
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnalysis of individual block length of amphiphilic di- & tri-block copolymers containing poly(ethylene oxide) and poly(methyl methacrylate)
Muhammad Khurram Tufail,
Rubina Abdul-Karim,
Sana Rahim,
Syed Ghulam Musharraf and
Muhammad Imran Malik*
and
Muhammad Imran Malik*
H.E.J. Research Institute of Chemistry, International Centre for Chemical and Biological Sciences (ICCBS), University of Karachi, Karachi 75270, Pakistan. E-mail: mimran.malik@iccs.edu
First published on 29th August 2017
Abstract
In this study, di- and tri-block copolymers composed of PEO and PMMA are synthesized and comprehensively characterized. Block copolymers are synthesized by ATRP of MMA using bromine-terminated PEO macro-initiators of varying molar masses ranging from 2000 to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1. End-group transformation of the macro-initiator is confirmed by NMR and FTIR. Initial feed ratios of macro-initiator and monomer are varied systematically in order to have block copolymers of different total molar mass, individual block lengths and compositions. Chemical composition of the block copolymers as obtained by NMR complements the expected values based on initial macro-initiator to monomer ratios. Block copolymers are characterized by size exclusion chromatography, and liquid chromatography at critical conditions on both RP and NP columns. The most complex distribution of block copolymers was obtained at the critical adsorption point of PMMA on a RP column. Fractions were collected at critical conditions of PMMA, and were subjected to detailed analysis by SEC, LCCC–PEG, and NMR in the second dimension. Two-dimensional analysis revealed details about the products that were not accessible by independent or stand-alone analysis using the same techniques.
000 g mol−1. End-group transformation of the macro-initiator is confirmed by NMR and FTIR. Initial feed ratios of macro-initiator and monomer are varied systematically in order to have block copolymers of different total molar mass, individual block lengths and compositions. Chemical composition of the block copolymers as obtained by NMR complements the expected values based on initial macro-initiator to monomer ratios. Block copolymers are characterized by size exclusion chromatography, and liquid chromatography at critical conditions on both RP and NP columns. The most complex distribution of block copolymers was obtained at the critical adsorption point of PMMA on a RP column. Fractions were collected at critical conditions of PMMA, and were subjected to detailed analysis by SEC, LCCC–PEG, and NMR in the second dimension. Two-dimensional analysis revealed details about the products that were not accessible by independent or stand-alone analysis using the same techniques.
Introduction
Covalent joining of hydrophilic and hydrophobic polymeric segments yields amphiphilic species. The hydrophobic part of such polymers does not like aqueous environments, hence it is shielded by the hydrophilic segment during self-assembly when dropped in water. This phenomenon produces core–shell type micellar structures. The size, structure and performance properties of these micelles can be manipulated at the molecular level by changes in chemical composition, total molar mass, individual block lengths, and architecture. Amphiphilic block copolymers have been employed for unique applications in various fields.1–3Poly(ethylene oxide) (PEO) is the most widely employed polymer as a hydrophilic segment owing to its extraordinary water solubility for the whole molar mass range, non-toxicity and biocompatibility.4 However, several different classes of polymers can be used as a hydrophobic segment that includes alkyl, aryl, polyether, polyester, polystyrene, polyacrylates etc. Selection of hydrophobic segment depends upon the intended application.1–3 For instance, biomedical applications require biodegradable non-polar segment such as lactones, lactides, carbonates etc.5,6 Some applications require extremely non-polar block such as polystyrene. Micelles formed by these blocks have dense and glassy core, highly swollen shell and extremely low CMC due to high hydrophobicity of PS block.7,8 Some other applications might require comparatively less non-polar segment such as poly(methyl methacrylate) (PMMA). PMMA is soft hydrophobic polymer that has lower glass transition temperature, and high optical properties. It is biocompatible and resistant to enzymatic attack.9–11 Unique hydrophilic/lipophilic balance, flexibility and crystallinity of block copolymers composed of PEO and PMMA make them alluring candidates for several applications. Degree and rate of crystallization of block copolymers of PMMA with PEO decreased with increasing PMMA block length.12,13 Potential applications of block copolymers of PMMA with PEO are found in the fields of keratoprosthesis modification, drug delivery, tissue engineering, and electronics.14–18 The performance properties are very much dependent upon the total molar mass, molar mass of individual blocks and extent of unwanted hompolymers of both types in the sample. These aspects of the materials under study have never been discussed.
Atom Transfer Radical Polymerization (ATRP) is a successful strategy to design and synthesize multifunctional, nanostructured materials. ATRP empowers control over macromolecular structure, order, and functionality, which are vital considerations for developing materials for any targeted application. ATRP allows the synthesis of block copolymers in facile, reproducible and controlled manner. End-group of commercially available polyethylene glycols (PEG) can easily be transformed to halogen group, which can then be utilized as macro-initiator for ATRP of MMA for synthesis of di- and tri-block copolymers.19–23
Properties of amphiphilic di-, and tri-block copolymers are strongly affected by their molecular characteristics such as total molar mass and its dispersity, molar mass and dispersity of individual blocks, relative block lengths, chemical structure, architecture, functionality and presence of residual parent homopolymers.1–3,24,25 In this context, a comprehensive and meticulous molecular characterization of block copolymers is imperative for optimization of synthesis methods and application properties. Spectroscopic techniques (such as NMR, MALDI-TOF-MS and FTIR) can only provide average values that are not sufficient for development of real structure–property correlations. Furthermore, spectroscopic techniques cannot differentiate between block copolymers and blends of their homopolymers. Presence or absence of unwanted homopolymers in block copolymer samples is another question that cannot be answered by these techniques in general. Several different types of block copolymers have been synthesized and exploited for various applications,26–34 however, the number of reports addressing analysis of individual block lengths and presence of unwanted homopolymers in the samples are rather limited.35–47 Nonetheless, the length of individual blocks and presence of homopolymers in block copolymer sample can enormously affect the final application properties, especially in case of amphiphilic block copolymers. The separation of homopolymers from amphiphilic block copolymer samples is a difficult task since products tend to make micelles when dropped in any solvent that has more compatibility for one block. Specifically block and graft copolymers of PEO and PMMA have been synthesized by employing several methods such as living anionic polymerization, ATRP, and coupling of individually synthesized homopolymers, without much emphasis on the extent of homopolymer formation and achieved length of the individual block.14,19–23,48–53
Chromatographic techniques can reveal more detailed and important information with regard to molecular composition of block copolymers. Three mode of liquid chromatography employed for polymer analysis include size exclusion chromatography (SEC), liquid absorption chromatography (LAC) or interaction chromatography (IC), and liquid chromatography under critical conditions (LCCC). SEC is most widely used mode for confirmation of synthesis of polymers. Determination of molar mass distribution and dispersity by SEC is simple, fast and economical. Ideally, there should not be any enthalpic interactions in SEC that results in separation of polymers on the basis of entropy induced by difference in the hydrodynamic volume of polymer species in dilute solution. Mostly, SEC reveals relative molar mass distribution with respect to calibration curve obtained by narrowly distributed polymer standards. In case of block copolymers, situation is more complex since different polymeric segments can have different hydrodynamic volume that lead to over- or under-estimation of the molar mass values. Another limitation of SEC is its limited resolution and parent homopolymers hardly get separated from block copolymers or sometimes even not detected.37,38,54,55
Detailed analysis of complex polymers can be realized by interaction modes of liquid chromatography namely liquid adsorption chromatography (LAC) or interaction chromatography (IC), and liquid chromatography at critical conditions (LCCC). Information beyond mere total molar mass can be obtained by interaction LC since separation is based on interaction strength of the repeat unit or end groups with the stationary phase. Solvent and temperature gradient LC have been employed for molar mass and chemical composition analysis of polymers.35–38,56–58 However, LAC or IC is not as universal as SEC and requires extensive experimentation to control strength of enthalpic interaction, in order to achieve a reproducible separation, and recovery of the samples.56,59,60
Liquid chromatography at critical conditions suppresses the effect of molar mass on elution pattern, and separation with regard to other variables such as molar mass of non-critical block or functionality can be realized. Critical conditions for a particular polymer can be realized by changing mobile phase composition and temperature.37,38 At critical conditions, homopolymers elute near the void volume of the column independent of their molar mass making them so called “chromatographically invisible”. Under these conditions separation with regard to end group61–65 or other block35–47 can be realized. LCCC reveals a reasonable estimate of the molar mass distribution of non-critical block, and information with regard to presence or absence of unwanted parent homopolymers in the sample.
In this study, di- and tri-block copolymers of PMMA with PEO are synthesized by ATRP of MMA in presence of bromine-terminated Me–PEO and PEG of varying molar masses as macro-initiator. Hydroxyl end-group of commercially available MeO–PEO and PEG are transformed into bromine, to be used as macro-initiator for ATRP. Ratios of macro-initiators and monomer are varied systematically in order to achieve different individual block lengths. Initial screening and augmentation in total molar mass is monitored by size exclusion chromatography. 1H-NMR has been used for chemical composition analysis of the block copolymers. Liquid chromatography at critical conditions was used for analysis of individual block lengths and presence of any unwanted homopolymers of both types. Fractions at critical conditions of PMMA are collected and analyzed by SEC, LCCC–NP, and 1H-NMR in second dimension. The study revealed several heterogeneities of the products that were not accessible by general methods used for this purpose. The novelty of current work lies in determination of individual block lengths of the block copolymers and information about presence/absence of unwanted homopolymers of both types in the sample. Furthermore, two-dimensional analyses revealed that growing chains get coupled during polymerization.
Experimental
Materials
2-Bromoisobutyryle bromide, copper(I) bromide (CuBr) were purchased from Sigma Aldrich and purified by adding in excess amount of acetic acid and stirred overnight. Thereafter they were washed with excess amount of ethanol and dried in vacuum. Methyl 2-bromopropionate and N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) were purchased from Merck (Germany). PEGs and MeO–PEOs of varying molar masses (PEG2k, PEG6k, PEG20k, MeO-PEO2k, and MeO-PEO5k) were purchased from Sigma-Aldrich (Germany) and were dried by azeotropic distillation with toluene before use. PMMA and PS SEC calibration standards were purchased from Polymer Standards Service (Mainz, Germany).Triethylamine (TEA) and methyl methacrylate (MMA) were purchased from Daejung Chemical & Material Co (Korea). Column of activated alumina of mesh size 120–160 was used to purify methyl methacrylate (MMA) in order to remove the inhibitor, stored over CaH2 and distilled. Tetrahydrofuran (THF) (>99%) (distilled over sodium metal and benzophenone for removal of moisture, oxygen and peroxide) and anhydrous ether (>99%) were purchased from RCI Labscan Ltd (Thailand).
HPLC grade solvents acetonitrile (ACN), tetrahydrofuran (THF), acetone, N,N-dimethylformamide (DMF) (Fisher Scientific, USA) and CHCl3 (RCI Labscan, Thailand) were used as received.
Synthetic part
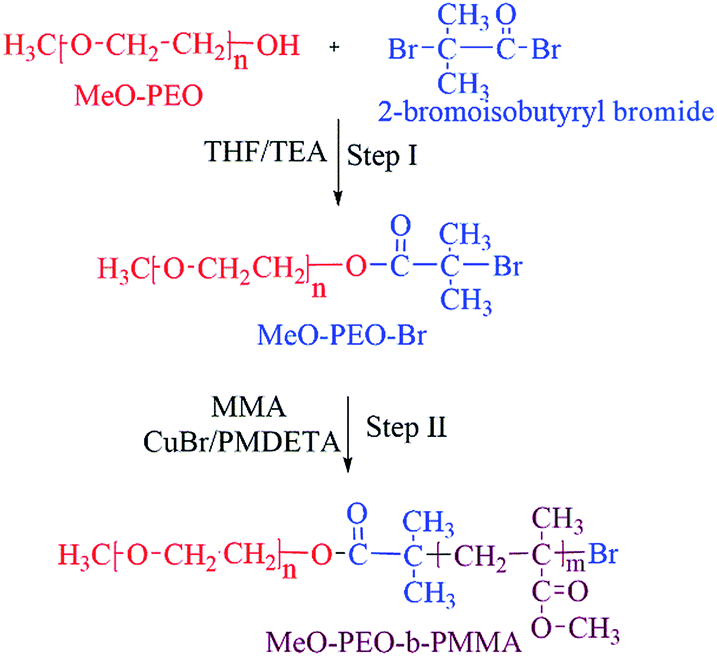
Synthesis of di-, and tri-block copolymers is accomplished by procedure reported in literature.23 A brief experimental procedure for synthesis of typical di-block copolymer by using MeO-PEO5k-Br is given below.An initiator with halogen end-group is required for atom transfer radical polymerization (ATRP). Therefore, the first step is transformation of hydroxyl end-group of commercially available MeO–PEOs and PEGs of varying molar masses, into bromine. As a typical case, 2.65 g of monomethoxy poly(ethylene oxide) (MeO-PEO5k)(terminal OH group = 0.53 mmol) were dissolved in 20 mL of dehydrated THF (312 mmol) in a 250 mL two-neck round bottom flask equipped with a rubber septum and thermometer. Thereafter 0.22 mL of triethylene amine (TEA) (1.59 mmol) was added and a homogenous solution was obtained under continuous stirring. 0.36 g of 2-bromoisobutyryl bromide (1.59 mmol) was added drop-wise in reaction mixture at 0 °C under stirring in 30 minutes. The reaction mixture was further stirred at room temperature for 18 hours. After completion of reaction, reaction mixture was poured into excess amount of hexane to obtain white precipitates of MeO-PEO5k-Br macro-initiator. Precipitates are separated by filtration and dried under vacuum.
MeO-PEO5k-b-PMMA di-block copolymers were synthesized by ATRP using MeO-PEO5k-Br as a macro-initiator. For synthesis of a di-block copolymer with 1:1 ratio of both blocks, initial feed is composed of monomer (MMA): macro-initiator (Meo-PEO5k-Br):catalyst (CuBr):ligand (PMDETA) = 3.3 g (33.3 mmol):3.0 g (0.60 mmol):0.08 g (0.60 mmol):0.10 g (0.60 mmol). All reagents were transferred into a 25 mL two neck round-bottom flask equipped with N2 gas inlet/outlet, thermometer and a magnetic stirrer. Flask was sealed under nitrogen after degassing by three freeze–thaw cycles and immersed in the oil bath at 90 °C under stirring for ATRP of MMA. After 90 min, the reaction mixture was cooled to room temperature. The gel like product was dissolved in THF and passed through activated Al2O3 packed column for removal of Cu complex with PMDETA. Hexane was poured into filtered solution to get white precipitate of MeO-PEO-b-PMMA copolymer. Precipitated product was filtered and dried under vacuum. Same procedure was followed for synthesis of PMMA-b-PEO-b-PMMA tri-block copolymers by using PEG of varying molar masses as macro-initiator. Ratios of monomer and macro-initiator are varied systematically in order to synthesize di-, and tri-block copolymers of varying molar masses, individual block lengths and chemical compositions. The ratio of macro-initiator and monomer shown for different products is initial feed ratio in mass. Complete synthetic scheme is shown in Scheme 1.
 | ||
| Scheme 1 Synthesis of di-block MeO-PEO-b-PMMA copolymer by using MeO–PEO macro-initiator. | ||
Liquid chromatography
An Agilent SEC instrument (infinity 1200 series) equipped with an isocratic pump, manual injector, column oven and a RI detector was used for SEC analysis. A set of three PLgel columns (300 × 7.5 mm, 5 μm, Mixed D) connected in front with a PLgel guard column (50 × 7.5 mm) was used. The column oven was maintained at 30 °C. Chloroform (HPLC grade) was used as an eluent at a flow rate of 1.0 mL min−1. The SEC system was calibrated with narrow molar mass PMMA standards by Polymer Standard Services (Germany). Therefore, all the molar mass values obtained by SEC analysis are relative to linear PMMA. The data was processed by Agilent OpenLAB ChemStation. Sample concentrations were kept between 3.0 to 10.0 mg mL−1, and 100 μL of sample solution was injected.Liquid chromatographic analyses at critical conditions were performed at an Agilent technologies chromatograph (infinity 1200 series). The system is equipped with a binary pump, a column oven, an auto-sampler and an evaporative light scattering detector. Column oven was maintained at 30 °C. Temperature of ELSD was set at 40 °C and nitrogen gas was used as carrier gas at a pressure of 50 psi. Mobile phase was delivered at 0.5 mL min−1. Sample concentrations were kept between 3.0 and 10.0 mg mL−1, and 20 μL of sample solution was injected. Data was processed by Agilent Open LAB ChemStation for LC system. Following columns were used in this study.
Jupiter C18 column, pore size 300 Å, particle size 5 μm, and column dimensions 250 × 4.6 mm (Phenomenex, USA).
Mobile phase used for LCCC–RP was a mixture of acetonitrile (ACN), tetrahydrofuran (THF) and trimethylamine (TEA).
PerfectSil column, pore size 300 Å, particle size 5 μm, and column dimensions 250 × 4.6 mm (MZ Analysentechnik, Germany).
Mobile phase for LCCC–NP was a mixture of tetrahydrofuran (THF) and N,N-dimethylformate (DMF).
Solvents were mixed by volume and vacuum degassed before used.
Results and discussion
ATRP requires halogenated end-group of initiator. Commercially available MeO–PEOs and PEGs of different molar masses are hydroxyl terminated. Hence, first step of synthesis of the di-, and tri-block copolymer is transformation of hydroxyl end-group of macro-initiator to bromine. Afterwards, the macro-initiator with bromine end-group is utilized for ATRP of MMA in order to synthesize di-, and tri-block copolymers containing PEO and PMMA, Scheme 1. As a typical example, the transformation of end-group for a MeO–PEO is shown in Scheme 1. Incomplete transformation of the end-group can be the first source of unreacted macro-initiator in the block copolymer sample. Therefore, it requires analysis at this step. For this purpose a low molar mass macro-initiator (MeO-PEO2k) was used in order to quantify the transformation. FT-IR spectra of MeO-PEO2k and MeO-PEO2k-Br are depicted in Fig. 1A. An extra band at 1735 cm−1 in the bromine-terminated MeO–PEO confirms successful transformation, originated from the carbonyl stretching for the macro-initiator MeO–PEO–Br formed by the acetylation reaction between 2-bromoisobutyryl bromide and MeO–PEO. 1H-NMR spectrum of the MeO–PEO–Br is shown in Fig. 1B. Signal at δ 2.45 ppm that corresponds to hydroxyl group of PEO disappeared after esterification, and two new signals appeared at δ 4.33 ppm (due to ethylene protons near to substituted group) and δ 1.94 ppm (due to methyl proton of the bromoisobutyryl groups). Ratio of peak areas of a, b and d confirms complete substitution.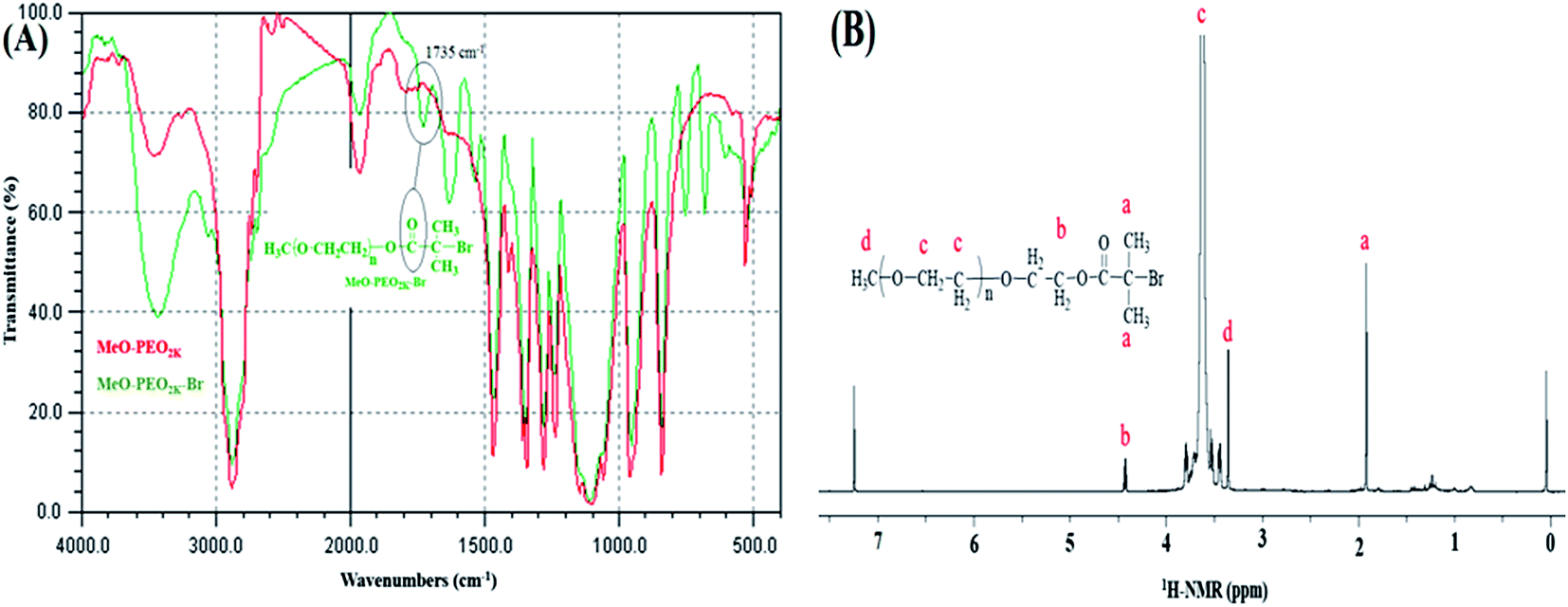 | ||
| Fig. 1 Transformation of end-group (A) FTIR spectra of MeO-PEO2k-OH and MeO-PEO2k-Br, (B) 1H-NMR spectrum of MeO-PEO2k-Br. | ||
After confirmation of successful quantitative transformation, ATRP of MMA is conducted by using macro-initiators of varying molar masses. Various mono- and bi-functional PEOs of varying molar masses were used as macro-initiators. The ratio of macro-initiator and monomer is varied systematically in order to obtain block copolymer of varying total molar mass, individual block lengths and chemical composition.
Correlation of chemical composition of block copolymers to the feed ratios is confirmed by 1H-NMR spectroscopy. As a typical example, 1H-NMR spectrum of MeO-PEO2k-PMMA di-block copolymer is depicted in Fig. 2. Signals at δ 0.8–1.0, 1.7–1.8, and 3.58 ppm correspond to –CH3 (e), –CH2 (f), and –OCH3 (g) group of MMA unit respectively. Characteristic signal of –CH2–CH2– of PEO is present at δ 3.63 ppm in 1H-NMR spectrum. The composition of di-, and tri-block copolymers can be determined by 1H-NMR spectroscopy by using following equation:
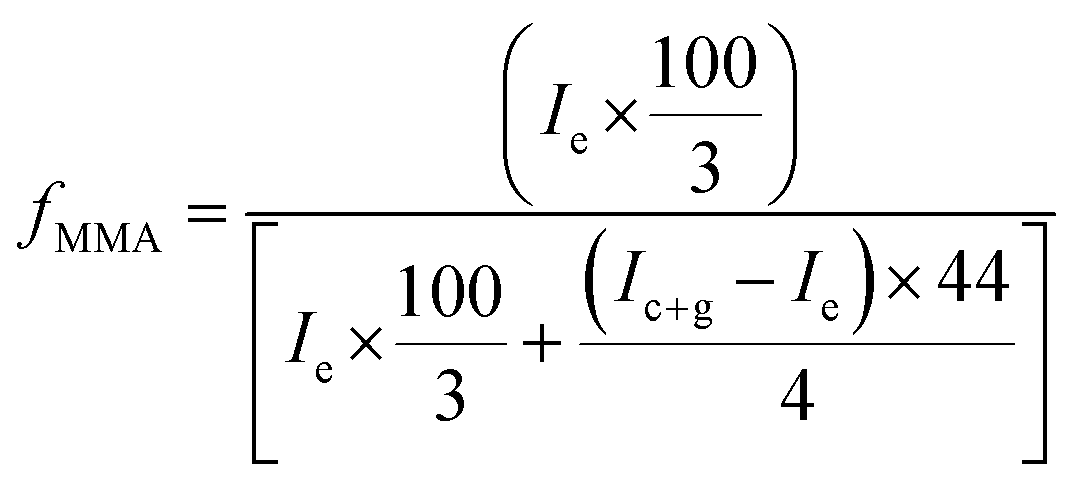
 | ||
| Fig. 2 1H-NMR spectrum of MeO-PEO2k-b-PMMA (1:1). | ||
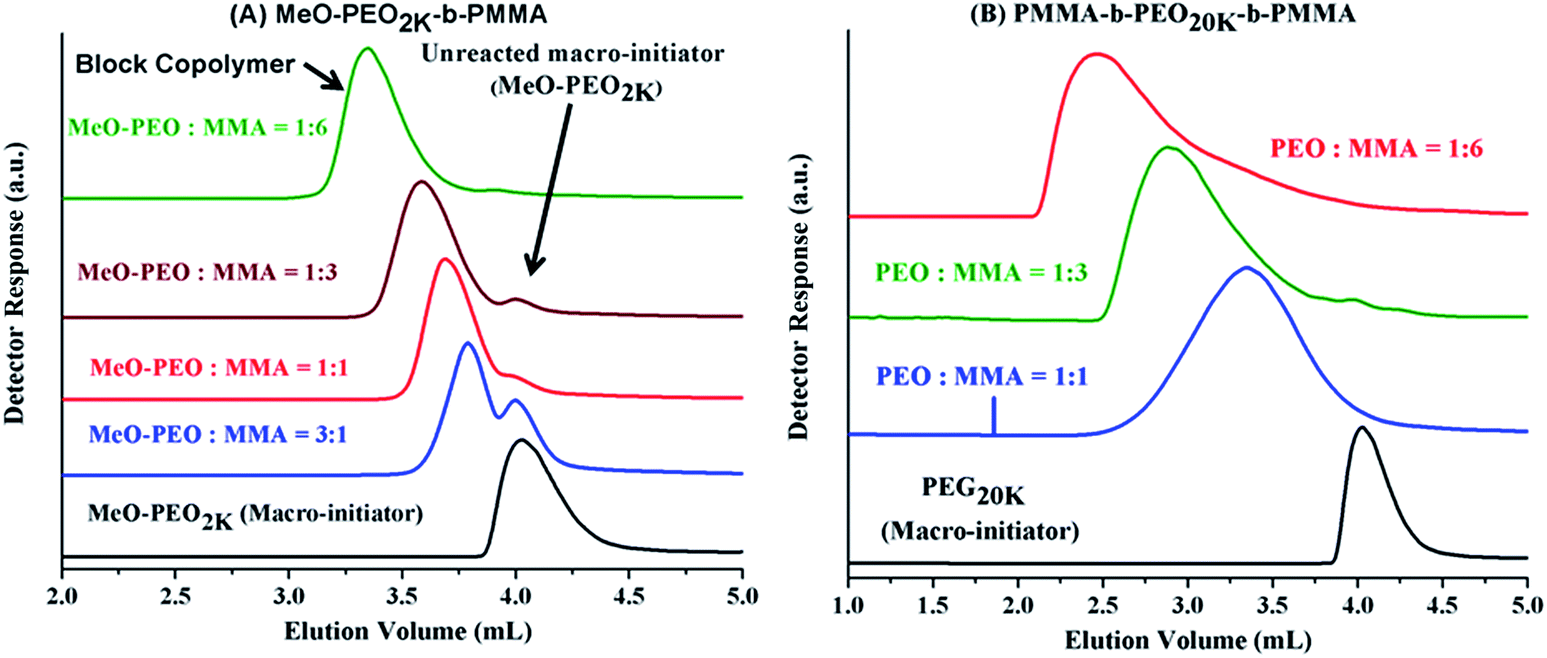
Next question is if molar mass of the block copolymer is increasing as per expectations based on initial feed ratio of macro-initiator and monomer. As typical examples, augmentation in the hydrodynamic size and in turn molar mass of block copolymers with increase in the relative monomer content in the reaction mixture is shown in Fig. 3. As can be noticed block copolymers elute earlier than the macro-initiator. Furthermore, order of elution of block copolymers is from higher to lower monomer content in the reaction mixture. Typical examples of one di-block and one tri-block copolymers are shown. SEC has limited resolution, hence, it is not possible to deduce if there is any unreacted macro-initiator or PMMA homopolymer is present in the sample. In case of tri-block copolymer, there is an indication of presence of macro-initiator in one of the sample. The source of unreacted macro-initiator can be incomplete transformation of end-group in first step. However, it is not possible to clearly conclude that if this bimodal distribution of some samples of tri-block copolymer is due to presence of PEO macro-initiator or PMMA homopolymer. Furthermore, total molar mass values obtained by SEC analysis are either over- or under-estimated depending upon the calibration curve used.41 Approximation of individual block length can also be unrealistic. The molar mass and its dispersity values of the products under study are given in Table 1. It is important to mention here that all the values are relative to PMMA calibration. The hydrodynamic volume of PEO macro-initiator is higher compared to PMMA in chloroform that results in earlier elution of PEO revealing higher molar mass than expected.
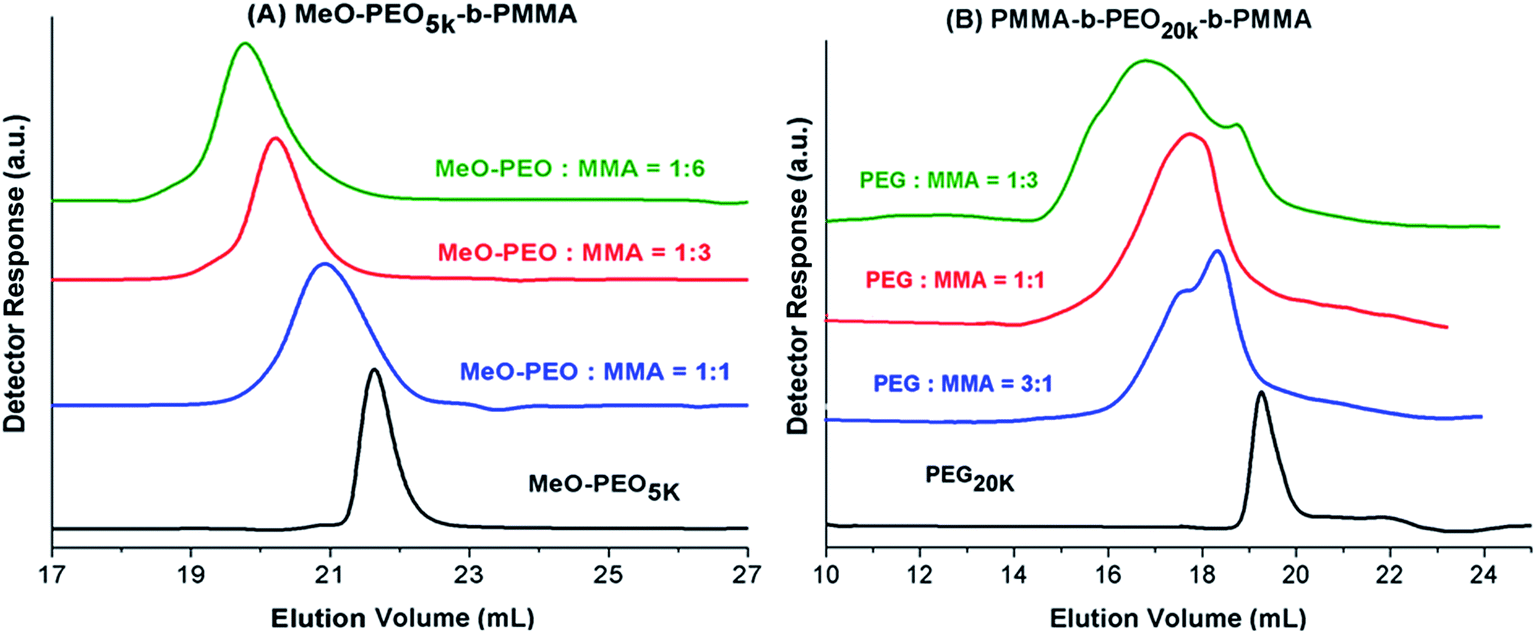 | ||
| Fig. 3 SEC of (A) MeO-PEO5k-b-PMMA, and (B) PMMA-b-PEO20k-b-PMMA with varying ratios of macro-initiator and monomer on a set of three PLgel columns (300 × 7.5 mm, 5 μm, Mixed D) and PLgel guard column (50 × 7.5 mm) in chloroform at flow rate of 1.0 mL min−1. Legend depicts the initial feed ratios in mass of macro-initiator and monomer (MMA). | ||
| Product | Mn (g mol−1) | Mw (g mol−1) | Mp (g mol−1) | PDI (Mw/Mn) |
|---|---|---|---|---|
| MeO-PEO5k macro-initiator | 8300 | 8600 | 8400 | 1.04 |
| MeO-PEO5k-b-PMMA (1:1) |
12800 |
14600 |
13000 |
1.14 |
| MeO-PEO5k-b-PMMA (1:3) |
20000 |
23000 |
21000 |
1.15 |
| MeO-PEO5k-b-PMMA (1:6) |
24500 |
28000 |
27000 |
1.14 |
| PEG20k macro-initiator | 36000 |
38000 |
36000 |
1.05 |
| PMMA-b-PEO20k-b-PMMA (3:1) |
42000 |
53000 |
43000 |
1.26 |
| PMMA-b-PEO20k-b-PMMA (1:1) |
48000 |
61000 |
49000 |
1.27 |
| PMMA-b-PEO20k-b-PMMA (1:3) |
70000 |
98100 |
81000 |
1.40 |
Most of the above-mentioned questions can be answered by liquid chromatography at critical conditions. The method is based on the concept of so called “chromatographic invisibility” that renders molar mass independent elution of a particular polymer. Under such conditions, separation and in turn characterization with regard to non-critical segments is possible. This is in fact the only way for reasonably reliable quantitative determination of individual block length and content of unwanted homopolymers in block copolymer samples. For the polymers under study, critical conditions or critical adsorption point (CAP) of both PEO and PMMA are required.
The CAP for PMMA has been reported using different mobile phases on various stationary phases of different nature.35–38 Selection of CAP for a particular polymer depends upon the segment that needs to be analyzed under such conditions. In current study, the other block PEO is polar than PMMA. On RP column, PEOs are expected to be excluded from the stationary phase at CAP of PMMA. Another aspect that needs to be considered is the solubility of all the segments of the polymer under study in the mobile phase. Taking into account above considerations, a ternary mixture of ACN, THF, and TEA has been selected. ACN is adsorption promoting solvent under such conditions whereas THF promotes desorption. Addition of 1.0% TEA is crucial in order to avoid unwanted polar interactions of ether groups with the unmodified silanol groups of the stationary phase.43 PMMA41k eluted later than PMMA2k while using mobile phase composed of ACN/THF/TEA: 94/5/1 v/v, showing slight adsorption of PMMA repeat unit, Fig. 4A. Increasing relative content of THF in the mobile phase reversed the elution order, PMMA41k eluted earlier than PMMA2k, a typical size exclusion behaviour, Fig. 4B. There must be a mobile phase composition between above-mentioned compositions that should result in molar mass independent elution of PMMAs. In mobile phase composition of ACN/THF/TEA: 93/6/1 v/v, all PMMAs eluted at the same elution volume irrespective of large difference in their molar masses, Fig. 4C. These conditions are termed as liquid chromatography at critical conditions (LCCC) or critical adsorption point (CAP). Under such conditions, other polymers show their own particular elution behaviour. The other block in current study, PEO is polar than PMMA hence elute in size exclusion regime earlier than the critical point of PMMAs, Fig. 4D. The conditions can be utilized for block copolymers of PMMA with any other polymer provided the noncritical polymer is soluble in the mobile phase. The block copolymer should be separated with regard to the other block, and PMMA homopolymer should be separated from block copolymer.35
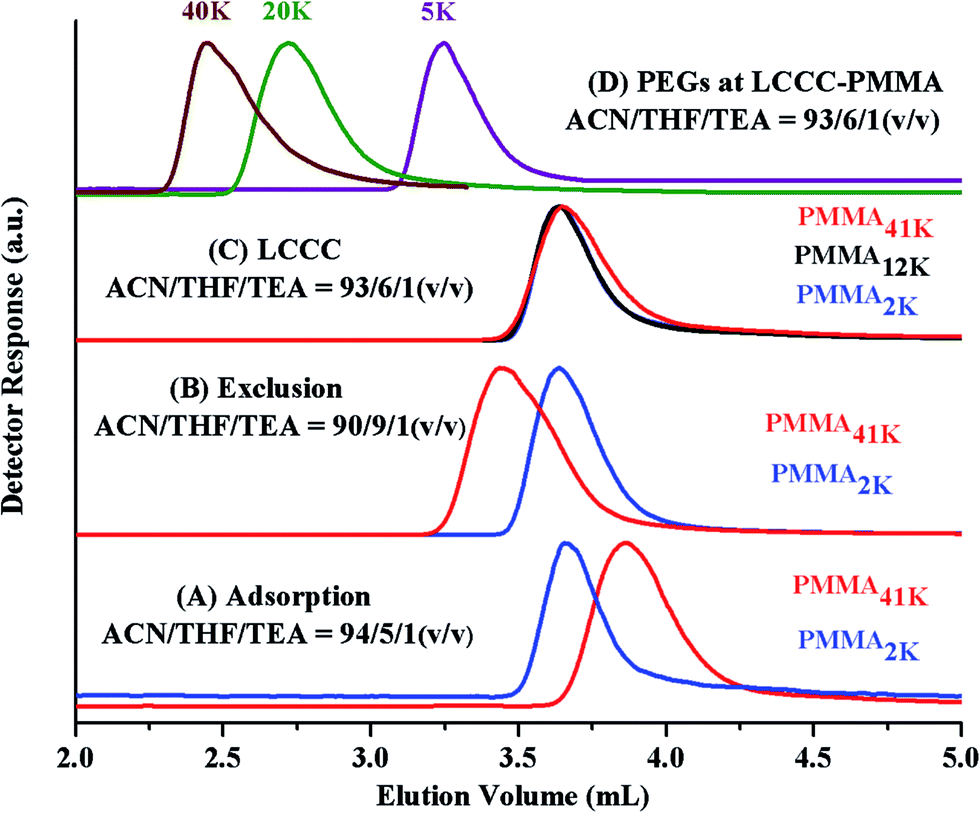 | ||
| Fig. 4 Elugrams of PMMAs of varying molar masses with different compositions of mobile phases; ACN/THF/TEA, (A) 94/5/1 (v/v) (adsorption), (B) 90/9/1 (v/v) (exclusion), (C) 93/6/1 (v/v) (critical adsorption point of PMMA); (D) elution behaviour of PEGs at CAP of PMMA – ACN/THF/TEA (v/v): 90/9/1 (v/v); column: Jupiter C18 300 Å, 5 μm, 250 × 4.6 mm column (Phenomenex), detector: ELS, column temperature: 30 °C. | ||
Fig. 5A shows the elugrams of the blends of similar molar mass samples of PMMA and PEG at CAP of PMMA. PMMAs eluted independent of their molar mass at void volume of the column, however, PEGs are excluded from the pores of the stationary phase and eluted in order of decreasing molar mass earlier than the critical point of PMMA. Fig. 5B demonstrates the plot of elution volume versus molar mass of PEG and PMMA at CAP of PMMA. The block copolymer under current study should show elution behaviour of PEO whereas PMMA homopolymers should be separated from the block copolymer at its critical point.
 | ||
| Fig. 5 (A) Elugrams of blends of similar molar mass PEG and PMMA at CAP of PMMA; (B) calibration curve of PEGs at CAP of PMMA; column: Jupiter C18 300 Å, 5 μm, 250 × 4.6 mm column, detector: ELS, column temperature: 30 °C; mobile phase ACN/THF/TEA: 93/6/1 (v/v). | ||
Analysis of di-, and tri-block copolymers at CAP of PMMA should reveal if there are any unwanted PMMA homopolymers in the sample, along with separation of block copolymers with regard to molar mass of PEO block. Macro-initiators of varying molar masses are used and ratio of monomer (MMA) is varied systematically in order to obtain variable block lengths. The products synthesized by same macro-initiator should be eluted at same point according to length of the macro-initiator irrespective of the monomer content in the initial feed ratio since no change in the molar mass of PEO is expected. This is the case as illustrated by di-, and tri-block copolymers with varying ratios of macro-initiator and monomer, Fig. 6. However, careful analysis of di-block copolymer synthesized by using MeO–PEO5k as macro-initiator revealed that elution of block copolymer is not completely independent of the length of critical block. In fact, elution volume increased with increase of the length of critical PMMA block, Fig. 6A. This is expected behaviour and is one of the limitations of LCCC; nevertheless, this is only possible method for reasonable estimation of individual block lengths of block copolymers.45,66 Absence of any PMMA homopolymers in the samples is confirmed by nonexistence of any peak at 3.7 mL, critical point of PMMA. Elugrams of tri-block copolymers initiated by PEG20k with varying macro-initiator to monomer ratio are shown in Fig. 6B. Tri-block copolymers have complex distribution. Major peak is found at elution volume of macro-initiator that is supposed to be the targeted product without any alteration in the block length of the macro-initiator. Tri-block copolymers also have PMMA homopolymers in the sample whose content increased with the increase of initial content of MMA in the feed (3.7 mL). However, most interesting and unexpected is the low elution volume shoulder of the major peak. Since the conditions are chromatographically invisible for PMMA and separate with regard to length of PEO block, the lower elution volume shoulder must have higher molar mass of PEO. Furthermore, maxima correspond to double molar mass of PEO macro-initiator under current conditions (Fig. 5B). Therefore, it might either be coupling product of macro-initiator or growing chains that needs to be explored further.
 | ||
| Fig. 6 Elugrams of (A) MeO-PEO5k-b-PMMA, and (B) PMMA-b-PEO20k-b-PMMA with varying ratios of macro-initiator and monomer at CAP of PMMA on a Jupiter C18 300 Å, 5 μm, 250 × 4.6 mm column, detector: ELS, column temperature: 30 °C; mobile phase ACN/THF/TEA: 93/6/1 (v/v%), (A) MeO-b-PEO5k-b-PMMA; (B) PMMA-b-PEO20k-b-PMMA. Legend depicts the initial feed ratios in mass of macro-initiator and monomer (MMA). | ||
Analysis by SEC and LCCC–RP revealed total molar mass distribution, estimate of block length of PEO macro-initiator, and presence or absence of PMMA homopolymers in the samples. However, there can be unreacted macro-initiator due to incomplete transformation of end-group in the first step (Scheme 1). The estimation of molar mass distribution of PMMA block is more important since it is the second block polymerized using PEO macro-initiator. In order to answer these questions, critical conditions of PEO are required. PEO block is polar compared PMMA block. Hence, at CAP of PEO on NP column, PMMAs should be excluded from the stationary phase. In current study a binary mixture of THF and DMF was used for establishment of CAP of PEO. THF and DMF are good solvents for both PEG and PMMA for fairly long molar mass range. THF promotes adsorption of PEGs on NP whereas DMF strongly promotes desorption. PEGs eluted in order of increasing molar mass while using mobile phase composed of 2% DMF and 98% THF, Fig. 7A. Increasing the DMF content in the mobile phase to 4% reversed the order of elution, higher molar mass eluted earlier than lower molar mass in a typical SEC regime, Fig. 7B. Chromatographic critical conditions for PEGs are realized in a mobile phase composition THF/DMF: 97.50/2.50 (v/v), Fig. 7C. As expected PMMAs eluted in size exclusion mode at CAP of PEG on NP column, Fig. 7D. The conditions can be utilized for block copolymers of PEO with any other block which is non-polar compared to PEO and is soluble in the mobile phase composition, such as block copolymers of PEO with PS.41
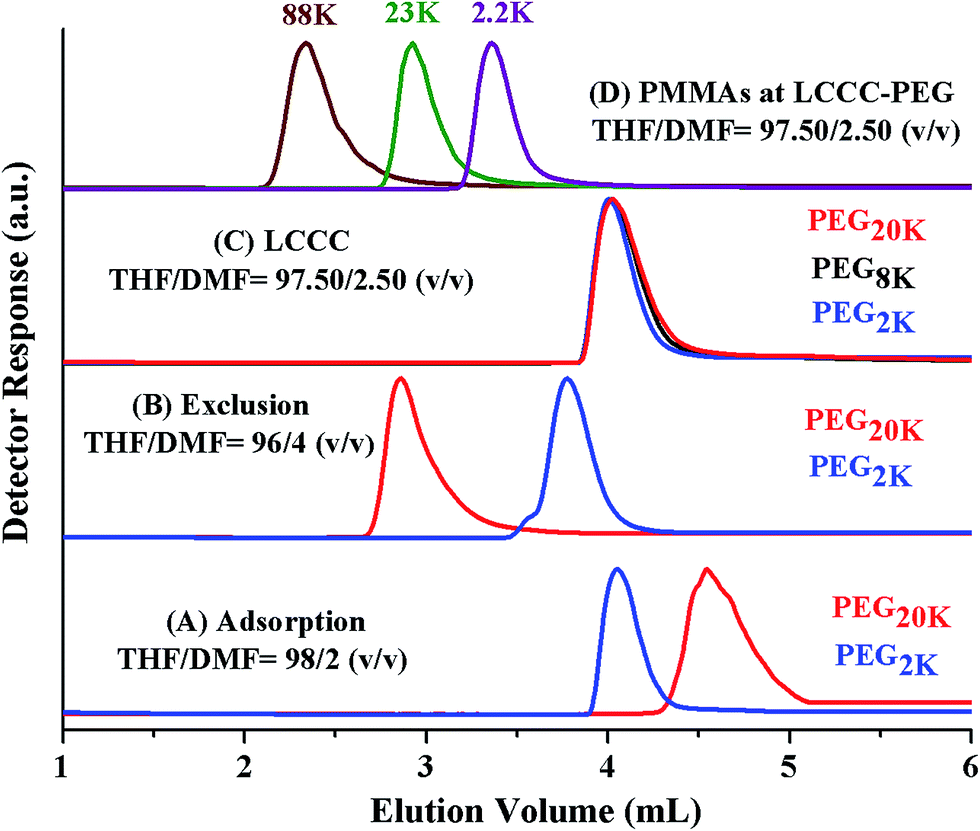 | ||
| Fig. 7 Elugrams of PEGs of varying molar masses with different compositions of mobile phases; THF/DMF (A) 98.0/2.0 (v/v) (adsorption mode), (B) 96.0/4.0 (v/v) (exclusion mode), (C) 97.5/2.5 (v/v) (critical adsorption point of PMMA); (D) elution behaviour of PMMAs at CAP of PEG – THF/DMF: 97.5/2.5 (v/v); column: PerfectSil 300 Å, 250 × 4.6 mm (MZ analytical), detector: ELS, column temperature: 30 °C. | ||
Fig. 8A presents the elution behaviour of blends of similar molar masses of PEG and PMMA at CAP of PEG. PEGs are chromatographically inseparable and whole molar mass range eluted at a sharp point (4.1 mL). However, PMMAs eluted in order of decreasing molar mass earlier than critical PEGs. Fig. 8B shows the calibration curve of PMMA at CAP of PEG that can be used to estimate the molar mass distribution of non-critical PMMA block in the block copolymer samples.
 | ||
| Fig. 8 (A) Elugrams of blends of similar molar mass PEGs and PMMAs at CAP of PEG; (B) calibration curve of PEGs at CAP of PMMA; column: PerfectSil 300 Å, 250 × 4.6 mm column (MZ analytical), detector: ELS, column temperature: 30 °C; mobile phase THF/DMF: 97.5/2.5 (v/v). | ||
Analyses of block copolymers at above-mentioned CAP of PEG should reveal a reasonable estimate of molar mass of non-critical block and presence or absence of any unreacted PEO macro-initiator. Fig. 9A presents chromatograms of di-block copolymers of PMMA initiated with same macro-initiator (MeO-PEO2k) by varying monomer to macro-initiator ratios at CAP of PEG. A small peak at 4.02 mL is present in most of the block copolymers, indicating presence of unreacted macro-initiator. The reason could be incomplete transformation of end-group in the first step of synthesis (Scheme 1). Elution volume of block copolymers decreased with increase in initial content of monomer, indicating longer PMMA block length. Analysis of tri-block copolymers synthesized by using PEO20k as macro-initiator by varying initial macro-initiator to monomer ratio, at CAP of PEG are shown in Fig. 9B. Absence of any peak at 4.04 mL confirms that polymerization initiated with all molecules of macro-initiator rendering no unreacted macro-initiator in the samples shown. However, broad peaks are obtained for block copolymers. The broadness of block copolymer peaks might be due to several reasons. The tri-block copolymers synthesized in current study are ABA type. The behaviour of invisible middle block could be somewhat different from invisible side blocks.61,67,68 Furthermore, the chromatographic behaviour of a single long block could be different from two short blocks of varying lengths. Higher elution volume tailing can be explained by presence of PMMA homopolymers in the sample that might have different length from the PMMA block in the block copolymers (see Fig. 6). However, there is nothing eluting at CAP of PEG (4.04 mL). The results at CAP of PMMA eliminate the assumption of coupling of PEO macro-initiator. It would have been eluted at its CAP around 4.04 mL if present in sample.
 | ||
| Fig. 9 Elugrams of (A) MeO-PEO2k-b-PMMA, and (B) PMMA-b-PEO20k-b-PMMA with varying ratios of macro-initiator and monomer at CAP of PEG on a PerfectSil 300 Å, 250 × 4.6 mm column (MZ analytical), detector: ELS, column temperature: 30 °C; mobile phase THF/DMF: 97.5/2.5 (v/v). Legend depicts the initial feed ratios in mass of macro-initiator and monomer (MMA). | ||
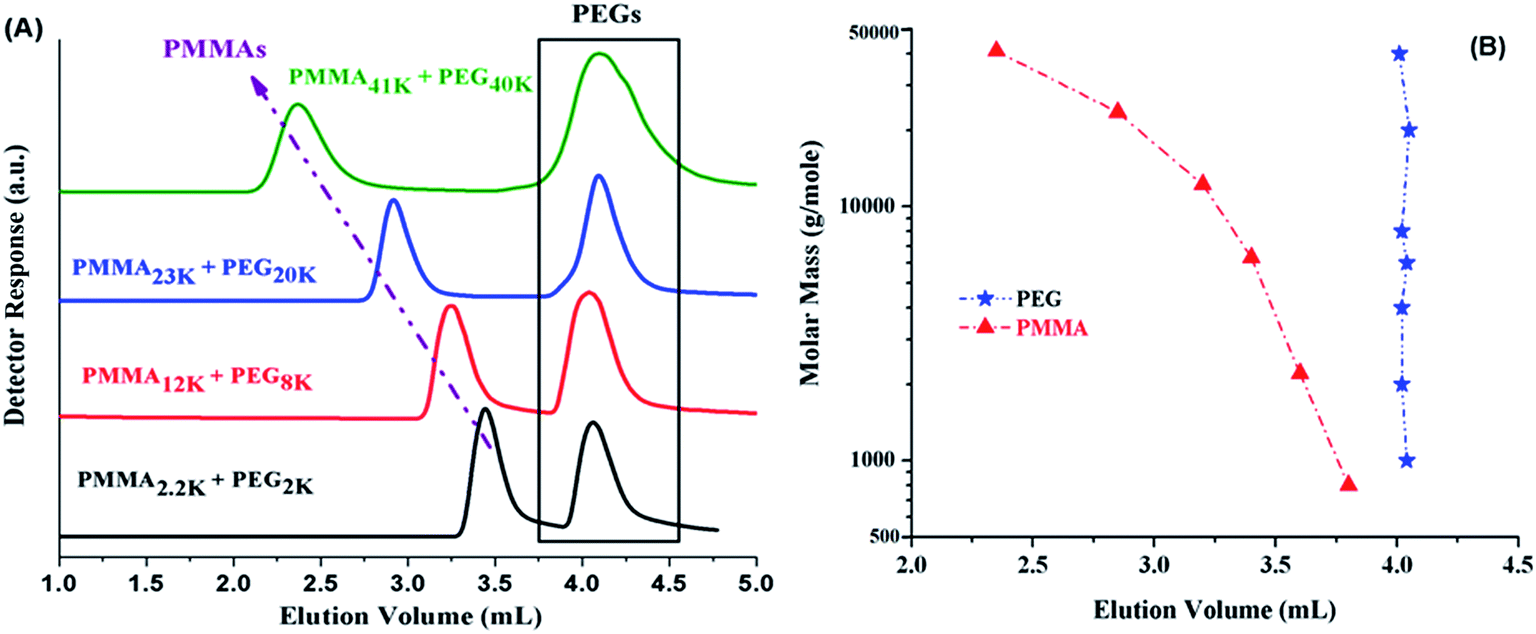
Most complex distribution of the block copolymers under study was obtained by analysis at LCCC–PMMA on a RP column (Fig. 6). The minor peak at 3.7 mL can be easily assigned to PMMA homopolymers in the sample. However, peak earlier than the elution volume of macro-initiator needs to be justified. Analysis at LCCC–PEO reveals the absence of any PEO homopolymers in the sample. In order to conclude about the nature of the early eluting peak at LCCC–PMMA, it is imperative to collect the fractions and analyze them thoroughly by chromatographic and spectroscopic techniques. The product selected for this purpose was PMMA-b-PEO20k-b-PMMA (3–1). This product was fractionated into three parts. The fraction limits are shown in Fig. 10. Mobile phase was evaporated and fractions are used for further analysis by SEC, LCCC–NP, and NMR.
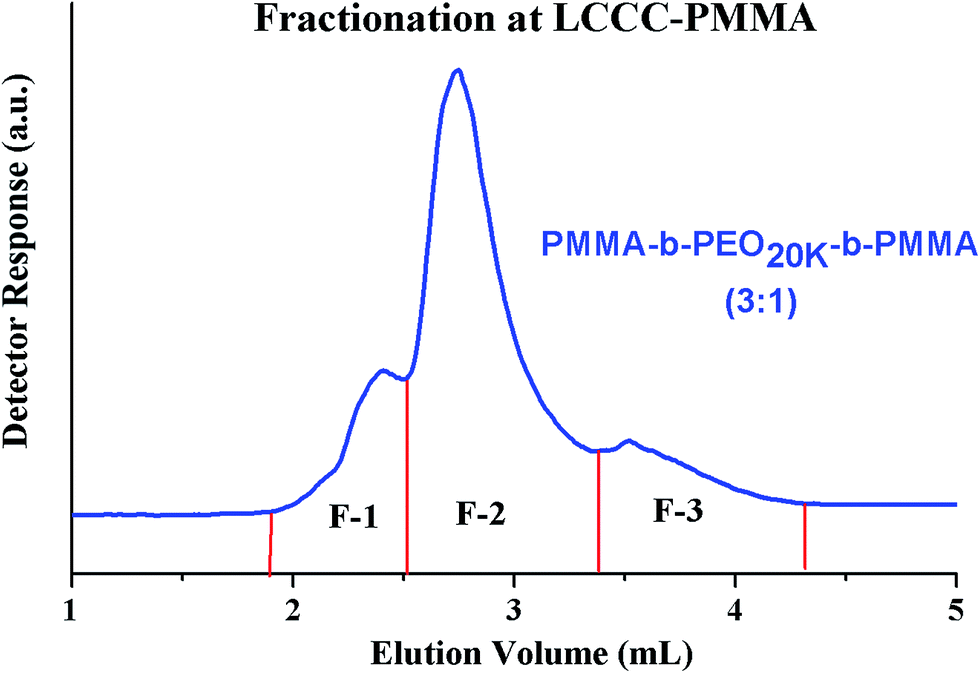 | ||
| Fig. 10 Fractionation of PMMA-b-PEO20k-b-PMMA (3:1) copolymer at the CAP of PMMA on a Jupiter C18 300 Å, 5 μm, 250 × 4.6 mm column, mobile phase: ACN/THF/TEA (93/6/1) v/v, detection: ELS. Legend depicts the initial feed ratios in mass of macro-initiator and monomer (MMA). | ||
Looking at the elution behaviour of PEO and PMMA under these conditions, it is not difficult to conclude that fraction-3 is PMMA homopolymers in the sample. Fraction-1 and fraction-2 belong to block copolymers and seems to be complex. The maximum of the early eluting shoulder corresponds to double value of molar mass of PEO macro-initiator under current chromatographic conditions.
NMR analysis of fractions revealed that the composition of the fractions F-1 and F-2 are not very different from the raw product, only slight variation is shown. Fraction-1 has somewhat more content of PEO compared to fraction-2. Furthermore, NMR analysis confirms that fraction F-3 is PMMA homopolymer. Fractions were further subjected to SEC analysis, Fig. 11A. As mentioned earlier, the raw product has bi-modal distribution with regard to total molar mass distribution (more accurately size distribution). Fraction-1 belongs to early eluting part of the peak that means it has larger size (higher total molar mass), fraction-2 has a bi-modal distribution similar to raw product. This fraction is actually a mixture because of incomplete separation in the first dimension. Fraction-3, PMMA homopolymers, belongs to higher elution volume of the peak of raw product (lower molar mass). It is expected as the content of MMA in the initial reaction mixture with macro-initiator is only 25%. Therefore, it can be concluded from SEC analysis of fractions that fraction-1 has highest total molar mass followed by fraction-2 and fraction-3.
 | ||
| Fig. 11 Analysis of fractions collected at LCCC–PMMA (RP) (A) SEC on on a set of three PLgel columns (300 × 7.5 mm, 5 μm, Mixed D) and PLgel guard column (50 × 7.5 mm) in chloroform at flow rate of 1.0 mL min−1; (B) LCCC–PEG (NP) on a PerfectSil 300 Å, 250 × 4.6 mm column (MZ analytical), detector: ELS, column temperature: 30 °C; mobile phase THF/DMF: 97.5/2.5 (v/v). Legend depicts the initial feed ratios in mass of macro-initiator and monomer (MMA). | ||
The collected fractions are also analyzed at CAP of PEO on NP column, Fig. 11B. As explained earlier, the analysis at CAP of PEO allows separation with regard to length of the PMMA segment in the block copolymer. Raw product under these conditions has a comparatively broad peak. Fraction-1 eluted a bit later than fraction-2. The earlier elution of fraction-2 from fraction-1 might be either due to shorter chain length of the PMMA or due to double length of macro-initiator. Fraction-3 has shortest chain length of PMMA compared to fraction-1 and fraction-2, this is understandable since these unwanted side reaction can initiate and terminate at any point during polymerization. Detailed analysis of the fractions collected at CAP of PMMA by NMR, SEC and LCCC–NP reveal that there is coupling of the growing chains during polymerization that resulted in double chain length of the macro-initiator and comparatively shorter PMMA segment. The coupled product is not a homopolymer as confirmed by analysis of the same product at LCCC–NP at CAP of PEO. Furthermore, the length of PMMA homopolymers is comparatively short compared to the PMMA block in the block copolymer.
Conclusion
In this study, di- and tri-block (A-B-A type) copolymer containing PMMA and PEO are synthesized and comprehensively characterized. Bromine end-group macro-initiators was required for ATRP of MMA. Quantitative transformation of lower molar mass macro-initiator (MeO–PEO) is confirmed by NMR and FTIR. Bromine terminated MeO–PEO and PEOs of varying molar masses are further used for ATRP of MMA. The chemical composition, molar mass of individual blocks, total molar mass are varied by using macro-initiators of different molar mass and different initial ratio of macro-initiator and monomer. The expected trend of chemical composition is confirmed by NMR analysis. SEC analysis revealed expected augmentation in the molar mass of the products. Individual block lengths and presence or absence of unwanted homopolymers of both types are monitored by liquid chromatography at critical conditions. LCCC at NP for PEO revealed the molar mass distribution of non-critical PMMA block. Similarly, LCCC–RP for PMMA revealed the molar mass distribution of PEO block of the block copolymers. Above-mentioned methods also gave information about presence or absence of critical homopolymers in the samples. Collection of fractions of multi-modal product at CAP of PMMA on RP column and its detailed analysis by NMR, SEC and LCCC–NP revealed more insight into the composition of the products. It has been shown that growing chains of PMMA were coupled during course of polymerization, and resulted in block copolymers with double length of the macro-initiator with comparatively shorter PMMA block. Two-dimensional analysis exposed several heterogeneities of the products that were not possible by independent or stand-alone analysis by these techniques.Conflicts of interest
Authors declare no conflict of interest.References
- S. B. Darling, Prog. Polym. Sci., 2007, 32, 1152–1204 CrossRef CAS.
- I. W. Hamley, Block Copolymers in Solution: Fundamentals and Applications, John Wiley & Sons, Ltd, Chichester, England, 2005 Search PubMed.
- N. Hadjichristidis, S. Pispas and G. Floudas, Block Copolymers: Synthetic Strategies, Physical Properties, and Applications, John Wiley & Sons, Hoboken, New Jersey, 2003 Search PubMed.
- A. D'Souza and R. Shegokar, Expert Opin. Drug Delivery, 2016, 13, 1257–1275 CrossRef PubMed.
- A. Rösler, G. W. M. Vandermeulen and H.-A. Klok, Adv. Drug Delivery Rev., 2012, 64, 270–279 CrossRef.
- M. L. Adams, A. Lavasanifar and G. S. Kwon, J. Pharm. Sci., 2003, 92, 1343–1355 CrossRef CAS PubMed.
- S. Sheiko and M. Schmidt, Adv. Polym. Sci., 2000, 151, 61–174 CrossRef CAS.
- V. H. Nguyen and J.-J. Shim, Colloid Polym. Sci., 2014, 293, 617–623 Search PubMed.
- M.-Q. Chen, A. Kishida, T. Serizawa and M. Akashi, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1811–1817 CrossRef CAS.
- D. Mu, J. Q. Li and S. Y. Feng, Phys. Chem. Chem. Phys., 2015, 17, 12492–12499 RSC.
- H. Desai, D. Varade, V. K. Aswal, P. S. Goyal and P. Bahadur, Eur. Polym. J., 2006, 42, 593–601 CrossRef CAS.
- D. J. Siegwart, W. Wu, M. Mandalaywala, M. Tamir, T. Sarbu, M. S. Silverstein, T. Kowalewski, J. O. Hollinger and K. Matyjaszewski, Polymer, 2007, 48, 7279–7290 CrossRef CAS.
- H. Kretzschmar, E.-J. Donth, H. Tanneberger, D. Garg and S. Höring, Thermochim. Acta, 1985, 93, 151–154 CrossRef CAS.
- S. Patel, R. G. Thakar, J. Wong, S. D. McLeod and S. Li, Biomaterials, 2006, 27, 2890–2897 CrossRef CAS PubMed.
- D. Mu, J. Q. Li, W. D. Li and S. Wang, J. Mol. Model., 2011, 17, 3027–3038 CrossRef CAS PubMed.
- Q. Xiao, X. Wang, W. Li, Z. Li, T. Zhang and H. Zhang, J. Membr. Sci., 2009, 334, 117–122 CrossRef CAS.
- K. Ishizu, M. Makino and S. Uchida, Macromol. Rapid Commun., 2007, 28, 882–887 CrossRef CAS.
- D. Mu, J.-Q. Li and S. Wang, J. Polym. Res., 2012, 19, 9910 CrossRef.
- P. J. Das, A. Barak, Y. Kawakami and T. Kannan, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1376–1386 CrossRef CAS.
- R. Krishnan and K. S. V. Srinivasan, Eur. Polym. J., 2003, 39, 205–210 CrossRef CAS.
- D. J. Siegwart, J. K. Oh and K. Matyjaszewski, Prog. Polym. Sci., 2012, 37, 18–37 CrossRef CAS PubMed.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- X. Sun, H. Zhang, X. Huang, X. Wang and Q.-F. Zhou, Polymer, 2005, 46, 5251–5257 CrossRef CAS.
- A. Panday, S. Mullin, E. D. Gomez, N. Wanakule, V. L. Chen, A. Hexemer, J. Pople and N. P. Balsara, Macromolecules, 2009, 42, 4632–4637 CrossRef CAS.
- G. Gaucher, M.-H. Dufresne, V. P. Sant, N. Kang, D. Maysinger and J.-C. Leroux, J. Controlled Release, 2005, 109, 169–188 CrossRef CAS PubMed.
- P. Georgopanos, T.-Y. Lo, R.-M. Ho and A. Avgeropoulos, Polym. Chem., 2017, 8, 843–850 RSC.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis, S. Pispas and A. Avgeropoulos, Prog. Polym. Sci., 2005, 30, 725–782 CrossRef CAS.
- G. Polymeropoulos, D. Moschovas, A. Kati, A. Karanastasis, S. Pelekanou, P. Christakopoulos, G. Sakellariou and A. Avgeropoulos, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 23–32 CrossRef CAS.
- W. Huang, Y. Wan, J. Chen, Q. Xu, X. Li, X. Yang, Y. Li and Y. Tu, Polym. Chem., 2014, 5, 945–954 RSC.
- P. Alexandridis and B. Lindman, Amphiphilic Block Copolymers, Self Assembly and Applications, Elsevier, Amsterdam, 2000 Search PubMed.
- V. Abetz, Adv. Polym. Sci., 2005, 189, 1–212 CrossRef.
- K. S. Pafiti, C. S. Patrickios, V. Filiz, S. Rangou, C. Abetz and V. Abetz, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 213–221 CrossRef CAS.
- M. L. Arnal, V. Balsamo, F. Lopez-Carrasquero, J. Contreras, M. Carrillo, H. Schmalz, V. Abetz, E. Laredo and A. J. Mueller, Macromolecules, 2001, 34, 7973–7982 CrossRef CAS.
- S. Dami, C. Abetz, B. Fischer, M. Radjabian, P. Georgopanos and V. Abetz, Polymer, 2017 DOI:10.1016/j.polymer.2017.05.024.
- T. Macko and D. Hunkeler, Adv. Polym. Sci., 2003, 163, 61–136 CrossRef CAS.
- A. Baumgaertel, E. Altuntaş and U. S. Schubert, J. Chromatogr. A, 2012, 1240, 1–20 CrossRef CAS PubMed.
- M. I. Malik and H. Pasch, Prog. Polym. Sci., 2014, 39, 87–123 CrossRef CAS.
- H. Pasch and B. Trathnigg, Multidimensional HPLC of Polymers, Springer, Berlin-Heidelberg-New York, 2013 Search PubMed.
- M. I. Malik, B. Trathnigg and C. O. Kappe, J. Chromatogr. A, 2009, 1216, 1167–1173 CrossRef CAS PubMed.
- M. I. Malik, B. Trathnigg and R. Saf, J. Chromatogr. A, 2009, 1216, 6627–6635 CrossRef CAS PubMed.
- M. I. Malik, G. W. Harding, M. E. Grabowsky and H. Pasch, J. Chromatogr. A, 2012, 1244, 77–87 CrossRef CAS PubMed.
- M. I. Malik, G. W. Harding and H. Pasch, Anal. Bioanal. Chem., 2012, 403, 601–611 CrossRef CAS PubMed.
- M. I. Malik, T. Mahboob and S. Ahmed, Anal. Bioanal. Chem., 2014, 406, 6311–6317 CrossRef CAS PubMed.
- R. Abdul-Karim, S. G. Musharraf and M. I. Malik, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1887–1893 CrossRef CAS.
- W. Lee, D. Y. Cho, T. Chang, K. J. Hanley and T. P. Lodge, Macromolecules, 2001, 34, 2353–2358 CrossRef CAS.
- W. Lee, S. Park and T. Chang, Anal. Chem., 2001, 73, 3884–3889 CrossRef CAS PubMed.
- S. Lee, H. Lee, L. Thieu, Y. Jeong, T. Chang, C. Fu, Y. Zhu and Y. Wang, Macromolecules, 2013, 46, 9114–9121 CrossRef CAS.
- T. Eisa and M. V. Sefton, Biomaterials, 1993, 14, 755–761 CrossRef CAS PubMed.
- H.-C. Chiu, C.-S. Chern, C.-K. Lee and H.-F. Chang, Polymer, 1998, 39, 1609–1616 CrossRef CAS.
- X. Zushun, F. Linxian, J. Jian, C. Shiyuan, C. Yongchun and Y. Changfeng, Eur. Polym. J., 1998, 34, 1499–1504 CrossRef.
- S. Guo, L. Shen and L. Feng, Polymer, 2001, 42, 1017–1022 CrossRef CAS.
- M. G. Dhara, D. Baskaran and S. Sivaram, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2132–2144 CrossRef CAS.
- X. Huang, S. Chen and J. Huang, J. Polym. Sci., Part A: Polym. Chem., 1998, 37, 825–833 CrossRef.
- A. M. Striegel, W. W. Yau, J. J. Kirkland and D. D. Bly, Modern Size-Exclusion Liquid Chromatography: Practice of Gel Permeation and Gel Filtration Chromatography, John Wiley and Sons Inc, Hoboken, New jersey, 2009 Search PubMed.
- D. Berek, J. Sep. Sci., 2010, 33, 315–335 CrossRef CAS PubMed.
- W. Radke, J. Chromatogr. A, 2014, 1335, 62–79 CrossRef CAS PubMed.
- W. Radke, S. Lee and T. Chang, J. Sep. Sci., 2010, 33, 3578–3583 CrossRef CAS PubMed.
- P. Jandera, M. Holčapek and L. Kolářová, J. Chromatogr. A, 2000, 869, 65–84 CrossRef CAS PubMed.
- E. Uliyanchenko, S. van der Wal and P. J. Schoenmakers, Polym. Chem., 2012, 3, 2313–2335 RSC.
- X. Jiang, A. van der Horst and P. J. Schoenmakers, J. Chromatogr. A, 2002, 982, 55–68 CrossRef CAS PubMed.
- I. Park, S. Park, D. Cho, T. Chang, E. Kim, K. Lee and Y. J. Kim, Macromolecules, 2003, 36, 8539–8543 CrossRef CAS.
- M. Adler, F. Rittig, S. Becker and H. Pasch, Macromol. Chem. Phys., 2005, 206, 2269–2277 CrossRef CAS.
- M. I. Malik, B. Trathnigg and C. O. Kappe, Eur. Polym. J., 2008, 44, 144–154 CrossRef CAS.
- M. Irfan, J. Oh, S. G. Musharraf, M. R. Shah, S. Ahmed and M. I. Malik, RSC Adv., 2016, 6, 98117–98127 RSC.
- M. I. Malik, M. Irfan, A. Khan, S. Rahim, R. Abdul-Karim and J. Hashim, J. Polym. Res., 2016, 23, 258 CrossRef.
- W. Jiang, S. Khan and Y. Wang, Macromolecules, 2005, 38, 7514–7520 CrossRef CAS.
- C. M. Guttman, E. A. Di Marzio and J. F. Douglas, Macromolecules, 1996, 29, 5723–5733 CrossRef CAS.
- B. Trathnigg, Polymer, 2005, 46, 9211–9223 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |