction (WAXD) and small angle X-ray scattering (SAXS) measurements were conducted at the BL40B2 beam line of SPring-8. The incident X-ray wavelength, λ, was 0.100 nm. Two-dimensional (2D) WAXD images were acquired on a flat-panel detector C9827DK-10 (Hamamatsu Photonics Co. Ltd., Shizuoka, Japan) consisting of 1024 × 1024 pixels (pixel size 50 μm). 2D-SAXS images were acquired on a 3000 × 3000 pixel imaging plate detector R-AXIS VII (Rigaku Corporation, Tokyo, Japan) whose pixel size is 100 × 100 μm2. The scattering vector, q = (4π/λ)sinθ, where 2θ is the scattering angle, was calibrated by peak positions of cerium oxide and silver behenate for the WAXD and SAXS measurements, respectively. The sample-to-detector distances of the WAXD and SAXS measurements were 51 mm and 2213 mm, respectively. The 2D-data were corrected by the background subtraction and X-ray absorption by the samples. The 2D-SAXS and 2D-WAXD patterns were approximately isotropic and the one-dimensional (1D) intensity profiles were obtained as a function of q after the circular average of the 2D patterns.
Dynamic mechanical analysis (DMA) was carried out with a dynamic viscoelastometer Rheovibron DDV-IIFP (Orientec A&D Co. Ltd., Tokyo, Japan). The measurements were performed at 1, 3.5, 11, 35, and 110 Hz frequency under a dry nitrogen atmosphere at a heating rate of 1 K min−1 over the temperature range of 123–393 K. The gauge length was 10 mm.
Thermogravimetric analysis (TGA) was performed on a EXSTAR TG/DTA-6200 thermobalance (Hitachi High-Tech Science Corporation, Tokyo, Japan) in a temperature range of 300 to 820 K under dry nitrogen stream at a heating rate of 10 K min−1.
Results and discussion
Synthesis of polymers with polycyclic aromatic diimide moieties
Characteristics of the synthesized polymers are summarized in Table 1. The weight fractions of the PCAD units and PPG in the polymers were determined by supposing the equimolar reaction of naphthalene or perylene tetracarboxylic dianhydrides and PPG-diamines. The condensation reaction proceeded smoothly at room temperature in couple of NTCDA and PPG-diamine. Both PI400-N and PI2000-N dissolved in THF. PI400-N provides a self-standing film, whereas PI2000-N was viscous fluid. In case of PTCDA, the condensation reaction hardly proceeded at room temperature to give low molecular weight oligomers. The poly(amic acid)s with large number of polycyclic aromatic rings would aggregate in the polymerization process to quench the chain extension. The condensation reaction of PTCDA progressed by heating the reaction vessel probably because the aggregates of polycyclic aromatic rings by π–π stacking interaction dissociate by thermal activation. Polycondensation of PTCDA and low molecular weight PPG-diamine produced insoluble precipitates. The precursor polymer was dissolved in chloroform, and the elastic films were obtained by solvent evaporation. The cohesive energy of the planar aromatic units is vital for network structure build-up rather than the weight fraction so that PTCDA-based polymers produce elastic films with low PCAD content. It should be noted that the relative molecular weight and the distribution were determined on the basis of polystyrene calibration, whereby they were overestimated or underestimated depending on the measurement condition because of the cohesive interaction of the polymers with PCAD units.| Sample ID | PCAD contenta (wt%) | Mnb | Mw/Mnb | Tgc (K) | Tmd (K) | ΔHme (J g−1) | Tcf (K) |
|---|---|---|---|---|---|---|---|
| a Calculated on the basis of weight of raw materials.b Determined by SEC against polystyrene standards in THF at 40 °C.c Glass transition temperature determined by DSC as a middle point temperature of slope region.d Melting temperature determined by DSC as an endothermic peak temperature.e Melting enthalpy determined by DSC.f Crystallization temperature determined by DSC as an exothermic peak temperature. | |||||||
| PI400-N | 39.0 | 20000 |
1.74 | 290 | — | — | — |
| PI2000-N | 11.8 | 32300 |
1.79 | 210 | — | — | — |
| PI2000-P | 16.4 | 18500 |
1.46 | 207 | 410 | 8.1 | 405 |
The polymer structure was verified by 1H-NMR and IR spectroscopies. A signal assigned to protons beside the imide rings was observed at 3.7–5.5 ppm in both NTCDA and PTCDA-based polymers, while the signals assigned to protons beside the poly(amic acid) disappeared (Fig. S1†).26 IR absorptions assigned to carbonyl stretching (1655 cm−1, symmetric; 1697 cm−1, asymmetric) and C–N stretching (1344 cm−1) derived from imide bonds were observed in the PTCDA-based polymer (PI2000-P), indicating the progress of thermal imidization by thermal annealing at 473 K (Fig. 2(b)).27 The NTCDA-based polymer (PI400-N) showed IR absorptions assigned to carbonyl stretching at 1667 cm−1 and 1708 cm−1, and C–N stretching at 1334 cm−1 (Fig. 2(a)). The peak shifts are attributed to the difference in the PCAD structure.
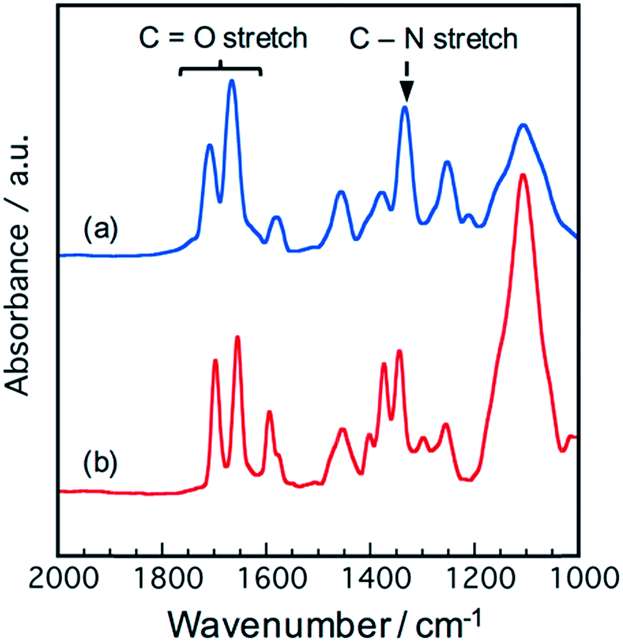 | ||
| Fig. 2 IR spectra of the polymers (a) PI400-N and (b) PI2000-P. | ||
Molecular aggregation structure of the polymers
Thermal properties of the polymers were measured by DSC measurement (Fig. 3). The PI2000-N, PI400-N and PI2000-P showed a baseline shift in the DSC curve at 210 K, 290 K and 207 K, respectively. The PPG-diamines showed a baseline shift at 190 K to 201 K (190 K for PPG400; 201 K for PPG2000). The difference in baseline shift temperature between the polymers and PPG-diamines indicates the degree of phase-separation. The polymers with NDI units showed a baseline shift at much higher temperature than the PPG-diamines indicating the phase-mixing. The polymers with PDI units (PI2000-P) showed almost identical baseline shift temperature with PPG-diamine. The PI2000-P showed an endothermic peak at 410 K in the heating scan, whereas the PI400-N and PI2000-N showed no endothermic peaks. The endotherm is assigned to the dissociation of the ordered stacking structure of the PDI units. These results indicate that the PDI units in PI2000-P are well segregated to build ordered structure through π–π stacking interaction in the PPG matrix, whereas the NDI units are compatible with PPG. The PPG chains in the PI2000-P receive any restraint in the segmental motion by segregation of the PDI units to show identical glass transition temperature (Tg) with PPG2000, whereas the NDI units restrict the segmental motion of PPG chains because of the phase-mixing resulting in the Tg boost. Because the PDI units consist of substantial conjugated π electrons, large cohesive energy for stacking is produced in the PDIs. In contrast, the NDI has less cohesive force due to the small planar aromatic structure. The association of PDI units assists the phase separation to keep the rubbery state of the PPG matrix. | ||
| Fig. 3 DSC thermograms observed for the PI2000-N on (a) the second cooling and (b) second heating scans, the PI400-N on (c) the second cooling and (d) second heating scans, and the PI2000-P on (e) the second cooling and (f) second heating sans. Scanning rate was 10 K min−1. | ||
SAXS is the appropriate approach to characterize the microphase-separated states of multiblock elastomers because transmission electron microscopy (TEM) observation of multiblock elastomers requires special techniques for preparation of ultra-thin cross-section films by cryo-microtome and observation by cryo-TEM. In case of room temperature observation, the staining condition have to be optimized to observe the phase separated structure. Meanwhile, we have to consider the surface rearrangement of the cross-section films by migration of thermally active polymer chains. The hydrophobic rubbery PPG segments spontaneously migrate to the outermost surface to minimize the interfacial free energy. Also, another advantage of SAXS is capability of in situ measurement under deformation. Fig. 4 shows azimuthally averaged SAXS intensity profiles of the polymer films. The PI400-N showed only diffuse scattering, indicating that no mesoscopic ordered structure exists in the film. In contrast, the PI2000-P showed clear scattering peaks derived from the periodic ordered mesoscopic structure. The scattering peaks were observed at 1.2 nm−1, and 2.4 nm−1. Multiple orders of Bragg reflection are observed in well-ordered samples, and the morphologies are identified from the positions of scattering peaks. For example, q/q* of the Bragg peaks, whose q* is scattering vector of the primary peak, are in the ratio: 1:2:3:4… for a lamellar structure, 1:31/2:2:71/2:3:121/2… for a hexagonally close packed structure.28 The scattering peak ratios in the PI2000-P were 1:2, so that the structure is identified to be lamellar structure. The thicknesses of the periodic lamellar phases were determined by curve fitting on the basis of a pseudo-two-phase model (see ESI†).29,30 The best fit Icalc(q) profile is shown in Fig. 4(b) as dotted line. The fitting curve deviates from Iobs(q) at high q region because of the overlap of the diffuse scattering from disordered aggregates as seen in PI400-N. The structure model with a lamellar period of 5.1 nm with high density layer thickness of 1.1 nm was provided by the best fit profile (Table S1†). The lamellar thickness ratio (high density phase: 1.1 nm/low density phase 4.0 nm = 0.28) is approximately consistent with the weight fraction ratio of PCAD units and PPG chains of 0.20 calculated from the mass balance in the synthesis. The discrepancy would be attributed to the density difference and partial phase-mixing in the interface.
 | ||
| Fig. 4 SAXS intensity profiles (open circles) against the scattering vector q for (a) PI400-N (blue circles) and (b) PI2000-P (red circles). The black dotted curve shows a calculated intensity profile based on the pseudo-two-phase model. | ||
Fig. 5 shows azimuthally averaged WAXD intensity profiles of the polymers. The PI400-N showed two diffraction peaks at 14.9 nm−1 (d: 0.42 nm) and 29.8 nm−1 (d: 0.21 nm). The diffraction peaks are derived from the NDI units. The PI2000-P showed almost identical diffraction peaks with PI400-N at 14.0 nm−1 (d: 0.45 nm) and 29.7 nm−1 (d: 0.21 nm), while an additional diffraction was observed at 18.4 nm−1 (d: 0.34 nm). The diffraction peaks at 14.0 nm−1 and 29.7 nm−1 are attributed to the PDI units. The inter-layer spacing of stacked aromatic rings is commonly observed at around 18.4 nm−1,26 so that the PDI units in the PI2000-P are ordered via π–π stacking interaction to build stacked structure with periodic order. The SAXS and WAXD results complementally demonstrated that the ordered stacked structure of the PCAD units was assembled in the case of PDI units, and the mesoscopic lamellar morphology composed of the stacked PDI and PPG phases was produced. However, the NDI units hardly produced the ordered stacked structure and mesoscopic ordered morphology.
 | ||
| Fig. 5 WAXD intensity profiles against the scattering vector q for (a) PI400-N (blue circles) and (b) PI2000-P (red circles). | ||
Viscoelastic characteristics of the polymer films
Temperature dependence of the dynamic storage modulus, E′, and dynamic loss modulus, E′′, of the polymer films were measured by dynamic mechanical analysis (Fig. 6). The PI2000-P exhibited typical viscoelastic characters as elastomers, a low Tg and a rubbery plateau, whereas the PI400-N deformed immediately after the glass transition at around 300 K. The PI400-N film shows absorptions at 158 K and 297 K in the E′′, while the PI2000-P film shows absorptions at 157 K and 223 K. The activation energies of the relaxations were obtained by Arrhenius plot (Fig. S2, Table S2†). On the basis of the activation energy, the first relaxations were assigned to the local motion in PPG (γ-relaxation), and the second relaxations were assigned to the segmental motion of the PPG (α-relaxation) where the polymer films transit from glassy state to rubbery state. The PI400-N has larger α-relaxation activation energy than the PI2000-P, indicating that NDI units restrained the segmental motion of PPG due to the phase-mixing. After the α-relaxation, PI2000-P showed a rubbery plateau until it failed at 350 K, indicating the domains of stacked PDI units act as physical cross-linking points to give network structure. The stacked PDI structure finally slipped because of the dissociation through thermal activation. Both the polymers showed two-step absorption in the α-relaxation process. Generally, the sign is attributed to various events including melting and recrystallization of the soft segment chains after the glass transition,31 microphase separation,32 or cold crystallization of the cohesive segments in the rubbery continuum.33 The PPG chains hardly produce crystalline ordered structure, so that the recrystallization of the PPG chains is unlikely to occur. The microstructure composed of two types of domains that PPG chains with different glass transition temperature are localized, is implausible. Therefore, it would be plausible to assume that the second absorptions are attributed to the rearrangement in stacking structure of the PCAD units as well as the promotion in the phase separation by thermal activation of PPG matrix. | ||
| Fig. 6 Temperature dependence of the dynamic storage modulus (E′) and the dynamic loss modulus (E′′) for the PI400-N and PI2000-P at frequency of 110 Hz. (a) E′ of PI400-N (blue filled circles), (b) E′′ of PI400-N (blue open circles), (c) E′ of PI2000-P (red filled circles), and (d) E′′ of PI2000-P (red open circles). | ||
Tensile tests for the polymer films were performed (Fig. S3†). The PI400-N films were too brittle to stretch. The films caused fracture at low strain. The PI2000-P films is stretchable but exhibits poor elastic properties. The elastic modulus and strain at break were 26.9 MPa and 0.16, respectively. The elastic modulus is in a range of common elastomers. The poor durability to large strain would be attributed to the defects and weak aggregation force in PCAD domains.
Thermal stability
Thermal stability of the polymer films was estimated by TGA (Fig. S4†). The PCAD units raised the thermal decomposition temperature of PPG chains in the elastomers. The 5% weight loss temperatures of the PI2000-P, PI400-N, and hydroxy-terminated PPG (Mw: 2500) were 650 K, 640 K, and 606 K, respectively. The PPG chain-end capping by aromatic diimide units prevents the thermal decomposition. The high bond-dissociation energy as well as the cohesive interaction in the PCAD units would promote the thermal stability. The weight losses were almost consistent with the weight fraction of PPG diamine in raw materials. Carbonized residues remained after heating to 820 K in PCAD elastomers, whereas the hydroxy-terminated PPG completely decomposed and produced no residue (Fig. S5†). The carbonized residues are identified to be a char of the PCAD units. The generation of inflammable carbonized char is valid for the flame retardancy.Conclusions
Polymers alternating PCAD moieties and flexible rubbery PPG chains were produced, then the molecular aggregation structure and viscoelastic characteristics were investigated. The PDI units associated by π–π interaction to produce domains of stacked PDI units in the PPG matrix. The domains worked as physical cross-linking points to give rubber elasticity. The larger PCAD units produced more ordered and thermally stable domain structure because of the strong cohesive interaction of the large conjugated aromatic planar structure, leading to favorable elastomers with wide rubbery plateau region. The thermal stability was improved, and the potential for flame-retardant elastomer was demonstrated. The stacked PDI domains can be regarded as a new type of physical cross-linking structure in thermoplastic elastomers, and the spontaneous anisotropic orientation of the planar aromatic groups under the stretched state is expected to be applied as anisotropic conductive elastic materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Photon and Quantum Basic Research Coordinated Development Program of the Ministry of Education, Culture, Sports, Science and Technology, Japan. Part of this work was funded by ImPACT Program of Council for Science, Technology and Innovation (Cabinet Office, Government of Japan). This work was performed under the Cooperative Research Program of “Network Joint Research Center for Materials and Devices”. This work was supported by the MEXT Project of “Integrated Research Consortium on Chemical Sciences”. This work was supported by Kyushu University Progress 100 program. The synchrotron radiation experiments were performed at BL40B2 (2013B1440, 2014A1249, 2014B1368) at SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI). Y. Seo was supported by MIKE of Korea.Notes and references
- K. Balakrishnan, A. Datar, T. Naddo, J. Huang, R. Oitker, M. Yen, J. Zhao and L. Zang, J. Am. Chem. Soc., 2006, 128, 7390–7398 CrossRef CAS PubMed.
- R. Schmidt, J. H. Oh, Y.-S. Sun, M. Deppisch, A.-M. Krause, K. Radacki, H. Braunschweig, M. Könemann, P. Erk, Z. Bao and F. Würthner, J. Am. Chem. Soc., 2009, 131, 6215–6228 CrossRef CAS PubMed.
- C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- Y. Y. Kim, B. J. Ree, M. Kido, Y. G. Ko, R. Ishige, T. Hirai, D. Wi, J. Kim, W. J. Kim, A. Takahara and M. Ree, Adv. Electron. Mater., 2015, 1, 1500197 CrossRef.
- F. Donati, A. Pucci, C. Cappelli, B. Mennucci and G. Ruggeri, J. Phys. Chem. B, 2008, 112, 3668–3679 CrossRef CAS PubMed.
- M. Hasegawa, N. Sensui, Y. Shindo and R. Yokota, Macromolecules, 1999, 32, 387–396 CrossRef CAS.
- M. Hasegawa, T. Matano, Y. Shindo and T. Sugimura, Macromolecules, 1996, 29, 7897–7909 CrossRef CAS.
- W. Rungswang, M. Kotaki, T. Shimojima, G. Kimura, S. Sakurai and S. Chirachanchai, Macromolecules, 2011, 44, 9276–9285 CrossRef CAS.
- H. Sun, C. Jiang, N. Ning, L. Zhang, M. Tian and S. Yuan, Polym. Chem., 2016, 7, 4072–4080 RSC.
- K. Y. Cho, A. Cho, H.-J. Kim, S.-H. Park, C. M. Koo, Y. J. Kwark, H. G. Yoon, S. S. Hwang and K.-Y. Baek, Polym. Chem., 2016, 7, 7391–7399 RSC.
- Y. Oniki, K. Suzuki, Y. Higaki, R. Ishige, N. Ohta and A. Takahara, Polym. Chem., 2013, 4, 3735–3743 RSC.
- K. Kojio, K. Matsuo, S. Motokucho, K. Yoshinaga, Y. Shimodaira and K. Kimura, Polym. J., 2011, 43, 692–699 CrossRef CAS.
- J. T. Koberstein and R. S. Stein, J. Polym. Sci., Polym. Phys. Ed., 1983, 21, 1439–1472 CrossRef CAS.
- J. T. Koberstein and T. P. Russell, Macromolecules, 1986, 19, 714–720 CrossRef CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. Folmer, J. H. Hirschberg, R. F. Lange, J. K. Lowe and E. W. Meijer, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- M. E. Rogers, T. E. Glass, S. J. Mecham, D. Rodrigues, G. Wilkes and J. E. McGrath, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 2663–2675 CrossRef CAS.
- R. M. Versteegen, R. Kleppinger, R. P. Sijbesma and E. W. Meijer, Macromolecules, 2006, 39, 772–783 CrossRef CAS.
- J. A. Syrett, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 978–987 RSC.
- Y. Chen, A. M. Kushner, G. A. Williams and Z. Guan, Nat. Chem., 2012, 4, 467–472 CrossRef CAS PubMed.
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. B. Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- L. R. Hart, J. L. Harries, B. W. Greenland, H. M. Colquhoun and W. Hayes, Polym. Chem., 2013, 4, 4860–4870 RSC.
- T. Narita, K. Mayumi, G. Ducouret and P. Hébraud, Macromolecules, 2013, 46, 4174–4183 CrossRef CAS.
- R. Ishige, G. A. Williams, Y. Higaki, N. Ohta, M. Sato, A. Takahara and Z. Guan, IUCrJ, 2016, 3, 211–218 CrossRef CAS PubMed.
- S. Yoshida, H. Ejima and N. Yoshie, Adv. Funct. Mater., 2017, 1701670 CrossRef.
- Z. Liang, R. A. Cormier, A. M. Nardes and B. A. Gregg, Synth. Met., 2011, 161, 1014–1021 CrossRef CAS.
- S. Rodrıguez-Llorente, R. Aroca and J. Duff, Spectrochim. Acta, Part A, 1999, 55, 969–978 CrossRef.
- T. Narayanan, in Soft Matter Characterization, Springer, Netherlands, 2008, ch. 17, pp. 899–952 Search PubMed.
- M. Shibayama and T. Hashimoto, Macromolecules, 1986, 19, 740–749 CrossRef CAS.
- R. Ishige, T. Ishii, M. Tokita, M. Koga, S. Kang and J. Watanabe, Macromolecules, 2011, 44, 4586–4588 CrossRef CAS.
- A. Takahara, J. Tashita, T. Kajiyama, M. Takayanagi and W. J. MacKnight, Polymer, 1985, 26, 987–996 CrossRef CAS.
- T. Takigawa, M. Oodate, K. Urayama and T. Masuda, J. Appl. Polym. Sci., 1996, 59, 1563–1568 CrossRef CAS.
- Y.-T. Shieh, Y.-S. Lin, Y.-K. Twu, H.-B. Tsai and R.-H. Lin, J. Appl. Polym. Sci., 2010, 116, 1334–1341 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra, SAXS data analysis, Arrhenius plots for relaxations in the DMA data, TGA profiles and appearance of residues. See DOI: 10.1039/c7ra08910d |
| This journal is © The Royal Society of Chemistry 2017 |