DOI:
10.1039/C7RA08832A
(Paper)
RSC Adv., 2017,
7, 49505-49517
A theoretical investigation of the two-photon absorption and fluorescent properties of coumarin-based derivatives for Pd2+ detection†
Received
10th August 2017
, Accepted 5th October 2017
First published on 24th October 2017
Abstract
Palladium is emitted due to automobile catalytic converters, and with rapid growth in the number of cars, palladium is thus a current threat to human health and the environment. Two-photon (TP) fluorescent probes are favorable and powerful molecular tools for palladium ion (Pd2+) detection due to localized excitation and reduced phototoxicity and photodamage. In the present study, a series of “turn-on” TP fluorescent dyes based on coumarin derivatives were designed for Pd2+ recognition. Our study revealed the origin of the peculiarly different fluorescence behaviors of the synthesized Pd2+ probe R1 and the product P′1 quantitatively and qualitatively from a theoretical perspective. Moreover, quantum-chemical calculations on electronic structures, one/two-photon absorption and fluorescence optical properties have first been carried out for these TP fluorescent chromophores using an ab initio approach. The calculated results demonstrate that chemical modifications of the coumarin core by the introduction of an electron-withdrawing group (–Cl or –CN) to its 4-position effectively increase the TP absorption cross-section per molecular weight more easily than extending the π-conjugated structure at the 3-position. In the present work, the product P′4, with a much smaller internal conversion rate (KIC = 1.28 × 106 s−1) than that of the Pd2+ probe R4 (KIC = 9.09 × 1011 s−1), possesses the largest TP absorption cross-section per molecular weight (3.91) and the longest fluorescence wavelength (590.3 nm) among all the studied molecules, which means it has better potential for Pd2+ detection. Consequently, we hope that this detailed study can provide guidance for the design and synthesis of new Pd2+ fluorescent probes.
1. Introduction
Significant amounts of palladium are emitted due to automobile catalytic converters and the rapid growth in the number of cars. As one of the platinum-group elements, palladium is hazardous to human health once it is taken up by the human body via contaminated water, medicine and food.1 In the natural environment, palladium has three states: Pd0 (metallic), Pd2+, and Pd4+. Its metallic form has little in vitro cytotoxicity, but Pd ions are capable of causing a series of cytotoxic effects, such as serious eye or primary skin irritation.2 Epidemiological studies have indicated that Pd ions, particularly from PdCl2, are the most common reactive sensitizers among metals (ranking second behind nickel). Moreover, proteins, DNA and other biomacromolecules, such as vitamin B6, have been reported to be able to strongly bind palladium ions, which may cause a variety of cellular dysfunction processes.3,4 It is noteworthy that governmental regulations on the levels of palladium (no more than 5–10 ppm) are fairly strict and the proposed dietary intake is less than 1.5–15 μg per person per day.5 Considering the serious threat of Pd2+ to the environment and human health, investigations of methodologies for the highly sensitive and selective detection of Pd2+ have attracted more and more attention from researchers.
Up to now, conventional methods have been developed for the efficient detection of palladium ions, including atomic absorption spectrometry, plasma emission spectroscopy, solid phase microextraction–high performance liquid chromatography, and X-ray fluorescence.2 These methods, however, usually require expensive instruments, complicated operation and highly skilled individuals. In contrast, colorimetric and fluorimetric methods can avoid these deficiencies while maintaining comparable efficiency and accuracy; therefore, they have been widely exploited by researchers for the detection of a variety of targets.6 Yet most of the reported Pd2+ fluorescent probes are excited by one-photon (OP) excitation with a short wavelength, which leads to obvious shortcomings, such as poor shallow tissue penetration depth, photobleaching and a high autofluorescence background.7–9 To overcome these problems, it is urgent to develop novel two-photon (TP) fluorescent probes that are excited by two photons with longer wavelength laser light to provide an improved three-dimensional imaging resolution with increased imaging depth (>500 μm) and little photo-damage to living tissue.
Coumarins have fascinated humans from the very beginning.10 Coumarin is a conformation-restricted form of a cis-cinnamic acid ester, thus it has high photostability, but coumarin itself shows a poor fluorescence quantum yield and TP response. However, in very recent years so-called expanded coumarins have been synthesized, effectively expanding the portfolio of existing architectures, and great efforts have been made so far to modulate their absorption from 300 nm to 550 nm, making coumarin derivatives emit strong fluorescence in the near-infrared (NIR) region and possess tunable TPA cross-sections.11 These π-expanded coumarin molecules have already been utilized in TP excited fluorescence microscopy.11 In addition, a unique family of rigid analogues P′1 (GCTPOC) possessing one coumarin moiety combined with a heterocyclic moiety was reported by Lin and coworkers.12 Importantly, P′1 as a product molecule has successfully been applied to detect a variety of biological species by TP fluorescence.12–16 The corresponding fluorescent probes with the product molecule P′1 show good photostability, high selectivity and sensitivity, low cytotoxicity and high biocompatibility. Additionally, another coumarin derivative, P′5 (3-benzothiazol-7-hydroxy-2H-chromen-2-one (TPF)), also displays excellent optical properties, such as a high fluorescence quantum yield (ϕ = 0.36), high photostability, and a large two-photon absorption (TPA) cross-section (δTmax = 217 GM).17 Consequently, the purpose of this work is to design a series of novel coumarin derivatives (based on the reported P′1 and P′5 molecules) with tunable absorption/emission spectra in the NIR region and relatively large TPA cross-section values, and more importantly, to obtain TP Pd2+ fluorescent probes for achieving the effective recognition of Pd2+.
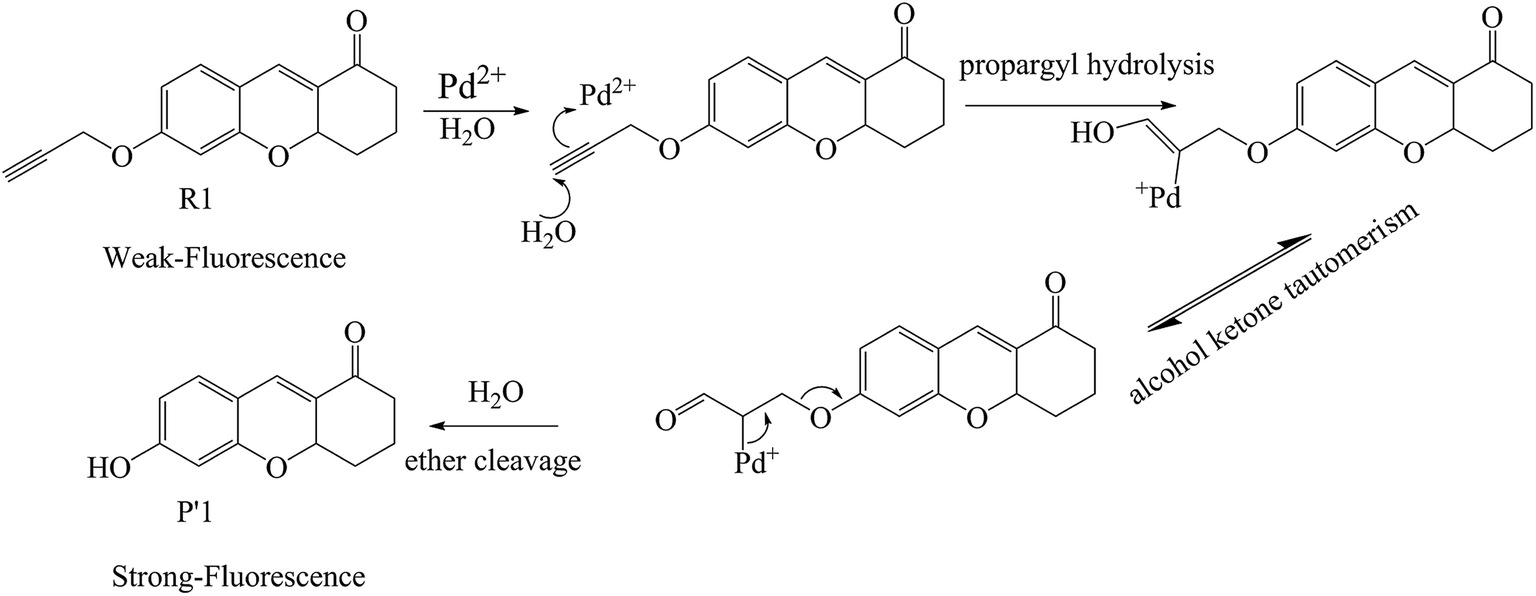
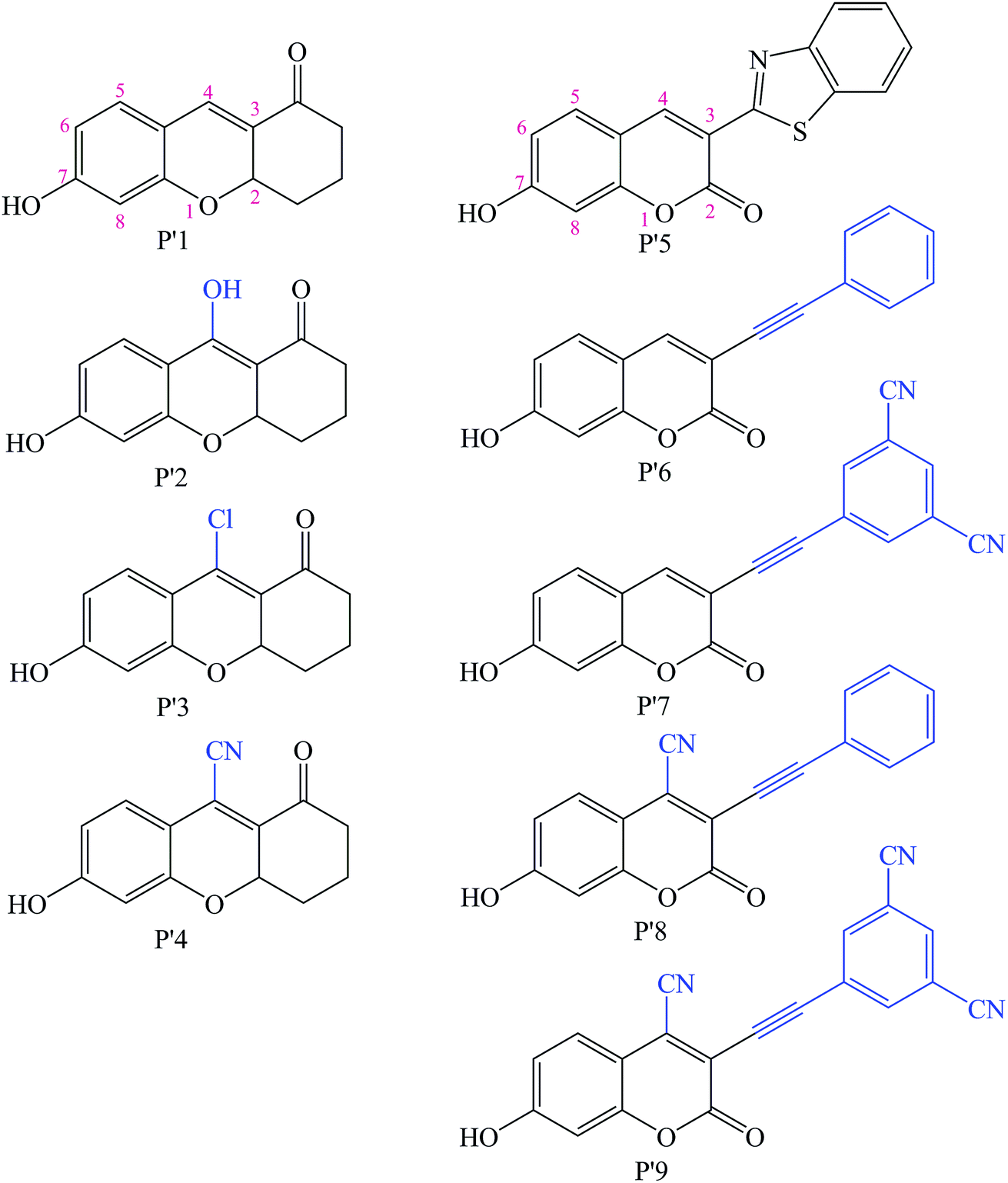
Additionally, so far, the detection mechanisms for Pd2+ are mostly based on the complexation of Pd2+ with ligands, or Pd2+-triggered chemical catalytic reactions.2 Yet most sensors based on the coordination mechanism have varying degrees of interference from other transition metal ions and strong fluorescence quenching due to the heavy atom effect.18 In contrast, chromophores built on Pd2+-triggered catalytic reaction mechanisms usually show better selectivity.8,9,19 The detailed palladium-catalyzed chemical conversion is displayed in Fig. 1.7,19–21 During the Pd2+ catalytic hydrolysis process, propargyl hydrolysis, alcohol ketone tautomerism and ether cleavage are important components and occur successively. In 2015, two TP Pd2+ fluorescent molecules (GCTPOC–Pd and GCTPOC) were reported by Lin’s group,14 based on this type of chemical reaction. The Pd2+ probe GCTPOC–Pd, which is termed R1 in this study for convenience, is synthesized by directly connecting a two-photon coumarin derivative fluorophore with an aryl propargyl ether recognition functional group, and is non-cytotoxic and has great biocompatibility. The Pd2+-responsive group (aryl propargyl ether) is distinguished by the high sensitivity (detection limit 22 nM), good selectivity, mild reaction temperature, irreversible nature and rapid response (45 min) and has been verified to be suitable for use with living cells and tissues.14 So far, although there are some theoretical and experimental reports for TP fluorescent probes,22–27 there have been no theoretical research reports of TP probes based on Pd2+-triggered chemical catalytic reactions and coumarin derivatives, and the relationship between the molecular structure and nonlinear absorption properties of coumarin derivatives is still unclear. This study is devoted to providing some strategies for designing a series of novel TP molecules based on coumarin cores, shown in Fig. S1† (Pd2+ probe molecules R1–9) and Fig. 2 (corresponding product molecules P′1–9). We further investigate their electronic structural properties and, more importantly, find out which structural modifications are beneficial for chromophores with longer excited wavelengths and stronger nonlinear responses. Additionally, our other goal is to clarify the microscopic mechanism for the “turned-on” optical signal during Pd2+ detection via quantum chemistry calculations.
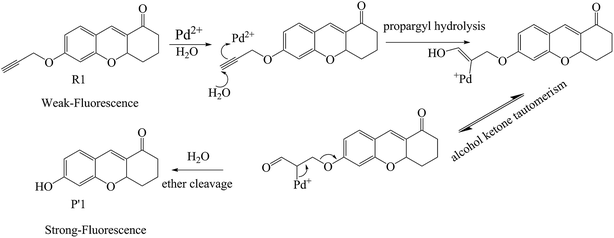 |
| | Fig. 1 The recognition mechanism of probe R1 by a Pd2+-induced depropargylation reaction. | |
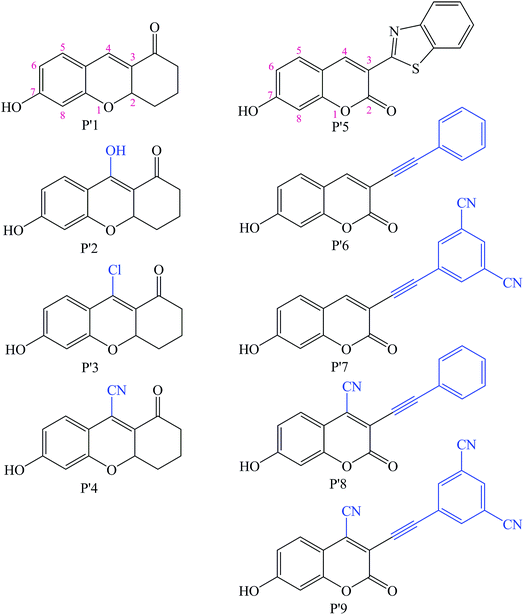 |
| | Fig. 2 Chemical structures of the modified Pd2+ reaction product molecules studied in this work based on the synthesized P′1 and P′5 molecules (blue groups are the modified parts). | |
2. Methodology
In this work, to select a suitable functional to optimize molecular geometries and calculate optical parameters in the ground state and excited state, we take the experimentally synthesized compounds P′1 (GCTPOC) and P′5 (TPF) as references to calculate their one-photon absorption (OPA) and emission features by means of a density functional theory (DFT) method with different functionals. The calculated OPA and emission wavelengths (λomax and λemax), as well as the corresponding oscillator strengths (fo and fe), are shown in Table S1.† Given the complexity of the practical environment and the fact that all the experimental measurements are recorded in PBS (phosphate buffered saline) solvents, complex solvation effects are considered. Taking the micro-solvation mode and research experience into consideration,28 three explicit water molecules were added around the strongest electrophilic carbonyl oxygen of each studied molecule in the calculations; meanwhile, the implicit solvent effect was considered by a Polarizable Continuum Model (PCM) approach.29 Comparing the calculated results of the explicit solvent model simulation with those of the implicit solvent by means of the common hybrid functional B3LYP (containing a 20% proportion of the Hartree–Fock exchange energy),30,31 the influence of the explicit solvent environment is crucial. As we know, the M06-2X hybrid functional is excellent for dealing with weak interaction and electron transition simulations, especially for electronic excitation energies; furthermore it has successfully been applied in theoretical computation.32,33 Thus, we performed calculations on the OPA and emission features of P′1 and P′5 using the M06-2X* functional (containing 44% HF exchange component); moreover, the calculated results are in good agreement with experimental data, shown in Table S1.† Therefore, the geometrical structures of all the studied molecules, as well as the OPA and emission spectra simulations, were fully optimized by using the M06-2X* (44% HF) hybrid functional along with the 6-31+G(d) basis set (the influence of the basis set on the spectra is displayed in Table S2†) in the implicit and explicit solvent models. Additionally, the fully optimized stationary point of a molecule can be characterized by vibrational frequency analysis to ensure that the real local minima without imaginary vibrational frequencies have been found. This vibrational frequency analysis has been successfully used in many reported investigations.23,33–35 The vibrational frequencies of every molecule (including the explicit water molecules) at the real local minima points had positive values (listed in Table S3†) at the M06-2X* (HF = 44%)/6-31+G(d) level, which confirmed the stability of the different studied geometries. In order to avoid the uncertainty that the density functional may bring to the pseudo-stability, the ωB97XD functional, which is good at dealing with weak interactions, was also used to calculate the vibrational frequencies of all the studied molecules (including the explicit water molecules). The calculated results are also listed in Table S3.† The results calculated by the ωB97XD functional were also positive values, which again implied the stability of the different geometries. All of the above calculations were carried out in the Gaussian 09 program package.36
Furthermore, the other main task of this work is to explore the two-photon absorption (TPA) features. The TPA cross-sections (δTmax), which can stand for the TPA transition probability, are evaluated by the quadratic response theory using the B3LYP functional with a 6-31+G(d) basis set, as implemented in the DALTON program.37
Theoretically, the TP transition matrix element Sαβ can be expressed as follows:38
| |
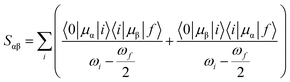 | (1) |
where
μα and
μβ are the electric dipole moment operators over the molecular
x,
y and
z axes,
ωi is the excitation energy from the ground state |0〉 to the excited state |
i〉, and
ωf/2 corresponds to half of the excitation energy associated with the transition from the ground to the final state |
f〉. With the help of
Sαβ, one can easily calculate the TP transition probability of a molecule for linearly polarized light, and the corresponding probability in the atomic unit can be written as follows:
39| | |
δTP = 6(Sxx2 + Syy2 + Szz2) + 8(Sxy2 + Sxz2 + Syz2) + 4(SxxSyy + SxxSzz + SyySzz)
| (2) |
The TPA cross-section value is directly comparable with the experimental measurement and is defined as:
| |
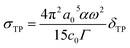 | (3) |
where
a0 denotes the Bohr radius,
α is the fine structure constant,
ω stands for the energy of the excited photons,
c0 represents the speed of light and
Γ represents the level broadening of the final state, which is assumed to be 0.10 eV.
40 The unit of the TPA cross-section is GM; 1 GM = 10
−50 cm
4 s per photon. In this work, the transition dipole moment, the state dipole moment and the excitation energy are calculated using the DALTON program.
Additionally, to judge the influence of charge-transfer during the TP transitions, we calculate the charge-transfer distance (dCT) and transferred charge (qCT) associated with the ground and excited states using a density-based approach, which has been applied in previously reported work.41 This density-based approach can yield more realistic charge transfer parameters, which can be used to describe the TPA properties.42
3. Results and discussion
3.1. Molecular design and geometry optimization of the ground state
As shown in Fig. S1† and Fig. 2, the molecules R1–9 are Pd2+ probes, and the molecules P′1–9 are the reaction products after Pd2+ treatment, respectively. In this work, the Pd2+ probes have a weak fluorescence signal; however, after reacting with Pd2+, the product molecules have strong fluorescence spectra. Thus, we focus on systematically exploring the optical properties of the reaction products P′1–9 in this study. In order to obtain molecules with better TPA properties, the strategies of changing the substituents at different positions in the coumarin core and extending the conjugation length are adopted. Additionally, substituting groups at the other end of the fluorophore to generate a long distance from the Pd2+-responsive group is very necessary, and can guarantee the high sensitivity of the probes.43 Therefore, the construction is mainly considered as follows: (a) introducing different substituents (electron-donating and -withdrawing groups), such as a hydroxyl moiety (–OH), a chlorine group (–Cl) and a cyano group (–CN) at the 4-position of the P′1 framework to access P′2, P′3 and P′4, respectively, which balance the optimum electron donor/acceptor; a literature survey revealed that 4-substituted coumarin derivatives could be synthesized and that their fluorescence spectral properties are promising,44–46 and, due to the strong electron-withdrawing nature and small structure size, –CN group substitution has been used in some biological fluorescent probes with low toxicity, especially for the successful tissue and cell imaging of organs.24,47,48 (b) Extending the π-conjugation structure at the 3-position with a phenylacetylene moiety, such as P′6, in order to gain NIR absorption and emission wavelength compounds for better applications; (c) introducing an electron-withdrawing group at the 4-position or the benzene ring of the P′5 framework; P′7, P′8 and P′9 are designed in this way to determine that introducing an electron-withdrawing unit into the P′5 framework can increase the TPA cross-sections. Meanwhile, due to the electronegativity of the oxygen atom in the carbonyl group under the polarization effect, weak hydrogen bond interactions are formed between the carbonyl group and water molecules, as shown in Fig. S2 and S3† in the explicit solvent model simulation, thus the solubility of all the studied molecules should be favorable.
The optimized molecular equilibrium geometries in the ground state are shown in Fig. S2 (Pd2+ probe molecules) and S3 (product molecules).† Frequency calculations suggest that the equilibrium geometries are stable. P′1 and P′5 are the synthesized product molecules.14,17 In our previous work, the explicit solvent model was considered,33 which revealed a significant influence on the OPA and TPA features. Thus, here explicit water molecules are also added to the studied molecules. For all the studied product compounds, the fluorophore frameworks are nearly planar, which is beneficial for reducing non-radiative transitions and increasing the fluorescence quantum yield. Furthermore, the greater rigidity of the structures of the studied compounds contributes significantly to the improvement of the nonlinear optical properties, which will be discussed in detail in the TPA properties section. Finally, considering the smaller diameter of the designed probes (maximum diameter 18.9 nm), we believe that the designed molecules have the potential to pass through the cell membrane to recognize Pd2+ like the synthesized probe R1.
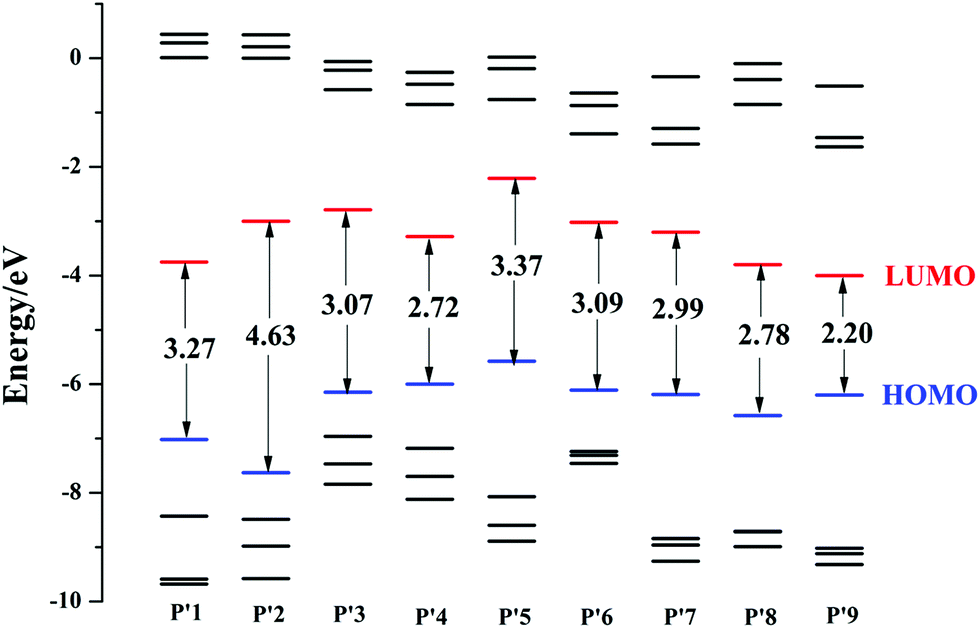
3.2. Electronic structural properties
The energies of the highest four occupied frontier molecular orbitals and the lowest four virtual frontier molecular orbitals, as well as the energy gap (ΔEH–L) between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), are schematically described in Fig. 3. The calculated results suggest that the ΔEH–L value of P′2 is larger than that of P′1, and is the largest among all the studied products. This shows that introducing an electron-donating group at the 4-position increases the transition energy, blue-shifts the optical wavelength and is inappropriate for designing NIR fluorescent probes here. Besides, introducing an electron-withdrawing unit at the 4-position using chlorine or cyano groups can reduce the ΔEH–L values, such as P′4 (2.72 eV) < P′3 (3.17 eV) < P′1 (3.27 eV), demonstrating that the electron-withdrawing group substitution at the 4-position is conducive to the decrease of ΔEH–L. Substituting an electron-withdrawing group on the P′5 core also has an obvious influence on the decrease of ΔEH–L. The energy gaps decrease in the order of P′5 (3.37 eV) > P′7 (2.99 eV) > P′8 (2.78 eV) > P′9 (2.20 eV), and similar changing trends also occur in their LUMO energy. Additionally, by modifying the molecular structures at the 3-position with an extended phenylacetylene π-conjugation moiety, the HOMO and LUMO energies are significantly decreased, for example P′6 (EHOMO/LUMO = −6.11/−3.02 eV) < P′5 (−5.58/−2.21 eV), and the ΔEH–L value is also reduced. Following cyano and phenylacetylene substitution, P′9 exhibits the smallest ΔEH–L value and largest OPA spectra (shown in the next section) compared to those of the other products P′1–8, due to the accelerated ICT process by the electron acceptor and the extended π-conjugation. The corresponding electron density plots of the HOMO and LUMO for all of the molecules are shown in Fig. S4.†
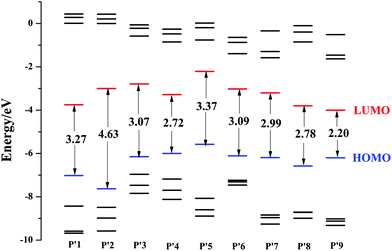 |
| | Fig. 3 Frontier molecular orbital energies of all the studied product molecules P′1–9 (The blue HOMO is the highest occupied molecular orbital, and the lower black lines are HOMO−1, HOMO−2 and HOMO−3, successively; the red LUMO is the lowest unoccupied molecular orbital, and the higher black lines are LUMO+1, LUMO+2, LUMO+3, successively. The ordinates are the corresponding energies of all the frontier molecular orbitals. The arrows represent the energy gap ΔEH–L between the HOMO and LUMO). | |
3.3. One-photon absorption (OPA) properties
The one-photon absorption (OPA) properties of all the studied molecules have been simulated in detail by the TDDFT//M06-2X* (HF% = 44%)/6-31+G(d) method on the basis of the equilibrium geometries. The calculated OPA wavelengths (λomax) and corresponding oscillator strengths (fo), as well as the corresponding transition natures and some experimental values, are listed in Table 1. The OPA wavelengths of all the studied coumarin derivatives are in the range of 360–480 nm, and these results are in good agreement with common photo-physical findings from the previous report.11 It can be seen from Table 1 that all the designed product chromophores (P′1–9) exhibit marked spectral variation following the proper modifications, demonstrating that the chemical modifications of the coumarin core in this work can effectively tune the absorption spectra to longer wavelengths. Notably, the OPA transitions of all the studied compounds are assigned as S0 → S1 and are composed of H → L. In addition, the main OPA band of P′2 (367.3 nm) with an electron-donor (hydroxyl) group substituted at the 4-position has a significant hypsochromic shift relative to that of P′1, while those of P′3/P′4 (428.6/451.0 nm) with electron-acceptor (chlorine/cyano) groups show a bathochromic shift. When the benzene ring at the 4-position is replaced by a stronger electron-withdrawing group unit (cyano), a larger bathochromic shift of the absorption is observed, such as P′1 (385.6 nm) < P′3 (428.6 nm) < P′4 (451.0 nm). On the other hand, the conjugated structure is also an important factor, as illustrated in Table 3. When benzothiazolium is replaced by the longer π-conjugation structure of phenylacetylene, an obvious bathochromic shift of λomax (P′5: 416.6 nm < P′6: 436.5 nm) occurs. This is ascribed to the fact that the extended π-conjugation structure can reinforce the charge delocalization in the whole molecule, resulting in a decreased ΔEH–L and red-shifted λomax. Besides, a slight bathochromic shift also takes place according to the substitution of cyano at the phenylacetylene ring, that is, P′6 (436.5 nm) < P′7 (452.2 nm). Of particular note is that introducing a cyano group only at the 4-position core (P′8) or the 4-position and phenylacetylene ring (P′9) at the same time still turns λomax to a longer wavelength. Eventually, P′9 has the longest red-shift among all the P′5 core derivatives (P′5, P′6, P′7, P′8 and P′9). It can be concluded from the above discussions that introducing an electron-acceptor cyano group or extending the π-conjugation structure is an efficient approach to obtain coumarin derivative molecules with longer excited wavelengths.
Table 1 The OPA spectra of the probes and products in the presence of Pd2+ by TDDFT//M06-2X* (HF = 44%)/6-31+G(d)a
| Mol. |
λomax/nm |
fo |
Transition character |
Mol. |
λomax/nm |
fo |
Transition character |
| H denotes the highest occupied molecular orbital, HOMO, and L is the lowest unoccupied molecular orbital, LUMO. |
| R1 |
445.0, 450 (ref. 14) |
0.47 |
S0 → S1 H → L 94.14% |
P′1 |
385.6, 386 (ref. 12) |
0.43 |
S0 → S1 H → L 93.37% |
| R2 |
419.3 |
0.53 |
S0 → S1 H → L 96.16% |
P′2 |
367.3 |
0.50 |
S0 → S1 H → L 95.74% |
| R3 |
460.9 |
0.45 |
S0 → S1 H → L 95.27% |
P′3 |
428.6 |
0.45 |
S0 → S1 H → L 91.55% |
| R4 |
476.6 |
0.28 |
S0 → S1 H → L 82.93% |
P′4 |
451.0 |
0.24 |
S0 → S1 H → L 89.35% |
| R5 |
379.8 |
1.05 |
S0 → S1 H → L 96.85% |
P′5 |
416.6, 450 (ref. 17) |
0.97 |
S0 → S1 H → L 97.04% |
| R6 |
376.0 |
1.22 |
S0 → S1 H → L 97.19% |
P′6 |
436.5 |
1.15 |
S0 → S1 H → L 97.63% |
| R7 |
373.5 |
1.39 |
S0 → S1 H → L 96.51% |
P′7 |
452.2 |
1.25 |
S0 → S1 H → L 96.54% |
| R8 |
433.7 |
1.00 |
S0 → S1 H → L 98.60% |
P′8 |
461.9 |
0.96 |
S0 → S1 H → L 98.69% |
| R9 |
422.6 |
1.00 |
S0 → S1 H → L 98.30% |
P′9 |
471.7 |
1.00 |
S0 → S1 H → L 98.43% |
Table 2 The calculated Kr, KIC, net-charge distribution, transferred charge, RMSD and vibrational relaxation energy of R1 and P′1a
| |
R1 |
P′1 |
| Weak fluorescence |
Strong fluorescence (at 514 nm) |
| Q is the calculated net-charge distribution of the acetylene ether group (–OCH2CCH) and hydroxyl group (–OH), respectively, shown in Fig. S5. qCT is the transferred charge of the whole molecule from S0 → S1. ΔEvib is the vibrational relaxation energy in the excited state, that is, the energy difference between the vertical excited state and the adiabatic excited state of S1. |
| Kr |
1.51 × 108 s−1 |
1.52 × 108 s−1 |
| KIC |
2.00 × 1012 s−1 |
3.05 × 107 s−1 |
| Q(S0) |
0.085 e |
0.291 e |
| Q(S1) |
−0.178 e |
0.191 e |
| qCT |
0.394 e |
0.520 e |
| RMSD |
0.228 Å |
0.191 Å |
| ΔEvib |
0.43 eV |
0.35 eV |
Table 3 The fluorescence emission properties of probes R1–9 and products P′1–9 in the presence of Pd2+ by TDDFT//M06-2X* (HF = 44%)/6-31+G(d)
| Mol. |
λemax/nm |
fe |
Transition character |
E0−0/eV |
τ/ns |
Kr/s−1 |
KIC/s−1 |
| R1 |
510.2, — |
0.59 |
S1 → S0 L → H 98.34% |
2.43 |
6.62 |
1.51 × 108 |
2.00 × 1012 |
| P′1 |
505.3, 514 (ref. 12) |
0.58 |
S1 → S0 L → H 98.34% |
2.45 |
6.58 |
1.52 × 108 |
3.05 × 107 |
| R2 |
455.7 |
0.32 |
S1 → S0 L → H 98.24% |
2.72 |
9.71 |
1.03 × 108 |
1.98 × 1011 |
| P′2 |
451.5 |
0.60 |
S1 → S0 L → H 98.24% |
2.75 |
5.10 |
1.96 × 108 |
1.05 × 1010 |
| R3 |
556.81 |
0.12 |
S1 → S0 L → H 98.81% |
2.23 |
38.5 |
0.26 × 108 |
4.38 × 1014 |
| P′3 |
575.9 |
0.38 |
S1 → S0 L → H 98.79% |
2.15 |
5.59 |
1.79 × 108 |
3.84 × 107 |
| R4 |
574.7 |
0.04 |
S1 → S0 L → H 98.18% |
2.16 |
125 |
0.08 × 108 |
9.09 × 1011 |
| P′4 |
590.3 |
1.23 |
S1 → S0 L → H 97.70% |
2.10 |
4.24 |
2.36 × 108 |
1.28 × 106 |
| R5 |
487.4 |
0.27 |
S1 → S0 L → H 97.18% |
2.54 |
13.2 |
0.76 × 108 |
9.89 × 1010 |
| P′5 |
483.6, 490 (ref. 17) |
1.20 |
S1 → S0 L → H 97.26% |
2.56 |
2.92 |
3.42 × 108 |
2.64 × 107 |
| R6 |
471.6 |
0.17 |
S1 → S0 L → H 97.75% |
2.63 |
19.6 |
0.51 × 108 |
5.96 × 109 |
| P′6 |
508.0 |
1.37 |
S1 → S0 L → H 97.81% |
2.44 |
2.82 |
3.54 × 108 |
3.00 × 106 |
| R7 |
462.6 |
0.07 |
S1 → S0 L → H 97.80% |
2.68 |
45.4 |
0.22 × 108 |
4.15 × 1010 |
| P′7 |
525.6 |
1.50 |
S1 → S0 L → H 98.26% |
2.36 |
2.76 |
3.62 × 108 |
7.51 × 106 |
| R8 |
555.1 |
0.15 |
S1 → S0 L → H 98.91% |
2.23 |
31.2 |
0.32 × 108 |
1.46 × 109 |
| P′8 |
552.7 |
1.09 |
S1 → S0 L → H 98.96% |
2.24 |
4.20 |
2.38 × 108 |
1.15 × 107 |
| R9 |
532.4 |
0.26 |
S1 → S0 L → H 98.85% |
2.33 |
16.4 |
0.61 × 108 |
1.32 × 1010 |
| P′9 |
579.7 |
1.19 |
S1 → S0 L → H 98.92% |
2.14 |
4.24 |
2.36 × 108 |
3.75 × 106 |
3.4. Validation of the “turn-on” fluorescence of the synthesized probe and product
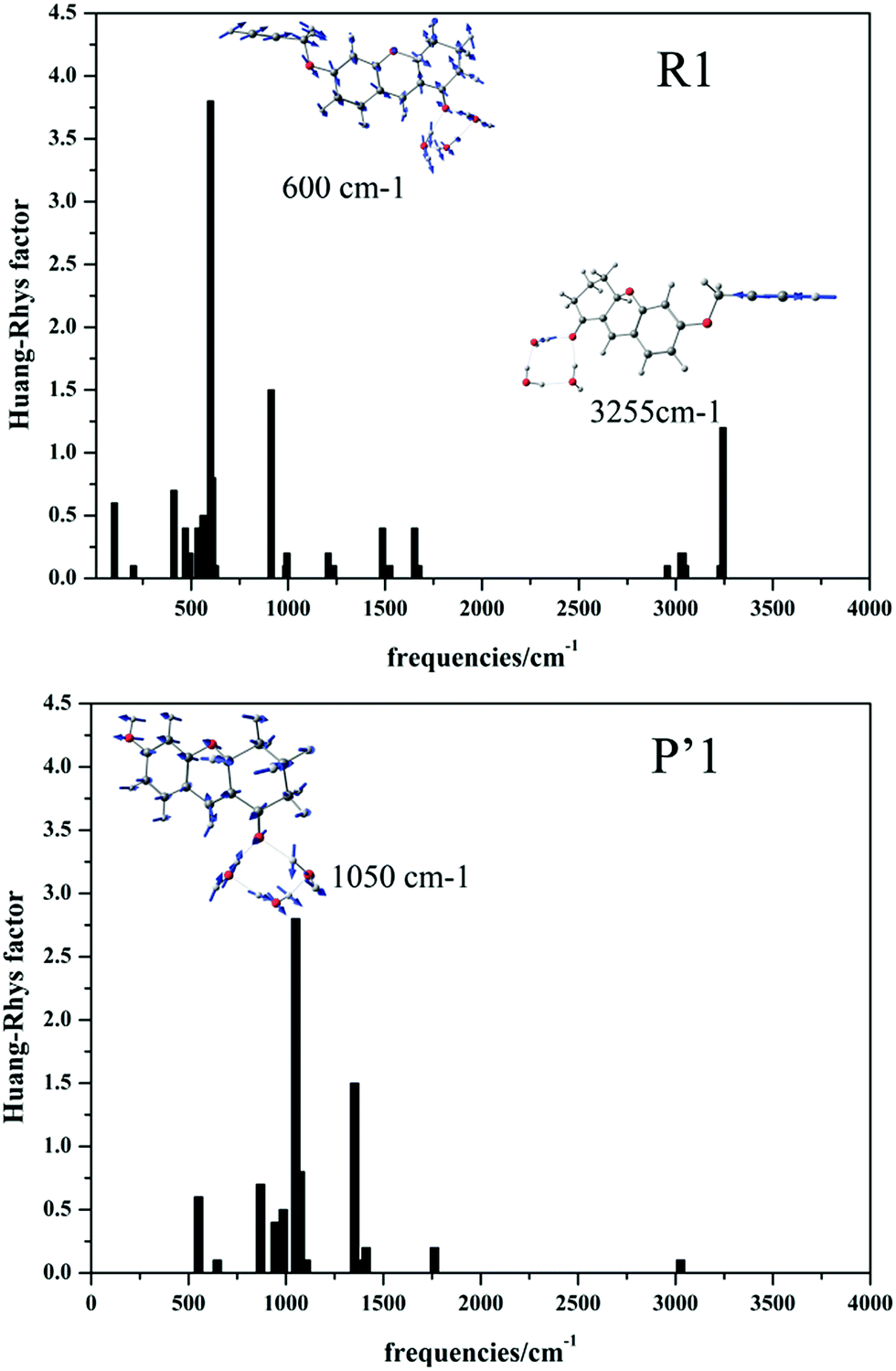
As reported by Lin’s group, the Pd2+ probe R1 shows weak fluorescence, while the recognition product P′1 possesses 100-fold fluorescence enhancement.14 To explain the “turn-on” fluorescence change mechanism, we obtained the radiative decay rate Kr and internal conversion rate KIC (the other non-radiative path could generally be ignored in the electronic transition) at a temperature of 298 K during the excited ground transition process by applying MOMAP software (detailed method description in the next section).49–52 The calculated Kr values of R1 and P′1 are 1.51 × 108 s−1 and 1.52 × 108 s−1, respectively, which are equal. However, the KIC values are 2.00 × 1012 s−1 and 3.05 × 107 s−1 for probe R1 and product P′1, respectively, indicating that in probe R1, there would be weak fluorescence because the radiationless rate (2.00 × 1012 s−1) is much larger than the radiative decay rate (1.51 × 108 s−1). To further clarify the origin of the “turn-on” fluorescence, we also analysed the differences in electron properties, geometry variations, reorganization energies, Huang–Rhys factors (HR) and vibrational modes.
Firstly, the net-charge distributions on the acetylene ether group (–OCH2CCH) and hydroxyl group (–OH), which are displayed in Table 2, are calculated based on nature bond orbital (NBO) analysis. It can be found that in the S1 state, the –OCH2CCH group exhibits electron-withdrawing character (−0.178 e); however, the –OH group shows electron-donating character (0.191 e). Meanwhile, the intramolecular charge transfer of the whole molecule associated with the ground and excited states was obtained by a density-based approach, and is also displayed in Table 2. In the presence of Pd2+, the –OCH2CCH group of probe R1 is hydrolyzed to the –OH of product P′1. The transferred charge was increased by the electron-donor –OH, such as P′1 (qCT = 0.52 e) > R1 (qCT = 0.39 e). The larger intramolecular charge transfer contributes to the easier electron transition and higher fluorescence efficiency. Additionally, the geometrical change of the stable ground state and lowest excited state for the two molecules was characterized by the root mean square displacement (RMSD) parameter, which can conveniently measure the overall degree of structural change. According to the geometry comparison simulation of the stable S0 and S1 states for R1 and P′1 shown in Fig. S6,† product P′1 is found to possess smaller geometrical change than probe R1, for example: P′1 (RMSD = 0.191) < R1 (RMSD = 0.228). Taking vertical excitation, the geometrical change mostly results from the geometrical relaxation of the excited state. The smaller geometrical relaxation of the excited state is beneficial for fluorescence enhancement by the reduction of radiationless energy. At the same time, the vibrational relaxation energies caused by the –OH group and the –OCH2CCH group were calculated. The results suggest that the –OH group of product P′1 caused smaller vibrational relaxation consumption than the –OCH2CCH group of probe R1 in the excited state, so largely reduced the vibrational relaxation energy, as displayed in Table 2 (ΔE(P′1): 0.35 eV < ΔE(R1): 0.43 eV), which alleviated the geometry distortion of product P′1 to achieve enhanced fluorescence efficiency. To the best of our knowledge, the Huang–Rhys factors (HR) can characterize the modifications of vibrational quanta (absorbed or emitted) when going from one electronic state to another, which are important for determining the KIC.53,54 It is calculated and clearly seen from Fig. 4 that (i) in general the HR factors for R1 (with a maximum value of 3.7) are much larger than those for P′1 (with a maximum value of 2.8) and (ii) the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bond stretching modes of probe R1 appear at the 3255 cm−1 high frequency region, which further indicates that R1 has larger vibrational relaxation consumption. As a result, probe R1 shows weak fluorescence compared to product P′1, which successfully explains the experimental results.
C bond stretching modes of probe R1 appear at the 3255 cm−1 high frequency region, which further indicates that R1 has larger vibrational relaxation consumption. As a result, probe R1 shows weak fluorescence compared to product P′1, which successfully explains the experimental results.
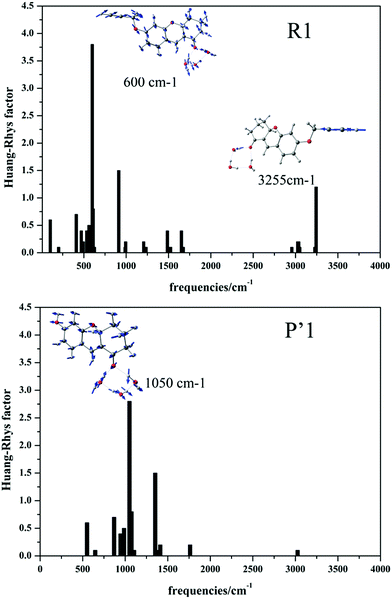 |
| | Fig. 4 Calculated Huang–Rhys factors versus the normal mode wave numbers and the normal mode displacement vectors for the strongest vibrations of R1 and P′1, respectively. | |
3.5. Fluorescence properties
Based on the stable excited-state geometry optimization using the TDDFT//M06-2X* (HF% = 44%)/6-31+G(d) method, the fluorescence properties of the studied molecules are calculated and are listed in Table 3, which also shows that the emission spectral scope is around 450–600 nm. The emission transitions of all the studied product compounds are assigned to S1 → S0, composed of L → H. During the chemical modifications based on the coumarin core in this work, the changing trends of the emission spectra are in accordance with the results of the OPA spectra.
The fluorescence lifetime (τ) and fluorescence radiation rate (Kr) for spontaneous emission are calculated by employing the Einstein transition probabilities according to the formulae in (4) and (5),55,56 and are also shown in Table 3:
| |
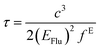 | (4) |
| |
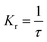 | (5) |
Here,
τ is the fluorescence lifetime,
c stands for the velocity of light,
EFlu denotes the transition energy,
fE represents the oscillator strength of the emission spectra, and
Kr is the rate of radiative decay.
In general, there are three major pathways for non-radiative decay for fluorescent molecules,50 i.e. (i) internal conversion (IC) from S1 to S0, (ii) an intersystem crossing process (ISC) from a singlet manifold to a triplet, and (iii) charge separation (CS) during the photoisomerization process occurring in the excited state, often assisted by solvent. According to the analysis of the molecular structure (not heavy-atom) and electronic transition (mainly the π → π* transition) of all the studied molecules, it would be difficult for the ISC process to happen. To further confirm the weak ISC process, we obtained the intersystem crossing rates (KISC) for S1 → T1, shown in Table S7,† and the small KISC values implied that the ISC progress would be much slower than the IC progress in this study. From Table S7,† we can also see that the ΔES1→T1 values were large enough to inhibit the ISC process. Furthermore, the small effective spin–orbit coupling (SOC) matrix elements (below 1 cm−1) reduced the KISC values significantly. The derivative details for KISC are shown in section B of the ESI.† Thus, here we focus on considering the influence of internal conversion non-radiative decay on the fluorescence efficiency of the studied molecules. Then, the final internal conversion rate (KIC) from S1 to S0 is calculated with the Gaussian 09 program package using the formulae given by Shuai:51
| |
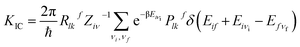 | (6) |
| |
Rlkf = 〈Φf|![[P with combining circumflex]](https://www.rsc.org/images/entities/i_char_0050_0302.gif) fl|Φi〉〈Φi|fk|Φf〉 fl|Φi〉〈Φi|fk|Φf〉
| (7) |
| | |
Plkf = 〈Θfνf|fl|Θiνi〉〈Θiνi|fk|Θfνf〉
| (8) |
where the non-radiative transition process is from an initial vibronic (excited) state |
iν〉 to the final (ground) state |
fν〉.
Rlkf represents the coupling between the electronic wave functions of the two states. |
Φi〉 and |
Φf〉 denote the initial and final electronic states, respectively; |
Θiνi〉 and |
Θfνf〉 denote the vibrational states of the system and are expressed as the products of the wave functions of each normal mode. The calculated
KIC result (2.00 × 10
12 s
−1) of probe R1 is much larger than its
Kr value (1.51 × 10
8 s
−1), which implied that the fluorescence of probe R1 is weak. After recognizing Pd
2+, the fluorescence radiation rate for the product P′1 (
Kr = 1.52 × 10
8 s
−1) is much larger than the non-radiation rate (
KIC = 3.05 × 10
7 s
−1). Thus, the product P′1 shows brighter fluorescence, achieving “turn-on” fluorescence detection for Pd
2+. Similarly, it can be forecasted that the synthesized product P′5 would emit stronger fluorescence. The simulated results are in good agreement with the strong fluorescence phenomena seen from experiments. In addition, the excellent designed product P′4 (1.28 × 10
6 s
−1) possesses a smaller
KIC than the corresponding probe R4 (9.09 × 10
11 s
−1), which demonstrates that R4 could detect Pd
2+ according to a “turn-on” fluorescence signal. Finally, for all the product molecules, except for P′2, the emission spectra are beyond 500 nm and possess strong fluorescent oscillator strengths and large
Kr values (≫
KIC), which demonstrate that these fluorescent molecules have the advantages of the smallest interference from autofluorescence and naked eye observation. These great fluorescent oscillator strengths and
Kr values of the products should be ascribed to the stronger electron-donating (–OH) groups and better rigid conformations of the coumarin cores with smaller Huang–Rhys factors, which increase the intramolecular net-charge transfer and reduce the vibrational relaxation energy in the excited states, respectively, leading to strong fluorescence compared to the probe molecules.
3.6. Two-photon absorption (TPA) properties
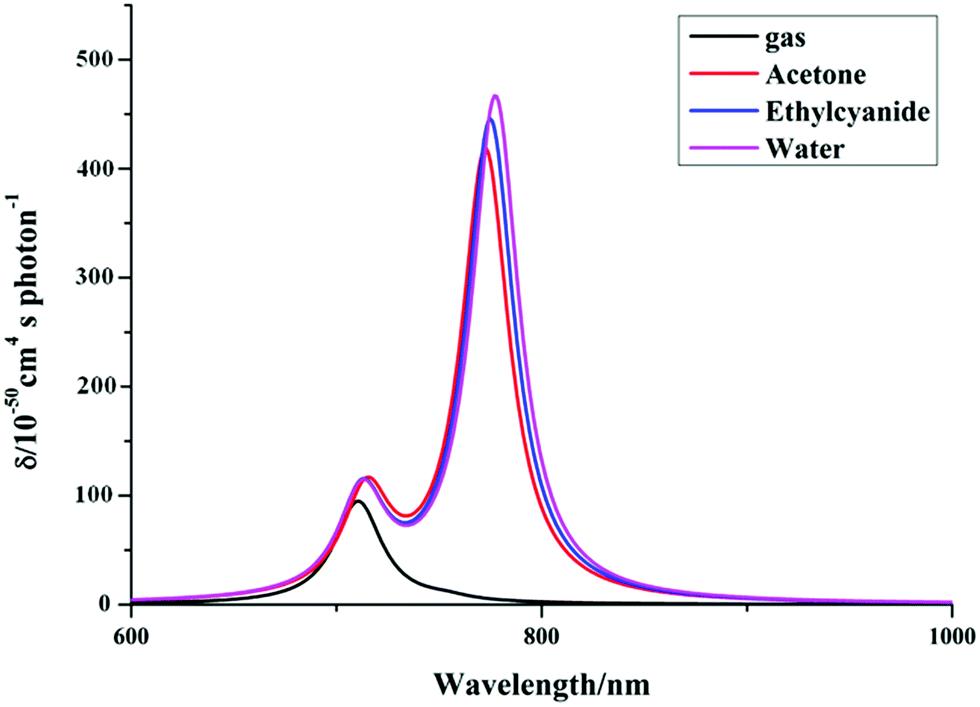
As we know, practical application of a TP fluorescent probe in a biological system requires that the TP excited wavelength is located in the NIR spectral region (650–1200 nm). In order to evaluate the TP response for the studied product molecules, we perform calculations of the two-photon absorption (TPA) properties of the molecules, including the maximum cross-sections (δTmax) and corresponding TPA wavelengths (λTmax), by a TDDFT method in the 700–1200 nm region. Firstly, we obtain the TPA spectra of the synthesized P′1 by three functionals implemented in the DALTON program, which are described in Fig. S7.† It can be found from Fig. S7† that while different functionals are accompanied by little change the TPA intensity, there is some effect on the TPA wavelength. The peak position of P′1 calculated by the B3LYP (20% HF) functional is longer than that calculated by the BhandHLYP (50% HF) functional, which is mainly ascribed to the smaller Hartree–Fock percentage with the B3LYP functional. Compared with CAM-B3LYP, the result of B3LYP has a better agreement with the experimental results, which may be due to some error compensation that counteracts the long-range-corrected effect on the molecule. Thus, the B3LYP functional is expected to be more suitable for describing the TPA properties of these studied molecules according to comparisons with the experimental TP excited wavelengths and TPA cross-sections. Secondly, since a proper account of the solvent polarity effect is crucial for direct forecasts of the TPA properties, we also performed calculations on the λTmax and δTmax values of the synthesized P′1 molecules in solutions of different polarity (acetone (ε = 4.2), ethylcyanide (ε = 36.34), water (ε = 78.39)) and in the gas phase. To make the comparison more intuitive, the relationships between λTmax and δTmax are illustrated in Fig. 5. It can clearly be seen that the λTmax and δTmax values are affected upon adding polar solvents, which suggests that the solvent effect is not negligible. For this reason, in addition to using the PCM, the explicit water solvent effect was also considered in the TPA calculations for the studied molecules.
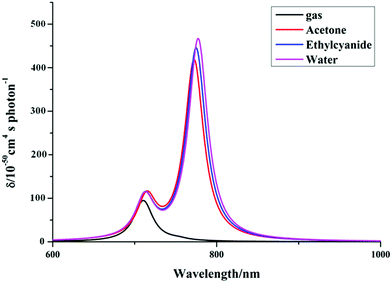 |
| | Fig. 5 Simulated TPA spectra of P′1 in solvents of different polarity by DALTON software (exp. λTmax = 820 nm, δTmax = 501 GM, order of solvent polarity: gas < acetone < ethylcyanide < water). | |
The calculations of the TPA properties of the studied compounds are implemented by applying the DALTON program at the same level, and the calculated results are listed in Table 4. The calculated δTmax values of P′1 and P′5 by the TDDFT method are 534.31 GM at 823.9 nm and 215.20 GM at 835.0 nm, respectively, which are in good agreement with the experimental data (501 GM at 820 nm and 217 GM at 800 nm, respectively). As shown in Table 4, the λTmax values of the studied molecules range from 700 to 1200 nm, and the corresponding δTmax values are as large as 210–3800 GM. Remarkably, for the studied compounds, the maximum TPA responses are from the first excited states, and the corresponding transition is S0 → S1.
Table 4 The TPA properties of the studied products P′1–9 calculated by the DALTON program
| Mol. |
λTmax/nm |
δTmax/GM |
Transition character |
σ |
| P′1 |
823.9, 820 (ref. 12) |
534.31, 501 (ref. 12) |
S0 → S1 |
2.47, 2.32 (ref. 12) |
| P′2 |
770.2 |
246.87 |
S0 → S1 |
1.06 |
| P′3 |
835.0 |
845.87 |
S0 → S1 |
2.95 |
| P′4 |
1112.6 |
946.56 |
S0 → S1 |
3.91 |
| P′5 |
835.0, 800 (ref. 17) |
215.20, 217 (ref. 17) |
S0 → S1 |
0.77, 0.78 (ref. 17) |
| P′6 |
839.0 |
437.49 |
S0 → S1 |
1.67 |
| P′7 |
840.7 |
613.32 |
S0 → S1 |
1.96 |
| P′8 |
988.0 |
700.58 |
S0 → S1 |
2.44 |
| P′9 |
1037.7 |
872.70 |
S0 → S1 |
2.59 |
According to the calculated results, P′4 bearing a cyano group as an acceptor at the 4-position has a much larger TPA cross-section than P′2 with a hydroxyl group as a donor at the 4-position, and P′3 containing a weaker electron-withdrawing chlorine at the 4-position, respectively. In addition to the effects of the electron-donating and electron-withdrawing influences, the π-conjugated structures also play important roles in the TPA properties of the studied compounds. From Table 4, it can be observed that P′6 with phenylacetylene as a π-conjugated structure displays 2-fold TPA cross-sections corresponding to those of P′5 with benzothiazolium. Furthermore, P′7 with a cyano group added on the phenylacetylene leads to a much longer TPA wavelength and larger cross-section than P′6. However, molecules with a large TPA cross-section per molecular weight (σ = δmax/MW) are essentially better for two-photon imaging detection.57 Although molecules with a long π-conjugation chain also have a strong TP response, the big molecular weight of the long π-conjugation chain cannot contribute to δmax/MW, such as in P′4 (σ = 3.91) > P′9 (σ = 2.59). What’s more, molecules with δmax/MW larger than 1.0 have been found to be useful for better applications.58–60 In this regard, we are devoted to increasing the δmax/MW values of the studied molecules to enable actual applications. As shown in Table 4, the two-photon cross-section per molecular weight σ values of all the designed molecules are over 1.5, except for that of P′2, indicating that the studied dyes have potential for Pd2+ recognition.
To further clarify the effects of internal factors on the TPA processes of these chromophores, in this subsection, we will focus on the main factors affecting the TPA process. Considering the strongest TPA transition, corresponding to S0 → S1, the two-state (2S) approximation was adopted for an explanation of the calculated results. The model uses the two-state expression:61
| |
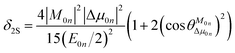 | (9) |
where
E is the corresponding excitation energy;
M represents the transition dipole moment from the ground state to the TPA final state; Δ
μ denotes the state dipole moment difference between the ground state and the TPA final state; and the subscripts 0 and
n stand for the ground state and the transition final state, respectively.
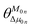
is the angle between these two vectors. The approximate proportional relationship between
δTmax and
δ2S during the TPA transition process (depicted in
Fig. 6) implies that
δ2S can predict the changing tendency of
δTmax in general.
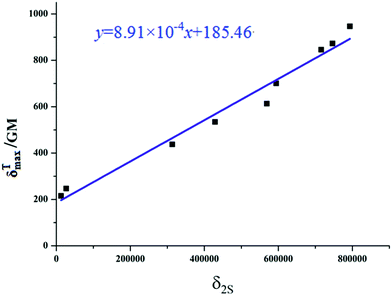 |
| | Fig. 6 Relationship plot of the maximum two-photon absorption cross-sections δTmax and δ2S 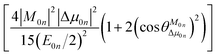 for all the studied product molecules. for all the studied product molecules. | |
According to the two-state expression, the δTmax values are largely determined by three parameters, which are calculated and the results are listed in Table 5. That is, (i) for all the studied compounds, the transition dipole moments M0n and the state dipole moment differences Δμ0n are critical factors influencing the δTmax values; the changing trends of the M01 and Δμ01 values are in accordance with those of the δTmax values. (ii) Compared with P′1 (534.31 GM), it can easily be observed that the M01 value of P′2 is much smaller than that of P′1, thus the decreased M01 value of P′2 should be responsible for its smaller δTmax value (246.87 GM). (iii) Adding an electron-withdrawing unit or increasing the electron-withdrawing ability at the 4-position leads to an obvious enhancement of both M0n and δTmax, which results in an enhanced TPA response, for instance, P′1 (6.70 Debye, 534.31 GM) < P′3 (7.92 Debye, 845.87 GM), P′1 (6.70 Debye, 534.31 GM) < P′4 (8.60 Debye, 946.56 GM). Notably, this similar changing trend also occurs in P′6 and P′8, so we can rationally elucidate that adding an acceptor unit or increasing the electron-withdrawing ability at the 4-position is favorable for the TPA response enhancement of coumarin derivative dyes. (iv) Comparing P′5 and P′6, it can be found that replacing the benzothiazolium moiety by a phenylacetylene group can obviously increase the δTmax values of the compounds, which is attributed to the largely increased Δμ01 and M01 values of the compounds with phenylacetylene units.
Table 5 Important related parameters of the TPA properties of the studied product molecules P′1–9
| Mol. |
M01 |
|Δμ01| |
θ/° |
E/eV |
δ2S |
qCT/e |
dCT/Å |
| P′1 |
6.70 |
5.89 |
25.52 |
2.95 |
428![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 934.2 934.2 |
0.521 |
0.851 |
| P′2 |
4.09 |
4.55 |
88.08 |
3.21 |
26604.86 |
0.503 |
1.694 |
| P′3 |
7.92 |
7.38 |
35.06 |
2.97 |
715805.3 |
0.609 |
1.859 |
| P′4 |
8.60 |
7.62 |
38.23 |
3.09 |
793553.7 |
0.708 |
2.461 |
| P′5 |
2.13 |
4.14 |
62.32 |
2.62 |
12808.38 |
0.447 |
0.036 |
| P′6 |
11.4 |
5.17 |
48.64 |
2.97 |
313567.6 |
0.435 |
0.834 |
| P′7 |
10.5 |
7.20 |
77.28 |
2.95 |
568973.5 |
0.528 |
1.750 |
| P′8 |
7.90 |
7.05 |
63.67 |
2.39 |
594192.7 |
0.521 |
0.805 |
| P′9 |
10.6 |
7.85 |
88.02 |
2.71 |
746409.8 |
0.769 |
1.201 |
Overall, the above results show that the state dipole moment difference Δμ0n plays a dominant role in the TPA properties. To the best of our knowledge, the state dipole moment difference Δμ0n is closely related to the charge-transfer distance (dCT) and the transferred charge (qCT), as presented in the expression:41
Here, Δ
μ is the difference of the dipole moments computed for the ground and the excited states;
dCT stands for the spatial distance between the two barycenters of the density distributions,
i.e. the charge-transfer distance.
qCT represents the transferred charge during the transition process. It has been shown that this density-based approach is much more meaningful than a charge-based approximation.
42 The calculated results of
dCT and
qCT are also shown in
Table 5. It is found that introducing an acceptor at the 4-position leads to larger
qCT and
dCT values, such as P′3 (0.609 e, 1.859 Å) > P′1 (0.521 e, 0.851 Å). Furthermore, the
qCT and
dCT values are larger for stronger acceptors, such as P′4 (0.708 e, 2.461 Å) > P′3 (0.609 e, 1.859 Å). Importantly, upon extending the conjugation length, the
dCT value is markedly increased, such as P′6 (0.834 Å) > P′5 (0.036 Å), which results in a TPA transition enhancement, P′6 (437.49 GM) > P′5 (215.20 GM). In conclusion, introducing an electron-withdrawing group or increasing the electron-withdrawing ability at the 4-position of the coumarin core can effectively enlarge the charge-transfer distance (
dCT) and transferred charge (
qCT); extending the conjugation length at the 3-position of the coumarin core can significantly increase the charge-transfer distance (
dCT), and all this leads to larger
δTmax values in the NIR region.
4. Conclusions
In the present study, we provided some strategies for designing a series of two-photon (TP) fluorescent palladium ion (Pd2+) probes and the corresponding product molecules by chemical modification, and firstly systematically and deeply investigated their electronic structures and one-photon absorption (OPA), fluorescence emission and two-photon absorption (TPA) optical properties from a theoretical viewpoint. Moreover, the radiative and non-radiative behaviors for the synthesized probe R1 (Kr: 1.51 × 108 s−1 ≪ KIC: 2.00 × 1012 s−1) and the synthesized product P′1 (Kr: 1.52 × 108 s−1 ≅ KIC: 3.05 × 107 s−1) have been revealed quantitatively and qualitatively. These are due to the presence of an auxochromic and electron-donating –OH group in the product molecule replacing a large –OCH2CCH group in the probe, reducing the geometrical relaxation and vibrational relaxation energies in the excited states and increasing intramolecular charge transfer. On the basis of the synthesized P′1 (GCTPOC) and P′5 (TPF) cores, it is easier to effectively increase the TP absorption cross-section per molecular weight by introducing an electron-withdrawing group (–Cl or –CN) to the 4-position than by extending the π-conjugated structure at the 3-position. Further investigations show that both introducing an electron-withdrawing group or increasing the electron-withdrawing ability at the 4-position, and extending the π-conjugated structure at the 3-position of the coumarin core with phenylethylene, result in small H → L energy gaps, a red-shift of the absorption and emission spectra, the enhancement of δTmax, and larger transition dipole moments. By introducing chlorine and cyano substituents at the 4-position in the central coumarin skeleton, the molecules P′3 and P′4 display obvious bathochromic shifts of λTmax and increased δTmax and transition dipole moments relative to those of P′1, which are ascribed to the strong electron-withdrawing ability increasing the charge transfer qCT. On the other hand, incorporating the π-extended structure of phenylethylene at the 3-position of the coumarin core (P′6) leads to a red-shift of the OPA and fluorescence emission bands, and a 222 GM enhancement of δTmax relative to P′5, which is attributed to the extended π-conjugated structure enlarging the charge-transfer distance dCT. For the fluorescence performances, except for product P′2, the emission spectra of the other products are beyond 500 nm and possess strong fluorescent oscillator strengths and large Kr values (≫KIC), which demonstrate that the designed Pd2+ TP probes could realize Pd2+ detection according to the “turn-on” fluorescence. Consequently, P′4, with a much smaller KIC (1.28 × 106 s−1) than R4 (9.09 × 1011 s−1), possesses the largest TPA cross-section per molecular weight (3.91) and the longest fluorescence wavelength (590.3 nm) among all the studied molecules. Considering these outstanding properties of product P′4, the Pd2+ probe R4 may be the most promising candidate for Pd2+ detection. Finally, we hope that this theoretical investigation of the coumarin-based TP strategy might also provide useful information for further studies on Pd2+ detection.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Natural Science Foundation of China (No. 21473071, 21173099, 20973078 and 11404041), the Major State Basis Research Development Program (Grant 2013CB 834801), and special funding for basic scientific research projects for Central Colleges.
References
- B. Liu, H. Wang, T. Wang, Y. Bao, F. Du, J. Tian, Q. Li and R. Bai, A new ratiometric ESIPT sensor for detection of palladium species in aqueous solution, Chem. Commun., 2012, 48, 2867–2869 RSC.
- B. Liu, Y. Bao, F. Du, H. Wang, J. Tian and R. Bai, Synthesis and characterization of a fluorescent polymer containing 2,6-bis(2-thienyl)pyridine moieties as a highly efficient sensor for Pd2+ detection, Chem. Commun., 2011, 47, 1731–1733 RSC.
- C. D. Spicer, T. Triemer and B. G. Davis, Palladium-mediated cell-surface labeling, J. Am. Chem. Soc., 2012, 134, 800–803 CrossRef CAS PubMed.
- R. M. Yusop, A. Unciti-Broceta, E. M. V. Johansson, R. M. Sanchez-Martin and M. Bradley, Palladium-mediated intracellular chemistry, Nat. Chem., 2011, 3, 239–243 CrossRef CAS PubMed.
- C. E. Garrett and K. Prasad, The Art of Meeting Palladium Specifications in Active Pharmaceutical Ingredients Produced by Pd-Catalyzed Reactions, Adv. Synth. Catal., 2004, 346, 889–900 CrossRef CAS.
- L. Zhou, Q. Wang, X. B. Zhang and W. Tan, Through-bond energy transfer-based ratiometric two-photon probe for fluorescent imaging of Pd2+ ions in living cells and tissues, Anal. Chem., 2015, 87, 4503–4507 CrossRef CAS PubMed.
- H. Chen, W. Lin and L. Yuan, Construction of a near-infrared fluorescence turn-on and ratiometric probe for imaging palladium in living cells, Org. Biomol. Chem., 2013, 11, 1938–1941 CAS.
- W. Liu, J. Jiang, C. Chen, X. Tang, J. Shi, P. Zhang, K. Zhang, Z. Li, W. Dou, L. Yang and W. Liu, Water-soluble colorimetric and ratiometric fluorescent probe for selective imaging of palladium species in living cells, Inorg. Chem., 2014, 53, 12590–12594 CrossRef CAS PubMed.
- B. Zhu, C. Gao, Y. Zhao, C. Liu, Y. Li, Q. Wei, Z. Ma, B. Du and X. Zhang, A 4-hydroxynaphthalimide-derived ratiometric fluorescent chemodosimeter for imaging palladium in living cells, Chem. Commun., 2011, 30, 8656–8658 RSC.
- R. O’Kennedy and R. D. Thornes, Coumarins: Biology, Applications and Mode of Actions, John Wiley and Sons, Chichester, 1997 Search PubMed.
- M. Tasior, D. Kim, S. Singha, M. Krzeszewski, K. H. Ahn and D. T. Gryko, π-Expanded coumarins: synthesis, optical properties and applications, J. Mater. Chem. C, 2015, 3, 1421–1446 RSC.
- L. Yuan, W. Lin, H. Chen, S. Zhu and L. He, A unique family of rigid analogues of the GFP chromophore with tunable two-photon action cross-sections for biological imaging, Angew. Chem., Int. Ed., 2013, 52, 10018–10022 CrossRef CAS PubMed.
- Y. H. Lee, W. X. Ren, J. Han, K. Sunwoo, J. Y. Lim, J. H. Kim and J. S. Kim, Highly selective two-photon imaging of cysteine in cancerous cells and tissues, Chem. Commun., 2015, 51, 14401–14404 RSC.
- H. Cui, H. Chen, Y. Pan and W. Lin, A two-photon fluorescent turn-on probe for palladium imaging in living tissues, Sens. Actuators, B, 2015, 219, 232–237 CrossRef CAS.
- K. Zheng, W. Lin, D. Cheng, H. Chen, Y. Liu and K. Liu, A two-photon fluorescent turn-on probe for nitroxyl (HNO) and its bioimaging application in living tissues, Chem. Commun., 2015, 51, 5754–5757 RSC.
- K. Zheng, W. Lin, L. Tan and D. Cheng, A two-photon fluorescent probe with a large turn-on signal for imaging hydrogen sulfide in living tissues, Anal. Chim. Acta, 2015, 853, 548–554 CrossRef CAS PubMed.
- Q. Chen, J. Yang, Y. Li, J. Zheng and R. Yang, Sensitive and rapid detection of endogenous hydrogen sulfide distributing in different mouse viscera via a two-photon fluorescent probe, Anal. Chim. Acta, 2015, 896, 128–136 CrossRef CAS PubMed.
- L. Duan, Y. Xu and X. Qian, Highly sensitive and selective Pd2+ sensor of naphthalimide derivative based on complexation with alkynes and thio-heterocycle, Chem. Commun., 2008, 47, 6339–6341 RSC.
- M. Kumar, N. Kumar and V. Bhalla, Highly selective fluorescent probe for detection and visualization of palladium ions in mixed aqueous media, RSC Adv., 2013, 3, 1097–1102 RSC.
- M. Santra, S. K. Ko, I. Shin and K. H. Ahn, Fluorescent detection of palladium species with an O-propargylated fluorescein, Chem. Commun., 2010, 46, 3964–3966 RSC.
- M. Pal, K. Parasuraman and K. R. Yeleswarapu, Palladium-catalyzed cleavage of O/N-propargyl protecting groups in aqueous media under a copper-free condition, Org. Lett., 2003, 5, 349–352 CrossRef CAS PubMed.
- S. Huang, L. Y. Zou, A. M. Ren, J. F. Guo, X. T. Liu, J. K. Feng and B. Z. Yang, Computational design of two-photon fluorescent probes for a zinc ion based on a salen ligand, Inorg. Chem., 2013, 52, 5702–5713 CrossRef CAS PubMed.
- X. Liu, J. Zhang, K. Li, X. Sun, Z. Wu, A. Ren and J. Feng, New insights into two-photon absorption properties of functionalized aza-BODIPY dyes at telecommunication wavelengths: a theoretical study, Phys. Chem. Chem. Phys., 2013, 15, 4666–4676 RSC.
- D. Wang, J. F. Guo, A. M. Ren, S. Huang, L. Zhang and J. K. Feng, Computational design of two-photon fluorescent probes for intracellular free zinc ions, J. Phys. Chem. B, 2014, 118, 10101–10110 CrossRef CAS PubMed.
- Z. Xu, A. M. Ren, D. Wang, J. F. Guo, J. K. Feng and X. Yu, A theoretical investigation on two latest two-photon pH fluorescent probes, J. Photochem. Photobiol., A, 2014, 293, 50–56 CrossRef CAS.
- R. Feng, Y. Sun, M. Tian, G. Zhang, R. Zhang, L. Guo, X. Li, X. Yu and N. Zhao, A membrane-permeable dye for living cells with large two-photon excited fluorescence action cross-sections for bioimaging, J. Mater. Chem. B, 2015, 44, 8644–8649 RSC.
- F. Miao, W. Zhang, Y. Sun, R. Zhang, Y. Liu, F. Guo, G. Song, M. Tian and X. Yu, Novel fluorescent probes for highly selective two-photon imaging of mitochondria in living cells, Biosens. Bioelectron., 2014, 55, 423–429 CrossRef CAS PubMed.
- C. Zhang, L. Y. Zou, D. Wang, L. Zhang and A. M. Ren, A theoretical investigation and design of fluorescent probes with large two-photon absorption cross-sections and dual signaling behavior for mercury ions in biological system, J. Photochem. Photobiol., A, 2017, 332, 440–452 CrossRef CAS.
- V. M. Aleksandr, J. C. Christopher and G. T. Donald, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- A. D. Becke, Density-functional thermochemistry. IV. A new dynamical correlation functional and implications for exact-exchange mixing, J. Chem. Phys., 1996, 104, 1040–1046 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- D. Wang, A. M. Ren, J. F. Guo, L. Y. Zou and S. Huang, Computational design of a two-photon excited FRET-based ratiometric fluorescent Cu2+ probe for living cell imaging, RSC Adv., 2015, 5, 98144–98153 RSC.
- N. S. Makarov, S. Mukhopadhyay, K. Yesudas, J. L. Brédas, J. W. Perry, A. Pron, M. Kivala, K. Mullen, A. Pron, M. Kivala and K. Müllen, Impact of electronic coupling, symmetry, and planarization on one and two-photon properties of triarylamines with one, two, or three diarylboryl acceptors, J. Phys. Chem. A, 2012, 116, 3781–3793 CrossRef CAS PubMed.
- X. Wang, F. Q. Bai, Y. Liu, Y. Wang, H. X. Zhang and Z. Lin, A computational way to achieve more effective candidates for photodynamic therapy, J. Chem. Inf. Model., 2017, 57, 1089–1100 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 09, revisions A.02 and B.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- DALTON, a molecular electronic structure program, Release Dalton2011, see http://daltonprogram.org/.
- Y. Luo, P. Norman, P. Macak and H. Ågren, Solvent-induced two-photon absorption of a push−pull molecule, J. Phys. Chem. A, 2000, 104, 4718–4722 CrossRef CAS.
- W. M. Mcclain, Excited state symmetry assignment through polarized two-photon absorption studies of fluids, J. Chem. Phys., 1971, 55, 2789–2796 CrossRef.
- M. Albota, D. Beljonne, J. L. Bredas, J. E. Ehrlich, J. Y. Fu, A. A. Heikal, S. E. Hess, T. Kogej, M. D. Levin and S. R. Marder, et al., Design of organic molecules with large two-photon absorption cross sections, Science, 1998, 281, 1653–1656 CrossRef CAS PubMed.
- T. L. Bahers, C. Adamo and I. Ciofini, A qualitative index of spatial extent in charge-transfer excitations, J. Chem. Theory Comput., 2011, 7, 2498–2506 CrossRef PubMed.
- B. L. Guennic, S. Chibani, A. Charaf-Eddin, J. Massue, R. Ziessel, G. Ulrich and D. Jacquemin, The NBO pattern in luminescent chromophores: unravelling excited-state features using TD-DFT, Phys. Chem. Chem. Phys., 2013, 15, 7534–7540 RSC.
- L. Yuan, L. Wang, B. K. Agrawalla, S. J. Park, H. Zhu, B. Sivaraman, J. Peng, Q. H. Xu and Y. T. Chang, Development of targetable two-photon fluorescent probes to image hypochlorous acid in mitochondria and lysosome in live cell and inflamed mouse model, J. Am. Chem. Soc., 2015, 137, 5930–5938 CrossRef CAS PubMed.
- H. Ammar, S. Abid and S. Fery-Forgues, Synthesis and spectroscopic study of new biscoumarin dyes based on 7-(4-methylcoumarinyl) diesters, Dyes Pigm., 2008, 78, 1–7 CrossRef CAS.
- K. Żamojć, W. Wiczk, B. Zaborowski, D. Jacewicz and L. Chmurzyński, Fluorescence quenching of 7-amino-4-methylcoumarin by different TEMPO derivatives, Spectrochim. Acta, Part A, 2015, 136, 1875–1880 CrossRef PubMed.
- A. Matta, V. Bahadur, T. Taniike, J. Van der Eycken and B. K. Singh, Synthesis, characterisation and photophysical studies of oxadiazolyl coumarin: a new class of blue light emitting fluorescent dyes, Dyes Pigm., 2017, 140, 250–260 CrossRef CAS.
- B. Zhu, W. Wang, L. Liu, H. Jiang, B. Du and Q. Wei, A highly selective colorimetric and long-wavelength fluorescent probe for Hg2+, Sens. Actuators, B, 2014, 191, 605–611 CrossRef CAS.
- Ł. G. Łukasiewicz, I. Deperasińska, Y. M. Poronik, Y. W. Jun, M. Banasiewicz, B. Kozankiewicz, K. H. Ahn and D. T. Gryko, Dipolar Dyes with a Pyrrolo[2,3-b]quinoxaline Skeleton Containing a Cyano Group and a Bridged Tertiary Amino Group: Synthesis, Solvatofluorochromism, and Bioimaging, Chem.–Asian J., 2016, 11, 1718–1724 CrossRef PubMed.
- Q. Peng, Y. Yi, Z. Shuai and J. Shao, Excited state radiationless decay process with Duschinsky rotation effect: formalism and implementation, J. Chem. Phys., 2007, 126, 114302 CrossRef PubMed.
- Q. Peng, Y. Yi, Z. Shuai and J. Shaooward, Quantitative Prediction of Molecular Fluorescence Quantum Efficiency: Role of Duschinsky Rotation, J. Am. Chem. Soc., 2007, 129, 9333–9339 CrossRef CAS PubMed.
- Y. Niu, Q. Peng and Z. Shuai, Promoting-mode free formalism for excited state radiationless decay process with Duschinsky rotation effect, Sci. China, Ser. B: Chem., 2008, 51, 1153–1158 CrossRef CAS.
- Y. Niu, Q. Peng, C. Deng, X. Gao and Z. Shuai, Theory of excited state decays and optical spectra: application to polyatomic molecules, J. Phys. Chem. A, 2010, 114, 7817–7831 CrossRef CAS PubMed.
- S. Yin, Q. Peng, Z. Shuai, W. Fang, Y. H. Wang and Y. Luo, Aggregation-enhanced luminescence and vibronic coupling of silole molecules from first principles, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 205409 CrossRef.
- G. Yu, S. Yin, Y. Liu, J. Chen, X. Xu, X. Sun, D. Ma, X. Zhan, Q. Peng, Z. Shuai, B. Tang, D. Zhu, W. Fang and Y. Luo, Structures, Electronic States, Photoluminescence, and Carrier Transport Properties of 1,1-Disubstituted 2,3,4,5-Tetraphenylsiloles, J. Am. Chem. Soc., 2005, 127, 6335–6346 CrossRef CAS PubMed.
- V. Lukeš, A. Aquino and H. Lischka, Theoretical Study of Vibrational and Optical Spectra of Methylene-Bridged Oligofluorenes, J. Phys. Chem. A, 2005, 109, 10232–10238 CrossRef PubMed.
- X. Liu, J. Guo, A. Ren, Z. Xu, S. Huang and J. Feng, Theoretical insight into linear optical and two-photon absorption properties for a series of N-arylpyrrole-based dyes, Org. Biomol. Chem., 2012, 10, 7527–7535 CAS.
- H. M. Kim and B. R. Cho, Two-photon materials with large two-photon cross sections: structure-property relationship, Chem. Commun., 2009, 2, 153–164 Search PubMed.
- H. M. Kim, C. Jung, B. R. Kim, S. Y. Jung, J. H. Hong, Y. G. Ko, K. J. Lee and B. R. Cho, Environment-sensitive two-photon probe for intracellular free magnesium ions in live tissue, Angew. Chem., Int. Ed., 2007, 46, 3460–3463 CrossRef CAS PubMed.
- H. M. Kim, B. R. Kim, J. H. Hong, J. S. Park, K. J. Lee and B. R. Cho, A two-photon fluorescent probe for calcium waves in living tissue, Angew. Chem., Int. Ed., 2007, 46, 7445–7448 CrossRef CAS PubMed.
- B. H. Cumpston, S. P. Ananthavel, S. Barlow, D. L. Dyer, J. E. Ehrlich, L. L. Erskine, A. A. Heikal, S. M. Kuebler, I. Y. S. Lee, D. McCord-Maughon, J. Qin, H. Rockel, M. Rumi, X. L. Wu, S. R. Marderk and J. W. Perry, Two-photon polymerization initiators for three dimensional optical data storage and microfabrication, Nature, 1999, 398, 51–54 CrossRef CAS.
- N. A. Murugan, R. Zaleśny, J. Kongsted, A. Nordberg and H. Ågren, Promising two-photon probes for in vivo detection of β amyloid deposits, Chem. Commun., 2014, 50, 11694–11697 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08832a |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a and
Dan Wangc
*a and
Dan Wangc
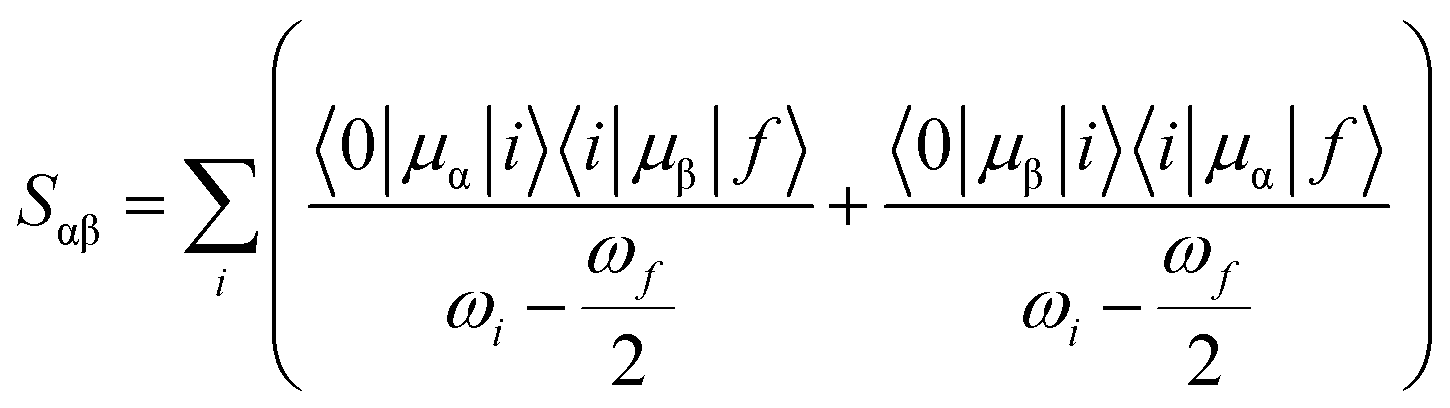
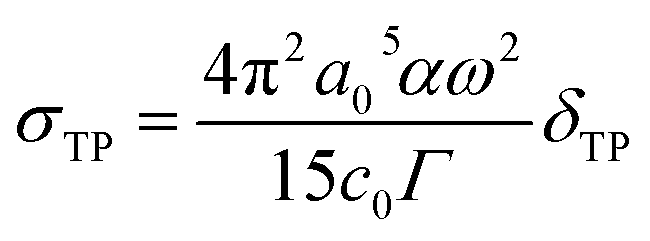
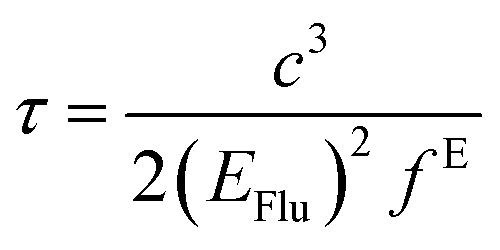
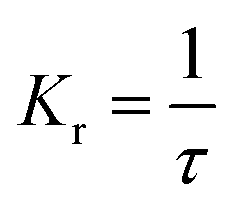
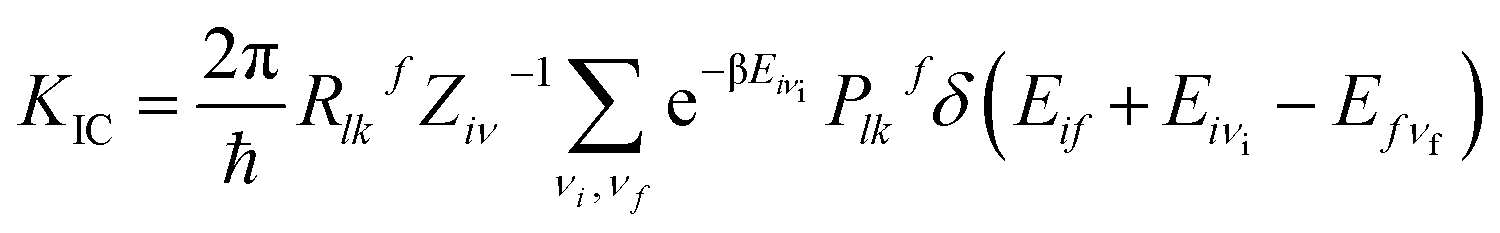
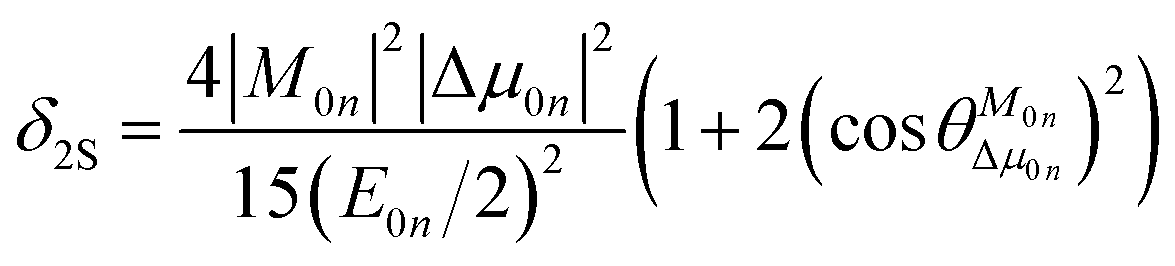
 is the angle between these two vectors. The approximate proportional relationship between δTmax and δ2S during the TPA transition process (depicted in Fig. 6) implies that δ2S can predict the changing tendency of δTmax in general.
is the angle between these two vectors. The approximate proportional relationship between δTmax and δ2S during the TPA transition process (depicted in Fig. 6) implies that δ2S can predict the changing tendency of δTmax in general.
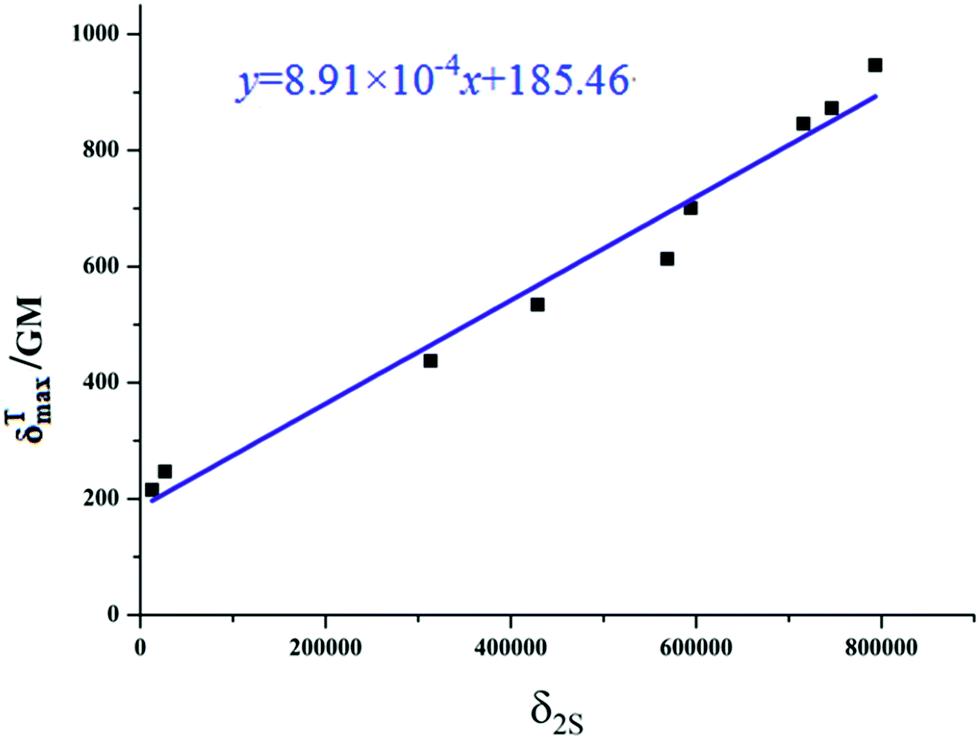
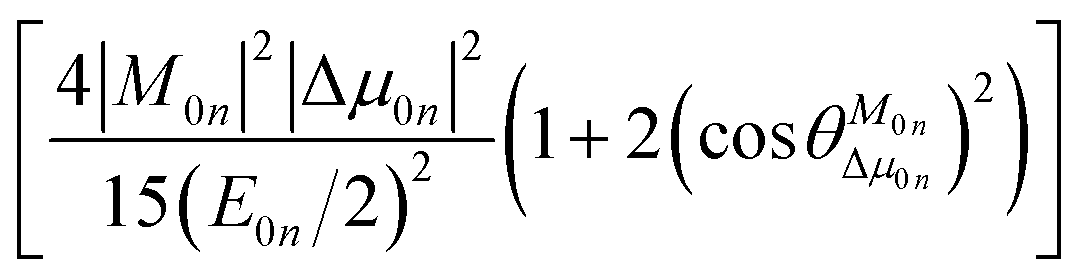 for all the studied product molecules.
for all the studied product molecules.