
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic and optical properties of metalloporphyrins of zinc on TiO2 cluster in dye-sensitized solar-cells (DSSC). A quantum chemistry study†
Fernando Mendizabal *a,
Raúl Mera-Adasmeb,
Wen-Hua Xucd and
Dage Sundholm*c
*a,
Raúl Mera-Adasmeb,
Wen-Hua Xucd and
Dage Sundholm*c
aDepartmento de Química, Facultad de Ciencias, Universidad de Chile, P.O. Box 653, Las Palmeras 3425, Ñuñoa, Santiago, Chile. E-mail: hagua@uchile.cl
bDepartamento de Ciencias del Ambiente, Facultad de Química y Biología, Universidad de Santiago de, Chile
cDepartment of Chemistry, University of Helsinki, A.I. Virtanens plats 1, P.O. Box 55, FI-00014 Helsinki, Finland. E-mail: Dage.Sundholm@helsinki.fi
dCollege of Chemistry and Materials Science, Northwest University, 710127, Xi'an, China
First published on 4th September 2017
Abstract
Dye-sensitized solar-cell (DSSC) systems have been investigated by calculating light-absorption and electron-injection processes of the LD13 ([5,15-bis(2,6-(1,1-dimethylethyl)-phenyl)-10-4-dimethylaminophenylethynyl-20-4-carboxy phenylethynyl porphyrinato]zinc-(II)) and YD2-o-C8 ([5,15-bis(2,6-dioctoxyphenyl)-10-(bis(4-hexylphenyl)amino-20-4-carboxyphenylethynyl)porphyrinato]zinc-(II)) dyes adsorbed on a TiO2 cluster simulating the semiconductor. The binding energy of the dyes with the TiO2 clusters has been calculated at the density functional theory (DFT) level using the B3LYP and CAM-B3LYP functionals. The electronic excitation energies have been calculated at the time-dependent DFT (TDDFT) level for the dyes in the gas and solvent phase employing the B3LYP, CAM-B3LYP and BHLYP functionals. The calculated excitation energies have been compared to values obtained at the algebraic diagrammatic construction through second order (ADC(2)) level of theory. The TDDFT calculations with the B3LYP in tetrahydrofuran solvent with the dye and dye–TiO2 models yield excitation energies that agree well with the transitions in the experimental absorption spectra. Changes in the free energy for electron injection support the better performance of the dyes on the TiO2 clusters.
Introduction
Dye Sensitized Solar Cells (DSSCs) were proposed in 1991 by Grätzel and O'Regan.1,2 The DSSCs consist of a light-absorbing dye attached to a semiconductor surface such as titanium dioxide (TiO2). The dye absorbs solar energy yielding to an electronic excited state that transfers an electron from the dye to the semiconductor.3,4 The TiO2 semiconductor has a large band gap, which can be excited only by ultraviolet radiation.5,6 The role of the dye is to extend the absorption range of the semiconductor to the visible and near-infrared region covering more of the solar spectrum. DSSCs are a cost-effective alternative to the currently commercialized solar cells made of silicon.7 The efficiency of the DSSCs can be improved by designing dyes with a broad absorption spectrum and good charge separation properties.A large number of experimental and computational research groups search for novel dyes with improved efficiency and stability to achieve high photoconversion efficiency.8–36 The efficiency of the DSSCs is determined by the photophysical properties of the semiconductor, the absorption spectrum of the dye and the ability of the electrolyte regenerator to transport charges. The solar-cell performance is measured by the power conversion efficiency (η), also called photo-conversion efficiency.2,3 DSSCs based on the sensitization of TiO2 by the classic ruthenium complexes dyes have a η between 7% and 13%, making practical devices and applications feasible.8–24 In recent years, ruthenium complexes have been replaced by other dyes that are cheaper as they are based compound with large π-aromatic moieties such as metalloporphyrins (MP).5 Metalloporphyrins have been shown to be good dye sensitizers.8–24 MP based zinc complexes are dyes with good photo-stability and large light-harvesting capabilities.
The η index varies between 6% and 15% depending on the porphyrin substituents which close to the η value of the best ruthenium complexes. The Zn-porphyrins denoted as YD2, YD2-o-C8, ZnPBAT, LD14, LD13, SM315 and SM135 are among the most efficient DSSCs. On the other hand, solar cells based on perovskites such as hybrid alkylammonium lead halide have been found to have a high power conversion efficiency of about 20%.37 However, the lead-containing perovskite is environmentally problematic.
In this work, we employ quantum chemistry methods for studying zinc-porphyrin based dye sensitizers that are attached to the TiO2 semiconductor surface through an anchor unit consisting of a carboxylic group (COOH).2–5 The excited state of the dye injects electrons into the semiconductor. Therefore, the energy of the lowest unoccupied molecular orbital (LUMO) of the dye must be above the lower limit of the conduction band of TiO2 to allow the electron transfer to the semiconductor. The energy of the highest occupied molecular orbital (HOMO) of the dye must be below the potential of the I3−/I− redox pair in order to allow an efficient regeneration of the oxidized dye.5,13 The interaction of the two zinc porphyrin-based dyes, LD13 and YD2-o-C8, with the (101) plane of TiO2 (anatase) or a TiO2 nanocluster has been previously modelled showing that the dyes are attached to TiO2 via the carboxylate group.25–29,33–36
The aim of this work is to extend the study to systems using the known coordination preference of the LD13 (η = 8.4%) and YD2-o-C8 (η = 12.1%) dyes. We use these two dyes because of their medium sized in comparison to other compounds and their high values of η. We employ computational methods for elucidating which anchor groups should be used for obtaining an improved conjugation of the MP ring with the TiO2 surface. Using state-of-the-art quantum chemistry methods, the largest molecular systems that we are able to study are about the same size as the light-receptor system of the Grätzel cells.8–24 The DSSC model systems considered in the present calculations consist of a dye attached to a TiO2 cluster representing the TiO2 surface.25–36 The molecular structures of the DSSC models have been optimized at density functional theory (DFT) levels for identifying and characterizing suitable light-capturing molecules. The ground-state calculations show how the dye binds to TiO2, whereas single-point quantum chemical calculations of the excitation energies provide information about the vertical excitation processes. The light-absorption processes in the DSSCs have been studied using time-dependent density functional theory (TDDFT) calculations and by performing ab initio correlated calculations at the algebraic diagrammatic construction through second order level (ADC(2)). By determining the properties of the interaction between the LD13 and YD2-o-C8 dyes and TiO2 one obtains a better understanding of the complete light-absorption mechanism of the DSSCs, which supports the design of future DSSC systems.
Models and computational details
The studied LD13 and YD2-o-C8 models are depicted in Fig. 1. The molecular structures of the ground state were fully optimized without any symmetry constraints at the density functional theory (DFT) level38 using the B3LYP and CAM-B3LYP functionals.39,40 The structure optimizations were performed with the molecules in a gas phase and for the molecules surrounded by tetrahydrofuran (THF) modelled by using the conductor-like screening model (COSMO) with a dielectric constant (ε) of 7.6.41 We have used this solvent since the experimental studies of both dyes have been performed with THF as solvent. Acetonitrile is though the most common solvent in DSSCs. Solvent effects must be considered in studies of the absorption energies of the chromophore in DSSCs because the electronic transitions are charge transfer transitions and the excited states are generally charge separated states.35 The fully relaxed structures of the dyes adsorbed on the TiO2 surface are shown in Fig. 2. The TiO2 structure was kept fixed in the structure optimization. In previous studies of the dye–TiO2 interactions, the dyes molecules were modelled with the –COO− group deprotonated.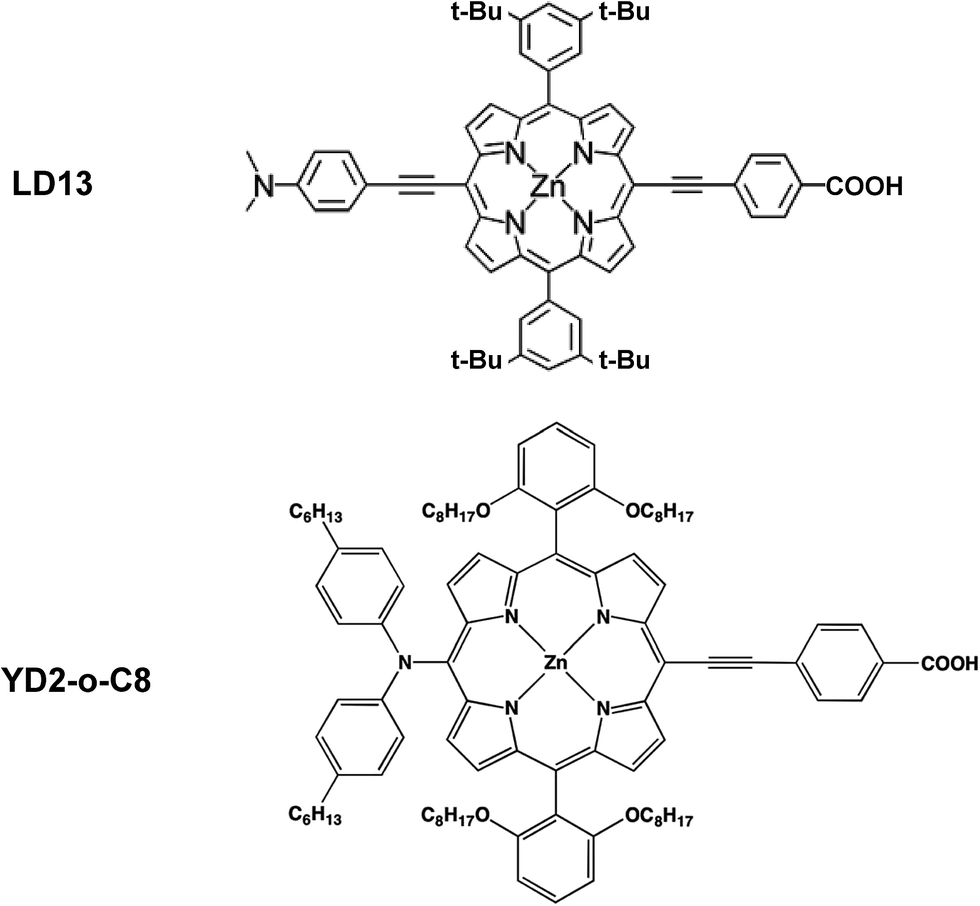 | ||
| Fig. 1 Molecular structures of LD13 and YD2-o-C8 dyes porphyrins. | ||
 | ||
| Fig. 2 LD13 and YD2-o-C8 on TiO2 models. | ||
Experimental and computational studies have shown that the carboxylic acid group of substituted porphyrins is bound to the TiO2 surface by monodentate or bidentate coordination depending on the dye.8–36 The [Ti16O34H4] cluster simulating the TiO2 surface was constructed by cutting a piece of titanium oxide out from the anatase crystal structure and saturating the dangling bonds with hydrogen atoms.36 TiO2 clusters of various shapes consisting of 5–80 titanium atoms have previously been used for modelling the TiO2 surface.25,26 Small TiO2 clusters have successfully been used in studies of the interactions between TiO2 and different dyes such as N749, N3, C101, J3, YD2, LD14, ZnPBAT, ZnPBA and WW3m-WW8m.25–36
The excitation energies have been calculated for the optimized structures at the DFT level using the time-dependent perturbation theory approach (TDDFT).42,43 The B3LYP,39 CAM-B3LYP40 and BHLYP44 functionals have been employed in the TDDFT calculations. Molecular structures optimized at the B3LYP level have been used in the BHLYP calculations of the absorption spectra. We have used several hybrid functionals because some absorption energies of the porphyrins have been found to be sensitive to the employed level of theory. We have not employed pure density functionals, because it has been shown that they yield an incorrect description of the optical properties of the complex metalloporphyrins.33–36 Excitation energies and oscillator strengths were also calculated for the LD13 dye in the gas and solvent phases at the ADC(2) level45,46 using the scaled opposite-spin (SOS) approximation.47,48 The hermitean ADC(2) method belongs to the family of approximate second-order coupled-cluster and configuration-interaction methods.46
The calculations were carried out using the Turbomole 7.0,49,50 Gaussian09 (ref. 51) and Orca 3.0.3 (ref. 52) program packages. We have used the functional B3LYP and ADC(2) with Turbomole. In addition, the B3LYP-CAM and BHLYP functionals were calculated with Gaussian09 and Orca, respectively. For Zn and Ti, the 10 core-electron pseudo-potentials (PP) of Andrae et al.53 were employed. Two d-type polarization and diffuse functions were added to the Zn and Ti basis sets.54 The 1s orbitals of C, N and O were also replaced by PPs. Double-zeta basis sets augmented with two d-type polarization and diffuse functions were used for the valence electrons.55 For the H atoms, a double-zeta basis set augmented with one p-type polarization function was used.56 The employed basis set for Zn consisted of 8s7p6d primitive functions contracted to 6s5p3d (8s7p6d/6s5p3d). The following basis sets were used for the other atoms: Ti (8s7p6d/6s5p3d), O (4s4p1d/2s2p2d), C (4s4p1d/2s2p2d), N (4s4p1d/2s2p2d), and H (4s1p/2s1p). Grimme's semi-empirical dispersion correction term (D3) has also been used.57
Results and discussion
Molecular structures
LD13 is a porphyrin with a phenylethynyl group in the meso position as the acceptor and bridging unit between the porphyrin and the carboxylic acid moiety binding to the TiO2 surface. The porphyrin has ortho-alkoxylated phenyl groups in the two meso positions next to the meso position with the acceptor and anchoring groups. The 4-dimethylaminophenylethynyl donor group is attached to the β position of one of the pyrrole rings on the opposite side of the porphyrin with respect to the acceptor group. The porphyrin chromophore function has a π-bridge in the donor π-conjugate-bridge acceptor (D–π–A) structure. The YD2-o-C8 dye has the same acceptor and anchor group as LD13. It has a 2,6-dioctoxyphenyl group in the two meso-positions closest to the anchor substituent. The donor group attached to the opposite meso position with respect to the anchor group consists of a bis(4-hexylphenyl)amino group. The porphyrin forms a π-bridge in the D–π–A structure. The molecular structures obtained at the B3LYP and CAM-B3LYP levels are very similar. Selected structural parameters and the Cartesian coordinates of the optimized molecular structures are given as ESI see Table S1.†The LD13 and YD2-o-C8 dyes are linked to the TiO2 model (see Fig. 2) by binding the carboxylate moiety to the Ti atoms of the (101) plane. The dyes do not undergo any large structural changes when they bind to TiO2. The largest structural changes occur at the carboxylate group when forming a bidentate bond to TiO2. The bond distance between the oxygen of the carboxylate group and the titanium atom is 217–221 pm depending on the employed level of theory. The second Ti–O distance of the bidentate bond of 212–215 pm is slightly shorter. The C5–O2 and C5–O1 distances are 127–130 pm, while the O2–C5–O1 angle of 128–129° is wider than for the free dye. The bonds of the COO− group are also slightly conjugated when the dyes are coordinated to the TiO2 surface. The most relevant geometrical parameters of the optimized structures and the Cartesian coordinates are reported in ESI see Table S2.†
The binding energy of the dye–TiO2 complex has been calculated at the B3LYP and CAM-B3LYP levels using the counterpoise (CP) method to correct for basis-set superposition errors (BSSE).58 The CP corrected B3LYP energies for the LD13–TiO2 bond are −70.1 kcal mol−1 and −66.8 kcal mol−1 as obtained in the gas-phase and COSMO calculations, respectively. Since the dye binds to two titanium atoms, the average binding energy of the dye with Ti is 35 kcal mol−1, which is weaker than for a typical covalent bond. Similar results have been previously obtained for the YD2 and LD14 dyes.33,34 At the CAM-B3LYP level, the corresponding binding energies are −68.3 kcal mol−1 and −65.5 kcal mol−1, respectively. B3LYP calculations on the YD2-o-C8–TiO2 complex yielded equivalent binding energies of −70.4 kcal mol−1 and −66.7 kcal mol−1. The corresponding CAM-B3LYP binding energies for YD2-o-C8–TiO2 are −68.2 kcal mol−1 and −65.1 kcal mol−1, respectively. The binding energies are practically the same for the two dyes, presumably because they have the same anchoring unit.
Natural Population Analyses (NPA) based on the B3LYP and CAM-B3LYP density matrices for the LD13, YD2-o-C8, TiO2, LD13–TiO2 and YD2-o-C8–TiO2 models in the gas and solvent phases show that 0.4–0.6 electrons are transferred from mainly the carboxylate group of the dyes to the two nearest Ti atoms when binding to the TiO2 cluster. The most relevant NPA charges are given in the ESI see Tables S3 and S4.† The NPA results and the binding energies show that there is a coordinating bond between the dye and the TiO2 cluster.
Excitation energy calculations
The UV-Vis spectra have been calculated at the TDDFT level using the B3LYP, CAM-B3LYP and BHLYP functionals. The excitation energies for LD13 were also calculated at the ADC(2) level to assess whether spurious charge transfer states occur for the investigated molecules. Since the electronic coupling between the dye and the semiconductor determines the electron injection rate and the power conversion efficiency (η), it is necessary to study properties of the excited states of the free dye and of the dye attached to TiO2. For porphyrins, the UV-Vis absorption spectra in the visible region usually consist of a weak Q band in the red part of the spectrum and a strong B (Soret) band at higher energies. Substituted porphyrins can also have a weak T band between the Q and B bands in addition to the vibrational band of the Q band.59 The vertical excitation energies calculated for the dyes and the dye–TiO2 complexes are reported in Table 1. The experimental spectra of LD13 and YD2-o-C8, free and adsorbed on TiO2, show a Q band maximum between 645 and 672 nm, while the maximum of the B band is between 448 and 472 nm (ref. 14 and 22) in THF solvent.| System | Method | B | T | Q |
|---|---|---|---|---|
| a Calculated using a [Ti16O34H4] cluster to simulate TiO2.b Measured in THF. | ||||
| LD13 | B3LYP | 427 | 615 | |
| LD13 | B3LYP (solv) | 438 | 632 | |
| LD13–TiO2 | B3LYPa | 434 | 602 | |
| LD13–TiO2 | B3LYP (solv)a | 445 | 606 | |
| LD13 | CAM-B3LYP | 384 | 597 | |
| LD13 | CAM-B3LYP (solv) | 397 | 615 | |
| LD13–TiO2 | CAM-B3LYPa | 388 | 583 | |
| LD13–TiO2 | CAM-B3LYP (solv)a | 389 | 591 | |
| LD13 | BHLYP (solv) | 361 | 557 | |
| LD13–TiO2 | BHLYP (solv)a | 359 | 540 | |
| LD13 | ADC(2) | 386 | 603 | |
| LD13 | ADC(2) (solv) | 401 | 612 | |
| YD2-o-C8 | B3LYP | 408 | 533 | 640 |
| YD2-o-C8 | B3LYP (solv) | 443 | 545 | 690 |
| YD2-o-C8–TiO2 | B3LYPa | 415 | 610 | |
| YD2-o-C8–TiO2 | B3LYP (solv)a | 454 | 621 | |
| YD2-o-C8 | CAM-B3LYP | 392 | 594 | |
| YD2-o-C8 | CAM-B3LYP (solv) | 404 | 605 | |
| YD2-o-C8–TiO2 | CAM-B3LYPa | 391 | 595 | |
| YD2-o-C8–TiO2 | CAM-B3LYP (solv)a | 393 | 611 | |
| YD2-o-C8 | BHLYP (solv) | 359 | 549 | |
| YD2-o-C8–TiO2 | BHLYP (solv)a | 404 | 557 | |
| YD2-o-C8 | CAM-B3LYP60 | 397 | 592 | |
| YD2-o-C8 | LC-ωPBE60 | 399 | 632 | |
| YD2-o-C8 | PBE60 | 477 | 879 | |
| YD2-o-C8 | HSE06 (ref. 60) | 420 | 671 | |
| LD13 | Expb22 | 458 | 672 | |
| LD13–TiO2 | Expb22 | 461 | 668 | |
| YD2-o-C8 | Expb14 | 448 | 581 | 645 |
| YD2-o-C8–TiO2 | Expb14 | 470 | 650 | |
Similar excitation energies were obtained for the Q band of LD13 at the DFT and ADC(2) levels. The first Q band transition calculated at the B3LYP level is slightly red shifted as compared to the ADC(2) value, whereas the lowest excitation energy of LD13 obtained in the CAM-B3LYP calculation is slightly blue shifted as compared to the ADC(2) one (see ESI†). The second excitation energy obtained at 580 nm in the B3LYP calculation agrees well with the corresponding ADC(2) excitation energy. The oscillator strength for the transition at 580 nm is small implying that it might be hidden in the experimental spectra among the vibrational bands. The B band consists of the third and fourth strong transition at 430 and 427 nm at the B3LYP level. At the ADC(2) level, the corresponding transition wave lengths are 386 and 381 nm, which are almost the same as obtained at the CAM-B3LYP level. At the ADC(2) and CAM-B3LYP levels, there are no other strong transitions above 350 nm, whereas B3LYP calculations yield two strong transitions at 406 and 398 nm and a weak transition at 410 nm.
The comparison of the excitation energies and oscillator strengths calculated at the four levels of theory shows that the excitation energies calculated at the B3LYP level are generally smaller than those obtained in the CAM-B3LYP and ADC(2) calculations. More states are also obtained at the B3LYP level suggesting that there are charge transfer problems in the high-energy region of the visible part of the absorption spectrum. Very similar absorption spectra were obtained at the CAM-B3LYP and ADC(2) levels in the studied energy range. The oscillator strengths calculated at the CAM-B3LYP and ADC(2) levels qualitatively agree for the first four excited states, whereas at the B3LYP level two strong transitions appear at about 400 nm. Such states cannot be identified at the CAM-B3LYP and ADC(2) levels. The same trends are obtained when solvent effects are considered by using COSMO. The excitation energies calculated with COSMO are slightly red shifted as compared to the values obtained in the gas-phase calculations.
The Q band of the LD13 dye attached to the TiO2 cluster is blue shifted as compared to the Q band for the free LD13 dye. The energy of the third electronic transition corresponding to the B band calculated at the B3LYP level is about 41 nm smaller than obtained at the CAM-B3LYP level. The excitation energies calculated at the B3LYP level are about 62 nm larger than the experimental values for LD13 and LD13 attached to TiO2. The excitation energies calculated at the BHLYP level are 200 nm larger than the corresponding B3LYP values.
For YD2-o-C8, the Q band calculated at the B3LYP level is redshifted relatively to the value obtained at using the CAM-B3LYP functional (see ESI†). The same holds for the other studied excitation energies. Similar oscillator strengths were obtained at the two levels of theory. At the B3LYP level, the strong B band consists of the fifth to the eighth transitions between 425 and 389 nm, whereas at the CAM-B3LYP level, the third to the sixth transitions between 428 and 392 nm form the B band as two apparently spurious weak states at the B3LYP level are not obtained in the CAM-B3LYP calculations. Solvent effects shift the B band to lower energies. The calculations are summarized in Table 1, where the obtained wave lengths of the Q, T and B bands are compared to available literature data.
The first strong transition is assigned as the Q band and the strong transitions at higher energy form the B band. Comparisons of the excitation energies for YD2-o-C8 attached to the TiO2 cluster calculated at the B3LYP and CAM-B3LYP levels show that four spurious states are obtained between the Q and B band in the gas-phase calculations. When solvent effects are considered, the Q and B bands are redshifted and seven spurious states are obtained between the Q and B bands at the B3LYP level. However, the transition wave lengths of the strong transitions calculated at the B3LYP level are in better agreement with the experimental values than those obtained at the other computational levels.
We consider only the strong transitions obtained at the B3LYP level in the discussion of the properties of excited states, since they agree rather well with experimental data and the spurious states are easily identified by comparing the excitation energies with the ones calculated at the other levels of theory. We discuss only the properties of the dye–TiO2 systems, whereas the corresponding data for the dyes are given in the ESI.† The simulated spectra for LD13–TiO2 and YD2-o-C8–TiO2 in THF solution calculated at the B3LYP level are shown in Fig. 3 and 4, respectively. The most important molecular orbitals for describing the electronic transitions are shown in Fig. 5 for LD13–TiO2 and in Fig. 6 for YD2-o-C8–TiO2.
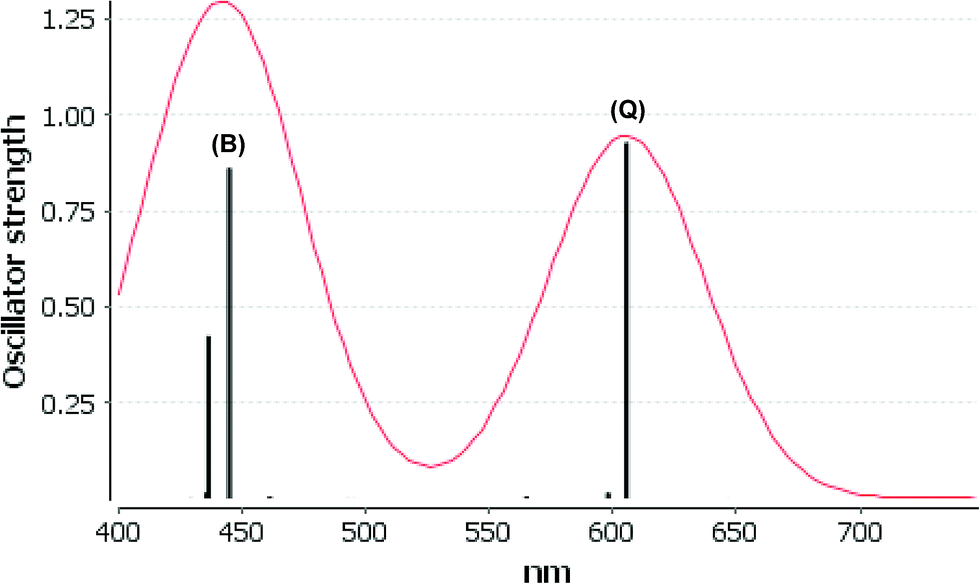 | ||
| Fig. 3 Electronic spectra at the B3LYP level calculated by LD13–TiO2 in THF. | ||
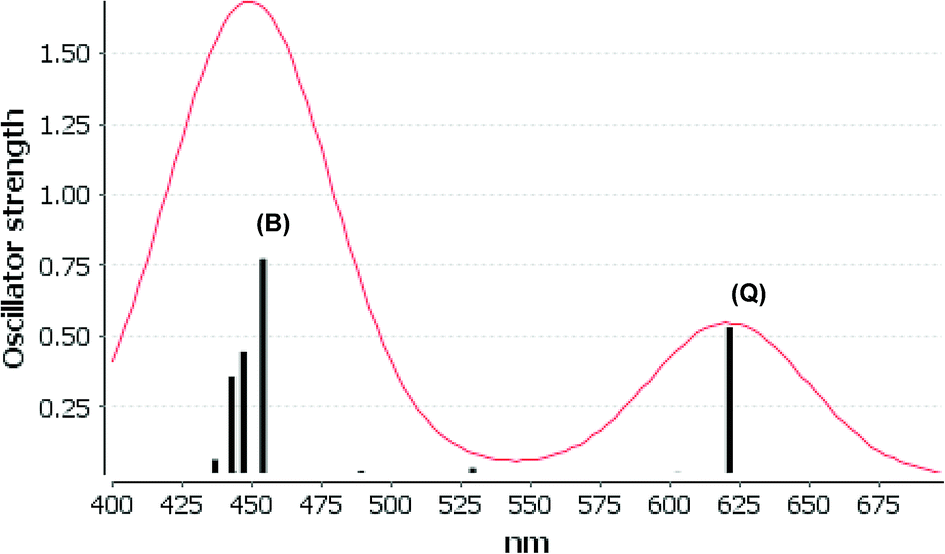 | ||
| Fig. 4 Electronic spectra at the B3LYP level calculated by YD2-o-C8–TiO2 in THF. | ||
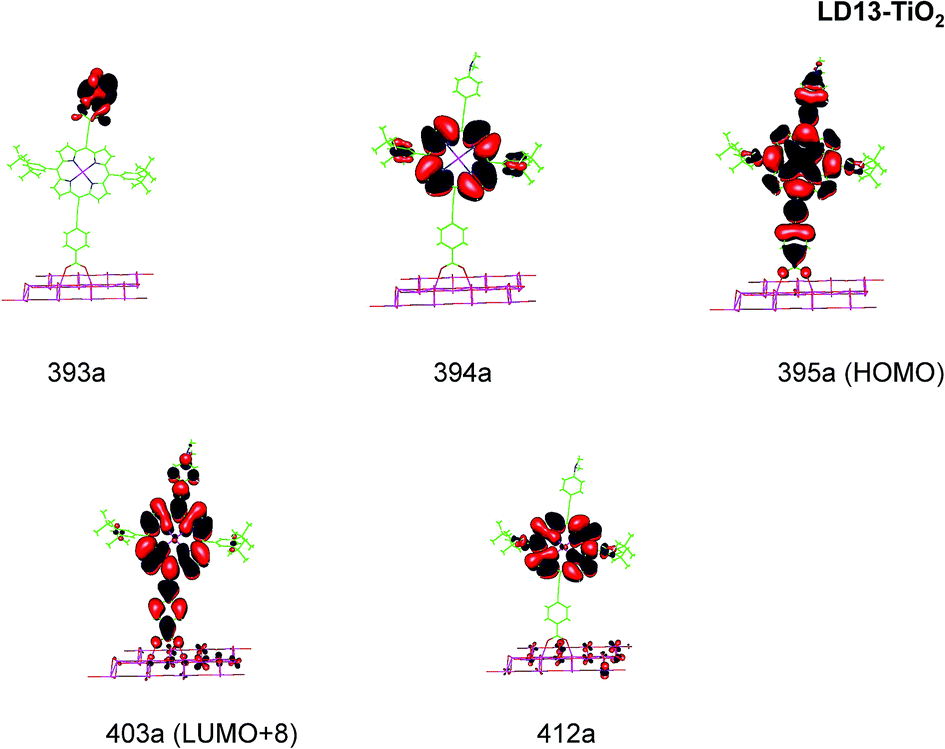 | ||
| Fig. 5 Most important active molecular orbitals in the electronic transitions of LD13–TiO2 models at the B3LYP level in THF. | ||
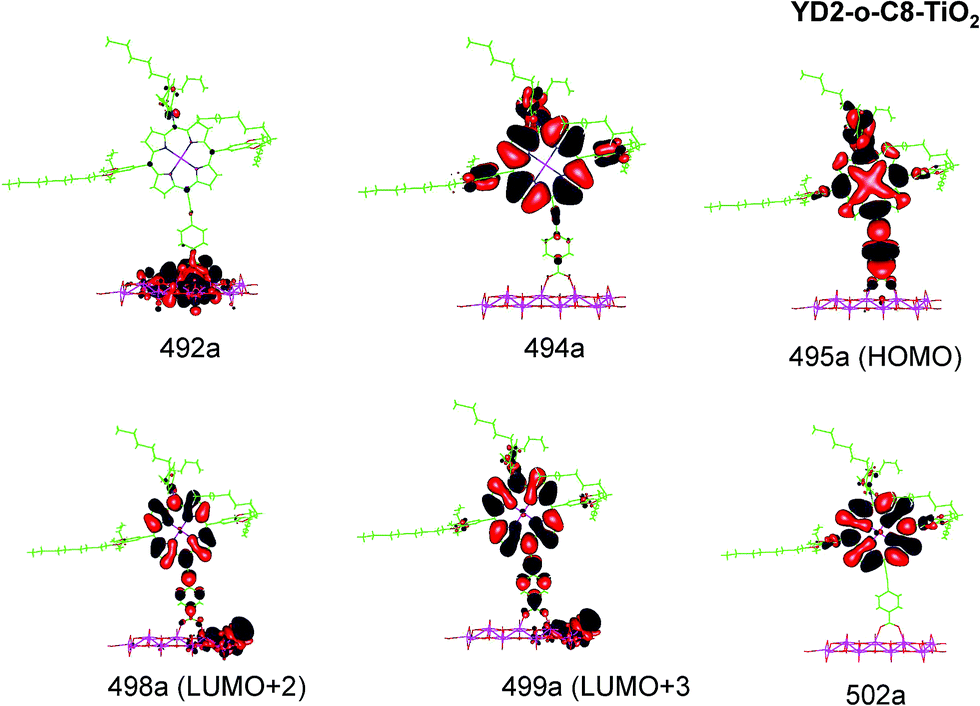 | ||
| Fig. 6 Most important active molecular orbitals in the electronic transitions of YD2-o-C8–TiO2 models at the B3LYP level in THF. | ||
LD13–TiO2
The electronic excitations of the LD13–TiO2 complex are assigned to transitions from the ground state to ligand-to-ligand (LL), metal-ligand-to-ligand (MLL), metal-ligand-to-metal-ligand (MLML) and ligand-to-metal-ligand (LML) charge transfers states using the expansion coefficients obtained in the TDDFT calculation and the composition of the most important molecular orbitals that participate in the absorption process. The excitation character of the strong bands are given in Table 2. Theoretical calculations show good agreement with the experimental results.22 We have used the wave function analysis to study the composition of each molecular orbital involved in electronic sates in the process of absorption.| System | λcalc | λexp | fa | Contributionb | Transition type |
|---|---|---|---|---|---|
| a Oscillator strength.b The reported values are |coefficient|2 × 100%. | |||||
| LD13–TiO2 | 606 (Q) | 668 | 0.929 | 395a →403a (84%) | MLMLCT (dxz/dyz + π → π* + dxz) |
| 445 (B) | 461 | 0.862 | 393a → 403a (60%) | LMLCT (π → π* + dxz) | |
| 394a → 412a (35%) | LLCT (π → π*) | ||||
| 437 (B) | 0.425 | 394a → 403a (42%) | LMLCT (π → dxz + π*) | ||
| 393a → 412a (24%) | LLCT (π → π*) | ||||
| YD2-o-C8–TiO2 | 621 (Q) | 650 | 0.532 | 495a → 499a (53%) | MLLMCT (dxz/dyz + π → π* + dTi) |
| 495a → 498a (29%) | MLLMCT (dxz/dyz + π → π* + dTi) | ||||
| 454 (B) | 470 | 0.774 | 494a → 502a (32%) | LLCT (π → π*) | |
| 492a → 499a (21%) | MLLM (π + dTi → π* + dTi) | ||||
| 492a → 498a (12%) | MLLM (π + dTi → π* + dTi) | ||||
| 447 (B) | 0.446 | 494a → 499a (30%) | LLMCT (π → π* + dTi) | ||
| 495a → 502a (19%) | MLLCT (dxz/dyz + π → π*) | ||||
| 494a → 498a (14%) | LLM (π → π* + dTi) | ||||
For the LD13–TiO2 complex, the absorption bands of the LD13–TiO2 complex are slight blue shifted as compared to the corresponding absorption bands of the free dye as also observed experimentally. The transition wave lengths, oscillator strengths and transition characters of the strong Q and B transitions are given in Table 2. The corresponding data for the free dye are reported in Table S9 in the ESI.† The calculated spectrum in Fig. 3 and the obtained TiO2 shifts are in reasonable agreement with experimental data.22 The Q band at 606 nm consists mainly of the transition between HOMO and LUMO+8 on the LD13 dye. The 395a → 403a (dxz/dyz + π → π* + dxz) transition can be assigned as an MLMLCT excitation. The 395a orbital (HOMO) has contributions from porphyrin π orbitals, Zn orbitals, and orbitals of phenylethynyl carboxylic acid moiety. The 403a orbital (LUMO+8) has mainly contributions from porphyrin and the phenylethynyl carboxylic acid moiety. The orbitals are shown in Fig. 5.
The B band at 445 nm also consists of two main orbital transitions. The first one is from 393a to 403a (π → π* + dxz), which is of LMLCT type. The two orbitals have a π character on the porphyrin ring with a small contribution from TiO2. The second significant orbital transition from 394a to 412a has LLCT character. The strong contribution to the B band at 437 nm is dominated by the 394a → 403a (π → π* + dxz) and 394a → 412a orbital transition, which are of the LMLCT and LLCT character, respectively.
YD2-o-C8–TiO2
The Q band of the calculated absorption spectrum of the YD2-o-C8–TiO2 complex is slightly redshifted as compared to LD13–TiO2, whereas in the experimental spectra YD2-o-C8–TiO2 absorbs at 650 nm as compared to 668 nm for LD13–TiO2.14,34 The absorption wave length of the B band of YD2-o-C8–TiO2 is about 10 nm longer than for LD13–TiO2. The calculated and measured absorption wave lengths are compared in Table 2. The B band is red shifted and the Q band is blue shifted with attaching YD2-o-C8 to TiO2, whereas experimentally TiO2 red shifts the Q and B bands. Since the shifts are small, there is though a reasonable agreement between calculated and experimental absorption spectra for YD2-o-C8 and YD2-o-C8–TiO2.14 The band assigned as T band for the free YD2-o-C8 dye disappears in the calculated and experimental spectra when attaching the dye to TiO2.The Q band at 621 nm consists mainly of the orbital transition from HOMO (495a) to LUMO+3 (499a) of dxz/dyz + π → π* + dTi character associated with MLLMCT. The second important orbital transition is from HOMO to LUMO+2 (498a), which is also of MLLMCT type. Orbital 495a (HOMO) has a porphyrin π character, Zn orbitals, diarylamino, and contributions from the phenylethynyl carboxylic acid moiety. Orbital 499a (LUMO+3) has the main contributions centered on the porphyrin, phenylethynyl carboxylic acid and contributions from the d orbitals of Ti on TiO2. The Zn orbitals do not contribute much to the 498a and 499a orbitals. The frontier orbitals of YD2-o-C8–TiO2 are shown in Fig. 6.
The band B is formed by electronic transitions to two excited states. The electronic excitation at 454 nm consists mainly of the 494a → 502a (π → π*) orbital transition of LLCT type. The 494a and 502a orbitals have both π orbital character on the porphyrin ring. However, the electronic transition has also significant MLLM character due to orbital transitions from 492a to 499a and to 498a. The second electronic transition of the B band at 447 nm has the largest contribution from the orbital transition between 494a and 499a (π → π* + dTi), which is of LLMCT type. Other significant orbital transitions of the second contribution to the B band are 495a → 502a and 494a → 498a, which are of MLLCT and LLM type, respectively.
The calculations show that for the dye–TiO2 complexes, orbital centered on TiO2 cluster contribute to the excited states. The orbitals on TiO2 are the electron accepting of the TiO2 electrode. The same behaviour has been observed for YD2–TiO2 (ref. 33) and LD14–TiO2 (ref. 34) complexes that we have also studied.36 Calculations of the free-energy for the electron injection process are discussed in the next section.
Absorption properties
The electron injection in DSSCs can be understood as a charge transfer (CT) process.60 Thus, the Marcus theory can be used for studying the electron transfer in DSSCs.61 The CT has been associated with the free-energy change for electron injection (ΔGinject).62 This free-energy change is a measure of the electron injection rate in DSSCs. In general, the larger the ΔGinject, the greater is the electron-injection efficiency (Φinject). The injection energy can be calculated as ΔGinject = Edye* − ECB,63 where ECB is the reduction potential of the conduction band (CB) of the TiO2. The conduction band is −3.66 eV for the used Ti16O34H4 cluster of this work. The oxidation potential of the excited state of the dye (Edye*) is determined by the redox potential of the ground state of the dye (Edye) and the vertical transition energy (λmax): Edye* = Edye − λmax. There are two methods to calculate Edye. The first is from the orbital energy of the HOMO by applying Koopmans' theorem IP ≈ −EHOMO.64 The second is the energy of the orbital that generates the transition.65,66 The calculated Edye* and ΔGinject for the Q and B bands of LD13 and YD2-o-C8 dyes are summarized in Table 3.| System | Transition | Edye* | ΔGinject | ||
|---|---|---|---|---|---|
| Q | B | Q | B | ||
| a Edye is the energy of the orbital that generates the transition in the band.b Edye is the absolute value of the HOMO energy. | |||||
| LD13 | Typea | 3.17 | 2.76 | −0.49 | −0.90 |
| LD13 | Typeb | 3.17 | 2.24 | −0.49 | −1.42 |
| YD2-o-C8 | Typea | 3.18 | 2.18 | −0.48 | −1.48 |
| YD2-o-C8 | Typeb | 3.18 | 2.18 | −0.48 | −1.48 |
The results for the YD2-o-C8 dye show that Edye* for the Q and B bands do not depend on whether the energy of the transition orbital or the HOMO energy is used. The ΔGinject values are negative, which means that the excited states of YD2-o-C8 with an effective charge transfer excitation character lies above the conduction band edge of TiO2. The calculated Edye* and ΔGinject energies of the Q and B bands for the LD13 dye are listed in Table 3. Even though the magnitudes vary, the favorable electron injection to TiO2 remains. The YD2-o-C8 dye (η = 12.1%)14 has a larger charge injection efficiency than to LD13 (η = 8.4%),22 which is also obtained in the calculations. The large free energy for the electron injection implies that the electron injection is fast as also obtained experimentally.13,65
Conclusions
In this work, we have presented a study of the electronic structures and spectroscopic properties of the dyes LD13 and YD2-o-C8, both free and adsorbed on TiO2 clusters. The coordination energy between the –COO− anchor group and TiO2 lies between 66 and 70 kcal mol−1 depending on the calculation level, showing a coordinative bond between dyes and cluster of TiO2. The charge transfer analysis shows that dye injects electrons to the TiO2 cluster. The obtained spectra at the B3LYP-D3 TDDFT level demonstrate that this is the most suitable functional for describing the Q and B bands of porphyrins in the dye free and dye–TiO2 models. The absorption energy is centered on the dye. The LUMO of the dye is delocalized in the region of the TiO2 cluster conduction band. Finally, using the Marcus theory for electron transfer, the charge transfer can be associated with the free energy change for electron injection (ΔGinject): i.e. dye and dye–TiO2 models show a large absolute value, generating greater value for YD2-o-C8 in the same experimental trend as the value η.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of this work under Conicyt-Aka-ERNC-001 and Fondecyt 1140503, is gratefully acknowledged. The authors thank CSC – IT Center for Science in Finland for computational resources. This work has also been supported by Magnus Ehrnrooth Foundation and the Academy of Finland through projects 266227 and 275845. RM-A Acknowledges financial support from CONICYT-USACH under Proyecto de Atracción e Inserción de Capital Humano Avanzado en la Academia No. 79150043.References
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef PubMed.
- G. K. Singh, Energy, 2013, 53, 1–350 CrossRef.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- H. Imahori, T. Umeyama and S. Ito, Acc. Chem. Res., 2009, 42, 1809–1818 CrossRef CAS PubMed.
- K. S. Gallagher, Daedalus, 2013, 142, 59–77 CrossRef.
- F. De Angelis, S. Fantacci, E. Mosconi, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. C, 2011, 115, 8825–8831 CAS.
- F. Gajardo, M. Barrera, R. Vargas, I. Crivelli and B. Loeb, Inorg. Chem., 2011, 50, 5910–5924 CrossRef CAS PubMed.
- M. Pastore, S. Fantacci and F. De Angelis, J. Phys. Chem. C, 2013, 117, 3685–3700 CAS.
- D. Casanova, F. P. Rotzinger and M. Grätzel, J. Chem. Theory Comput., 2010, 6, 1219–1227 CrossRef CAS.
- J. Feng, Y. Jiao, W. Ma, M. K. Nazeeruddin, M. Grätzel and S. Meng, J. Phys. Chem. C, 2013, 117, 3772–3778 CAS.
- M. Urbani, M. Grätzel, M. K. Nazeeruddin and T. Torres, Chem. Rev., 2014, 114, 12330–12396 CrossRef CAS PubMed.
- A. Yella, H. W. Lee, H. N. Tsea, C. Y. Yi, A. K. Chandirian, M. K. Nazeeruddin, E. W. G. Diau, C. Y. Yeh, S. M. Zakeeruddin and M. Grätzet, Science, 2011, 334, 629–634 CrossRef CAS PubMed.
- Y. Chang, C. Wang, Y.-T. Pan, H. Kuo, C. Lo, H. Hsu, C. Lin and E. W. G. Diau, Chem. Commun., 2011, 47, 8910–8912 RSC.
- N. K. Subbaiyan, J. P. Hill, K. Ariga, S. Fukuzumi and F. D'Souza, Chem. Commun., 2011, 47, 6003–6005 RSC.
- T. Bessho, S. M. Zakeeruddin, C.-Y. Yeh, E. W.-G. Diau and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6646–6649 CrossRef CAS PubMed.
- L.-L. Li and W. W.-G. Eiau, Chem. Soc. Rev., 2013, 42, 291–304 RSC.
- K. Kurotobi, Y. Toude, K. Kawamoto, Y. Fujimori, S. Ita, P. Chabera, V. Sundström and H. Imahori, Chem.–Eur. J., 2013, 19, 17075–17081 CrossRef CAS PubMed.
- Y. Chi, K.-L. Wu and T.-C. Wei, Chem.–Asian J., 2015, 10, 1098–1115 CrossRef CAS PubMed.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 1–6 CrossRef PubMed.
- T. Le Bahers, E. Brémond, I. Ciofini and C. Adamo, Phys. Chem. Chem. Phys., 2014, 16, 14435–14444 RSC.
- C. L. Wang, C. M. Lan, S. H. Hong, Y. F. Wang, T. Y. Pan, C. W. Chang, H. H. Kuo, M. Y. Kuo, E. W. G. Diau and C. Y. Lin, Energy Environ. Sci., 2012, 5, 6933–6940 CAS.
- T. Le Bahers, T. Pauporté, P. P. Lainé, F. Labat, C. Adamo and I. Ciofini, J. Phys. Chem. Lett., 2013, 4, 1044–1050 CrossRef CAS PubMed.
- F. Risplendi, G. Cicero, G. Mallia and N. H. Harrison, Phys. Chem. Chem. Phys., 2013, 15, 235–243 RSC.
- J. Chen, F.-Q. Bai, J. Wang, L. Hao, Z.-F. Xie, Q.-J. Pan and H.-X. Zhang, Dyes Pigm., 2012, 94, 459–468 CrossRef CAS.
- S. Liu, H. Fu, Y. Cheng, S. Wu, S. Ho, Y. Chi and P. Chou, J. Phys. Chem. C, 2012, 116, 16338–16345 CAS.
- X. Zarate, E. Schott, T. Gomez and R. Arratía-Pérez, J. Phys. Chem. A, 2013, 117, 430–438 CrossRef CAS PubMed.
- E. Mosconi, J. Yum, C. Gómez, M. Nazeeruddin, M. Grätzel and F. De Angelis, J. Am. Chem. Soc., 2012, 134, 19438–19453 CrossRef CAS PubMed.
- T. Gomez, X. Zarate, E. Schott and R. Arratia-Perez, RSC Adv., 2014, 4, 9639–9646 RSC.
- W. Li, Z. Liu, H. Wu, Y. B. Cheng, Z. Zhao and H. He, J. Phys. Chem. C, 2015, 119, 5265–5273 CAS.
- S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242–247 CrossRef CAS PubMed.
- N. Klein-Kedem, D. Cahen and G. Hodes, Acc. Chem. Res., 2016, 49, 347–354 CrossRef CAS PubMed.
- F. Mendizabal, A. Lopéz, R. Arratia-Pérez, N. Inostroza and C. Linarres-Flores, J. Mol. Model., 2015, 21, 226–236 CrossRef PubMed.
- F. Mendizabal, A. Lopéz, R. Arratia-Pérez and G. Zapata-Torres, Comput. Theor. Chem., 2015, 1070, 117–125 CrossRef CAS.
- R. Mera-Adasme, W. H. Xu, D. Sundholm and F. Mendizabal, Phys. Chem. Chem. Phys., 2016, 18, 27877–27884 RSC.
- K. Paredes-Gil, F. Mendizabal, D. Páez-Hernández and R. Arratia-Pérez, Comput. Mater. Sci., 2017, 126, 514–527 CrossRef CAS.
- J. Burschka, N. Pellet, S. J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Grätzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 2014, 140, 18A301 CrossRef PubMed.
- T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- A. Klamt and G. Schüürman, J. Chem. Soc., Perkin Trans. 2, 1993, 5, 799–805 RSC.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS.
- M. E. Casida and M. Huix-Rotllant, Annu. Rev. Phys. Chem., 2012, 63, 287–323 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- J. Schirmer, Phys. Rev. A, 1982, 26, 2395–2416 CrossRef CAS.
- C. H. Hatting, Adv. Quantum Chem., 2005, 50, 37–60 CrossRef.
- Y. Jung, C. Rohini, D. Anthony and M. Head-Gordon, J. Chem. Phys., 2004, 121, 9793–9802 CrossRef CAS PubMed.
- Y. M. Rhee and M. Head-Gordon, J. Phys. Chem. A, 2007, 111, 5314–5326 CrossRef CAS PubMed.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- F. Furche, R. Ahlrichs, C. Häting, W. Klopper, M. Sierka and F. Weigend, Wire, 2014, 4, 91–100 CAS.
- Gaussian 09, Inc., Pittsburgh PA, 2003 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- T. Dunning and P. Hay, in Modern Theoretical Chemistry, ed. H. Schaefer, Plenum Press, 1997, vol. 3, pp. 1–28 Search PubMed.
- A. Bergner, M. Dolg, W. Küchle, H. Stoll and H. Preuss, Mol. Phys., 1993, 80, 1431–1441 CrossRef CAS.
- S. Huzinaga, J. Chem. Phys., 1965, 52, 1293–1301 CrossRef.
- W. Hujo and S. Grimme, J. Chem. Theory Comput., 2011, 7, 3866–3871 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- D. Sundholm, Phys. Chem. Chem. Phys., 2000, 2, 2275–2281 RSC.
- L.-H. Han, C.-R. Zhang, J. W. Zhe, N.-Z. Jin, Y.-L. Shen, W. Wang, J. J. Gong, Y.-H. Chen and Z.-J. Liu, Int. J. Mol. Sci., 2013, 14, 20171–20188 CrossRef PubMed.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599–610 CrossRef CAS.
- W. Fan, D. Tan and W.-Q. Deng, ChemPhysChem, 2012, 13, 2051–2060 CrossRef CAS PubMed.
- J. Preat, C. Michaux, D. Jacquemin and E. A. Perpete, J. Phys. Chem. C, 2009, 113, 16821–16833 CAS.
- T. Koopmans, Physica, 1934, 1, 104–113 CrossRef.
- P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793–1874 CrossRef CAS PubMed.
- M. Torrent-Sucarrat, F. De Proft, P. Geerlings and P. W. Ayers, Chem.–Eur. J., 2008, 14, 8652–8660 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1 and S2 contain main geometric parameters of the LD13, YD2-o-C8, LD13–TiO2 and YD2-o-C8–TiO2. Tables S3 and S4 show Natural Population Analysis (NPA) charge on the systems. Tables S5–S8 show the electronic transitions to different methodologies (B3LYP, CAM-B3LYP, BHHLYP and ADC(2)) in gas and solvent phases. Table S9 described the singlet excitation energies calculated for LD13 and YD2-o-C8 in THF. Fig. S1 and S2 calculated B3LYP electronic spectra of LD13 and YD2-o-C8 in THF. Fig. S3 and S4 active molecular orbitals in the electronic transitions of LD13 and YD2-o-C8 at the B3LYP level in a solvent. Fig. S5 and S6 shows frontier orbitals on TiO2 cluster in systems LD13–TiO2 and YD2-o-C8–TiO2 at the B3LYP level. Tables S10–S17 Cartesian coordinates (in angstroms) for the optimized geometries of the systems studied in this work at the B3LYP level. See DOI: 10.1039/c7ra08648b |
| This journal is © The Royal Society of Chemistry 2017 |