
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The modular synthesis of multivalent functionalised glycodendrons for the detection of lectins including DC-SIGN†
Stefan Munnekea,
Kristel Kodara,
Gavin F. Painter bc,
Bridget L. Stocker*ab and
Mattie S. M. Timmer*ab
bc,
Bridget L. Stocker*ab and
Mattie S. M. Timmer*ab
aSchool of Chemical and Physical Sciences, Victoria University of Wellington, P.O. Box 600, Wellington, New Zealand. E-mail: mattie.timmer@vuw.ac.nz
bCentre for Biodiscovery, Victoria University of Wellington, P.O. Box 600, Wellington, New Zealand
cFerrier Research Institute, Victoria University of Wellington, P.O. Box 600, Wellington, New Zealand
First published on 22nd September 2017
Abstract
Glycodendrons are excellent tools for the biological evaluation of lectins. Herein, we present the expedient and efficient synthesis of a versatile second generation dendron scaffold using double exponential growth methodology. The dendron scaffold can be rapidly functionalised, as illustrated by the conjugation of the dendron scaffold to biotin or a fluorescent probe, followed by oxyamine glycan conjugation to the glycan of choice. The use of a fluorescent LewisX glycodendron for the detection of the C-type lectin DC-SIGN on macrophages is then demonstrated.
Introduction
Glycoconjugates, such as glycolipids and glycoproteins, are essential to all forms of life. These fundamental cellular constituents are involved in a variety of biological processes, such as host cell recognition, inflammation, cell signalling and proliferation,1,2 with the carbohydrate portion of the glycoconjugate often interacting with lectins (carbohydrate binding proteins) that are expressed on the surface of cells. Lectins, however, generally have a weak affinity for their carbohydrate ligands and therefore require multivalent interactions in order to induce a biological response.3 Thus, to study carbohydrate–lectin binding, the glycan epitope is typically conjugated to a substrate that allows for multivalent presentation.4 To this end, dendrimers and dendrons have both been used for the multivalent presentation of glycans. One advantage of the use of a glycodendron is that its core can be functionalised with reporter groups (e.g., fluorescent groups, biotin) or with toxins, which can allow for the detection, isolation or elimination of cells that expresses the target lectin.Given the application of glycodendrons for the study of carbohydrate–lectin interactions, we became interested in developing a highly efficient synthesis of a dendron scaffold that could be readily functionalised with the molecular probe of choice and with glycans using our recently developed oxyamine linker methodology.5,6 This oxyamine methodology is not only highly efficient and allows for the conjugation of naturally occurring (isolated) glycans, but it also leads to the formation of β-linked GlcNAc residues, which are commonly found conjugated to proteins in the form of N-glycans.7 To this end, we proposed that the target glycodendron 1 could be prepared via peptide coupling of the carboxy-functionalised dendron 2 to the amine-functionalised neoglycoside prepared via conjugation of glycan 3 to 3-(methoxyamino)propan-1-amine (4) (Scheme 1). The dendron scaffold 2 would in turn contain the molecular probe of choice. The oxyamine ligation methodology has not been used for the conjugation of multiple copies of a glycan to a molecular scaffold, so we first envisioned synthesizing a glycodendron containing GlcNAc as proof-of-concept, before extending our methodology to include the glycan LewisX. LewisX is a ligand for the C-type lectin DC-SIGN [also known as Cluster of Differentiation 209 (CD209)],8 which is expressed on the cell wall of several cells types,9–11 with alternatively-activated (‘M2-like’) macrophages expressing higher levels of DC-SIGN compared to their classically-activated (‘M1-like’) counterparts.12 Moreover, DC-SIGN can be used by the human immunodeficiency virus (HIV), Ebola, hepatitis C, and non-viral pathogens, such as Mycobacterium tuberculosis, to infect host cells.13,14 Accordingly, DC-SIGN is an attractive target for glycodendron/glycodendrimer therapeutics.15–18
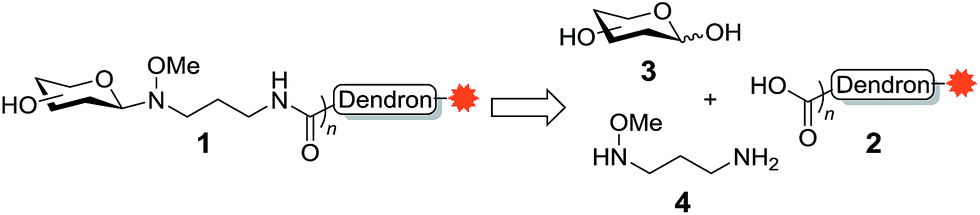 | ||
| Scheme 1 Assembly of glycodendrons using an amine-functionalised oxyamine linker. | ||
To synthesise the dendron scaffold 2 we proposed a double exponential growth strategy.19 For the double exponential growth methodology, a protected first-generation dendron is synthesised and then converted to a second-generation dendron in a parallel synthesis. This strategy has merit since it allows for the easy purification of the first-generation dendron, minimises the number of steps to assemble larger dendrons, and therefore facilitates the rapid assembly of second-, third- and fourth-generation dendrons.20 Depending on the functional groups at the periphery of the dendron, a variety of strategies can then be employed for the conjugation of glycans, including copper-catalysed ‘click’ reactions,21 and reductive amination.22 We envisioned that dendron 2 could be obtained from precursor 5 via Staudinger reduction of the azide in 5 to an amine and subsequent peptide coupling with a probe equipped with a carboxylic acid, followed by hydrolysis of the terminal t-butyl esters groups (Scheme 2). The protected dendron 5 could in turn be prepared from the first generation dendron 6 via double exponential growth methodology. Finally, it was envisioned that dendron 6 could be synthesised in two steps from the highly versatile branching unit tris(hydroxymethyl)aminomethane (TRIS, 7)23 using amide formation and tri-O-alkylation with tert-butyl bromoacetate.
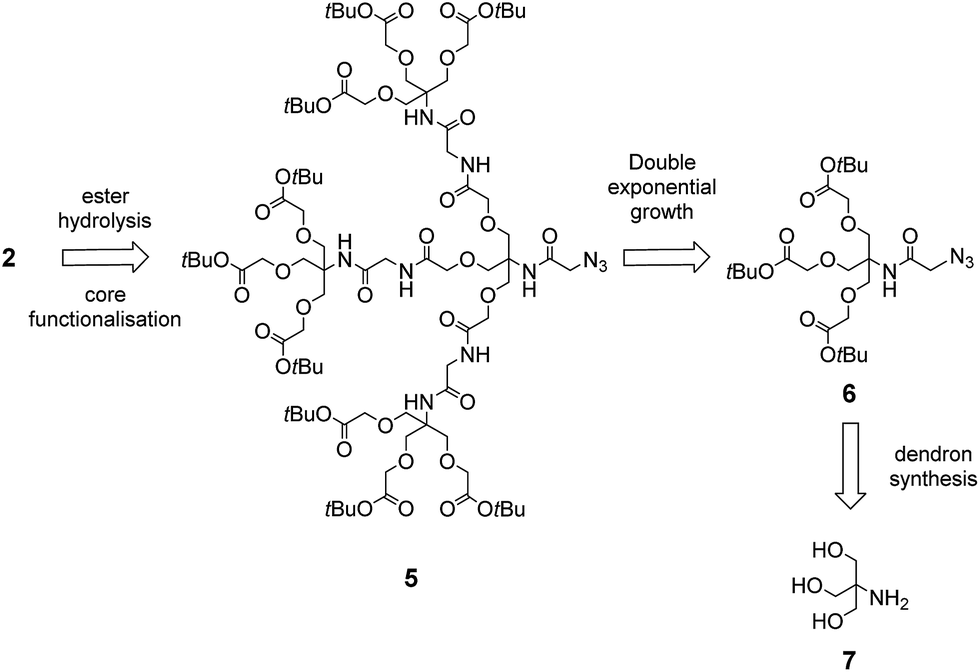 | ||
| Scheme 2 Retrosynthesis of the dendron scaffold. | ||
Results
The synthesis of the first generation dendron commenced with azide substitution of bromoacetic acid (8) to give azide derivative 9, which in turn was converted into the activated ester 10 via a DCC-mediated coupling with N-hydroxysuccinimide (Scheme 3). Conjugation of 10 with excess TRIS (7) then gave triol 11 in 80% yield over three steps. With the triol in hand, alkylation using NaH and tert-butyl bromoacetate in DMF then gave the first generation dendron 6. Here it was determined that the rapid addition of NaH to a mixture of triol 11 and tert-butyl bromoacetate in DMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :toluene (1:1) at room temperature minimised the formation of partially alkylated intermediates and afforded dendron 6 in 47% yield. While this alkylation yield may appear modest, the remaining steps for the synthesis of our target dendron scaffold 5 are very efficient (vide infra). Moreover, similar yields have been reported for the tri-O-alkylation of TRIS using analogous substrates, as illustrated by Dupuy et al. who reported a 53% yield for the tri-O-alkyl-ation of N,N-diphenyl-TRIS with tert-butyl bromoacetate using a two-phase system,24 while Newkome et al. prepared a tri-acid functionalised dendron core in two-steps via the Michael addition of TRIS to acrylonitrile followed by nitrile hydrolysis to give the ethyl esters in 54% over two steps.25 It should also be noted that our route was readily amenable to synthesis on the gram scale, and following purification of dendron 6 via silica gel chromatography followed by reverse phase (C18 beads) chromatography, the first-generation dendron 6 was obtained in excellent purity.
:toluene (1:1) at room temperature minimised the formation of partially alkylated intermediates and afforded dendron 6 in 47% yield. While this alkylation yield may appear modest, the remaining steps for the synthesis of our target dendron scaffold 5 are very efficient (vide infra). Moreover, similar yields have been reported for the tri-O-alkylation of TRIS using analogous substrates, as illustrated by Dupuy et al. who reported a 53% yield for the tri-O-alkyl-ation of N,N-diphenyl-TRIS with tert-butyl bromoacetate using a two-phase system,24 while Newkome et al. prepared a tri-acid functionalised dendron core in two-steps via the Michael addition of TRIS to acrylonitrile followed by nitrile hydrolysis to give the ethyl esters in 54% over two steps.25 It should also be noted that our route was readily amenable to synthesis on the gram scale, and following purification of dendron 6 via silica gel chromatography followed by reverse phase (C18 beads) chromatography, the first-generation dendron 6 was obtained in excellent purity.
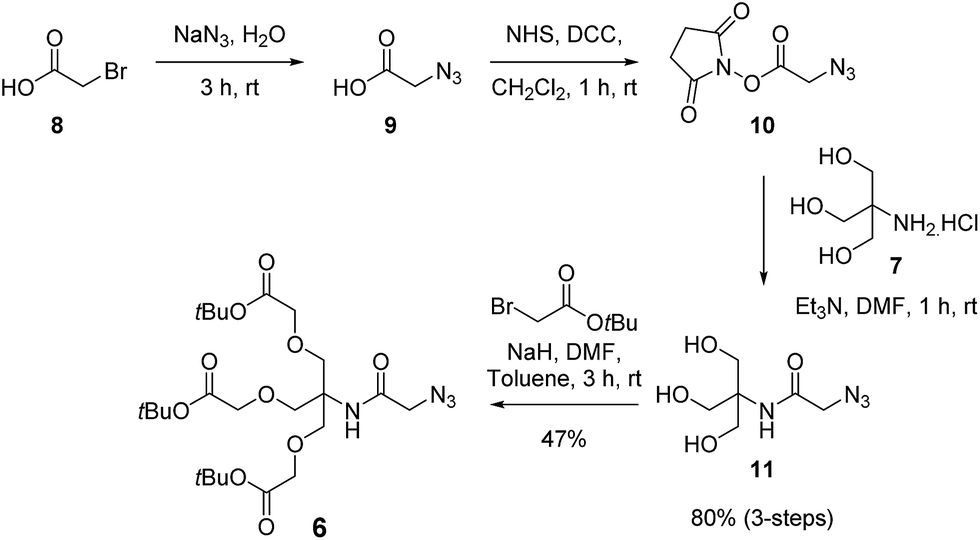 | ||
| Scheme 3 Synthesis of the first generation dendron 8. | ||
With the first generation dendron in hand, the synthesis of the second-generation dendron was then explored using the double exponential growth approach. Accordingly, the first generation dendron 6 was divided into two batches for conversion into the two reactive substrates (Scheme 4). Deprotection of the tert-butyl esters of 6 via the agency of TFA/CH2Cl2 (1/1, v/v) gave the tri-valent acid 12 in quantitative yield, while azide reduction of 6, using RANEY® nickel mediated hydrogenation, afforded primary amine 13, again in quantitative yield. Next, N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU)-mediated coupling of tri-acid 12 with amine 13 was undertaken to give the second-generation dendron 5 in 81% yield and in >95% purity following silica gel chromatography followed by reverse phase (C18 beads) chromatography. Again, these reactions could be performed on the gram scale.
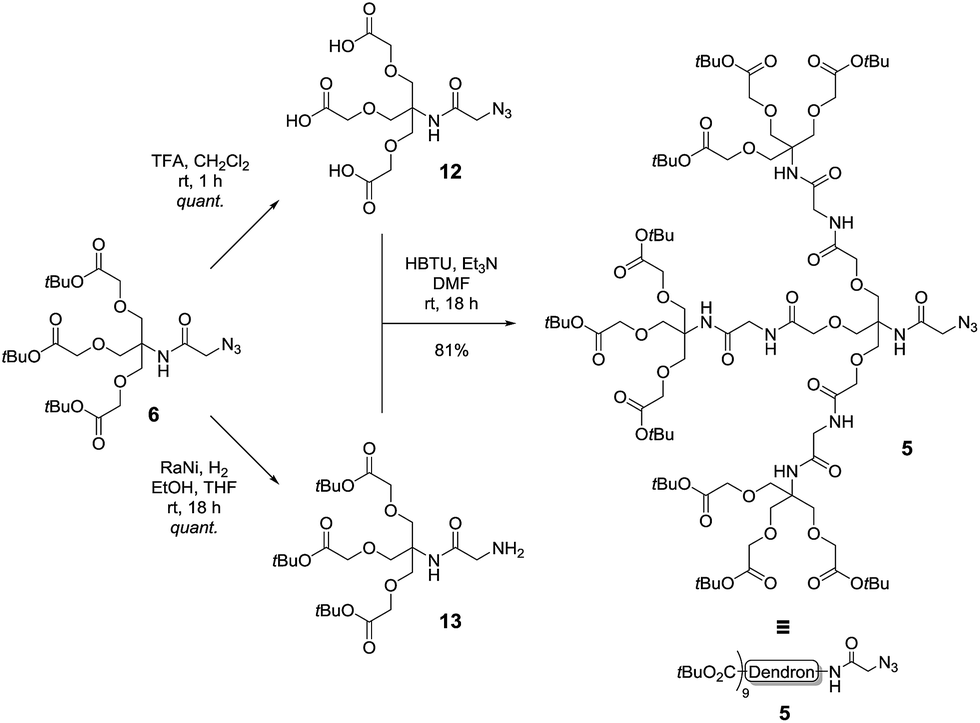 | ||
| Scheme 4 Synthesis of second generation dendron 5 using a double exponential growth coupling strategy. | ||
To demonstrate the versatility of our synthetic methodology, we then sought to functionalise the dendron scaffold 5 with either biotin or a fluorescent group. First, proof-of-concept was established via the synthesis of a second-generation biotin-functionalised dendron scaffold containing GlcNAc (Scheme 5). To this end, the azide in dendron 5 was converted into an amide via RANEY® nickel hydrogenation and the crude product coupled to D-biotin using an HBTU-mediated peptide ligation to give the biotinylated dendron 14 in 95% yield over two steps (Scheme 5). Removal of the tert-butyl esters using TFA in CH2Cl2 then occurred smoothly to afford carboxy dendron 15 in excellent yield, which was subsequently conjugated to oxyamine glycoside 16, itself prepared in 81% yield via the conjugation of GlcNAc to 3-(methoxyamino)propanyl-amine hydrochloride 4 according to previously published procedures.5 To facilitate the conjugation, excess oxyamine-functionalised glycan 16 (2 equiv. per carboxyl group) was used and the reaction mixture was stirred at room temperature overnight. Dialysis (cellulose ester dialysis membrane, 500 Da molecular weight cut-off) for 48 hours with a 0.1 M Na2HPO4 buffer at pH 7.5 to prevent oxyamine linker hydrolysis,26 followed by lyophilisation and reverse phase chromatography (C8) then gave the biotinlyated glycodendron 17 in very good yield (72%).
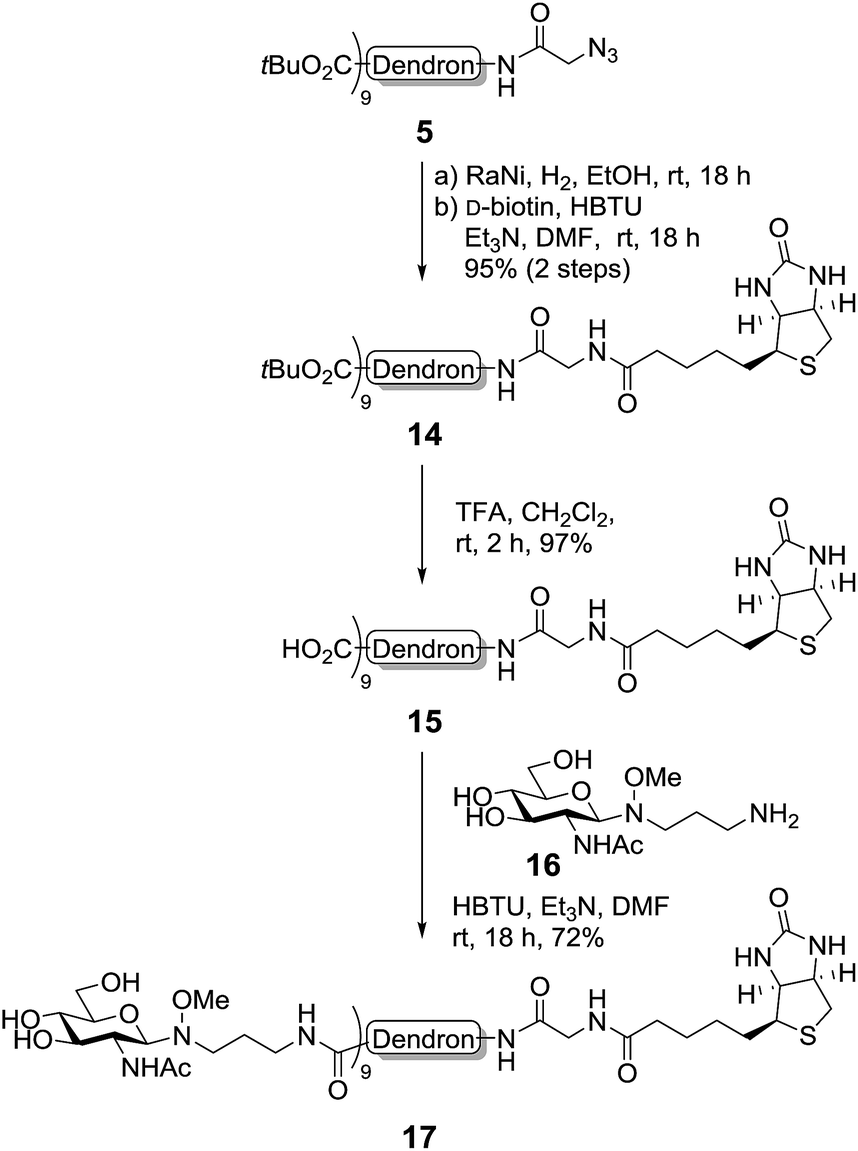 | ||
| Scheme 5 Functionalisation of the second-generation dendron with biotin and GlcNAc. | ||
To synthesise the LewisX glycodendron, a similar reaction route to that employed for the biotinylation of the dendrimer core was undertaken. Accordingly, the azide-group in the tert-butyl ester-functionalised dendron 5 was first reduced to the corresponding amine and then coupled to fluorescein isothiocyanate (FITC) to yield fluorescein derivative 18 in good yield (71%, 2 steps, Scheme 6). Treatment of 18 with TFA however, resulted in the Edman degradation27 products 19 and 20. To prevent this reaction, the core dendron 5 was first converted into the corresponding amine via Staudinger reduction and the tert-butyl esters were then removed via treatment with TFA to give the amine-functionalised carboxy dendron 21 in excellent yield (99%, 2 steps). Next, conjugation with FITC under basic conditions (NEt3 in DMF) gave the target fluorescent dendron 22, also in very good (74%) yield. The fluorescent dendron was initially obtained as the triethylamine salt after size exclusion chromatography, however, as this salt hampered ligation to the neoglycoconjugates, ion exchange (Dowex-H+) was performed to obtain the dendron as the nona-valent acid.
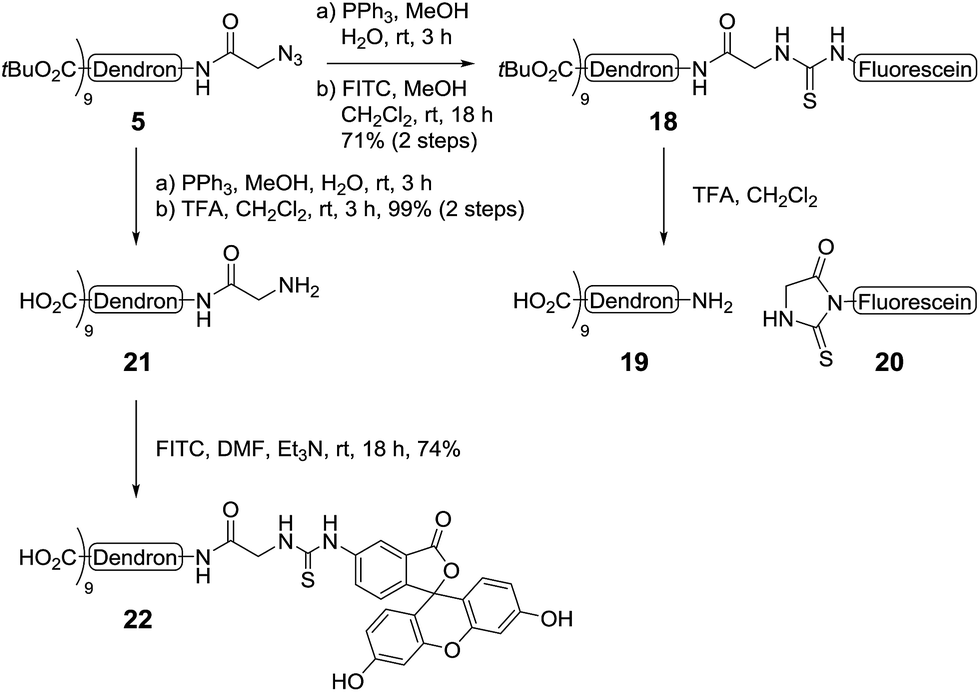 | ||
| Scheme 6 Synthesis of fluorescent dendron. | ||
For the assembly of the target LewisX glycodendron, LewisX (23)28 was condensed with amine-functionalised oxyamine linker 410 (Scheme 7). When the reaction was performed at room temperature, only a small amount of glycoconjugate was observed, however, heating the reaction at 40 °C for 36 hours saw complete conversion to the desired oxyamine-linked N-glycan. Purification by size exclusion chromatography (BioGel P2) then allowed for the isolation of the target oxyamine-linked glycan as the ammonium formate salt. As the presence of this salt could lead to the formation of formamide by-products in the subsequent peptide coupling reaction, the oxyamine-linked glycan was treated with ion exchange resin (Dowex OH−) to give the free amine 24 in excellent yield (88%). Next, conjugation with the FITC-labelled second-generation dendron 22 was performed using an HBTU-mediated peptide coupling reaction. Here, the order of addition of reagents was found to be important with the fluorescent dendron 22 being dissolved in freshly distilled DMF and then added to the LewisX glycan 23 followed by the addition of HBTU and Et3N, as the glycan itself had poor solubility in DMF alone, though completely dissolved after the addition of HBTU and Et3N. The reaction mixture was then stirred at room temperature overnight, upon which analysis by HRMS indicated that the desired nona-valent glycodendron was formed (m/z for [C285H476N36O168S]4+ calcd: 1781.7393, obsd: 1781.7380). Purification of the glycodendron using size exclusion chromatography (Sephadex CM C-25, 0.1 M aq. NH4HCO2) then allowed for the isolation of the fluorescent LewisX-functionalised glycodendron 25 in 51% yield.
 | ||
| Scheme 7 Synthesis of the LewisX functionalized glycodendron via conjugation of LewisX to the fluorescein-derived second-generation dendron using the bi-functional oxyamine linker. | ||
To demonstrate the potential of glycodendron 25 as a chemical tool, we then assessed whether the glycodendron could be used to detect DC-SIGN on the human macrophage cell line THP-1. To this end, human THP-1 cells were converted to the M2-like phenotype via treatment with phorbol-12-myristate-13-acetate (PMA) and interleukin (IL)-4,29 and the presence of DC-SIGN on the cells confirmed through use of an anti-DC-SIGN antibody (Fig. 1A). Glycodendron 25 was then made up in a stock-solution of 0.66 mg mL−1 (0.1 μM) and assessed for its ability to detect DC-SIGN on the THP-1 + PMA + IL-4 cells, with the unglycosylated dendron 22 being used as a negative control (Fig. 1B). As illustrated, both the anti-DC-SIGN antibody and the glycodendron 25 bound DC-SIGN in a concentration dependent manner (Fig. 1A and B). The unglycosylated dendron 22 did not show binding to the macrophages at low μM concentrations, with only the highest concentration tested (100 μM) giving a slight increase in mean fluorescence due to non-specific binding. Next, co-staining of the glycodendron with the anti-DC-SIGN antibody and 25 was conducted (Fig. 1C and D). Here, it is important to note that the co-staining with the antibody did not interfere with the glycan-mediated binding.
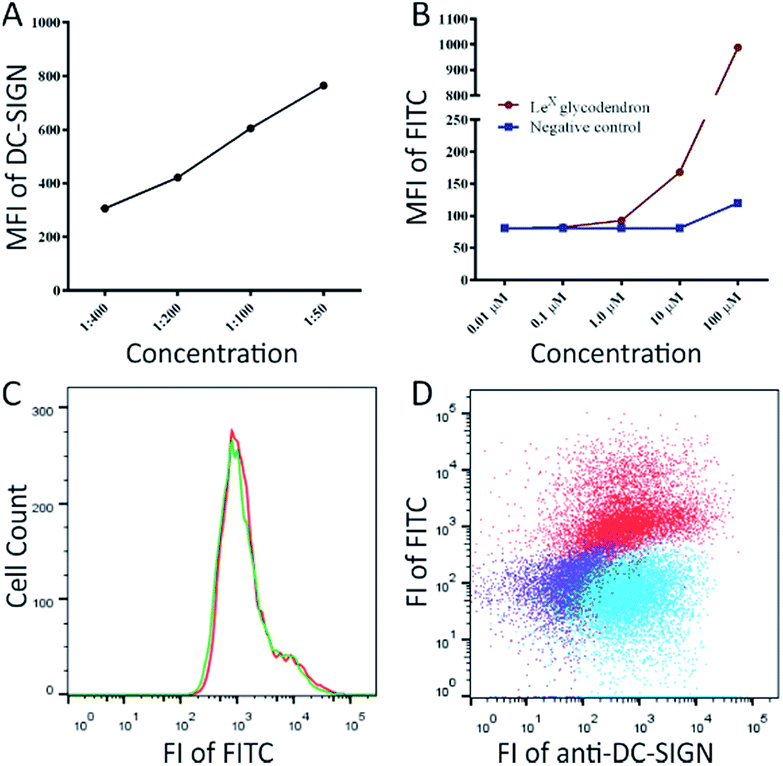 | ||
| Fig. 1 The expression of DC-SIGN on THP-1 + PMA + IL-4 cells measured with (A) anti-DC-SIGN antibody and (B) unglycosylated dendron 21 (negative control) and the fluorescent LewisX glycodendron 27. (C) The co-staining with anti-DC-SIGN antibody does not affect glycodendron binding to THP-1 + PMA + IL-4 cells, single staining with 100 μM glycodendron (green), co-staining of the anti-DC-SIGN antibody with 100 μM glycodendron (red); (D) the expression of DC-SIGN visualised with glycodendron and anti-DC-SIGN antibody on THP-1 + PMA + IL-4 cells, using single staining with the anti-DC-SIGN antibody (blue), co-staining of the antibody with 100 μM glycodendron (red) and unstained cells (purple). | ||
Conclusions
In conclusion, we have presented the design and synthesis of a dendron scaffold that can be readily functionalised at both its core and periphery. The synthesis of the second-generation dendron scaffold 5 was achieved in six linear steps and in an excellent overall yield of 30%. Moreover, the ease of functionalisation of the dendron core was demonstrated via its rapid and efficient conversion to both biotinylated and fluorescently labelled derivatives. The use of oxyamine ligation methodology to efficiently conjugate multiple glycans to a dendron scaffold was presented via the conjugation of GlcNAc to the biotinylated dendron scaffold and the conjugation of LewisX to a fluorescently labelled dendron scaffold, whereby the fluorescently-labelled LewisX glycodendron was used to detect DC-SIGN on macrophages. Given the efficiency and versatility of our procedures, we envision undertaking future carbohydrate–lectin binding studies employing glycodendron 25 and related glycodendrons in due course.Experimental
General procedures
Prior to use, THF was distilled from sodium and benzophenone, CH2Cl2 was distilled from P2O5, DMF was distilled from BaO, Et3N was distilled from KOH. All other reagents were used as received. 3-(Methoxyamino)propanyl-amine hydrochloride 4,5 oxyamine-linked GlcNAc 16,5 and LewisX (23)27 were synthesised as previously described. All solvents were removed by evaporation under reduced pressure. Reactions were monitored by TLC-analysis on Macherey-Nagel silica gel coated plastic sheets (0.20 mm, with fluorescent indicator UV254) with detection by UV-absorption (short wave UV – 254 nm; long wave UV – 366 nm), by dipping in 10% H2SO4 in EtOH followed by charring at ∼150 °C, by dipping in I2 in silica, or by dipping into a solution of ninhydrin in EtOH followed by charring at ∼150 °C. Column chromatography was performed on Pure Science silica gel (40-63 micron). AccuBOND II ODS-C18 (Agilent) was used for reverse phase chromatography. Infrared spectra were recorded as thin films using a Bruker Tensor 27 FTIR spectrometer equipped with an Attenuated Total Reflectance (ATR) sampling accessory and are reported in wave numbers (cm−1). Nuclear magnetic resonance spectra were recorded at 20 °C in D2O, CD3OD, CDCl3, or pyridine-d5 using either a Varian INOVA operating at 500 MHz or Varian VNMRS operating at 600 MHz. Chemical shifts are given in ppm (δ) relative to solvent residues. NMR peak assignments were made using COSY, HSQC and HMBC experiments.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 13C NMR (125 MHz, CDCl3) δ 168.6 (2 × N–CO), 164.3 (O–CO), 48.1 (CH2–N3), 25.7 (2 × αCH2).:EtOAc, 50/50 → 0/100, v/v) yielded the title compound 11 as a white foam (7.84 g, 38.4 mmol, 86%). Rf = 0.50 (CH2Cl2/EtOH/MeOH/NH3 (aq. 33%), 5/2/2/1, v/v/v/v); IR (film) 3359, 2979, 2946, 2890, 2113, 1739, 1650, 1540, 1454, 1367, 1282, 1229, 1217, 1053 cm−1; 1H NMR (500 MHz, D2O) δ 4.00 (s, 2H, CH2–N3), 3.76 (s, 6H, 3 × CH2–O); 13C NMR (125 MHz, D2O) δ 170.4 (CO amide), 62.1 (Cq tris) 60.1 (3 × CH2–O), 51.9 (CH2–N3); HRMS(ESI) m/z calcd for [C6H13N4O4]+: 205.0931, obsd: 205.0930.:EtOAc, 85/15 → 75/25, v/v) and reverse phase column chromatography (C18, H2O/MeOH, 50/50 → 20/80, v/v) yielded tri-alkylated 6 as a white foam (464 mg, 0.85 mmol, 47%). Rf = 0.44 (PE/EtOAc, 70/30, v/v); IR (film) 3316, 2979, 2935, 2107, 1745, 1684, 1536, 1473, 1428, 1393, 1368, 1229, 1158, 1119, 1037, 971, 915, 845, 731, 647 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.86 (s, 1H, NH), 3.95 (s, 6H, 3 × CH2CO), 3.92 (s, 6H, 3 × CH2–O), 3.85 (s, 2H, CH2–N3), 1.45 (s, 27H, CH3 tBu); 13C NMR (125 MHz, CDCl3) δ 170.2 (3 × CO ester), 167.5 (1 × CO amide), 82.0 (3 × Cq tBu), 70.1 (3 × CH2–O), 68.9 (3 × CH2CO), 60.1 (Cq tris), 52.9 (CH2–N3), 28.2 (9 × CH3 tBu); HRMS(ESI) m/z calcd for [C24H43N4O10]+: 547.2974, obsd: 547.2981.O), 3.98 (s, 6H, 3 × CH2–O), 3.90 (s, 2H, CH2–N3); 13C NMR (125 MHz, CDCl3) δ 174.3 (3 × CO), 170.2 (1 × CO amide), 69.6 (3 × CH2–O), 68.1 (3 × CH2CO), 59.9 (Cq tris), 52.0 (CH2–N3); HRMS(ESI) m/z calcd for [C12H19N4O10Na]+: 401.0915, obsd: 401.0920.O), 3.88 (s, 6H, 3 × CH2–O), 3.30 (s, 2H, CH2–NH2), 2.07 (bs, 2H, NH2) 1.42 (s, 27H, 9 × CH3 tBu); 13C NMR (125 MHz, CDCl3) δ 173.2 (1 × CO amide), 170.3 (3 × CO ester), 81.8 (3 × Cq tBu), 70.2 (3 × CH2–O), 69.0 (3 × CH2CO), 59.5 (Cq tris), 45.6 (CH2–NH2), 28.2 (9 × CH3 tBu); HRMS(ESI) m/z calcd for [C24H45N2O10Na]+: 543.2888, obsd: 543.2891.O), 4.01 (d, 6H, JCH2,NH = 4.5 Hz, 3 × CH2–N), 3.98 (s, 2H, CH2–N3), 3.95 (s, 18H, 9 × CH2CO), 3.93 (s, 6H, 3 × CH2–O), 3.89 (s, 18H, 9 × CH2–O), 1.45 (s, 54H, 18 × CH3 t-Bu); 13C NMR (125 MHz, CDCl3) δ 170.3 (9 × CO ester), 169.6 (3 × CO amide), 168.5 (3 × CO amide), 168.2 (1 × CO amide), 82.0 (Cq tBu), 70.7 (3 × CH2–O), 70.6 (3 × CH2CO), 70.2 (9 × CH2–O), 68.9 (9 × CH2CO), 60.0 (3 × Cq tris), 59.8 (1 × Cq tris) 52.5 (CH2–N3), 42.2 (3 × CH2–NH), 28.3 (27 × CH3 tBu); HRMS(ESI) m/z calcd for [C84H146N10O37]2+: 943.4920, obsd: 943.4929.O, 3 × CH2CO, CH2N), 3.08 (dt, 1H, J5,6 = 4.8 Hz, J4,5 = 7.0 Hz, H5-biotin), 2.85 (dd, 1H, J7,8a = 5.3 Hz, J8a,8b = 13.0 Hz, H8a-biotin), 2.69 (d, 1H, J8a,8b = 12.8 Hz, H8b-biotin), 2.28–2.16 (m, 2H, CH2-1-biotin), 1.71–1.50 (m, 6H, CH2-2, CH2-3, CH2-4-biotin), 1.41 (s, 81H, 27 × CH3 tBu); 13C NMR (125 MHz, CDCl3) δ 174.4 (CO biotin), 170.6 (CO dendron), 170.1 (9 × CO), 170.0 (3 × CO), 169.0 (3 × CO), 163.9 (CO urea biotin), 81.9 (9 × Cq tBu), 70.6 (3 × CH2CO), 70.4 (3 × CH2–O), 70.0 (9 × CH2–O), 68.8 (9 × CH2CO), 61.8 (C6-biotin), 60.1 (C7-biotin), 59.9 (3 × Cq tris), 59.5 (1 × Cq tris), 55.5 (C5-biotin), 43.7 (1 × CH2–N), 42.2 (3 × CH2–N), 40.3 (C8-biotin), 35.2 (C1-biotin), 28.2 (C2-biotin), 28.0 (27 × CH3), 27.9 (C4-biotin), 25.3 (C3-biotin); HRMS(ESI) m/z calcd for [C94H162N10O39S]2+: 1043.5355, obsd: 1043.5351.O biotin), 174.2 (9 × CO), 172.8 (3 × CO), 171.3 (1 × CO), 170.7 (3 × CO), 165.2 (CO urea biotin), 70.0 (3 × CH2CO), 69.8 (3 × CH2–O), 69.7 (9 × CH2–O), 68.0 (9 × CH2CO), 62.0 (C6-biotin), 60.2 (C7-biotin), 59.8 (3 × Cq tris), 59.6 (1 × Cq tris), 55.2 (C5-biotin), 43.0 (1 × CH2–N), 42.4 (3 × CH2–N), 39.6 (C8-biotin), 35.1 (C1-biotin), 27.9 (C2-biotin), 27.6 (C4-biotin), 25.0 (C3-biotin); HRMS(ESI) m/z calcd for [C58H90N10O39S]2+: 791.2538, obsd: 791.2536.O dendron), 4.07 (s, 18H, 9 × CH2CO dendron), 3.96 (s, 6H, 3 × CH2–N dendron), 3.92–3.83 (m, 37H, 1 × CH2N dendron, 9 × H-2 GlcNAc, 9 × H-6a GlcNAc, 9 × CH2-dendron), 3.72 (dd, 9H, J5,6a = 5.5 Hz, J6a,6b = 12.4 Hz, 9 × H-6b GlcNAc), 3.51 (dd, 9H, J2,3 = 8.8 Hz, J3,4 = 9.8 Hz, 9 × H-3 GlcNAc), 3.48 (s, 27H, 9 × NOCH3), 3.43–3.28 (m, 28H, 9 × CH2a-3 linker, 9 × H-4 GlcNAc, 9 × H-5 GlcNAc, H5-biotin), 3.24 (dt, 9H, J2,3 = 7.2 Hz, J3a,3b = 13.4 Hz, 9 × CH2b-3 linker), 3.01–2.87 (m, 19H, 9 × CH2-1 linker, H8a-biotin), 2.75 (d, 1H, J8a,8b = 12.9 Hz, H8b-biotin), 2.30 (t, 2H, J1,2 = 7.3 Hz, CH2-1-biotin), 2.03 (s, 27H, 9 × N–Ac), 1.88–1.78 (m, 1H, H4a-biotin), 1.77–1.69 (m, 18H, 9 × CH2-2 linker), 1.69–1.58 (m, 1H, H4a-biotin) 1.58–1.50 (m, 2H, CH2-2-biotin), 1.44–1.36 (m, 2H, CH2-3-biotin); HRMS(ESI) m/z calcd for [C166H298N37O84S]3+: 1395.6641, obsd: 1395.6648.O), 4.00 (s, 6H, 3 × CH2CO), 3.83 (s, 6H, 3 × CH2N), 3.77 (s, 6H, 3 × CH2O), 3.75 (s, 18H, 9 × CH2O), 3.71 (s, 2H, 1 × CH2NH2); 13C NMR (125 MHz, D2O) δ 174.1 (9 × CO CO2H), 172.6 (3 × CONH), 170.6 (3 × CO), 166.7 (1 × CO), 162.8, 162.5, 162.2, 161.9 (CO TFA), 119.4, 117.1, 114.8, 112.5 (CF3 TFA), 69.83, 69.77 (3 × CH2O, 3 × CH2CO), 69.6 (9 × CH2O), 67.9 (9 × CH2CO), 59.9 (1 × Cq tris), 59.7 (3 × Cq tris), 42.3 (3 × CH2NH), 40.6 (1 × CH2NH2); HRMS(ESI) m/z calcd for [C48H76N8O37]2+: 678.2150, obsd: 678.2136.O, 9 × CH2CO), 4.02–3.92 (m, 12H, 3 × CH2O, 3 × CH2NH), 3.92–3.86 (m, 18H, 9 × CH2O); 13C NMR (125 MHz, CD3OD) δ 183.5 (CS), 174.5 (9 × CO), 173.0 (3 × CO), 172.0 (1 × CO glyc), 171.4 (3 × CO glyc), 171.2 (CO C-1), 161.5, 154.2 (C-12, C14), 149.7 (C-7), 142.2 (C-2/4/8), 132.1 (C-5), 130.4 (C-13), 130.3 (C-2/4/8), 128.9 (C-2/4/8), 125.8 (C-6), 120.3 (C-3), 113.8 (C-11), 111.5 (C-9), 103.6 (C-10), 71.4 (3 × CH2–O), 71.0 (9 × CH2O), 69.3 (3 × CH2CO), 69.2 (9 × CH2CO), 61.3 (3 × Cq tris), 61.2 (1 × Cq tris), 48.9 (1 × CH2NH), 43.5 (3 × CH2NH); HRMS(ESI) m/z calcd for [C69H87N9O42S]2+: 872.7329, obsd: 872.7335.117, 1647, 1586, 1466, 1451, 1415, 1380, 1350, 1302, 1233, 1193, 1085, 1026, 968, 917 cm−1; 1H NMR (500 MHz, D2O) δ 5.12 (d, 1H, J1′′′,2′′′ = 3.9 Hz, H-1′′′), 4.84 (q, 1H, J5′′′,6′′′ = 6.7 Hz, H-5′′′), 4.49–4.42 (m, 1H, H-1′, H-1′′), 4.05 (m, 2H, H-2′, H-6a′), 3.93–3.81 (m, 5H, H-3′′′, H-4′′, H-4′, H-3′, H-6b′), 3.80 (d, 1H, J3′′′,4′′′ = J4′′′,5′′′ = 2.9 Hz, H-4′′′), 3.76 (m, 3H, H-6a′′, H-6b′′, H-2′′′), 3.64 (dd, 1H, J3′′,4′′ = 3.2 Hz, J2′′,3′′ = 9.9 Hz, H-3′′), 3.60–3.55 (m, 1H, H-5′′), 3.54–3.46 (m, 1H, H-5′′), 3.54–3.46 (m, 5H, OCH3, H-5′, H-2′′), 3.11–2.92 (m, 4H, CH2-1, CH2-3), 2.02 (s, 3H, CH3 Ac), 1.95 (m, 2H, CH2-2), 1.17 (d, 3H, J5′′′,6′′′ = 6.7 Hz, H-6′′′); 13C NMR (125 MHz, D2O) 170.5 (CO), 101.8 (C-1′′), 98.7 (C-1′′′), 90.5 (C-1′), 76.9 (C-5′), 76.1 (C-3′), 74.8 (C-5′′), 73.2 (C-4′), 72.4 (C-3′′), 71.8 (C-4′′′), 70.9 (C-2′′), 69.1 (C-3′′′), 68.3 (C-4′′), 67.6 (C-2′′′), 66.7 (C-5′′′), 61.4 (C-6′′), 60.9 (OCH3), 59.7 (C-6′), 52.5 (C-2′), 47.2 (C-1), 37.5 (C-3), 24.5 (C-2), 22.1 (CH3 Ac), 15.2 (C-6′′′); HRMS(ESI) m/z calcd for [C24H46N3O15]+: 616.2923, obsd: 616.2938.
O); 13C NMR (125 MHz, CDCl3) δ 168.6 (2 × N–CO), 164.3 (O–CO), 48.1 (CH2–N3), 25.7 (2 × αCH2).:EtOAc, 50/50 → 0/100, v/v) yielded the title compound 11 as a white foam (7.84 g, 38.4 mmol, 86%). Rf = 0.50 (CH2Cl2/EtOH/MeOH/NH3 (aq. 33%), 5/2/2/1, v/v/v/v); IR (film) 3359, 2979, 2946, 2890, 2113, 1739, 1650, 1540, 1454, 1367, 1282, 1229, 1217, 1053 cm−1; 1H NMR (500 MHz, D2O) δ 4.00 (s, 2H, CH2–N3), 3.76 (s, 6H, 3 × CH2–O); 13C NMR (125 MHz, D2O) δ 170.4 (CO amide), 62.1 (Cq tris) 60.1 (3 × CH2–O), 51.9 (CH2–N3); HRMS(ESI) m/z calcd for [C6H13N4O4]+: 205.0931, obsd: 205.0930.:EtOAc, 85/15 → 75/25, v/v) and reverse phase column chromatography (C18, H2O/MeOH, 50/50 → 20/80, v/v) yielded tri-alkylated 6 as a white foam (464 mg, 0.85 mmol, 47%). Rf = 0.44 (PE/EtOAc, 70/30, v/v); IR (film) 3316, 2979, 2935, 2107, 1745, 1684, 1536, 1473, 1428, 1393, 1368, 1229, 1158, 1119, 1037, 971, 915, 845, 731, 647 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.86 (s, 1H, NH), 3.95 (s, 6H, 3 × CH2CO), 3.92 (s, 6H, 3 × CH2–O), 3.85 (s, 2H, CH2–N3), 1.45 (s, 27H, CH3 tBu); 13C NMR (125 MHz, CDCl3) δ 170.2 (3 × CO ester), 167.5 (1 × CO amide), 82.0 (3 × Cq tBu), 70.1 (3 × CH2–O), 68.9 (3 × CH2CO), 60.1 (Cq tris), 52.9 (CH2–N3), 28.2 (9 × CH3 tBu); HRMS(ESI) m/z calcd for [C24H43N4O10]+: 547.2974, obsd: 547.2981.O), 3.98 (s, 6H, 3 × CH2–O), 3.90 (s, 2H, CH2–N3); 13C NMR (125 MHz, CDCl3) δ 174.3 (3 × CO), 170.2 (1 × CO amide), 69.6 (3 × CH2–O), 68.1 (3 × CH2CO), 59.9 (Cq tris), 52.0 (CH2–N3); HRMS(ESI) m/z calcd for [C12H19N4O10Na]+: 401.0915, obsd: 401.0920.O), 3.88 (s, 6H, 3 × CH2–O), 3.30 (s, 2H, CH2–NH2), 2.07 (bs, 2H, NH2) 1.42 (s, 27H, 9 × CH3 tBu); 13C NMR (125 MHz, CDCl3) δ 173.2 (1 × CO amide), 170.3 (3 × CO ester), 81.8 (3 × Cq tBu), 70.2 (3 × CH2–O), 69.0 (3 × CH2CO), 59.5 (Cq tris), 45.6 (CH2–NH2), 28.2 (9 × CH3 tBu); HRMS(ESI) m/z calcd for [C24H45N2O10Na]+: 543.2888, obsd: 543.2891.O), 4.01 (d, 6H, JCH2,NH = 4.5 Hz, 3 × CH2–N), 3.98 (s, 2H, CH2–N3), 3.95 (s, 18H, 9 × CH2CO), 3.93 (s, 6H, 3 × CH2–O), 3.89 (s, 18H, 9 × CH2–O), 1.45 (s, 54H, 18 × CH3 t-Bu); 13C NMR (125 MHz, CDCl3) δ 170.3 (9 × CO ester), 169.6 (3 × CO amide), 168.5 (3 × CO amide), 168.2 (1 × CO amide), 82.0 (Cq tBu), 70.7 (3 × CH2–O), 70.6 (3 × CH2CO), 70.2 (9 × CH2–O), 68.9 (9 × CH2CO), 60.0 (3 × Cq tris), 59.8 (1 × Cq tris) 52.5 (CH2–N3), 42.2 (3 × CH2–NH), 28.3 (27 × CH3 tBu); HRMS(ESI) m/z calcd for [C84H146N10O37]2+: 943.4920, obsd: 943.4929.O, 3 × CH2CO, CH2N), 3.08 (dt, 1H, J5,6 = 4.8 Hz, J4,5 = 7.0 Hz, H5-biotin), 2.85 (dd, 1H, J7,8a = 5.3 Hz, J8a,8b = 13.0 Hz, H8a-biotin), 2.69 (d, 1H, J8a,8b = 12.8 Hz, H8b-biotin), 2.28–2.16 (m, 2H, CH2-1-biotin), 1.71–1.50 (m, 6H, CH2-2, CH2-3, CH2-4-biotin), 1.41 (s, 81H, 27 × CH3 tBu); 13C NMR (125 MHz, CDCl3) δ 174.4 (CO biotin), 170.6 (CO dendron), 170.1 (9 × CO), 170.0 (3 × CO), 169.0 (3 × CO), 163.9 (CO urea biotin), 81.9 (9 × Cq tBu), 70.6 (3 × CH2CO), 70.4 (3 × CH2–O), 70.0 (9 × CH2–O), 68.8 (9 × CH2CO), 61.8 (C6-biotin), 60.1 (C7-biotin), 59.9 (3 × Cq tris), 59.5 (1 × Cq tris), 55.5 (C5-biotin), 43.7 (1 × CH2–N), 42.2 (3 × CH2–N), 40.3 (C8-biotin), 35.2 (C1-biotin), 28.2 (C2-biotin), 28.0 (27 × CH3), 27.9 (C4-biotin), 25.3 (C3-biotin); HRMS(ESI) m/z calcd for [C94H162N10O39S]2+: 1043.5355, obsd: 1043.5351.O biotin), 174.2 (9 × CO), 172.8 (3 × CO), 171.3 (1 × CO), 170.7 (3 × CO), 165.2 (CO urea biotin), 70.0 (3 × CH2CO), 69.8 (3 × CH2–O), 69.7 (9 × CH2–O), 68.0 (9 × CH2CO), 62.0 (C6-biotin), 60.2 (C7-biotin), 59.8 (3 × Cq tris), 59.6 (1 × Cq tris), 55.2 (C5-biotin), 43.0 (1 × CH2–N), 42.4 (3 × CH2–N), 39.6 (C8-biotin), 35.1 (C1-biotin), 27.9 (C2-biotin), 27.6 (C4-biotin), 25.0 (C3-biotin); HRMS(ESI) m/z calcd for [C58H90N10O39S]2+: 791.2538, obsd: 791.2536.O dendron), 4.07 (s, 18H, 9 × CH2CO dendron), 3.96 (s, 6H, 3 × CH2–N dendron), 3.92–3.83 (m, 37H, 1 × CH2N dendron, 9 × H-2 GlcNAc, 9 × H-6a GlcNAc, 9 × CH2-dendron), 3.72 (dd, 9H, J5,6a = 5.5 Hz, J6a,6b = 12.4 Hz, 9 × H-6b GlcNAc), 3.51 (dd, 9H, J2,3 = 8.8 Hz, J3,4 = 9.8 Hz, 9 × H-3 GlcNAc), 3.48 (s, 27H, 9 × NOCH3), 3.43–3.28 (m, 28H, 9 × CH2a-3 linker, 9 × H-4 GlcNAc, 9 × H-5 GlcNAc, H5-biotin), 3.24 (dt, 9H, J2,3 = 7.2 Hz, J3a,3b = 13.4 Hz, 9 × CH2b-3 linker), 3.01–2.87 (m, 19H, 9 × CH2-1 linker, H8a-biotin), 2.75 (d, 1H, J8a,8b = 12.9 Hz, H8b-biotin), 2.30 (t, 2H, J1,2 = 7.3 Hz, CH2-1-biotin), 2.03 (s, 27H, 9 × N–Ac), 1.88–1.78 (m, 1H, H4a-biotin), 1.77–1.69 (m, 18H, 9 × CH2-2 linker), 1.69–1.58 (m, 1H, H4a-biotin) 1.58–1.50 (m, 2H, CH2-2-biotin), 1.44–1.36 (m, 2H, CH2-3-biotin); HRMS(ESI) m/z calcd for [C166H298N37O84S]3+: 1395.6641, obsd: 1395.6648.O), 4.00 (s, 6H, 3 × CH2CO), 3.83 (s, 6H, 3 × CH2N), 3.77 (s, 6H, 3 × CH2O), 3.75 (s, 18H, 9 × CH2O), 3.71 (s, 2H, 1 × CH2NH2); 13C NMR (125 MHz, D2O) δ 174.1 (9 × CO CO2H), 172.6 (3 × CONH), 170.6 (3 × CO), 166.7 (1 × CO), 162.8, 162.5, 162.2, 161.9 (CO TFA), 119.4, 117.1, 114.8, 112.5 (CF3 TFA), 69.83, 69.77 (3 × CH2O, 3 × CH2CO), 69.6 (9 × CH2O), 67.9 (9 × CH2CO), 59.9 (1 × Cq tris), 59.7 (3 × Cq tris), 42.3 (3 × CH2NH), 40.6 (1 × CH2NH2); HRMS(ESI) m/z calcd for [C48H76N8O37]2+: 678.2150, obsd: 678.2136.O, 9 × CH2CO), 4.02–3.92 (m, 12H, 3 × CH2O, 3 × CH2NH), 3.92–3.86 (m, 18H, 9 × CH2O); 13C NMR (125 MHz, CD3OD) δ 183.5 (CS), 174.5 (9 × CO), 173.0 (3 × CO), 172.0 (1 × CO glyc), 171.4 (3 × CO glyc), 171.2 (CO C-1), 161.5, 154.2 (C-12, C14), 149.7 (C-7), 142.2 (C-2/4/8), 132.1 (C-5), 130.4 (C-13), 130.3 (C-2/4/8), 128.9 (C-2/4/8), 125.8 (C-6), 120.3 (C-3), 113.8 (C-11), 111.5 (C-9), 103.6 (C-10), 71.4 (3 × CH2–O), 71.0 (9 × CH2O), 69.3 (3 × CH2CO), 69.2 (9 × CH2CO), 61.3 (3 × Cq tris), 61.2 (1 × Cq tris), 48.9 (1 × CH2NH), 43.5 (3 × CH2NH); HRMS(ESI) m/z calcd for [C69H87N9O42S]2+: 872.7329, obsd: 872.7335.117, 1647, 1586, 1466, 1451, 1415, 1380, 1350, 1302, 1233, 1193, 1085, 1026, 968, 917 cm−1; 1H NMR (500 MHz, D2O) δ 5.12 (d, 1H, J1′′′,2′′′ = 3.9 Hz, H-1′′′), 4.84 (q, 1H, J5′′′,6′′′ = 6.7 Hz, H-5′′′), 4.49–4.42 (m, 1H, H-1′, H-1′′), 4.05 (m, 2H, H-2′, H-6a′), 3.93–3.81 (m, 5H, H-3′′′, H-4′′, H-4′, H-3′, H-6b′), 3.80 (d, 1H, J3′′′,4′′′ = J4′′′,5′′′ = 2.9 Hz, H-4′′′), 3.76 (m, 3H, H-6a′′, H-6b′′, H-2′′′), 3.64 (dd, 1H, J3′′,4′′ = 3.2 Hz, J2′′,3′′ = 9.9 Hz, H-3′′), 3.60–3.55 (m, 1H, H-5′′), 3.54–3.46 (m, 1H, H-5′′), 3.54–3.46 (m, 5H, OCH3, H-5′, H-2′′), 3.11–2.92 (m, 4H, CH2-1, CH2-3), 2.02 (s, 3H, CH3 Ac), 1.95 (m, 2H, CH2-2), 1.17 (d, 3H, J5′′′,6′′′ = 6.7 Hz, H-6′′′); 13C NMR (125 MHz, D2O) 170.5 (CO), 101.8 (C-1′′), 98.7 (C-1′′′), 90.5 (C-1′), 76.9 (C-5′), 76.1 (C-3′), 74.8 (C-5′′), 73.2 (C-4′), 72.4 (C-3′′), 71.8 (C-4′′′), 70.9 (C-2′′), 69.1 (C-3′′′), 68.3 (C-4′′), 67.6 (C-2′′′), 66.7 (C-5′′′), 61.4 (C-6′′), 60.9 (OCH3), 59.7 (C-6′), 52.5 (C-2′), 47.2 (C-1), 37.5 (C-3), 24.5 (C-2), 22.1 (CH3 Ac), 15.2 (C-6′′′); HRMS(ESI) m/z calcd for [C24H46N3O15]+: 616.2923, obsd: 616.2938.THP-1 derived macrophage differentiation
The THP-1 acute monocytic lymphoma cell line was cultured in RPMI 1640 medium supplemented with 10% Fetal Calf Serum (FCS), 1% Glutamax and 1% Penstrep, and seeded 2.5 × 105 cells per mL. Differentiation of the THP-1 cells was induced via the treatment with PMA (50 ng mL−1, 48 h.) followed by addition of IL-4 (20 ng mL−1, 24 h) to obtian ‘M2-like’ Mϕs according to the procedures of Puig-Kröger et al.29 For subsequent analysis, the differentiated cells were detached from the tissue culture plates by incubating the cells in PBS on ice.Flow cytometry
The macrophages were stained by incubating for 1 hour at 4 °C with either FITC-labelled glycodendron 25, negative control (non-glycosylated dendron 22) or anti-DC-SIGN antibody, washed twice with PBS, and DC-SIGN expression measured by flow cytometry. The fluorescent LewisX glycodendron and the negative control were used in various concentrations (10−1, 101, 102, 103, 104 and 105 nM, diluted in 10% FCS), where the human anti-DC-SIGN antibody (phycoerythrin-labelled) positive control was diluted by a factor 2 (1/50 → 1/1600, v/v).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Wellington Medical Research Foundation for financial support and the Health Research Council of New Zealand (Hercus Fellowship, BLS).Notes and references
- S. Weinbaum, J. M. Tarbell and E. R. Damiano, Annu. Rev. Biomed. Eng., 2007, 9, 121–167 CrossRef CAS PubMed.
- R. D. Cummings, Mol. BioSyst., 2009, 5, 1087–1104 RSC.
- (a) M. Mammen, S.-K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef; (b) J. J. Lundquist, S. D. Debenham and E. J. Toone, J. Org. Chem., 2000, 65, 8245–8250 CrossRef CAS PubMed.
- C. Müller, G. Despras and T. K. Lindhorst, Chem. Soc. Rev., 2016, 45, 3275–3302 RSC.
- S. Munneke, J. R. Prevost, G. F. Painter, B. L. Stocker and M. S. M. Timmer, Org. Lett., 2015, 17, 624–627 CrossRef CAS PubMed.
- S. Munneke, E. M. Dangerfield, B. L. Stocker and M. S. M. Timmer, Glycoconjugate J., 2017, 34, 633–642 CrossRef CAS PubMed.
- P. Stanley, H. Schachter and N. Taniguchi, N-Glycans, in Essentials of Glycobiology, ed. A. Varki, R. D. Cummings and J. D. Esko, et al., Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), 2nd edn, 2009, ch. 8 Search PubMed.
- T. B. Geijtenbeek, R. Torensma, S. J. van Vliet, G. C. van Duijnhoven, G. J. Adema, Y. van Kooyk and C. G. Figdor, Cell, 2000, 100, 575–585 CrossRef CAS PubMed.
- E. J. Soilleux, L. S. Morris, G. Leslie, J. Chehimi, Q. Luo, E. Levroney, J. Trowsdale, L. J. Montaner, R. W. Doms, D. Weissman, N. Coleman and B. Lee, J. Leukocyte Biol., 2002, 71, 445–457 CAS.
- W. K. Lai, P. J. Sun, J. Zhang, A. Jennings, P. F. Lalor, S. Hubscher, J. A. McKeating and D. H. Adams, Am. J. Pathol., 2006, 169, 200–208 CrossRef CAS PubMed.
- L. Tailleux, N. Pham-Thi, A. Bergeron-Lafaurie, J.-L. Herrmann, P. Charles, O. Schwartz, P. Scheinmann, P. H. Lagrange, J. de Blic, A. Tazi, B. Gicquel and O. Neyrolles, PLoS Med., 2005, 2, e381 Search PubMed.
- A. Domínguez-Soto, E. Sierra-Filardi, A. Puig-Kröger, B. Pérez-Maceda, F. Gómez-Aguado, M. T. Corcuera, P. Sánchez-Mateos and A. L. Corbí, J. Immunol., 2011, 186, 2192–2200 CrossRef PubMed.
- B. J. Appelmelk, I. van Die, S. J. van Vliet, C. M. J. E. Vandenbroucke-Grauls, T. B. H. Geijtenbeek and Y. van Kooyk, J. Immunol., 2003, 170, 1635–1639 CrossRef CAS.
- Y. van Kooyk and T. B. H. Geijtenbeek, Nat. Rev. Immunol., 2003, 3, 697–709 CrossRef CAS PubMed.
- J. J. Garcia-Vallejo, N. Koning, M. Ambrosini, H. Kalay, I. Vuist, R. Sarrami-Forooshani, T. B. H. Geijtenbeek and Y. van Kooyk, Int. Immunol., 2013, 25, 221–233 CrossRef CAS PubMed.
- R. Ribeiro-Viana, M. Sánchez-Navarro, J. Luczkowiak, J. R. Koeppe, R. Delgado, J. Rojo and B. G. Davis, Nat. Commun., 2012, 3, 1–8 Search PubMed.
- F. Lasala, E. Arce, J. R. Otero, J. Rojo and R. Delgado, Antimicrob. Agents Chemother., 2003, 47, 3970–3972 CrossRef CAS PubMed.
- R. Ribeiro-Viana, J. J. García-Vallejo, D. Collado, E. Pérez-Inestrosa, K. Bloem, Y. van Kooyk and J. Rojo, Biomacromolecules, 2012, 13, 3209–3219 CrossRef CAS PubMed.
- T. Kawaguchi, K. L. Walker, C. L. Wilkins and J. S. Moore, J. Am. Chem. Soc., 1995, 117, 2159–2165 CrossRef CAS.
- M. V. Walter and M. Malkoch, Chem. Soc. Rev., 2012, 41, 4593–4609 RSC.
- W. Kowalczyk, A. Mascaraque, M. Sánchez-Navarro, J. Rojo and D. Andreu, Eur. J. Org. Chem., 2012, 4565–4573 CrossRef CAS.
- A. Nelson and J. F. Stoddart, Carbohydr. Res., 2004, 339, 2069–2075 CrossRef CAS PubMed.
- For some representative glycodendron syntheses using TRIS as the branching core, see: (a) Y. M. Chabre, C. Contino-Pépin, V. Placide, T. C. Shiao and R. Roy, J. Org. Chem., 2008, 73, 5602–5605 CrossRef CAS PubMed; (b) R. Kaplánek, T. Bříza, M. Havlík, P. Martásek and V. Král, J. Fluorine Chem., 2007, 128, 179–183 CrossRef; (c) P. R. Ashton, S. E. Boyd, C. L. Brown, N. Jayaraman, S. A. Nepogodiev and J. F. Stoddart, Chem.–Eur. J., 1996, 2, 1115–1128 CrossRef CAS.
- C. Dupuy, R. Viguier and A. Dupraz, Synth. Commun., 2001, 31, 1307–1313 CrossRef CAS.
- G. R. Newkome and X. Lin, Macromolecules, 1991, 24, 1443–1444 CrossRef CAS.
- S. Munneke, J. C. Hill, M. S. M. Timmer and B. L. Stocker, Eur. J. Org. Chem., 2017, 3722–3728 CrossRef CAS.
- P. Edman, E. Högfeldt, L. G. Sillén and P.-O. Kinell, Acta Chem. Scand., 1950, 4, 283–293 CrossRef CAS.
- S. Munneke, G. Gainsford, G. F. Painter, B. L. Stocker and M. S. M. Timmer, Carbohydr. Res., 2015, 414, 1–7 CrossRef CAS PubMed.
- A. Puig-Kröger, D. Serrano-Gómez, E. Caparrós, A. Domínguez-Soto, M. Relloso, M. Colmenares, L. Martínez-Muñoz, N. Longo, N. Sánchez-Sánchez, M. Rincon, L. Rivas, P. Sánchez-Mateos, E. Fernández-Ruiz and A. L. Corbí, J. Biol. Chem., 2004, 279, 25680–25688 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08872h |
| This journal is © The Royal Society of Chemistry 2017 |