
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Protecting group-free use of alcohols as carbon electrophiles in atom efficient aluminium triflate-catalysed dehydrative nucleophilic displacement reactions†
Adam Cullena,
Alfred J. Mullera and
D. Bradley G. Williams *ab
*ab
aResearch Centre for Synthesis and Catalysis, University of Johannesburg, P.O. Box 564, Auckland Park, 2006, South Africa
bSchool of Mathematical and Physical Sciences, University of Technology Sydney, P.O. Box 123, Broadway, Sydney, NSW 2007, Australia. E-mail: Bradley.williams@uts.edu.au
First published on 30th August 2017
Abstract
Benzylic and allylic alcohols are rendered electrophilic without chemical modification by the use of aluminium triflate as catalyst. The reaction succeeds with alcohol, thiol, carbon and nitrogen nucleophiles. When phenols are employed as nucleophiles, C-alkylation ensues. An advanced application of the method is demonstrated in the synthesis of 2H-chromenes and their N and S analogues.
Introduction
Many of the problems relating to regio- or chemoselective synthesis have not yet been solved.1 As such, organic synthesis usually requires protecting group strategies to deliver selectivity. In multistep syntheses protection–deprotection strategies are not only critical to the success of the synthesis, they often comprise a significant proportion of the steps involved.1 Because of these deficiencies, atom economic chemistry remains a touchstone by which the efficiency of synthetic organic chemistry is measured. Alcohols have been used for decades as a source of electrophiles once they have been converted into halides or other leaving groups such as tosylates or triflates.2 Alternatively, it is known that propargylic and benzyl alcohols can form stabilised carbocations under appropriate conditions, usually in the presence of aluminium- and gallium triflates, respectively.3 Aluminium triflate has been shown to be a highly active yet selective catalyst for a range of reactions, including some highly atom economic processes where 100% of the atoms are incorporated into the products.4–7 Aluminium triflate is believed to exert its function in many cases as a Lewis acid-assisted Brønsted acid.8–10 It is also useful as a co-catalyst in the Pd-catalysed hydromethoxycarbonylation reaction, where it takes the place of traditional Brønsted acids.6,7 Importantly, it tolerates protic conditions and so can be readily recycled by simply extracting it into water and recovered by subsequent removal of the water.11When investigating epoxide ring-opening reactions,12,13 we became interested in the possibility of employing chiral alcohols as nucleophiles by which to potentially desymmetrise the epoxides. During that study, we employed (S)-1-phenylethanol (1) as the nucleophile and obtained the S,R (meso) and S,S diastereomers 2 of di(1-phenylethyl) ether as the major products.13 The scrambling of the stereogenic centre implied that a benzylic carbocation had formed in the process, under the action of Al(OTf)3, and could be captured by a nucleophile present in the reaction mixture (Scheme 1). This manuscript details the direct use of benzylic and allylic alcohols as electrophiles in high yielding C–O, C–S, C–N and C–C bond-forming reactions, where water is the by-product, in intramolecular and intermolecular reactions.
 | ||
| Scheme 1 Conversion of (S)-1-phenethyl alcohol into di(1-phenylethyl) ether. | ||
Results and discussion
Benzhydrol 3 was subjected to reaction with a range of nucleophiles in the presence of Al(OTf)3 in nitroethane solvent at 70 °C, affording the corresponding elaborated products in very high yields (Table 1) [nitroethane demonstrates low toxicity to a range of organisms, does not bioaccumulate and readily biodegrades14]. Notably, the reaction is successful with O, C, N and S nucleophiles, requires no manipulation of the heteroatoms and it can be performed solvent-free without loss of yield or selectivity (Table 1, entries 7 and 11). Furthermore, reactions involving phenols yield C-alkylated aromatics as products (Table 1, entries 8 and 9). It is possible that the reaction initially yields the analogous O-alkylated product which converts into the C-alkylated thermodynamic product. To test this hypothesis, analogues 5 (Scheme 2) of the putative benzyl ether intermediates were subjected to reaction with Al(OTf)3 under identical reaction conditions, affording the C-alkylated products in quantitative (>98%) yield. This result supports the notion that a kinetically produced intermediate converts into the thermodynamically preferred C-alkylated product. Interestingly, the ortho product was the exclusive product for both substrates (Scheme 2) in this reaction. While this is expected for the p-cresol-derived ether 5b, it was somewhat unexpected for the phenol-derived substrate 5a. In contrast, the product of the reaction of phenol and benzhydrol is the para product 4h (Table 1, entry 8). The difference in outcome may be accounted for by the relative stability of the diphenylmethyl carbocation that forms (compared to benzyl cation), allowing it to migrate to the less hindered para position of the phenol ring. This is in contrast to O-diphenylmethyl trichloroacetimidate which affords O-alkylated aromatics from 2-naphthol and several phenols.15 PdCl2 (10 mol%) as catalyst in dichloroethane also affords O-alkylated aromatics from benzhydrol.16 Usefully, ribose derivative 4k can be selectively deprotected of the diphenylmethane group in high yield (90%) by a simple reaction involving Al(OTf)3 and 2 equivalents of EtOH. This compares favourably to PdCl2-catalysed (10 mol%) deprotection of monosaccharides with EtOH (10 equivalents) in dichloroethane.
|
|||
|---|---|---|---|
| Entry | Nucleophile | Product | Yield |
| a Solvent free, 40 °C, performed under vacuum (5 mm Hg). | |||
| 1 | Ethanol | 4a | 98% |
| 2 | Allyl alcohol | 4b | 94% |
| 3 | Propargyl alcohol | 4c | >98% |
| 4 | Acetyl acetone | 4d | 96% |
| 5 | Ethyl acetoacetate | 4e | 87% |
| 6 | Thiophenol | 4f | 98% |
| 7a | L-Menthol | 4g | 85% |
| 8 | Phenol | 4h | 84% |
| 9 | 2-Naphthol | 4i | 98% |
| 10 | Indole | 4j | 80% |
| 11a | Methyl 2,3-isopropyl-idene-β-D-riboside | 4k | 68% |

 | ||
| Scheme 2 Conversion of benzyl ethers into C-alkylated products. | ||
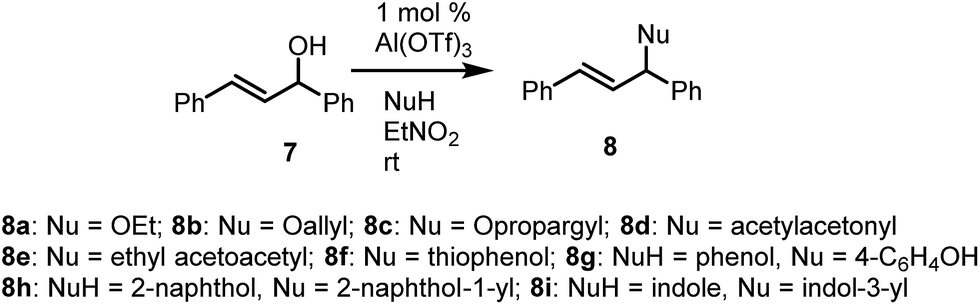
trans-1,3-Diphenylprop-2-en-1-ol (7) was subjected to a similar set of reactions (Table 2). Once again, high yields of products were obtained and the reaction also tolerated a range of nucleophiles, including O, C, N and S nucleophiles (Table 2). As was the case with benzhydrol, phenols and indole underwent C-alkylation while thiophenol underwent S-alkylation in otherwise smooth transformations. The reactions were performed at ambient temperature, affording very high yields of products across the range of nucleophiles employed.
|
|||
|---|---|---|---|
| Entry | Nucleophile | Product | Yield |
| 1 | Ethanol | 8a | 85% |
| 2 | Allyl alcohol | 8b | 84% |
| 3 | Propargyl alcohol | 8c | 96% |
| 4 | Acetyl acetone | 8d | 98% |
| 5 | Ethyl acetoacetate | 8e | 98% |
| 6 | Thiophenol | 8f | 86% |
| 7 | Phenol | 8g | 82% |
| 8 | 2-Naphthol | 8h | 81% |
| 9 | Indole | 8i | 87% |
Reactions with benzhydrol or trans-1,3-diphenylprop-2-en-1-ol did not proceed with unprotected anilines, acetanilide or their p-tosylamide analogues. This is consistent with other work17 with electron-deficient amines, where higher catalyst loadings (1–5 mol%) and (often) microwave heating were required before successful substitution reactions were observed. However, successful N-alkylation could be achieved in the present instance when using p-toluenesulphonyl amide (Scheme 3), with which the C–N bond formation proceeded quite readily to produce the N-alkylated products in quantitative (>98%) yields. Nitroethane remained the solvent of choice.
 | ||
| Scheme 3 Reactions of activated alcohols with p-toluenesulphonyl amide. | ||
We next turned our attention to the synthesis of chromenes via intramolecular cyclisation reactions. The chromene sub-structure is present in a wide range of physiologically active molecules which demonstrate anti-HIV,18 antitumour,19 antibacterial20 and antiprotozoal21 activity. Significant efforts have been made towards the synthesis of the 2H-chromene motif,22 including sugar-derived chiral systems.23 In addition, there has been continuing interest in heteroatom analogues of chromenes such as dihydroquinolines24 and thiochromenes,25 which are also associated with biological activity. To set up the intramolecular cyclisation that would build the chromene motif in our case, it was necessary to synthesise phenylpropenols 10, which was accomplished in four steps in good yields (see ESI for details†). Propenols 10 were subjected to the action of 1 mole percent Al(OTf)3 in nitroethane at ambient temperature in an effort to produce the corresponding 2H-chromenes (Scheme 4). Upon addition of the Al(OTf)3 to the solution of the propenols in nitroethane a deep purple colour ensued immediately, presumably the consequence of carbocation formation, and the starting alcohol was fully consumed (by tlc analysis) within a few minutes. Al(OTf)3 is known to produce carbocations in solution from retinyl acetate, for example, which has been used to probe the likely mechanism by which Al(OTf)3 functions.8 Unfortunately, the propenol reaction produced an intractable mixture of products. To tame the reactivity of the system, the reaction was performed in dichloromethane, which allowed the desired 2H-chromenes to be prepared in acceptable to good yields in a dehydrative endo cyclisation step (Scheme 4). 2H-Chromene 11a could also be prepared from 12 (Scheme 5; see ESI† for details on the synthesis of 12), a regioisomer of 10a, under the same conditions in comparable yield for the endo-cyclisation step of its regioisomer of 10a. This anticipated ability of the reaction to proceed as desired regardless of the position of the leaving OH group imparts significant flexibility in the synthesis protocols that might lead to the functionalised diphenylpropenol substrates. In a previous study, this type of intramolecular cyclisation reaction of phenylpropenols in the presence of 5 mole percent of a (t-Bu)2P(2-C6H4-Ph)-AuCl/AgOTf catalyst at elevated temperatures has been noted.26 The Au catalyst tolerates some functional groups but fails outright in some instances.
 | ||
| Scheme 4 Synthesis of 2H-chromenes in Al(OTf)3-catalysed cyclisation reactions. | ||
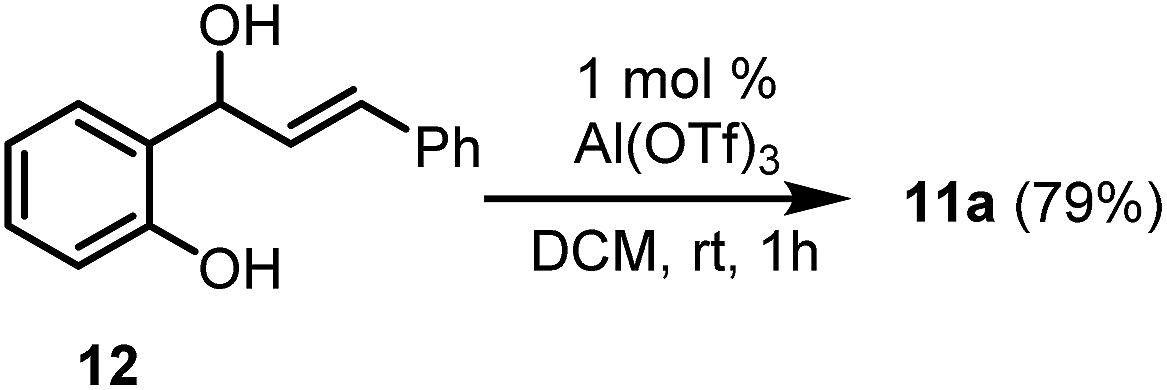 | ||
| Scheme 5 Synthesis of 2H-chromene from an isomeric substrate. | ||
Finally, propenol 13 could also be made to undergo Al(OTf)3-catalysed dehydrative cyclisation, to afford the desired dihydroquinoline 14 in a pleasing yield of 94% (Scheme 6), using only 0.05 mol% of the catalyst. While the use of DCM is less desirable, the overall high yields on the one hand, and the fact that this method enables alcohols to be used directly in nucleophilic displacement reactions, cannot be overlooked. Further investigations will most likely unveil other benign solvents which are compatible with this chemistry.
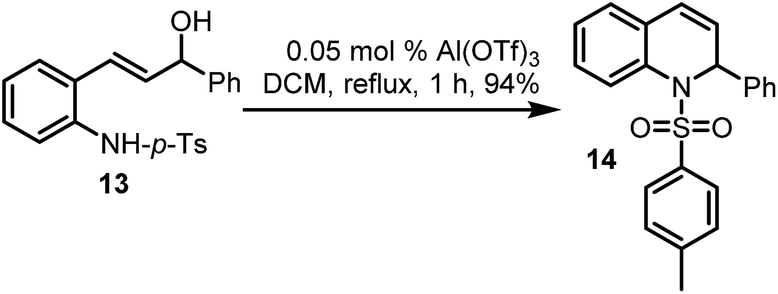 | ||
| Scheme 6 Synthesis of dihydroquinoline in an Al(OTf)3-catalysed dehydrative cyclisation. | ||
Conclusions
Al(OTf)3 catalyses the direct nucleophilic displacement of OH groups in ‘activated’ systems with various O, N, S and C nucleophiles. This efficient process allows the ready synthesis of a wide range of product types in very high yields with water as the by-product. Furthermore, the displacement reaction is usefully employed in the synthesis of 2H-chromenes and dihydroquinolines in endo cyclisation reactions. This facile and high yielding dehydrative procedure is expected to be useful in the synthesis of a wide range of organic materials and biologically active molecules. We are currently investigating the applications of this innovation to other ring structures.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge Sasol Ltd (South Africa), NRF, THRIP, the University of Johannesburg and University of Technology Sydney for generous funding.Notes and references
- K. C. Nicolaou and J. S. Chen, Classics in total synthesis III, Wiley-VCH, Weinheim, 2011 Search PubMed.
- R. C. Larock, Comprehensive organic transformations, Wiley-VCH, Weinheim, 2010 Search PubMed.
- (a) M. Gohain, C. Marais and B. C. B. Bezuidenhoudt, Tetrahedron Lett., 2012, 53, 4704–4707 CrossRef CAS; (b) X. Han and J. Wu, Org. Lett., 2010, 12, 5780–5782 CrossRef CAS PubMed.
- D. B. G. Williams and M. L. Lawton, Green Chem., 2008, 10, 914–917 RSC.
- D. B. G. Williams, A. Cullen, A. Fourie, H. Henning, M. Lawton, W. Mommsen, P. Nangu, J. Parker and A. Renison, Green Chem., 2010, 12, 1919–1921 RSC.
- D. B. G. Williams and T. Bredenkamp, ChemCatChem, 2012, 4, 206–208 CrossRef CAS.
- B. M. Makume, T. Bredenkamp and D. B. G. Williams, ChemCatChem, 2014, 6, 2801–2804 CrossRef CAS.
- D. B. G. Williams and M. Lawton, J. Mol. Catal. A: Chem., 2010, 317, 68–71 CrossRef CAS.
- D. B. G. Williams, M. S. Sibiya and P. S. van Heerden, Fuel Process. Technol., 2012, 94, 74–79 CrossRef.
- D. B. G. Williams, S. B. Simelane, M. Lawton and H. H. Kinfe, Tetrahedron, 2010, 66, 4573–4576 CrossRef CAS.
- D. B. G. Williams, M. Shaw and T. Hughes, Organometallics, 2011, 30, 4968–4973 CrossRef CAS.
- D. B. G. Williams and M. Lawton, Tetrahedron Lett., 2006, 47, 6557–6560 CrossRef CAS.
- A. Cullen, Aluminium triflate mediated organic synthesis, Ph.D thesis, University of Johannesburg, 2010.
- https://toxnet.nlm.nih.gov/cgi-bin/sis/search/a?dbs+hsdb:@term+@DOCNO+105, accessed 16 March 2017.
- K. T. Howard, B. C. Duffy, M. R. Linaburg and J. D. Chisholm, Org. Biomol. Chem., 2016, 14, 1623–1628 CAS.
- Y. Bikard, R. Mezaache, J.-M. Weibel, A. Benkouider, C. Sirlin and P. Pale, Tetrahedron, 2008, 64, 10224–10232 CrossRef CAS.
- T. Ohshima, J. Ipposhi, Y. Nakahara, R. Shibuya and K. Mashima, Adv. Synth. Catal., 2012, 354, 2447–2452 CrossRef CAS.
- P. Cos, L. Maes, D. VandenBerghe, N. Hermans, L. Pieters and A. Vlietinck, J. Nat. Prod., 2004, 67, 284–293 CrossRef CAS PubMed.
- A. Mohammad, K. Mehdi, R. Ali, E. Saeed, Z. Abdolhossein, F. Omidreza, M. Ramin and S. Abbas, Eur. J. Med. Chem., 2013, 59, 15–22 CrossRef PubMed.
- M. S. Nermien, M. M. Hany, E. A. H. K. Essam Shawky, S. M. Shymaa and M. E. A. Ahmed, Eur. J. Med. Chem., 2011, 46, 765–772 CrossRef PubMed.
- D. Harel, S. A. Khalid, M. Kaiser, R. Brun, B. Wünsch and T. J. Schmidt, J. Ethnopharmacol., 2011, 137, 620–625 CrossRef CAS PubMed.
- Comprehensive heterocyclic chemistry, ed. A. R.Katritzky, C. W. Rees, A. J. Boulton and A. Mckillop, Pergamon Press, Oxford, 1984, vol. 3 Search PubMed.
- S. B. Simelane, H. H. Kinfe, A. Muller and D. B. G. Williams, Org. Lett., 2014, 16, 4543–4545 CrossRef CAS PubMed.
- Z. Wang, S. Li, B. Yu, H. Wu, Y. Wang and X. Sun, J. Org. Chem., 2012, 77, 8615–8620 CrossRef CAS PubMed.
- X.-S. Wang, J.-R. Wu, J. Zhou and M.-M. Zhang, J. Heterocycl. Chem., 2011, 48, 1056–1060 CrossRef CAS.
- A. Aponick, B. Biannic and M. R. Jong, Chem. Commun., 2010, 46, 6849–6851 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and analytical data; NMR spectra. See DOI: 10.1039/c7ra08784e |
| This journal is © The Royal Society of Chemistry 2017 |