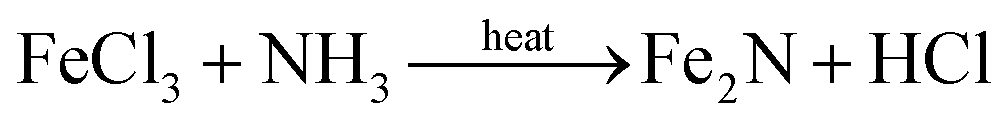DOI:
10.1039/C7RA08704G
(Paper)
RSC Adv., 2017,
7, 44619-44625
Nanocasting synthesis of an iron nitride-ordered mesopore carbon composite as a novel electrode material for supercapacitors†
Received
7th August 2017
, Accepted 1st September 2017
First published on 15th September 2017
Abstract
Herein, a series of novel iron nitride (Fe2N)/cubic-ordered mesoporous carbon (OMC) composites (Fe2N@OMC) were synthesized via a facile nanocasting route and ammonia calcination. Physical characterization results indicate that small non-aggregated Fe2N nanoparticles were evenly distributed on the external surface and in the pores of the carbon material with a linear array of mesopores in a regular interval arrangement. An electrochemical test was carried out using Fe2N@OMC as a negative electrode of a supercapacitor in 6 mol L−1 KOH aqueous electrolytes. The results indicated that the obtained Fe2N@OMC-2 exhibited a superior specific capacitance of 547 F g−1 at 1 mV s−1 and 520 F g−1 at 0.5 A g−1, an excellent rate capability (398 F g−1 at 20 A g−1 with capacitance retention of 76%), and an outstanding cycling stability. After 1000 cycles, the specific capacitance retention of Fe2N@OMC-2 remained 85%, which was much higher than 28% of the bare Fe2N.
1. Introduction
In recent years, as there is an increasing requirement for the storage and utilization of a mass of energy, electrochemical devices have been developing rapidly. Among them, supercapacitors have attracted significant attention due to their high energy density, great rate capacity, and cycling stability.1–3 Thus, many researchers are engaged in the investigation and research of electrode materials for supercapacitors. Transition metal oxides (Fe2O3, Co3O4, and MnO2), bimetallic oxides, and hydroxides are always used as electrode materials because of high capacitance of transition metal compounds.4–8 For instance, Yu et al. presented the design and development of novel Ag/PEDOT:PSS-nanopillar/MnO2 layers by a layer structure for high-performance flexible supercapacitors. This electrode provided excellent specific capacitances of 862 F g−1 (based on MnO2) at a current density of 2.5 A g−1.9 Thomas et al. have reported an all-solid-state, energy harvesting and storing ribbon that integrates a solar cell and a supercapacitor. In this device, MnO2 deposited on Cu(OH)2 nanotube arrays was used as an electrode, and the capacitance of the final electrodes was increased to 145.15 mF cm−2.10 However, the high electrical resistivity of transition metal compounds affects the power density; this results in a significant reduction in capacitance under operation at high-intensity current, which is not expected for electrode materials used in energy utilization and storage systems.11–14 Previous reports have proved that transition metal nitrides, such as chromium nitride, cobalt nitride, vanadium nitride, and iron nitride, used as electrode materials for supercapacitors have more advantages than transition metal oxides due to their superior electrical conductivity (4000–55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 S cm−1) and outstanding pseudocapacitive behaviour.15–18 For example, Lu et al. demonstrated a novel core–shell structure in which TiN nanowires were covered with ultrathin amorphous carbon protective layers. This TiN solid-state supercapacitor demonstrated a remarkable 91.3% retention in aqueous electrolytes after 15000 cycles without any oxidation.19 Yu et al. synthesized tungsten oxynitride (WON) nanowires on carbon cloths, and the WON asymmetric supercapacitor anode showed a high volumetric capacitance of 4.95 F cm−3 at 12.5 mA cm−3.20 Metal nitrides are considered to be the most promising representatives of new generation of electrode materials for electrochemical storage device such as lithium-ion batteries (LIBs), fuel cells, sodium-ion batteries, and supercapacitors.21–24 The unique nature of M (metal)–N (nitride) bonding can provide simultaneous multiple model contributions from metallic, covalent, and ionic bonding.25 Moreover, transition metal nitrides may be able to provide similar properties to the noble metals due to their similar electronic structures.26
500 S cm−1) and outstanding pseudocapacitive behaviour.15–18 For example, Lu et al. demonstrated a novel core–shell structure in which TiN nanowires were covered with ultrathin amorphous carbon protective layers. This TiN solid-state supercapacitor demonstrated a remarkable 91.3% retention in aqueous electrolytes after 15000 cycles without any oxidation.19 Yu et al. synthesized tungsten oxynitride (WON) nanowires on carbon cloths, and the WON asymmetric supercapacitor anode showed a high volumetric capacitance of 4.95 F cm−3 at 12.5 mA cm−3.20 Metal nitrides are considered to be the most promising representatives of new generation of electrode materials for electrochemical storage device such as lithium-ion batteries (LIBs), fuel cells, sodium-ion batteries, and supercapacitors.21–24 The unique nature of M (metal)–N (nitride) bonding can provide simultaneous multiple model contributions from metallic, covalent, and ionic bonding.25 Moreover, transition metal nitrides may be able to provide similar properties to the noble metals due to their similar electronic structures.26
Among many kinds of metal nitrides, Fe2N is recognized as the best choice as a pseudocapacitive electrode material due to its outstanding electrical conductivity, low cost, low resistance, and environmental friendliness.27,28 However, in the synthesis of transition metal nitrides, products always form a large number of bulks of transition metal nitrides; this leads to low utilization rate of their high capacitances in a strong current system. To overcome this problem and enhance the electrochemical performance, carbon material and transition metal nitride composites have been synthesized by researchers. The carbon materials such as ordered mesoporous carbons (OMC), activated carbons, carbon nanofibers, and carbon nanotubes29–32 with a high specific surface area can increase the electric conductivity of composite materials and provide a carbon skeleton to support the uniform deposition of metal nitride particles, thus increasing the utilization efficiency of transition metal nitrides.33–36
There are only a few investigations and research on the use of iron nitride as an electrode material for supercapacitors. Zhu et al. reported that Fe2N/graphene was synthesized via deposition of ZnO on a graphene nanosheet surface. Then, ZnO and Fe3+ were subjected to ion-exchange and finally annealed under an atmosphere of ammonia. The synthetic composite tested as an electrode in an electrolyte of 1 M LiCl showed a specific capacitance of 170 F g−1 at a scan rate of 10 mV s−1 and 120 F g−1 at 100 mV s−1 in a three-electrode system.37 A Fe2N/activated carbon composite was fabricated via hydrothermal and ammonia treatments by Agata Śliwaka et al. The specific capacitance of this composite used as an electrode active material for supercapacitors operating in 6 M KOH is 498 and 371 F g−1 at the scan rates of 10 and 100 mV s−1, respectively.38 In this study, we first report the synthesis of a series of novel Fe2N@ordered cubic mesoporous carbon composites (Fe2N@OMC), and the resultant best composite as an active material of the electrode for supercapacitors operating in 6 M KOH shows a superior capacitance values of 506 F g−1 at the scan rates of 10 mV s−1 and 435 F g−1 at the scan rates of 100 mV s−1.
2. Experimental
2.1 Materials
Triblock copolymer Pluronic P123 (MW = 5800, PEG–PPG–PEG) was purchased from Macklin Biochemical Co. Ltd (Shanghai, China). Iron(III) chloride hexahydrate (FeCl3·6H2O), HCl water solution (AR, 36.0–38.0%), and other chemicals were supplied by Kemiou Chemical Reagent Co. Ltd (Tianjin, China).
2.2 Synthesis of the KIT-6 template and OMC
The cubic three-dimensional (3D) ordered mesoporous silica template KIT-6 was fabricated using the synthesis procedures reported in the literature,29 and the 3D-ordered mesoporous carbon (OMC) materials were prepared using sucrose as a carbon source and KIT-6-ordered mesoporous silica as a hard template. As shown in Fig. 1, 1.00 g of sucrose precursor and 1.00 g of KIT-6 were added to 20 mL DI water and stirred to obtain a homogeneous mixture. The sucrose precursor was made to continuously flow into a mesoporous silica template by vacuum impregnation at room temperature. The obtained mixture was dried at 80 °C, and then, the dried solid was ground to form a fine powder. This powder was then calcined at 800 °C under an inert atmosphere at a temperature ramp rate of 2 °C min−1 and kept for 3 h. The calcined product was then removed out of the silica template using 10 wt% HF solution, and pure OMC was obtained.
 |
| | Fig. 1 Schematic for the nanocasting synthesis of OMC and Fe2N@OMC composites. | |
2.3 Synthesis of Fe2N, Fe2N@OMC-1, Fe2N@OMC-2, and Fe2N@OMC-3
Fe2N was prepared by a straightforward route. First, 1.00 g FeCl3·6H2O was dried at 80 °C in a vacuum oven, and then anhydrous FeCl3 was ground to form a fine powder. Subsequently, the powder sample was tiled into a thin layer at the bottom of a quartz boat and calcined at 800 °C for 2 h at a heating rate of 5°C min−1 under the flow of ammonia at a rate of 100 mL min−1. The calcined product was Fe2N. Fe2N@OMC-1, Fe2N@OMC-2, and Fe2N@OMC-3 were prepared using a vacuum nanocasting method, as shown in Fig. 1. OMC weighing 0.10 g was first dispersed in 20 mL of DI water under sonication for 2 h, and different amounts (0.25 g, 0.50 g, and 1.00 g) of FeCl3·6H2O were added to 20 mL DI water under stirring for 8 h to obtain a homogeneous mixture. The subsequent steps (drying, grinding, and calcination) are the same as those in the procedure for preparing Fe2N. The obtained products were Fe2N@OMC-1, Fe2N@OMC-2, and Fe2N@OMC-3.
2.4 Material characterization
The sample materials were characterized by X-ray diffraction (XRD, D8 FOCUS, Bruker, Germany) with Cu Kα 1.5406 Å radiation. The microstructure of the sample materials was observed using a transmission electron microscope (TEM, H-7650, Hitachi, Japan) at a voltage of 100 kV, a high-resolution transmission electron microscope (HRTEM, Tecnai G2 F20, FEI, USA) at a voltage of 200 kV, and a scanning electron microscope (SEM, S-3400, Hitachi, Japan). Thermogravimetric analysis (TGA) was performed using a Pyris1TGA (PerkinElmer Instruments) under an air atmosphere at a heating rate of 5 °C min−1. Nitrogen sorption analysis was conducted at 77 K using a physical adsorption instrument (AUTOSORB-1, Quantachrome, USA). The specific surface area was determined by the Brunauer–Emmett–Teller (BET) method, and the pore size distribution calculation was derived from the adsorption branches of the isotherms by the Barrett–Joyner–Halenda (BJH) theory. X-ray photoelectron spectroscopy (XPS) was performed using a spectrometer (ESCALAB 250Xi, Thermo Fisher, USA) with Al Kα radiation (1486.6 eV). The shifts in energy (charging) of the XPS spectra were calibrated using the C 1s peak at 284.6 eV as a reference peak.
2.5 Electrochemical characterization
The electrochemical measurements were carried out using a standard three-electrode cell, where Pt foil and Hg/HgO electrode were used as the counter and reference electrodes, respectively, in a 6 M KOH aqueous solution. The working electrodes were prepared as follows:
The clean nickel foams (1 × 10 cm2) were prepared in advance. Then, 1 × 1 cm2 nickel foams were coated by mixing 80 wt% active material (OMC, Fe2N, Fe2N@OMC-1, Fe2N@OMC-2, or Fe2N@OMC-3) with 10 wt% polytetrafluoroethylene (PTFE) as a binding agent and 10 wt% acetylene black conductor. The mass of the active materials on each electrode was approximately 4.00 mg. Cyclic voltammetry (CV) and galvanostatic charge/discharge measurements were carried out by an electrochemical work station (CHI660E). The electrochemical impedance spectroscopy (EIS) measurement was performed using an electrochemical work station (PAR VersaSTAT 4) at an open circuit potential from 100 kHz to 0.01 Hz with an alternating current amplitude of 5 mV. The specific capacitance (C, F g−1) of the electrode materials can be calculated from the galvanostatic discharged curves and the cyclic voltammograms according to the following equations:
| |
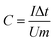 | (1) |
| |
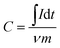 | (2) |
where
I is the current (A),
U is the operating cell voltage (V),
t is the time (s),
ν is the scan rate (V s
−1), and
m is the mass of the active material in the electrode (g).
3. Results and discussion
X-ray diffraction (XRD) analysis confirmed the presence of Fe2N in all the samples except OMC, as shown in Fig. 2. The peaks at 2θ = 37.4 (021), 40.9 (200), 43.0 (121), 56.9 (221), 68.0 (023), and 76.0° (321) are consistent with the orthorhombic Fe2N phase (JC-PDS #50-0958). No iron oxide phase was observed. These results support the complete conversion of FeCl3 into Fe2N by NH3 annealing, as shown in eqn (3). The two broad peaks at approximately 23.8 and 43.6° in the XRD pattern of OMC arise from the (002) and (100) planes of graphite-type carbon, respectively.| |
 | (3) |
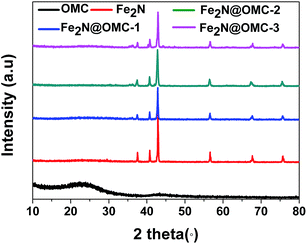 |
| | Fig. 2 The XRD patterns of OMC, Fe2N, Fe2N@OMC-1, Fe2N@OMC-2, and Fe2N@OMC-3. | |
Transmission electron microscopy (TEM) was performed to further observe the microstructure and the distribution of Fe2N particles for the synthesized products. The TEM image of OMC shows a highly ordered mesoporous structure with a linear array of mesopores in a regular interval arrangement (Fig. 3a).39 The TEM image of Fe2N@-OMC-2 indicates a homogeneous distribution of small and non-agglomerative Fe2N nanoparticles (with a diameter of 10–30 nm) on the surface of OMC, as shown in Fig. 3c. By contrast, the TEM image of Fe2N@OMC-1 shows that the well-ordered structure is still retained and the presence of Fe2N particles almost cannot be observed in Fig. 3b. On the other hand, the image of Fe2N@OMC-3 revealed an uneven distribution of big Fe2N particles on the surface of OMC, and the ordered structure was severely damaged, as shown in Fig. 3d. The high-resolution TEM (HRTEM) image (Fig. 3e) exhibits well-resolved lattice fringes, again confirming the high-crystalline nature of the Fe2N nanoparticles for Fe2N@OMC-2. The lattice fringe spacings are measured to be 0.21 and 0.44 nm, which are assigned to the d-spacing of (102) and (200) planes of the orthorhombic Fe2N phase (JC-PDS #50-0958), respectively, and the result is consistent with that of the analysis of XRD.40 X-ray energy-dispersive spectrometer (EDS) elemental mapping was performed to further confirm the element distribution. The SEM image (Fig. S1a ESI†) of Fe2N@OMC-2 and its EDS elemental mapping data (Fig. S1c and d ESI†) show that the particles are mainly C (red), Fe (purple), and N (green), which are uniformly distributed in Fe2N@OMC-2.
 |
| | Fig. 3 TEM images of OMC (a), Fe2N@OMC-1 (b), Fe2N@OMC-2 (c), and Fe2N@OMC-3 (d). HRTEM image of Fe2N@OMC-2 (e). | |
The content of Fe2N calculated from thermogravimetric analysis (Fig. S2 and Table S1, ESI†) was 28.1, 40.3, and 57.1 wt% in Fe2N@OMC-1, Fe2N@OMC-2, and Fe2N@OMC-3, respectively. For further characterization of the porous structure of the prepared composites, the N2 adsorption–desorption isotherm (Fig. 4a) and pore size distribution curves (Fig. 4b) were obtained by nitrogen adsorption–desorption analysis. The samples OMC, Fe2N@OMC-1, and Fe2N@OMC-2 exhibit type IV curves with an H1 hysteresis loop, which indicates the mesoporous nature of the synthesized composite materials due to the presence of OMC. In comparison, the H1 hysteresis loop cannot be observed in the N2 adsorption–desorption isotherm of Fe2N and Fe2N@OMC-3 (Fig. 4a). The textural properties and parameters, such as the BET surface area (SBET), the total pore volume (VT), and the average pore diameter (DBJH), are estimated, and the data value are summarized in Table 1. The BET surface area, total pore volume, and average pore diameter of initial OMC are 1609 m2 g−1, 2.13 cm3 g−1, and 5.29 nm, respectively. The porosity development in the composite materials is gradually reduced with the increasing deposition of Fe2N on the carbon surface. Particularly, when the content of Fe2N increases to 57 wt% of Fe2N@OMC-3, the BET surface area significantly decreases from 1609 to 583 m2 g−1 from OMC to Fe2N@OMC-3 due to the blockage of the pore system by the big Fe2N particles deposited on the external carbon surface, which can be detected in the TEM image in Fig. 3d. This finding is also supported by a significant decrease in the total pore volume from 2.13 cm3 g−1 for OMC to 0.44 cm3 g−1 for Fe2N@OMC-3. For Fe2N@OMC-1, the porosity of OMC is preserved to a greater extent (SBET: 1232 m2 g−1, VT: 1.55 cm3 g−1), but the content of Fe2N is only 28 wt% of Fe2N@OMC-1. The BET surface area of Fe2N@OMC-2 is much higher than that of Fe2N@OMC-3 (919 vs. 583 m2 g−1). The total pore volume of Fe2N@OMC-2 is 0.99 cm3 g−1, which is only two-fold lower than that of OMC, 2.13 cm3 g−1. Moreover, the content of Fe2N can reach 40.3 wt% of Fe2N@OMC-2. The pore size distribution calculated from DBJH reveals that the widths of pores are 5.29, 14.2, 5.03, 4.88, and 2.62 nm in OMC, Fe2N, Fe2N@OMC-1, Fe2N@OMC-2, and Fe2N@OMC-3, respectively.
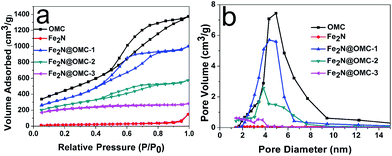 |
| | Fig. 4 The nitrogen adsorption–desorption isotherms (a) and pore diameter distribution (b) of OMC, Fe2N, Fe2N@OMC-1, Fe2N@OMC-2, and Fe2N@OMC-3. | |
Table 1 Porous property of composite materials identified by BET and BJH
| Sample name |
SBET (m2 g−1) |
VT (cm3 g−1) |
DBJH (nm) |
| OMC |
1609 |
2.13 |
5.29 |
| Fe2N |
62 |
0.02 |
14.2 |
| Fe2N@OMC-1 |
1232 |
1.55 |
5.03 |
| Fe2N@OMC-2 |
919 |
0.99 |
4.88 |
| Fe2N@OMC-3 |
583 |
0.44 |
2.62 |
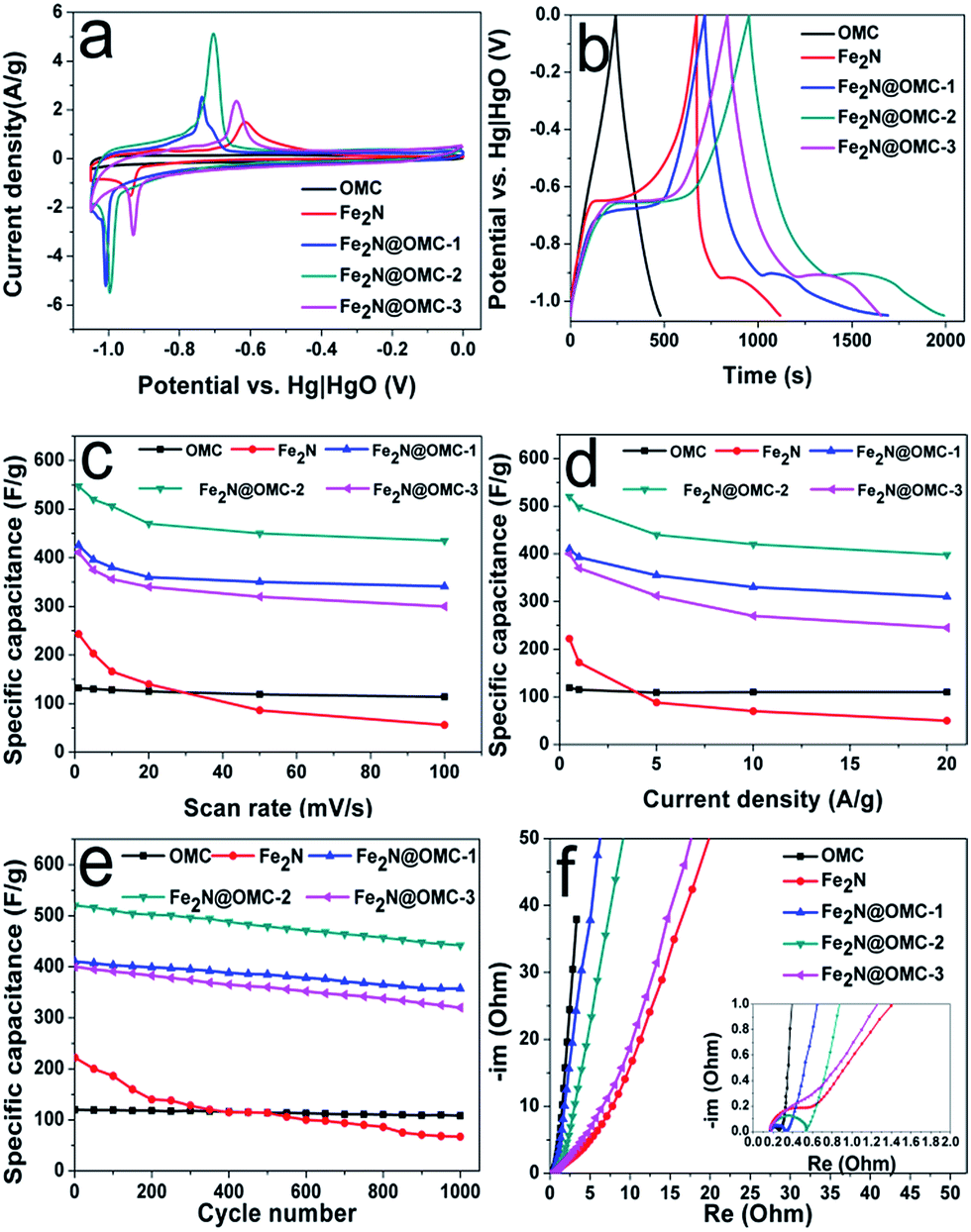
X-ray photoelectron spectroscopy (XPS) measurements were performed to analyze the surface information of the synthesized products. The surface iron concentration of Fe2N@OMC-1, Fe2N@OMC-2, and Fe2N@OMC-3 is 1.7, 6.2, and 14.6 at%, respectively (Table S1, ESI†). The N 1s XPS spectra of Fe2N@OMC-2 (Fig. 5c) and Fe2N@OMC-3 (Fig. 5e) were divided into five component peaks, which corresponded to nitrogen bonded with metal nitride (∼397.6 eV), pyridinic-N (∼398.4 eV), pyrrolic-N (∼399.8 eV), graphitic-N (∼400.8 eV), and nitrogen oxide (∼401.9 eV) functionalities.29,38 The N 1s XPS spectrum of Fe2N@OMC-1 indicates no metal nitride functionality (Fig. 5a) as compared to that of Fe2N@OMC-2 and Fe2N@OMC-3, whereas Fe2N really exists in the Fe2N@OMC-1, as demonstrated by XRD analysis. This finding indicated that iron nitride also exists in the pores of OMC and this part of iron nitride cannot be detected via XPS, which also applies to Fe2N@OMC-2 and Fe2N@OMC-3. The high-resolution XPS spectrum of the as-made samples for the Fe 2p level exhibits main peaks at around 707.5 and 721.4 eV that correspond to the Fe 2p3/2 and Fe 2p1/2 levels, respectively. They are assigned to Fe(II) in iron nitride. Moreover, the peaks appear at 710.0, 715.6, 723.8, and 729.1 eV, which are attributed to the presence of Fe–O bonds.41,42 The electrochemical performance of the obtained samples was analyzed in a standard three electrode system. The cyclic voltammogram of OMC shows a rectangular and symmetric shape, indicating a typical electric double-layer (EDL) behaviour, and all cyclic voltammograms of Fe2N-containing samples exhibit undoubtedly the reversible faradaic redox reactions attributing to Fe2N, as shown in Fig. 6a. All the composites of Fe2N@OMC-1, Fe2N@OMC-2, and Fe2N@OMC-3 have sharper and higher redox peaks as compared to bare Fe2N; this indicates that the presence of OMC can effectively enhance the electrochemical performance of these composites due to better utilization of Fe2N during charging and discharging when Fe2N is evenly distributed on the surface of OMC. The higher specific surface area of Fe2N@OMC-2, which implies a better distribution of Fe2N particles on the surface of OMC as compared to that of Fe2N@OMC-3, is a reason for the stronger pseudocapacitive behavior reflected by the voltammetry curve. However, excessive specific surface area of Fe2N@OMC-1 will sacrifice the content of Fe2N, thereby decreasing the electrochemical pseudocapacity, which is not expected. For Fe2N@OMC-1, Fe2N@OMC-2, and Fe2N@OMC-3 composites, both the charge storage mechanism of the electric double-layer behavior and faradaic redox reactions derived from OMC and Fe2N, respectively, must be investigated and studied. In Fig. 6b, a symmetrical triangular can be observed remarkably in the galvanostatic charge–discharge curves of OMC, which demonstrates that OMC has an outstanding double-layer capacitance and the platforms of the triangular shape shown by the galvanostatic charge–discharge curves of Fe2N, Fe2N@OMC-1, Fe2N@OMC-2, and Fe2N@OMC-3 correspond to the redox peaks of cyclic voltammogram at −0.75 ÷ −0.62 V and −1.05 ÷ −0.94 V. Moreover, it was obvious that the discharge time curves (Fig. S3 ESI†) of synthetic products were in the order of OMC < Fe2N < Fe2N@OMC-3 < Fe2N@OMC-1 < Fe2N@OMC-2. For Fe2N@OMC-2, excellent capacitance values of 547 F g−1 at 1 mV s−1 and 520 F g−1 at 0.5 A g−1 can be calculated by voltammetry and galvanostatic charge–discharge. Fe2N@OMC-2 has better specific capacitance as compared to those reported in other studies,37,38 as shown in Table S2.† In the application of supercapacitors, the material of the OMC-based Fe2N composite with very high specific capacitance has never been synthesized before. The capacitance of Fe2N@OMC-2 is more than two times that of bare Fe2N at 1 mV s−1 and 0.5 A g−1 and dramatically decreases in the strong current and high scan rate regions (Fig. 6c and d). The specific capacitances of Fe2N@OMC-1, Fe2N@OMC-2, and Fe2N@OMC-3 were 310, 398, and 245 F g−1 at a high current density of 20 A g−1, maintaining 75, 76, and 61% of the initial capacitance at 0.5 A g−1. For comparison, bare Fe2N exhibits a specific capacitance of only 50 F g−1 at a current density of 20 A g−1; this fully illustrates the significance of OMC of carbon-based Fe2N composite in improving the energy density. In addition, for supercapacitors, the cyclic stability is another indispensable performance indicator. All the prepared products were run for 1000 charge–discharge cycles at a current density of 1 A g−1, as shown in Fig. 6e. It can be seen that all carbon-based Fe2N composites show excellent cyclic stability after 1000 cycles due to a high specific surface area and the appropriate porous size. Particularly, Fe2N@OMC-2 shows a much higher retention of 85% of the initial capacitance as compared to bare Fe2N, which exhibits only 28% retention. Moreover, it was clearly observed that only a very small amount of Fe2N nanoparticles of Fe2N@OMC-2 accumulated after 1000 cycles (Fig. S4b ESI†) compared to those before 1000 cycles (Fig. S4a ESI†). The stable nanostructure made the Fe2N@OMC-2 electrode have an outstanding cyclic stability. The nanocasting route can achieve a homogeneous dispersion of small non-aggregated Fe2N nanoparticles on the external surface and in the pores of OMC; this has been demonstrated by nitrogen sorption analysis, SEM, TEM, HRTEM, and XPS results. This feature can improve the transport of ions from the bulk electrolyte to the composite material. The collective effect of the two factors, OMC providing more active sites due to the high specific surface area and the high pseudocapacitance of Fe2N, accounts for the outstanding electrochemical performance of the Fe2N@OMC-2 composite as an electrode material for supercapacitors.38
 |
| | Fig. 5 High-resolution XPS spectra of N 1s of Fe2N@OMC-1 (a), Fe2N@OMC-2 (c), and Fe2N@OMC-3 (e). Fe 2p spectra of Fe2N@OMC-1 (b), Fe2N@OMC-2 (d), and Fe2N@OMC-3 (f). | |
 |
| | Fig. 6 Cyclic voltammograms of the synthetic products at 1 mV s−1 (a), galvanostatic charge/discharge curves performed at 0.5 A g−1 (b), specific capacitance as a function of the scan rate (c) and current density (d), cycle stability at a current density of 1 A g−1 (e), and Nyquist plots with an inset representing the high-frequency region (f). | |
The resistive characteristics of electrode materials were analyzed by EIS (Fig. 6f). In the Nyquist plots of OMC, Fe2N@OMC-1, and Fe2N@OMC-2, a small semicircular line corresponding to the interfacial charge-transfer resistance is noticed in the high-frequency region, which indicates that all the samples OMC, Fe2N@OMC-1, and Fe2N@OMC-2 have a small charge-transfer resistance. The slope lines at an angle of 80–90° are observed markedly in the lower frequency region of OMC, Fe2N@OMC-1, and Fe2N@OMC-2, which show an excellent charge–discharge capacitive behavior because of the fast charge-transfer rate from the active electrode material to the current collector.40,43 By contrast, no obvious semicircle is detected for bare Fe2N and Fe2N@OMC-3; this demonstrates the presence of charge-transfer resistance, resulting in a poor rate capability.44 In addition, the slope lines at an angle near 45° for Fe2N and Fe2N@OMC-3 reveal the occurrence of the Warburg impedance, standing for ion-diffusion resistance.40,45 The ion-diffusion resistance of Fe2N and Fe2N@OMC-3 is higher than that of OMC, Fe2N@OMC-1, and Fe2N@OMC-2; this may be caused by the aggregated and bulky particles of Fe2N, making ion diffusion in the electrolyte become more difficult.46 The EIS measurements can further prove that the composite of OMC and small non-aggregated Fe2N particles is advantageous in ion-transport, which plays a significant role in rate performance and specific capacitance.
Different electrolytes can better test the performance of the electrodes.47 The Fe2N@OMC-2 electrode was tested in 1 M LiOH, NaOH, and KOH electrolytes, as shown in Fig. S5 ESI.† It is clear that the redox peaks of CV curves increase in turn as the size of the cations in the electrolytes decreases (K+ > Na+ > Li+). This observation indicates enhanced pseudocapacitive reactions with smaller cations as they are easier to get intercalated into the pores of Fe2N@OMC-2. Moreover, we tested the cyclic stability in different electrolytes (LiOH, NaOH, and KOH). After 1000 cycles, the specific capacitance retention of Fe2N@OMC-2 remained to be 87% in KOH, which was much higher than that in LiOH (67%) and NaOH (72%). This is mainly because the smaller cation is difficult to desorb from the pore of the as-made electrode materials.
4. Conclusions
A novel Fe2N@OMC-2 composite was successfully synthesized via a nanocasting route and NH3 annealing. The vacuum impregnation of OMC with iron chloride resulted in the deposition of small and non-aggregated Fe2N nanoparticles both on the external surface and in the pores of OMC. Owing to the synergistic effect between the mesoporous property of OMC and an excellent pseudocapacitance of Fe2N, Fe2N@OMC-2 exhibits an eye-catching high capacitance of 547 F g−1 at 1 mV s−1. A great rate capability of 76% was retained when the current densities increased from 0.5 to 20 A g−1, and a superior cyclic stability of 85% was retained after 1000 cycles. This study constitutes the first demonstration of employing the composite of Fe2N and OMC as an electrode material for supercapacitors, which can effectively enhance the performance of energy storage devices.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We gratefully acknowledge the support for this study provided by the National Natural Science Foundation of China (No. 21501104), the Natural Science Foundation of Heilongjiang Province (B2015014), and the University Nursing Program for Young Scholars with Creative Talents in Heilongjiang Province (UNPYSCT-2016088).
Notes and references
- S. L. Chiam, H. N. Lim, C. Y. Foo, A. Pandikumar and N. M. Huang, Electrochim. Acta, 2017, 246, 1141–1146 CrossRef CAS.
- Y. Liu and X. Peng, Applied Materials Today, 2017, 8, 104–115 CrossRef.
- H. Yang and Y. Zhang, J. Power Sources, 2016, 312, 165–171 CrossRef CAS.
- K.-T. Lee, C.-B. Tsai, W.-H. Ho and N.-L. Wu, Electrochem. Commun., 2010, 12, 886–889 CrossRef CAS.
- X. Meng and R. Wang, Energy Procedia, 2012, 17, 1585–1590 CrossRef CAS.
- J. Lv, Z. Wang, T. Liang, Y. Meng, K. Suzuki and H. Miura, J. Electroanal. Chem., 2017, 799, 595–601 CrossRef CAS.
- L. Liu, H. Zhang, L. Fang, Y. Mu and Y. Wang, J. Power Sources, 2016, 327, 135–144 CrossRef CAS.
- X. Liu, A. Zhou, T. Pan, Y. Dou, M. Shao, J. Han and M. Wei, J. Mater. Chem. A, 2016, 4, 8421–8427 CAS.
- Z. Yu, C. Li, D. Abbitt and J. Thomas, J. Mater. Chem. A, 2014, 2, 10923–10929 CAS.
- C. Li, M. M. Islam, J. Moore, J. Sleppy, C. Morrison, K. Konstantinov, S. X. Dou, C. Renduchintala and J. Thomas, Nat. Commun., 2016, 7, 13319 CrossRef CAS PubMed.
- D. Yang, J. Power Sources, 2011, 196, 8843–8849 CrossRef CAS.
- H. Chang and H. Wu, Energy Environ. Sci., 2013, 6, 3483–3507 CAS.
- M. S. Javed, C. Zhang, L. Chen, Y. Xi and C. Hu, J. Mater. Chem. A, 2016, 4, 8851–8859 CAS.
- L. Ma, L. Su, J. Zhang, D. Zhao, C. Qin, Z. Jin and K. Zhao, J. Electroanal. Chem., 2016, 777, 75–84 CrossRef CAS.
- M. Idrees, S. M. Abbas, R. Ata Ur, N. Ahmad, M. W. Mushtaq, R. A. Naqvi, K.-W. Nam, B. Muhammad and Z. Iqbal, J. Chem. Eng., 2017, 327, 361–370 CrossRef CAS.
- B. Das, M. V. Reddy and B. V. Chowdari, Nanoscale, 2013, 5, 1961–1966 RSC.
- P. J. Hanumantha, M. K. Datta, K. Kadakia, C. Okoli, P. Patel and P. N. Kumta, Electrochim. Acta, 2016, 207, 37–47 CrossRef CAS.
- N. Choudhary, C. Li, J. Moore, N. Nagaiah, L. Zhai, Y. Jung and J. Thomas, Adv. Mater., 2017, 29, 1605336 CrossRef PubMed.
- X. Lu, G. Wang, T. Zhai, M. Yu, S. Xie, Y. Ling, C. Liang, Y. Tong and Y. Li, Nano Lett., 2012, 12, 5376–5381 CrossRef CAS PubMed.
- M. Yu, Y. Han, X. Cheng, L. Hu, Y. Zeng, M. Chen, F. Cheng, X. Lu and Y. Tong, Adv. Mater., 2015, 27, 3085–3091 CrossRef CAS PubMed.
- N. Mosavati, S. O. Salley and K. Y. S. Ng, J. Power Sources, 2017, 340, 210–216 CrossRef CAS.
- S. Dong, X. Chen, X. Zhang and G. Cui, Coord. Chem. Rev., 2013, 257, 1946–1956 CrossRef CAS.
- S. Liu, J. Liu, W. Wang, L. Yang, K. Zhu and H. Wang, RSC Adv., 2016, 6, 86131–86136 RSC.
- Y. Yu, W. Gao, Z. Shen, Q. Zheng, H. Wu, X. Wang, W. Song and K. Ding, J. Mater. Chem. A, 2015, 3, 16633–16641 CAS.
- L. Jiang, L. Lin, F. Yan, B. Fan, Y. Chen and Y. Qiu, Ceram. Int., 2017, 43, 9226–9234 CrossRef CAS.
- G. R. Li, J. Song, G. L. Pan and X. P. Gao, Energy Environ. Sci., 2011, 4, 1680 CAS.
- H. Huang, S. Gao, A.-M. Wu, K. Cheng, X.-N. Li, X.-X. Gao, J.-J. Zhao, X.-L. Dong and G.-Z. Cao, Nano Energy, 2017, 31, 74–83 CrossRef CAS.
- D. Di Lecce, R. Verrelli and J. Hassoun, Green Chem., 2017, 19, 3442–3467 RSC.
- T. Panja, D. Bhattacharjya and J.-S. Yu, J. Mater. Chem. A, 2015, 3, 18001–18009 CAS.
- Y. Teng, E. Liu, R. Ding, K. Liu, R. Liu, L. Wang, Z. Yang and H. Jiang, Electrochim. Acta, 2016, 194, 394–404 CrossRef CAS.
- J.-G. Wang, Y. Yang, Z.-H. Huang and F. Kang, Carbon, 2013, 61, 190–199 CrossRef CAS.
- Y. Xie, L. Lu, Y. Tang, F. Zhang, C. Shen, X. Zang, X. Ding, W. Cai and L. Lin, Mater. Lett., 2017, 186, 70–73 CrossRef CAS.
- L. Feng, G. Li, S. Zhang and Y. X. Zhang, Ceram. Int., 2017, 43, 8321–8328 CrossRef CAS.
- X. Zhang, J. Luo, P. Tang, X. Ye, X. Peng, H. Tang, S.-G. Sun and J. Fransaer, Nano Energy, 2017, 31, 311–321 CrossRef CAS.
- V. C. Lokhande, A. C. Lokhande, C. D. Lokhande, J. H. Kim and T. Ji, J. Alloys Compd., 2016, 682, 381–403 CrossRef CAS.
- L.-Q. Fan, G.-J. Liu, J.-H. Wu, L. Liu, J.-M. Lin and Y.-L. Wei, Electrochim. Acta, 2014, 137, 26–33 CrossRef CAS.
- C. Zhu, P. Yang, D. Chao, X. Wang, X. Zhang, S. Chen, B. K. Tay, H. Huang, H. Zhang, W. Mai and H. J. Fan, Adv. Mater., 2015, 27, 4566–4571 CrossRef CAS PubMed.
- A. Sliwak, A. Moyseowicz and G. Gryglewicza, J. Mater. Chem. A, 2017, 5, 5680–5684 CAS.
- J. Feng, W. Song, L. Sun and L. Xu, RSC Adv., 2016, 6, 110337–110343 RSC.
- M.-S. Balogun, M. Yu, Y. Huang, C. Li, P. Fang, Y. Liu, X. Lu and Y. Tong, Nano Energy, 2015, 11, 348–355 CrossRef CAS.
- P. Yu, L. Wang, F. Sun, D. Zhao, C. Tian, L. Zhao, X. Liu, J. Wang and H. Fu, Chem.–Eur. J., 2015, 21, 3249–3256 CrossRef CAS PubMed.
- B. P. Vinayan, T. Diemant, R. J. Behm and S. Ramaprabhu, RSC Adv., 2015, 5, 66494–66501 RSC.
- L. Ren, G. Zhang, Z. Yan, L. Kang, H. Xu, F. Shi, Z. Lei and Z. H. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 28294–28302 CAS.
- F. Miao, C. Shao, X. Li, K. Wang and Y. Liu, J. Mater. Chem. A, 2016, 4, 4180–4187 CAS.
- J. Huang, S. Yang, Y. Xu, X. Zhou, X. Jiang, N. Shi, D. Cao, J. Yin and G. Wang, J. Electroanal. Chem., 2014, 713, 98–102 CrossRef CAS.
- B. Liang, Z. Qin, T. Li, Z. Dou, F. Zeng, Y. Cai, M. Zhu and Z. Zhou, Electrochim. Acta, 2015, 177, 335–342 CrossRef CAS.
- N. Choudhary, C. Li, H. S. Chung, J. Moore, J. Thomas and Y. Jung, ACS Nano, 2016, 10, 10726–10735 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08704g |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Li Sun*,
Jian Feng,
Linlin Qi,
Ishaq Muhammad,
Jabeen Maher,
Xiaoyu Cheng and
Weiming Song*
,
Li Sun*,
Jian Feng,
Linlin Qi,
Ishaq Muhammad,
Jabeen Maher,
Xiaoyu Cheng and
Weiming Song*