
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Titania-coated gold nanorods as an effective carrier for gambogic acid†
Hong-Ye Wan ,
Jian-Li Chen,
Xiao-Yan Yu and
Xiao-Ming Zhu*
,
Jian-Li Chen,
Xiao-Yan Yu and
Xiao-Ming Zhu*
State Key Laboratory of Quality Research in Chinese Medicine, Macau Institute for Applied Research in Medicine and Health, Macau University of Science and Technology, Taipa, Macau, China. E-mail: xmzhu@must.edu.mo
First published on 25th October 2017
Abstract
Gambogic acid (GA), a natural product, exhibits potent anticancer effects. Unfortunately, further clinical application of this drug is limited by its poor solubility in aqueous solutions. In the present study, titanium dioxide (TiO2)-coated gold nanorod (GNR/TiO2) nanostructures were used as a carrier for GA. The GNR/TiO2 nanostructure-based delivery was found to provide a stable aqueous dispersion of GA. Compared with the free form, enhanced intracellular GA delivery was achieved by using the GNR/TiO2 nanostructures. An in vitro cytotoxicity study indicated that the GA-loaded GNR/TiO2 nanostructures were much more effective in inhibiting the proliferation of human glioblastoma U-87 MG cells than free GA, particularly at lower working concentrations. Furthermore, the GNR/TiO2 nanostructures displayed a high photothermal conversion efficiency, and irradiation at a low dose (5.3 W cm−2, 2 min) with an 808 nm laser also synergistically enhanced the anticancer effect of the GA-loaded GNR/TiO2 nanostructures. Therefore, our results suggest that GNR/TiO2 nanostructure-based drug delivery is a promising method to improve the therapeutic efficacy of GA.
1. Introduction
Gambogic acid (GA) is a xanthonoid compound isolated from the resin of Garcinia hanburyi. It has been identified as a potent anticancer agent that inhibits the proliferation of a variety of cancer cells in vitro and in vivo.1–5 GA has been approved by the Chinese Food and Drug Administration (CFDA) for a phase II clinical trial for treating lung cancer. Despite this great potential, a major problem encountered with GA is its poor water solubility (<1 μg mL−1), which limits its clinical application. This is because direct intravenous administration of hydrophobic drugs usually causes aggregation, which may block blood capillaries, or lead to an insufficient drug dose at the lesion site.6 Therefore, a critical challenge for furthering the application of GA is its pharmaceutical formulation.Nanotechnology holds great promise for the delivery of anticancer drugs, especially for hydrophobic drugs. Encapsulation of hydrophobic drugs in nanomaterials can increase the solubility of these drugs. The passive and active targeting effects of the nanocarriers also allow for preferential accumulation of drug at the tumor site.7 Moreover, the intracellular drug concentration is often higher when nanoparticles are used, as P-glycoprotein efflux pumps frequently do not recognize nanoparticles.8,9 Abraxane™ (Abraxis, Celgene), which is an albumin-bounded nano-formulation of paclitaxel, has been approved by the U.S. FDA for treating metastatic breast cancer.10,11 Recently, Cremophor EL,12 chitosan,13 polyethylene glycol (PEG),14 lipoproteins,15 and dendrimers,12 have been used as drug-delivery vehicles for GA. However, these drug-delivery systems still have some drawbacks. For example, Cremophor EL has several side effects including cardiotoxicity, nephrotoxicity, neurotoxicity, and hypersensitivity reactions.16 Therefore, developing novel delivery systems for GA is essential.
Recently, gold nanostructures have attracted much attention for their applications in cancer diagnosis and therapy.17–19 Among various gold nanostructures, gold nanorods (GNRs) are widely studied due to their unique plasmonic properties. Their localized surface plasmon resonance (LSPR) can be easily tuned to the near-infrared (NIR) region, and this property makes GNRs a candidate agent for in vivo imaging and therapy,19,20 as light in the NIR region can penetrate deeply into tissues.21 When irradiated with an NIR laser, GNRs are able to convert light energy into heat, which induces cancer cell death. GNRs show great potential for cancer photothermal therapy. When GNRs serve as anticancer drug carriers, the heat can trigger anticancer drug release or enhance the cytotoxicity.22–24
Titanium dioxide (TiO2) nanoparticles have many important applications in solar cells25 and photocatalysis.26 TiO2 shows a high binding affinity to carboxylic acids, and the types of binding exhibited by carboxylic groups on TiO2 include simple adsorption (electrostatic attraction and hydrogen bonding) and chemical adsorption (ester linkage, bridging, and chelating).27 In this study, TiO2-coated GNR (GNR/TiO2) nanostructures were firstly reported as a drug carrier for GA, as the GA molecule has a carboxylic acid group. The anticancer effect of GA-loaded GNR/TiO2 nanostructures was compared with that of the free drug. In addition, the synergistic anticancer effect of GNR/TiO2 nanostructure-based photothermal therapy and GA was investigated.
2. Experimental
2.1. Preparation of the GNR nanostructures
A cetyltrimethylammonium bromide (CTAB)-capped GNR (GNR/CTAB) sample was prepared through a seed-mediated growth method.28 Briefly, the seed solution was made by adding a freshly prepared, ice-cold NaBH4 solution (0.01 M, 0.6 mL) into a mixture of HAuCl4 (0.01 M, 0.25 mL) and CTAB (0.1 M, 9.75 mL). The resultant seed solution was kept at room temperature for 2 h before use. The CTAB growth solution was prepared by the sequential addition of HAuCl4 (0.01 M, 2 mL), AgNO3 (0.01 M, 0.4 mL), HCl (1.0 M, 0.8 mL) and ascorbic acid (0.1 M, 0.32 mL) into the CTAB solution (0.1 M, 40 mL). After the solution was well mixed, the seed solution (40 μL) was rapidly added into the growth solution. The resultant solution was mixed by stirring for 30 s and was then kept at 30 °C overnight. After centrifugation (7000g, 10 min), the obtained pellet was redispersed in deionized water (40 mL).2.2. Preparation of the GNR/TiO2 nanostructures
GNR/TiO2 nanostructures were prepared as described in a previous report.28 The CTAB-capped GNRs were first coated with poly(sodium 4-styrenesulfonate) (PSS). Briefly, the CTAB-capped GNR solution (10 mL) was added dropwise into a PSS solution (molecular weight at 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 2 g L−1, 6 mM NaCl, 10 mL) followed by stirring for 6 h. The excess PSS was removed by centrifugation (7000g, 10 min) twice, and the resultant pellet was redispersed into deionized water (0.2 mL). Then, a TiCl3 solution (15 wt%, 0.2 mL, containing 20–30 wt% HCl) was added into deionized water (6 mL) under stirring, followed by the dropwise addition of NaHCO3 (0.93 M, 1.35 mL) and immediate addition of the concentrated GNR/PSS solution (0.2 mL) into the mixture. After stirring for 30 min, the produced GNR/TiO2 nanostructures were precipitated by centrifugation (6000g, 10 min) and redispersed in deionized water (10 mL).
000, 2 g L−1, 6 mM NaCl, 10 mL) followed by stirring for 6 h. The excess PSS was removed by centrifugation (7000g, 10 min) twice, and the resultant pellet was redispersed into deionized water (0.2 mL). Then, a TiCl3 solution (15 wt%, 0.2 mL, containing 20–30 wt% HCl) was added into deionized water (6 mL) under stirring, followed by the dropwise addition of NaHCO3 (0.93 M, 1.35 mL) and immediate addition of the concentrated GNR/PSS solution (0.2 mL) into the mixture. After stirring for 30 min, the produced GNR/TiO2 nanostructures were precipitated by centrifugation (6000g, 10 min) and redispersed in deionized water (10 mL).
2.3. Nanostructure characterization
The extinction spectrum of the nanostructures was measured using a Hitachi U-3501 UV/visible/NIR spectrophotometer. The sizes and shapes of the synthesized nanostructures were characterized using an FEI Tecnai F20 microscope at 120 kV. The zeta potential and hydrodynamic size of the nanostructures were determined with a Malvern Zetasizer Nano ZS90 analyzer in deionized water. The concentration of the GNR/TiO2 nanostructures was measured by an Agilent inductively coupled plasma mass spectrometry (ICP-MS) 7500a system.2.4. Determination of the photothermal conversion efficiency
An aqueous GNR/TiO2 nanostructure solution (2 mL), with the optical density at 808 nm adjusted to 2.0, in a 1 cm square quartz cuvette was irradiated with an NIR laser (808 nm) at different power intensities (0.6, 1.0, or 1.4 W) for 30 min. The temperature of the solution was measured with a digital thermometer every 30 s. The probe of the thermometer was placed at such a position that the direct irradiation of the laser on the probe head was avoided. A cooling curve was obtained after the laser was switched off. The photothermal conversion efficiency was determined using a previously reported method.292.5. Cell culture
Human glioblastoma U-87 MG cells were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA), and were cultured in alpha-modified Minimum Essential Medium (α-MEM, Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum (FBS), 100 U mL−1 penicillin and 100 μg mL−1 streptomycin at 37 °C in a humidified 5% CO2 atmosphere.2.6. Cell viability assay
A CellTiter-Glo luminescent cell viability assay kit (Promega Corporation, Madison, WI, USA) was used to evaluate the cell viability. This is a homogeneous method to determine the number of viable cells, based on quantifying the adenosine triphosphate (ATP) present, which represents the presence of metabolically active cells. U-87 MG cells were seeded in the wells of a 96-well plate at a density of 5 × 103 cells per well in 100 μL α-MEM. After incubation for 12 h, the medium in the wells was replaced with a fresh medium containing GNR/TiO2 nanostructures, GA or GA-loaded GNR/TiO2 nanostructures. After incubation for 48 h, the cells were washed with phosphate buffered saline (PBS), and a fresh medium (100 μL) was added into each well. CellTiter-Glo reagent (100 μL) was then added to the wells, followed by shaking for 2 min on an orbital shaker. After further incubation for 10 min at room temperature to allow for stabilization of the luminescent signals, the solution from the wells was centrifuged (10000g, 10 min). The collected supernatant (180 μL) was then transferred into each well of a 96-well white plate, and the luminescent signals were detected using a Molecular Devices SpectraMax Paradigm multi-mode microplate reader. The cell viability (%) was calculated for each sample relative to a control.
2.7. Cellular uptake observations
Eight thousand cells were seeded into each well of a 24-well plate. After incubation for 12 h, the culture medium was replaced with a fresh medium containing the GNR/TiO2 nanostructures (30 μg Au mL−1, 0.5 mL). After the cells were further incubated for 24 h, they were washed with PBS extensively to remove any nanoparticles adsorbed on the cell surface. The cellular uptake of the GNR/TiO2 nanostructures was observed using an Olympus IX71 microscope under bright field.2.8. Drug loading
The GNR/TiO2 nanostructures (750 μg Au mL−1, 0.1 mL) were centrifuged (6000g, 5 min) and washed with ethanol (0.2 mL). After centrifugation, the resulting pellet was redispersed in a 0.1 mL ethanol solution containing GA (200 μM, Sigma-Aldrich, St. Louis, MO, USA), followed by overnight stirring. The GA-loaded GNR/TiO2 nanostructures were then obtained by centrifugation, and dispersed in the medium (0.1 mL) for further cell studies.2.9. Drug loading capacity calculations
After centrifugation (6000g, 10 min), the pellet of the GA-loaded GNR/TiO2 nanostructure solution was mixed with dimethyl sulfoxide (DMSO, 0.1 mL), and sonicated for 30 min to dissolve the loaded drug. After centrifugation (12000g, 10 min) of this solution, the GA concentration of the supernatant was determined according to the standard curve using an Agilent 6230 time-of-flight (TOF) liquid chromatography-mass spectrometry (LC/MS) system. The drug loading capacity was then calculated.
2.10. Drug desorption tests
GA desorption from GA-loaded GNR/TiO2 nanostructures was studied in NaOH (0.1 M), PBS (pH 7.4), or citrate buffer (20 mM, pH 4.5). The GA-loaded GNR/TiO2 nanostructures (15 μg Au mL−1) were dispersed in the buffer (1 mL) at 37 °C for 24 h. After centrifugation (12000 rpm, 10 min), the supernatant was collected, and the released GA in the supernatant was extracted three times using chloroform (0.5 mL). After the solvent was evaporated, the residual GA was dissolved with methanol (50 μL), and the GA concentration was determined using LC/MS. The drug desorption percentage was then calculated.
2.11. Intracellular GA content assay
U-87 MG cells were cultured in 100 mm Petri dishes. When the cells reached 80% confluence, the culture medium was replaced with a fresh medium containing the GA-loaded GNR/TiO2 nanostructures or free GA at the equal GA concentration of 0.27 μM. After incubation for 24 h, the cells were washed with PBS, collected and counted. The cells were subsequently centrifuged at 4500g for 5 min. DMSO (0.1 mL) was used to extract the GA from the cell pellets. After centrifugation (12000g, 10 min), the supernatant was collected and the GA concentration was determined using LC/MS. The GA amount per cell was then calculated.
2.12. Nuclei staining
After the U-87 MG cells were incubated with the GA-loaded GNR/TiO2 nanostructures or free GA for 24 h, the cells were washed with PBS to remove any nanoparticles adsorbed on the cell surface. The nuclei of the cells were subsequently stained with Hoechst 33342 (100 ng mL−1, Thermo Fisher Scientific) for 30 min, and the cells were observed on an Olympus IX71 microscope.2.13. Photothermal therapy
Five thousand U-87 MG cells were seeded into each well of a 96-well plate. After incubation for 24 h, the culture medium was changed with phenol red-free α-MEM (100 μL) containing GA (0.27 μM), GNR/TiO2 nanostructures (12.5 μg Au mL−1), or GA loaded GNR/TiO2 nanostructures (0.27 μM GA, 12.5 μg Au mL−1), followed by further incubation for 24 h. The designated wells were exposed to a continuous-wave semiconductor diode laser (808 nm, MDL-N-808-10W, Changchun New Industries Optoelectronics Tech. Co., Ltd., China) for 2 min. The laser power density was 4.4, 5.3, 6.2 or 7.9 W cm−2, and the laser spot diameter was ∼6 mm, which is equal to the diameter of the bottom of a well on the 96-well plate. The cells were incubated for another 24 h, and ATP assay and calcein acetoxymethyl ester (calcein AM) staining were separately performed to determine the cell viability. After the treatment, the medium in the wells was replaced with serum-free α-MEM containing calcein AM (1 μM, Thermo Fisher Scientific). After incubation for 30 min, the cells were washed with fresh medium, and an Olympus IX71 microscope was used to capture images.2.14. Statistical analysis
Each experiment was repeated at least 3 times. Data are expressed as mean ± standard error of the mean (S.E.M.). The statistical difference was evaluated with one-way analysis of variance (ANOVA) followed by Tukey's post hoc test. A P value <0.05 was considered as statistically significant.3. Results and discussion
3.1. Synthesis and characterization of the GNR/TiO2 nanostructures
GNR/CTAB nanostructures were prepared using a seed-mediated growth method.28 Transmission electron microscopy (TEM) imaging showed rod shapes with a narrow size distribution (Fig. 1a). The length and diameter of the GNRs were determined to be 90 ± 4 nm and 29 ± 2 nm, respectively (Fig. 1a). The zeta potential of the GNR/CTAB nanostructures was found to be +47 ± 7 mV. The GNRs were found to support two plasmon modes, a longitudinal and a transverse mode.20 The extinction spectrum of the GNR/CTAB in aqueous solution (Fig. 1c) shows that the longitudinal and transverse plasmon resonance peaks are located at 742 nm and 526 nm, respectively. The GNR/TiO2 nanostructures were prepared by employing TiCl3 as the precursor.28 The zeta potential of the GNR/TiO2 sample was determined to be −2 ± 2 mV. The TiO2 coating with a thickness of 12 ± 1 nm (Fig. 1b) induces a considerable red shift of the longitudinal plasmon resonance wavelength (LPRW) to 806 nm (Fig. 1c), which is within the biological window of 650–900 nm.21 In addition, the hydrodynamic diameter of the GNR/TiO2 nanostructures, measured by the dynamic light scattering technique, is 102 ± 18 nm. After incubation with the culture medium for 12 h, the size was determined to be 105 ± 21 nm, thus indicating that no particle aggregation occurred in the culture medium.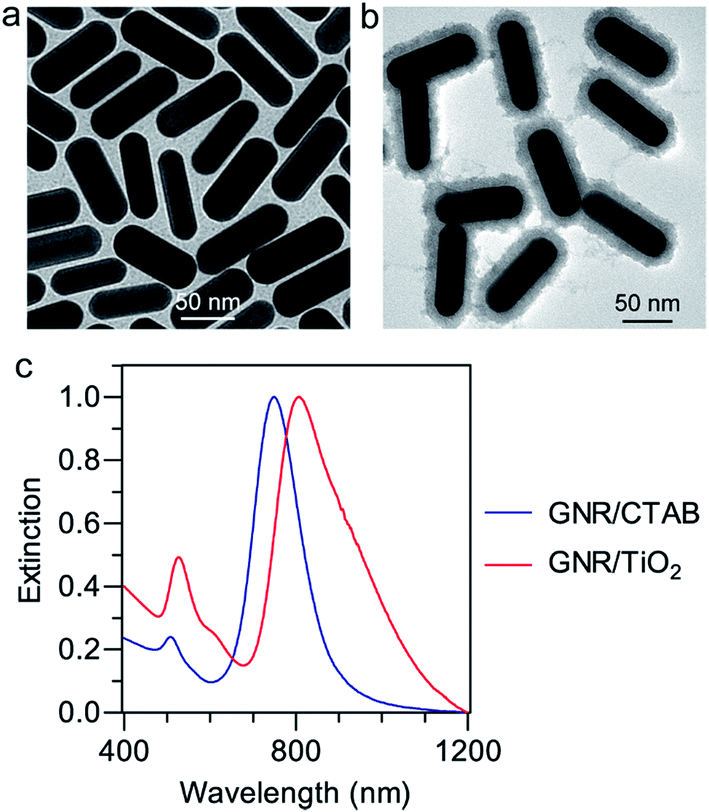 | ||
| Fig. 1 TEM images of GNR/CTAB (a) and GNR/TiO2 (b) nanostructures, and (c) extinction spectra of the synthesized nanostructures. | ||
3.2. Photothermal conversion of the GNR/TiO2 nanostructures
Plasmonic gold nanostructures can absorb light and convert it into heat when irradiated with an NIR laser. This heat is thereafter transferred to the surrounding environment to cause a temperature increase. Gold nanostructure-based photothermal therapy has been previously employed to selectively ablate cancer cells,30 and trigger drug31 or gene release.32,33 Generally, due to their higher photothermal conversion efficiency, gold nanostructures can cause an equal degree of cancer cell death at a lower concentration, a shorter irradiation time, and a lower laser power density. A high photothermal conversion efficiency of gold nanostructures is essential for achieving photothermal therapeutic effects.To measure the photothermal conversion performance of the GNR/TiO2 nanostructures, radiation from an 808 nm laser was sent through a quartz cuvette with a 1 cm path length containing an aqueous GNR/TiO2 nanostructure dispersion (50 μg Au mL−1, 2 mL). The optical extinction of the dispersion at 808 nm was adjusted to 2.0, and a thermocouple was inserted into the aqueous dispersion at such a position that the direct irradiation of the laser on the probe was avoided. The temperature was recorded every 30 s after the laser had been turned on for 30 min until the temperature of the GNR/TiO2 nanostructure dispersion solution reached a steady state. The solution was kept stirring during the measurements. The laser was then switched off and the temperature decrease profile was recorded to determine the rate of the heat transfer from the system to the environment. Fig. 2a shows the temperature rise traces of the GNR/TiO2 nanostructures under 808 nm laser irradiation. The temperature increases rapidly in the beginning and then reaches a plateau after about 20 min of irradiation. After irradiation for 30 min with the 808 nm laser at a power of 1.4 W, the end temperature reaches 57.1 °C (Fig. 2b).
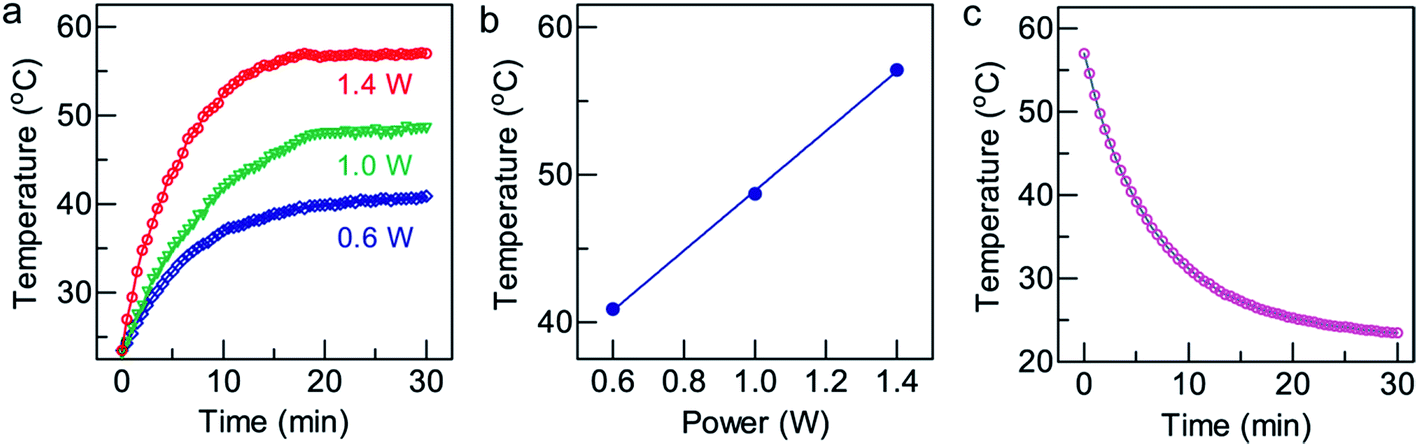 | ||
| Fig. 2 Photothermal conversion of GNR/TiO2 nanostructures. (a) Temperature rise curves of the GNR/TiO2 nanostructures in water (50 μg Au mL−1, 2 mL) acquired under 808 nm laser irradiation at various laser powers for 30 min. (b) Plateau temperatures reached as a function of the laser power. (c) Temperature decay. The data points were measured during the cooling process after the GNR/TiO2 nanostructure solution (50 μg Au mL−1, 2 mL) was irradiated with the 808 nm laser at 1.4 W for 30 min. The blue line was obtained from fitting. | ||
A previously-reported theoretical model29 was used to determine the photothermal conversion efficiency η according to the following eqn (1):
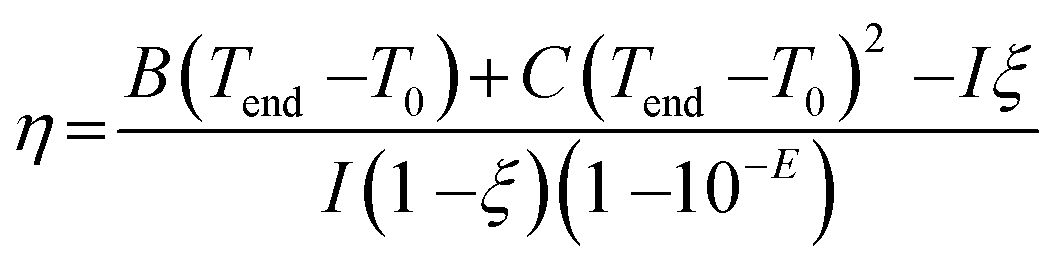 | (1) |
3.3. Cellular uptake and cytotoxicity of the GNR/TiO2 nanostructures
As an anticancer drug carrier, efficient internalization by cancer cells and biocompatibility should be considered to be of utmost importance.34,35 Glioblastoma is one of the most aggressive cancers, and the anticancer effect of GA on glioblastoma has attracted increasing attention.1 In this study, cellular uptake and cytotoxicity of GNR/TiO2 nanostructures were studied in human glioblastoma U-87 MG cells. Bright field imaging under an inverted microscope was used as a simple method to evaluate the cellular uptake of GNR/TiO2 nanostructures, and they show a dark red color. As shown in Fig. 3a, the GNR/TiO2 nanostructures are readily internalized by U-87 MG cells, and most of the nanostructures were observed in the cytoplasm around the nuclei. In addition, endosome/lysosome staining with LysoSensor Green (Fig. S1†) shows that internalized GNR/TiO2 nanostructures are mainly located in the endosomes/lysosomes. After treatment for 48 hours with the GNR/TiO2 nanostructures (0–150 μg Au mL−1), no obvious cytotoxicity was observed in the U-87 MG cells (Fig. 3b), indicating that the GNR/TiO2 nanostructures are biocompatible.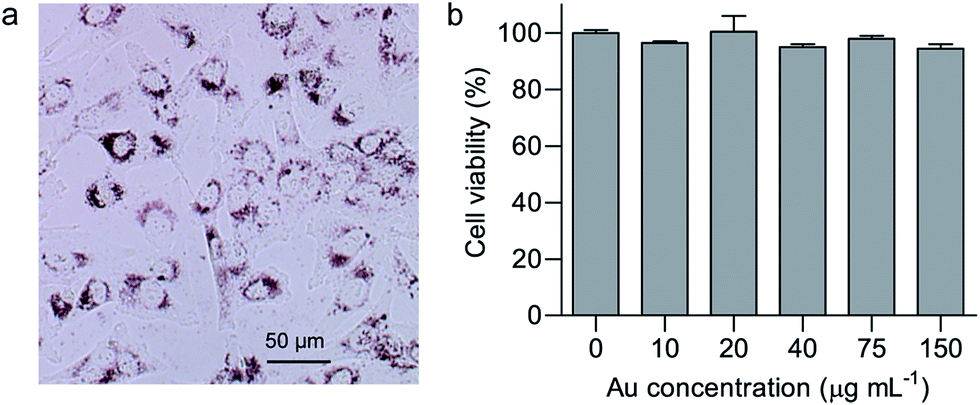 | ||
| Fig. 3 Cellular uptake and cytotoxicity of GNR/TiO2 nanostructures. (a) Bright field imaging of U-87 MG cells after incubation with GNR/TiO2 nanostructures (30 μg Au mL−1). Dark red GNR/TiO2 granules are evident in the cytoplasm around the nuclei of cells. (b) Effect of GNR/TiO2 nanostructures on U-87 MG cell viability. U-87 MG cells were exposed to varying concentrations (0–150 μg Au mL−1) of GNR/TiO2 nanostructures for 48 h, followed by an ATP assay. The data shown represent the mean ± S.E.M. | ||
3.4. Photothermal therapy study
Hyperthermia induces cell death at temperatures above 43 °C.36 Compared with common hyperthermia therapy, photothermal therapy is able to noninvasively and selectively ablate cancer cells. Efficient light absorption and photothermal conversion in the NIR region enables GNRs to denature cell proteins and therefore cause irreversible cell death upon light irradiation.37 Nanostructure-based photothermal therapeutic effects have previously been found to be highly dependent on the irradiation power density and duration. In this study, solo irradiation with the 808 nm laser at the power density of 7.9 W cm−2 for 2 min did not affect the cell viability of U-87 MG cells. Irradiation with the 808 nm laser for 2 min at the power density of 6.2 and 7.9 W cm−2 in the presence of the GNR/TiO2 nanostructures reduced the cell viability of U-87 MG cells to 48.7 ± 5.2% and 25.0 ± 6.1%, respectively (Fig. 4a). In addition, calcein AM staining was also used to confirm this result. Calcein AM can reveal the ubiquitous intracellular esterase activity inside live cells, and therefore only live cells are stained with green fluorescence.38 Most cells in the control group display green fluorescence and maintain their normal morphology. In contrast, rounding of the cell morphology was clearly observed for the cells after laser irradiation at 6.2 W cm−2 in the presence of the GNR/TiO2 nanostructures. The green fluorescence was almost undetectable after laser irradiation at 7.9 W cm−2, indicating that the irradiation power density of 7.9 W cm−2 is able to kill most of the U-87 MG cells (Fig. 4b). A low irradiation dose (5.3 W cm−2, 2 min) was chosen in the following experiments.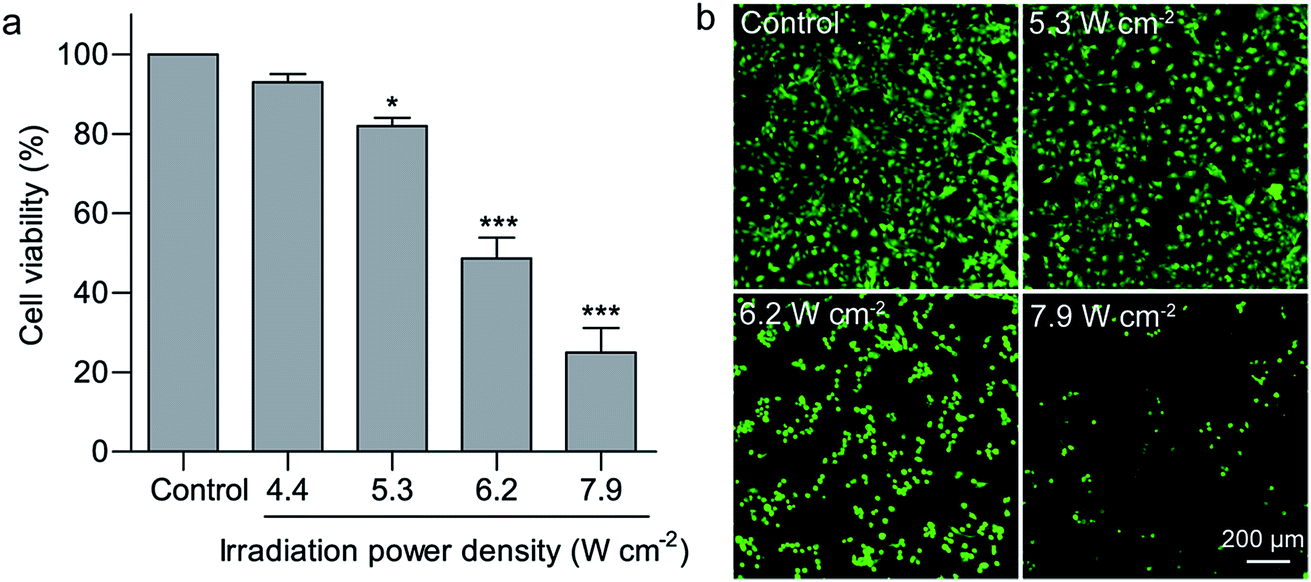 | ||
| Fig. 4 Cell viability of U-87 MG cells upon photothermal therapy. U-87 MG cells were incubated with the GNR/TiO2 nanostructures at the concentration of 12.5 μg Au mL−1 for 24 h, followed by 808 nm NIR laser irradiation at 4.4, 5.3, 6.2 or 7.9 W cm−2 for 2 min. The cell viability was determined by (a) ATP assay or (b) calcein AM staining after further incubation for 24 h. Live cells were stained with green fluorescence by calcein AM. The data shown represent the mean ± S.E.M., *P < 0.05, ***P < 0.001. | ||
3.5. Loading GA onto GNR/TiO2 nanostructures
Carboxylic acid has a high binding affinity to TiO2,27 so we then tried to use the GNR/TiO2 nanostructures as the delivery carrier for the GA molecule, which has a carboxylic group (Fig. 5a). The drug loading was performed in ethanol. The obtained GA-loaded GNR/TiO2 nanostructures were well dispersible in water (Fig. 5b). The GA loading capacity of the GNR/TiO2 nanostructures was calculated to be 1.33 ± 0.1%, according to the following eqn (2).
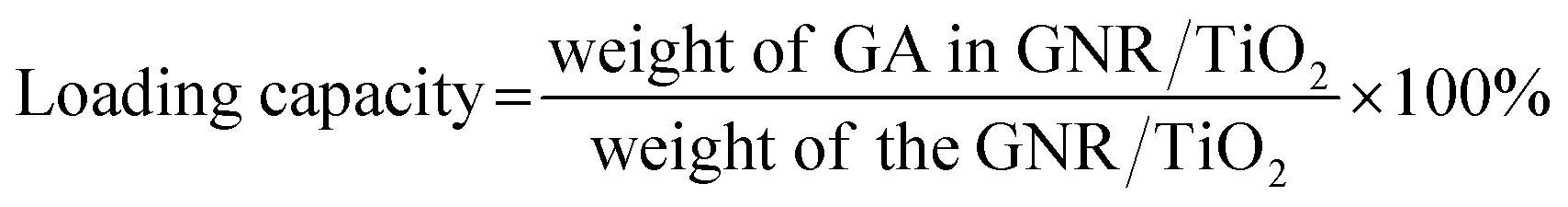 | (2) |
 | ||
| Fig. 5 GA loading. (a) Chemical structure of GA. (b) GA and GA-loaded GNR/TiO2 suspensions in water. (c) Desorption of GA in NaOH and PBS. GA-loaded GNR/TiO2 nanostructures (15 μg Au mL−1) were dispersed in 1 mL of NaOH (0.1 M) or PBS (pH 7.4) for 24 h. The drug desorption percentage was calculated by measuring the drug concentration of the supernatant. The data shown represent the mean ± S.E.M. | ||
Alkaline solutions are effective agents for desorbing carboxylic acid molecules from the surface of TiO2.39,40 To evaluate the binding between GA and TiO2, GA-loaded GNR/TiO2 nanostructures were immersed in a basic aqueous solution (NaOH, 0.1 M) for 24 h. 79.6 ± 4.3% of GA was stripped from the TiO2 surfaces as shown in Fig. 5c. However, only 6.8 ± 3.7% of GA desorbed from the GNR/TiO2 nanostructures in PBS (pH 7.4). We assumed that the dramatic GA desorption in the NaOH solution is due to the carboxylic acid group of GA reacting with NaOH and it being converted into a water-soluble salt. As the internalized nanoparticles are mainly distributed in the acidic endosomes/lysosomes, we also studied the GA desorption in endosome/lysosome mimicking buffer (20 mM citrate, pH 4.5).41 18.6 ± 3.1% of GA released from the GA-loaded GNR/TiO2 nanostructures was observed after incubation for 24 h. This result is attributed to the fact that citric acid is able to competitively displace GA from the GNR/TiO2 nanostructures, as it also has carboxylic acid groups.
3.6. Enhanced cytotoxicity of the GA-loaded GNR/TiO2 nanostructures
As the GNR/TiO2 nanostructures are readily internalized by U-87 MG cells, we speculated that the GNR/TiO2 nanostructures are able to carry more GA into these cells than free GA. We compared the intracellular GA content of the cells treated with free GA and those treated with the GA-loaded GNR/TiO2 nanostructures at the same drug concentration. After GA was extracted from the cells with the organic solvent, the GA concentration was analyzed using LC/MS, which is suitable for detecting GA at a very low concentration range. As shown in Fig. 6a, after incubation of 0.27 μM GA for 24 h, the intracellular GA content of cells that underwent the free-form treatment was (9.6 ± 2.2) × 10−17 g per cell. On the other hand, this value was determined to be (6.0 ± 0.7) × 10−16 g per cell for the GA-loaded GNR/TiO2 nanostructure treatment group (P < 0.001). This result indicated that the GNR/TiO2 nanostructures can increase the concentration of GA in the cancer cells compared with the free form, and therefore, they offer the possibility of higher therapeutic efficacy.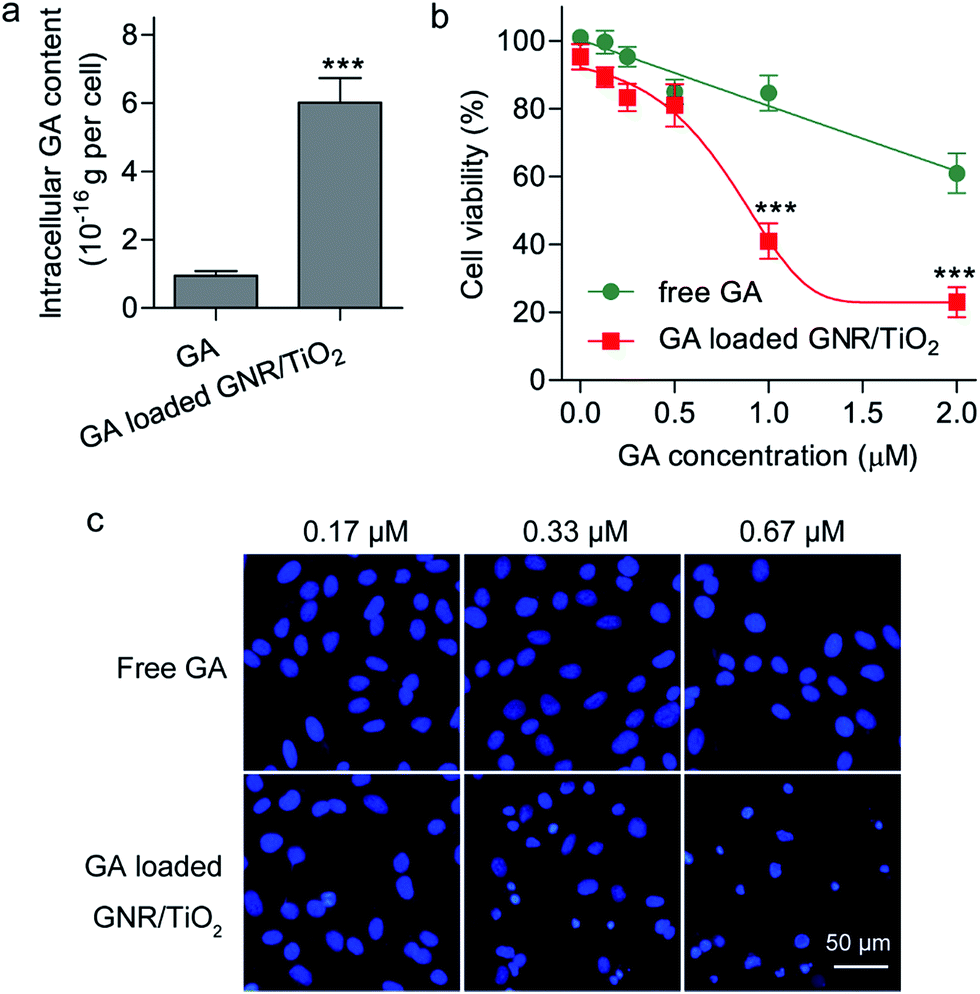 | ||
| Fig. 6 Comparison of the cytotoxic effects of free GA and GA-loaded GNR/TiO2 nanostructures. (a) Intracellular GA content. U-87 MG cells were treated with free GA or GA-loaded GNR/TiO2 nanostructures at a GA concentration of 0.27 μM for 24 h, and the intracellular GA content was determined using LC/MS. (b) Effect of GA-loaded GNR/TiO2 nanostructures and free GA on cell viability of U-87 MG cells. U-87 MG cells were treated with free GA or GA-loaded GNR/TiO2 nanostructures at various GA concentrations for 48 h, followed by ATP assay. (c) Effect of GA-loaded GNR/TiO2 nanostructures and free GA on DNA fragmentation in U-87 MG cells. After incubation for 24 h, the cells were stained with 100 ng mL−1 Hoechst 33342. The data shown represent the mean ± S.E.M., ***P < 0.001. | ||
The cytotoxic effect of the GA-loaded GNR/TiO2 nanostructures was also compared with that of the free GA. Both GA and the GA-loaded GNR/TiO2 nanostructures showed an apparent dose-dependent cytotoxicity in U-87 MG cells, as shown in Fig. 6b. Enhanced cytotoxicity was achieved by using GNR/TiO2 nanostructure-based intracellular delivery. After incubation for 48 h with 1 μM GA, the cell viability of the U-87 MG cells was 84.7 ± 5.2%. However, for the cells treated with the GA-loaded GNR/TiO2 nanostructures at the same drug concentration, the cell viability was reduced to 41.0 ± 5.2% (P < 0.001). We further used a cell-permeable DNA dye, Hoechst 33342, to stain the DNA of the cells. The GA-loaded GNR/TiO2 nanostructures (0.33 μM GA, 24 h) caused cell shrinkage with an intact cell membrane, chromosome condensation and DNA fragmentation (Fig. 6c). No lactate dehydrogenase (LDH) was detected in the culture medium. These results indicate that cell apoptosis rather than necrosis was induced by the GA-loaded GNR/TiO2 nanostructures. However, U-87 MG cells treated with free GA (0.67 μM) showed an intact nuclear architecture (Fig. 6c). A similar result was obtained in one of our previous studies, where enhanced cellular uptake and cytotoxicity of hydrophobic anticancer drugs were achieved by using hollow superparamagnetic iron oxide (SPIO) nanoshells as the drug carrier.41 These results suggest that owing to the ability of the GNR/TiO2 nanostructures to carry more drug into the cells, they significantly enhance the cytotoxicity of GA.
3.7. Photothermal therapy-enhanced cytotoxicity of GA
In some cases, hyperthermia therapy is performed at a moderately elevated temperature,42 and it synergistically enhances the anticancer effects of chemotherapeutic agents.43,44 The results from a recent clinical study indicate that a combination of hyperthermia and chemotherapy results in a higher survival rate for the non-muscle-invasive bladder cancer patients.45 The photothermal therapy-mediating agents encapsulated with anticancer drugs can deliver both drugs and heat simultaneously to the tumor. This multi-modal treatment strategy can result in additionally enhanced anticancer efficacy.46 The mechanisms for chemo-sensitization with hyperthermia include enhanced reactive oxygen species production, inhibition of DNA repair, and increased intracellular drug accumulation.42In this regard, we examined the efficacy of GNR/TiO2 nanostructure-mediated photothermal therapy in combination with GA. Here, a low irradiation dose (5.3 W cm−2, 2 min) and a low GA dose (0.27 μM, 48 h) were used in this experiment. The cell viabilities after these two treatments were 96.7 ± 0.9% and 97.3 ± 0.9%, respectively (Fig. 7a). The GA-loaded GNR/TiO2 nanostructures (0.27 μM GA, 48 h) slightly reduced the viability to 78.0 ± 3.8%. GNR/TiO2 nanostructure-based photothermal therapy at the same irradiation dose (5.3 W cm−2, 2 min) also reduced the cell viability to 83.3 ± 0.6%. However, a combination of the GA-loaded GNR/TiO2 nanostructures and photothermal therapy reduced the cell viability to 42.7 ± 1.8% in a synergistic manner (Fig. 7a). This result was also confirmed by calcein AM staining. Almost all of the cells became rounded and green fluorescence was not detectable for most cells after the combined treatment. In contrast, most cells after receiving the mono-therapy alone displayed green fluorescence and maintained their normal morphology (Fig. 7b). Our results indicate that the application of GNR/TiO2 nanostructure-based photothermal therapy synergistically enhances the anticancer effect of GA.
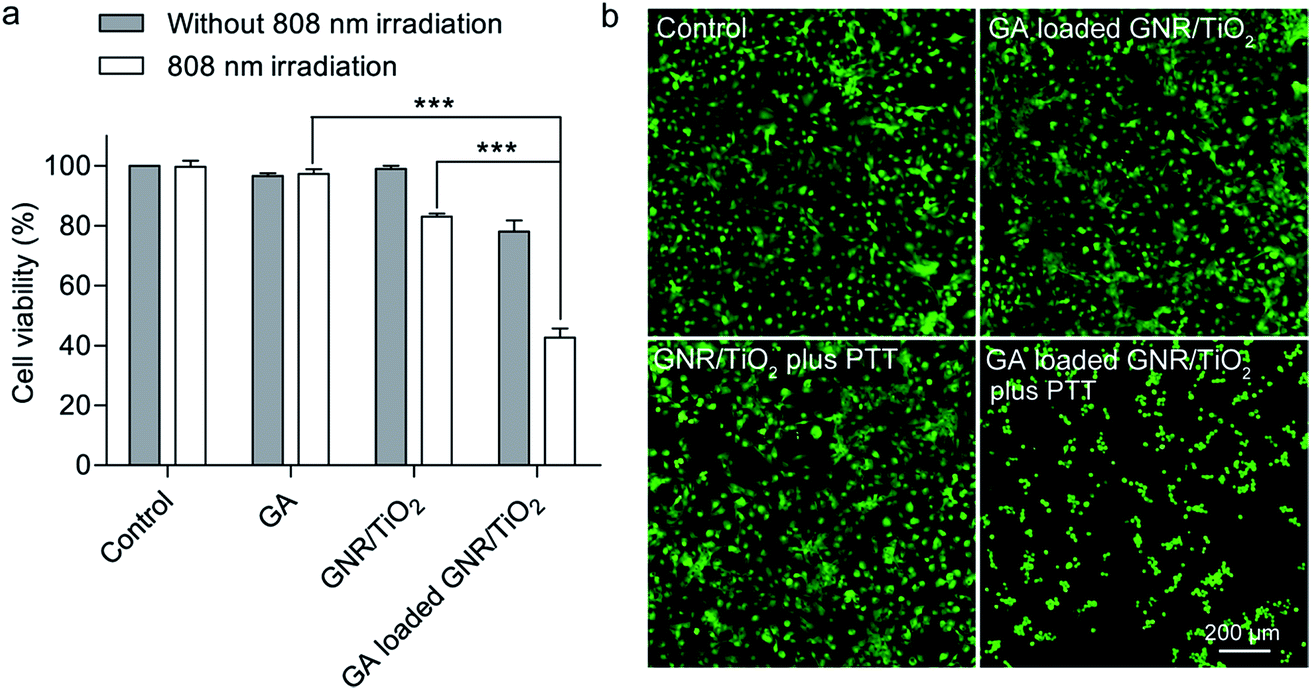 | ||
| Fig. 7 Enhanced cytotoxic effect of GA by using GNR/TiO2 nanostructure-mediated photothermal therapy. U-87 MG cells were incubated with GNR/TiO2 or GA-loaded GNR/TiO2 nanostructures at the GA concentration of 0.27 μM for 24 h, followed by 808 nm NIR irradiation (5.3 W cm−2) for 2 min. After incubation for 24 h, cell viability was determined using an ATP assay (a) and calcein AM staining (b). Live cells were stained with green fluorescence by calcein AM. The data shown represent the mean ± S.E.M., ***P < 0.001. | ||
Chemotherapy dose has a direct impact on cancer patient outcomes.47 Although the risk of toxicity from intensive chemotherapy should be considered, inadequate treatment that might induce tumor recurrence should be avoided. In view of this fact, the incorporation of GNR/TiO2 nanostructure-mediated photothermal therapy with chemotherapy will be useful for improving the efficacy of GA. This combined treatment provides the possibility of reducing the GA dosage or achieving enhanced anticancer effects.
4. Conclusions
In summary, GNR/TiO2 nanostructures as an intracellular carrier for GA were reported for the first time. This delivery approach provides a stable aqueous dispersion of GA. Compared with the free GA, the GA-loaded GNR/TiO2 nanostructures induce a higher intracellular GA content and superior cytotoxicity. In addition, the GNR/TiO2 nanostructures show a high photothermal conversion efficiency under 808 nm NIR laser irradiation. Photothermal therapy at a low irradiation dose (5.3 W cm−2, 2 min) further synergistically enhances the anticancer effects of the GA-loaded GNR/TiO2 nanostructures. Our results suggest that the GNR/TiO2 nanostructures will offer great potential for GA delivery.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by Macau Science and Technology Development Fund (Project No. 014/2014/A1).References
- L. Qiang, Y. Yang, Q.-D. You, Y.-J. Ma, L. Yang, F.-F. Nie, H.-Y. Gu, L. Zhao, N. Lu, Q. Qi, W. Liu, X.-T. Wang and Q.-L. Guo, Biochem. Pharmacol., 2008, 75, 1083–1092 CrossRef CAS PubMed
.
- L.-H. Wang, Y. Li, S.-N. Yang, F.-Y. Wang, Y. Hou, W. Cui, K. Chen, Q. Cao, S. Wang, T.-Y. Zhang, Z.-Z. Wang, W. Xiao, J.-Y. Yang and C.-F. Wu, Br. J. Cancer, 2014, 110, 341–352 CrossRef CAS PubMed
- D. Z. Duan, B. X. Zhang, J. Yao, Y. P. Liu, J. Y. Sun, C. P. Ge, S. J. Peng and J. G. Fang, Free Radical Biol. Med., 2014, 69, 15–25 CrossRef CAS PubMed
- L. L. Liang and Z. X. Zhang, Cell. Physiol. Biochem., 2016, 38, 1618–1630 CrossRef CAS PubMed
- X. P. Shi, X. Chen, X. F. Li, X. Y. Lan, C. Zhao, S. T. Liu, H. B. Huang, N. N. Liu, S. Y. Liao, W. B. Song, P. Zhou, S. Q. Wang, L. Xu, X. J. Wang, Q. P. Dou and J. B. Liu, Clin. Cancer Res., 2014, 20, 151–163 CrossRef CAS PubMed
- P. van Hoogevest, X. L. Liu and A. Fahr, Expert Opin. Drug Delivery, 2011, 8, 1481–1500 CrossRef CAS PubMed
- N. Bertrand, J. Wu, X. Y. Xu, N. Kamaly and O. C. Farokhzad, Adv. Drug Delivery Rev., 2014, 66, 2–25 CrossRef CAS PubMed
- I.-P. Huang, S.-P. Sun, S.-H. Cheng, C.-H. Lee, C.-Y. Wu, C.-S. Yang, L.-W. Lo and Y.-K. Lai, Mol. Cancer Ther., 2011, 10, 761–769 CrossRef CAS PubMed
- X. W. Dong, C. A. Mattingly, M. T. Tseng, M. J. Cho, Y. Liu, V. R. Adams and R. J. Mumper, Cancer Res., 2009, 69, 3918–3926 CrossRef CAS PubMed
- M. R. Green, G. M. Manikhas, S. Orlov, B. Afanasyev, A. M. Makhson, P. Bhar and M. J. Hawkins, Ann. Oncol., 2006, 17, 1263–1268 CrossRef CAS PubMed
- E. Miele, G. P. Spinelli, E. Miele, F. Tomao and S. Tomao, Int. J. Nanomed., 2009, 4, 99–105 CrossRef CAS
- W. Z. Huang, X. Wang, C. Y. Shi, D. D. Guo, G. F. Xu, L. L. Wang, A. Bodman and J. T. Luo, Mol. Pharmaceutics, 2015, 12, 1216–1229 CrossRef CAS PubMed
- G. W. Qu, X. Zhu, C. Zhang and Q. N. Ping, Drug Delivery, 2009, 16, 363–370 CrossRef CAS PubMed
- L. L. Cai, N. Qiu, M. L. Xiang, R. S. Tong, J. F. Yan, L. He, J. Y. Shi, T. Chen, J. L. Wen, W. W. Wang and L. J. Chen, Int. J. Nanomed., 2014, 9, 243–255 Search PubMed
- Y. Ding, Y. Z. Wang, Y. Opoku-Damoah, C. Wang, L. J. Shen, L. F. Yin and J. P. Zhou, Biomaterials, 2015, 72, 90–103 CrossRef CAS PubMed
- H. Gelderblom, J. Verweij, K. Nooter and A. Sparreboom, Eur. J. Cancer, 2001, 37, 1590–1598 CrossRef CAS PubMed
- S. Her, D. A. Jaffray and C. Allen, Adv. Drug Delivery Rev., 2017, 109, 84–101 CrossRef CAS PubMed
- A. M. Alkilany, L. B. Thompson, S. P. Boulos, P. N. Sisco and C. J. Murphy, Adv. Drug Delivery Rev., 2012, 64, 190–199 CrossRef CAS PubMed
- C. M. Cobley, J. Y. Chen, E. C. Cho, L. V. Wang and Y. N. Xia, Chem. Soc. Rev., 2011, 40, 44–56 RSC
- H. J. Chen, L. Shao, Q. Li and J. F. Wang, Chem. Soc. Rev., 2013, 42, 2679–2724 RSC
- R. A. Weissleder, Nat. Biotechnol., 2001, 19, 316–317 CrossRef CAS PubMed
- J. B. Song, X. Y. Yang, O. Jacobson, L. S. Lin, P. Huang, G. Niu, Q. J. Ma and X. Y. Chen, ACS Nano, 2015, 9, 9199–9209 CrossRef CAS PubMed
- J. F. Liao, W. T. Li, J. R. Peng, Q. Yang, H. Li, Y. Q. Wei, X. N. Zhang and Z. Y. Qian, Theranostics, 2015, 5, 345–356 CrossRef PubMed
- N. Larson, A. Gormley, N. Frazier and H. Ghandehari, J. Controlled Release, 2013, 170, 41–50 CrossRef CAS PubMed
- J. Nowotny, M. A. Alim, T. Bak, M. A. Idris, M. Ionescu, K. Prince, M. Z. Sahdan, K. Sopian, M. A. Mat Teridi and W. Sigmund, Chem. Soc. Rev., 2015, 44, 8424–8442 RSC
- J. Schneider, M. Matsuoka, M. Takeuchi, J. L. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed
- Q. Y. Qu, H. W. Geng, R. X. Peng, Q. Cui, X. H. Gu, F. Q. Li and M. T. Wang, Langmuir, 2010, 26, 9539–9546 CrossRef CAS PubMed
- X.-M. Zhu, C. H. Fang, H. L. Jia, Y. Huang, C. H. K. Cheng, C.-H. Ko, Z. Y. Chen, J. F. Wang and Y.-X. J. Wang, Nanoscale, 2014, 6, 11462–11472 RSC
- H. J. Chen, L. Shao, T. Ming, Z. H. Sun, C. M. Zhao, B. C. Yang and J. F. Wang, Small, 2010, 6, 2272–2280 CrossRef CAS PubMed
- X. H. Huang, I. H. El-Sayed, W. Qian and M. A. El-Sayed, J. Am. Chem. Soc., 2006, 128, 2115–2120 CrossRef CAS PubMed
- W.-H. Chen, C.-X. Yang, W.-X. Qiu, G.-F. Luo, H.-Z. Jia, Q. Lei, X.-Y. Wang, G. Liu, R.-X. Zhuo and X.-Z. Zhang, Adv. Healthcare Mater., 2015, 4, 2247–2259 CrossRef CAS PubMed
- J. Song, K. Im, S. Hwang, J. Hur, J. Nam, G.-O. Ahn, S. Hwang, S. Kim and N. Park, Nanoscale, 2015, 7, 9433–9437 RSC
- A. M. Goodman, N. J. Hogan, S. Gottheim, C. Li, S. E. Clare and N. J. Halas, ACS Nano, 2017, 11, 171–179 CrossRef CAS PubMed
- F. Zhao, Y. Zhao, Y. Liu, X. L. Chang, C. Y. Chen and Y. L. Zhao, Small, 2011, 7, 1322–1337 CrossRef CAS PubMed
- B. Yameen, W. I. Choi, C. Vilos, A. Swami, J. J. Shi and O. C. Farokhzad, J. Controlled Release, 2014, 190, 485–499 CrossRef CAS PubMed
- B. Hildebrandt, P. Wust, O. Ahlers, A. Dieing, G. Sreenivasa, T. Kerner, R. Felix and H. Riess, Crit. Rev. Oncol. Hematol., 2002, 43, 33–56 CrossRef PubMed
- E. B. Dickerson, E. C. Dreaden, X. H. Huang, I. H. El-Sayed, H. H. Chu, S. Pushpanketh, J. F. McDonald and M. A. El-Sayed, Cancer Lett., 2008, 269, 57–66 CrossRef CAS PubMed
- D. Gaspar, J. M. Freire, T. R. Pacheco, J. T. Barata and M. A. Castanho, Biochim. Biophys. Acta, 2015, 1853, 308–316 CrossRef CAS PubMed
- H. K. Wayment-Steele, L. E. Johnson, F. Y. Tian, M. C. Dixon, L. Benz and M. S. Johal, ACS Appl. Mater. Interfaces, 2014, 6, 9093–9099 CAS
- D. H. Kim, M. D. Losego, K. Hanson, L. Alibabaei, K. Lee, T. J. Meyer and G. N. Parsons, Phys. Chem. Chem. Phys., 2014, 16, 8615–8622 RSC
- X.-M. Zhu, J. Yuan, K. C.-F. Leung, S.-F. Lee, K. W. Y. Sham, C. H. K. Cheng, D. W. T. Au, G.-J. Teng, A. T. Ahuja and Y.-X. J. Wang, Nanoscale, 2012, 4, 5744–5754 RSC
- M. Hurwitz and P. Stauffer, Semin. Oncol., 2014, 41, 714–729 CrossRef PubMed
- R. D. Issels, Eur. J. Cancer, 2008, 44, 2546–2554 CrossRef CAS PubMed
- X.-M. Zhu, H.-Y. Wan, H. L. Jia, L. Liu and J. F. Wang, Adv. Healthcare Mater., 2016, 5, 3165–3172 CrossRef CAS PubMed
- R. Colombo, A. Salonia, Z. Leib, M. Pavone-Macaluso and D. Engelstein, BJU Int., 2011, 107, 912–918 CrossRef CAS PubMed
- M.-C. Chen, Z.-W. Lin and M.-H. Ling, ACS Nano, 2016, 10, 93–101 CrossRef CAS PubMed
- G. H. Lyman, J. Natl. Compr. Cancer Network, 2009, 7, 99–108 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08560e |
| This journal is © The Royal Society of Chemistry 2017 |