
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConversion of glycals into vicinal-1,2-diazides and 1,2-(or 2,1)-azidoacetates using hypervalent iodine reagents and Me3SiN3. Application in the synthesis of N-glycopeptides, pseudo-trisaccharides and an iminosugar†
Ande Chennaiah,
Srijita Bhowmick‡
and
Yashwant D. Vankar *
*
Department of Chemistry, Indian Institute of Technology, Kanpur – 208016, India. E-mail: vankar@iitk.ac.in
First published on 29th August 2017
Abstract
Glycals were found to react with a reagent system comprising of phenyliodine bis(trifluoroacetate) (PIFA) and Me3SiN3 in the presence of TMSOTf as a catalyst to form the corresponding vicinal 1,2-diazides. On the other hand, they reacted with another reagent system phenyliodine diacetate (PIDA) and Me3SiN3, also in the presence of TMSOTf as a catalyst, to lead to the corresponding vicinal 1,2-azidoacetates. These azido derivatives were converted into a number of 2-azido-N-glycopeptides, pseudotrisaccharides, and a piperidine triol derivative, an iminosugar.
Introduction
The growing importance of aminosugars,1 N-glycopeptides2 and sugar derived triazoles3 (obtained via click-chemistry) in biological systems is apparent from the recent literature. Synthesis of such compounds has led to the development of a number of synthetic methodologies involving functionalization of monosaccharides, in particular glycals. In this regard, methodologies that allow attachment of an azido moiety4 at the anomeric center are frequently used in the synthesis of N-glycopeptides, although there are some other ways2e,5 of introducing a ‘nitrogen’ unit. The azide group also allows click-chemistry, to link with another sugar unit leading to pseudo di-, tri- or oligosaccharides, to be carried out.3 Apart from this, introduction of an amino functionality (or an azido group) at the C-2 position of a sugar moiety, with an appropriate functional group at the anomeric carbon for effecting glycosylation, is one of the most important reactions known in carbohydrate chemistry.1a,6 This allows access to amino sugars with the amino group at the C-2 position, a structural feature that is prevalent in a number of oligosaccharides including aminoglycoside antibiotics. Toward this endeavor, conversion of glycals into sugar derived 2-azido-1-nitrates using a combination of ceric ammonium nitrate and NaN3 (and its improvements) in CH3CN, a protocol developed by Lemieux et al.,6a is universally followed. At the same time, there exist many oligosaccharides that contain N-glycopeptide units in which the sugar moiety carries a C-2 amino functionality, and the glycopeptide part has an Asn-linkage at the anomeric carbon.2b,7 Interest in the synthesis of such molecules stems from the fact that protein N-glycosylation occurs during post-translational modifications in biological systems. This has led to newer approaches towards functionalization of glycals (or any other sugar derivative such as glycosamines) to introduce 1,2-diamino (or equivalent) functionalities that are useful in the synthesis of N-glycopeptides.8 Thus, improved or alternate approaches toward the introduction of a nitrogen based functional group at C-1 and/or C-2 of a sugar moiety are important. In view of our continued interest2f,4a–c,8a,9 in functionalizing glycals en route to the synthesis of iminosugars, aminosugars and glycopeptides, we herein report an easy access to 1,2-diazido sugars from glycals upon treatment with phenyliodine bis(trifluoroacetate) (PIFA) and Me3SiN3 reagent system. Also, we have developed conditions that allow conversion of glycals into 1-azido-2-acetoxy sugars or 2-azido-1-acetoxy sugars upon reaction with phenyliodine diacetate (PIDA) and Me3SiN3. Further, we have demonstrated the utility of these azido compounds in the synthesis of 2-amino-N-glycopeptides, pseudotrisaccharides, and an iminosugar.Results and discussion
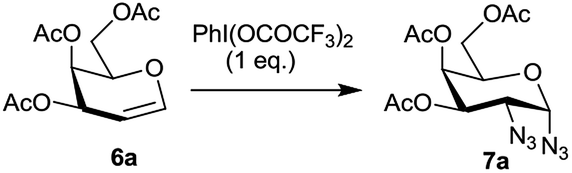
Hypervalent iodine reagents have gained enormous importance in recent years due to their low toxicity, ease of handling and ready availability.10 The use of hypervalent iodine reagents, most notably PIDA, in conjunction with a halide ion source has been reported on a few occasions in functionalizing olefins, including glycals, to form vicinal halo alkoxides in an intermolecular fashion.11 Introduction of a nitrogen based functional group at C-2 of a glucal derivative has also been realized in an intramolecular fashion from glucal 3-carbamate upon its reaction with PIDA along with Rh2(OAc)4.6c However, one of the most interesting reactions to stereoselectively convert glycals into sugar derived trans-1,2-diacetates involves their reaction with PIDA in presence of BF3·Et2O as a catalyst as reported by Gin et al.12 Further, the in situ formed trans-1,2-diacetates lead to O-glycosylation when treated with an alcohol (ROH) and TfOH (as a catalyst) in the same pot. The proposed mechanism suggests that PIDA acts as an electrophile making a π-complex with a glycal, followed by the attack of acetate ion at the anomeric carbon as well as at C-2 leading to the observed trans-1,2-diacetates. In view of this, and in view of the importance of amino sugars and N-glycopeptides (vide supra) we surmised that reaction of glycals with PIDA combined with Me3SiN3 may lead to 1,2-diazido sugars. Our initial experiments involving reaction of 3,4,6-tri-O-benzyl galactal 1 (Scheme 1) with both one equivalent of PIDA and Me3SiN3 in the presence of BF3·Et2O or TMSOTf (30 mol%) as a catalyst led to a mixture of four products. These were 1-azido-2-acetoxy sugar 2, 2-azido-1-acetoxy sugar 3; 1,2-diacetoxy sugar 4 and 1,2-diazido sugar 5. Increasing the amount of Me3SiN3 did not lead to increased formation of 1,2-diazides and there was always an incorporation of the acetate moiety onto galactal 1 due to competition between the azide and the acetate ions. Although, conversion of olefins into vicinal diazides is known with few reagents13a such as Mn(OAc)3–NaN3,13b azido iodine(III) reagent,13c NaIO4/NaN3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13d and Zhdankin reagent (a hypervalent iodine reagent) along with a copper catalyst,13e formation of sugar derived 1,2-diazides from glycals has been reported rarely.13b,14 Thus, for example, 2,4,6-tri-O-acetyl galactal has been reported to react with Me3SiN3–PIDA–PhSeSePh14a to give cis-1,2-diazido galactal albeit in low yield and as a side product. On the other hand, more recently Xu et al.14b demonstrated that the in situ prepared Zhdankin reagent in presence of an iron catalyst converts a broad range of olefins, including one example of a glucal derivative, to the corresponding 1,2-diazides. In order to find an alternate route to convert glycals into sugar derived 1,2-diazides, we explored their reactivity with PIFA–Me3SiN3 reagent system15 in presence of an acid catalyst. Such a reagent system has been used to introduce an azide group into aromatics and some heterocycles akin to aromatic substitution,15a,b for substitution of an azide moiety15c at benzylic positions, and more recently to perform C–H activation.15d,e Apart from this, an interesting intramolecular azidoarylation of alkenes using PIFA–Me3SiN3 combination has been reported by Antonchick et al.15f In these examples, PIFA–Me3SiN3 reagent system was proposed to lead to the formation of PhI(N3)2 or PhIN3(OCOCF3) as intermediate species which act as a source of azide radical to effect the observed reactions. We expected that if PhI(N3)2 is formed as an intermediate, in presence of an acid catalyst it may convert glycals to sugar-derived 1,2-diazides in an analogous manner as sugar derived 1,2-diacetates were obtained from glycals upon reaction with PIDA as reported by Gin et al.12 Towards this endeavor, initial experiments involved treatment of 3,4,6-tri-O-acetyl galactal 6a with PIFA (1.0 eq.) and 2 eq. of Me3SiN3 in presence of BF3·Et2O (30 mol%) at −30 °C which gave only 38% of the expected 1,2-diazide 7a as a single isomer. Further optimizations with respect to azide source and acid catalyst (Table 1) led to the use of 3 eq. of TMSN3 and 30 mol% of TMSOTf at −30 °C as the best condition forming 7a in 61% yield (entry 7, Table 1). With this optimized condition we carried out the remaining studies. Thus, a variety of differently protected glycal derivatives led to the corresponding 1,2-diazides in moderate yields (Scheme 2).
13d and Zhdankin reagent (a hypervalent iodine reagent) along with a copper catalyst,13e formation of sugar derived 1,2-diazides from glycals has been reported rarely.13b,14 Thus, for example, 2,4,6-tri-O-acetyl galactal has been reported to react with Me3SiN3–PIDA–PhSeSePh14a to give cis-1,2-diazido galactal albeit in low yield and as a side product. On the other hand, more recently Xu et al.14b demonstrated that the in situ prepared Zhdankin reagent in presence of an iron catalyst converts a broad range of olefins, including one example of a glucal derivative, to the corresponding 1,2-diazides. In order to find an alternate route to convert glycals into sugar derived 1,2-diazides, we explored their reactivity with PIFA–Me3SiN3 reagent system15 in presence of an acid catalyst. Such a reagent system has been used to introduce an azide group into aromatics and some heterocycles akin to aromatic substitution,15a,b for substitution of an azide moiety15c at benzylic positions, and more recently to perform C–H activation.15d,e Apart from this, an interesting intramolecular azidoarylation of alkenes using PIFA–Me3SiN3 combination has been reported by Antonchick et al.15f In these examples, PIFA–Me3SiN3 reagent system was proposed to lead to the formation of PhI(N3)2 or PhIN3(OCOCF3) as intermediate species which act as a source of azide radical to effect the observed reactions. We expected that if PhI(N3)2 is formed as an intermediate, in presence of an acid catalyst it may convert glycals to sugar-derived 1,2-diazides in an analogous manner as sugar derived 1,2-diacetates were obtained from glycals upon reaction with PIDA as reported by Gin et al.12 Towards this endeavor, initial experiments involved treatment of 3,4,6-tri-O-acetyl galactal 6a with PIFA (1.0 eq.) and 2 eq. of Me3SiN3 in presence of BF3·Et2O (30 mol%) at −30 °C which gave only 38% of the expected 1,2-diazide 7a as a single isomer. Further optimizations with respect to azide source and acid catalyst (Table 1) led to the use of 3 eq. of TMSN3 and 30 mol% of TMSOTf at −30 °C as the best condition forming 7a in 61% yield (entry 7, Table 1). With this optimized condition we carried out the remaining studies. Thus, a variety of differently protected glycal derivatives led to the corresponding 1,2-diazides in moderate yields (Scheme 2).
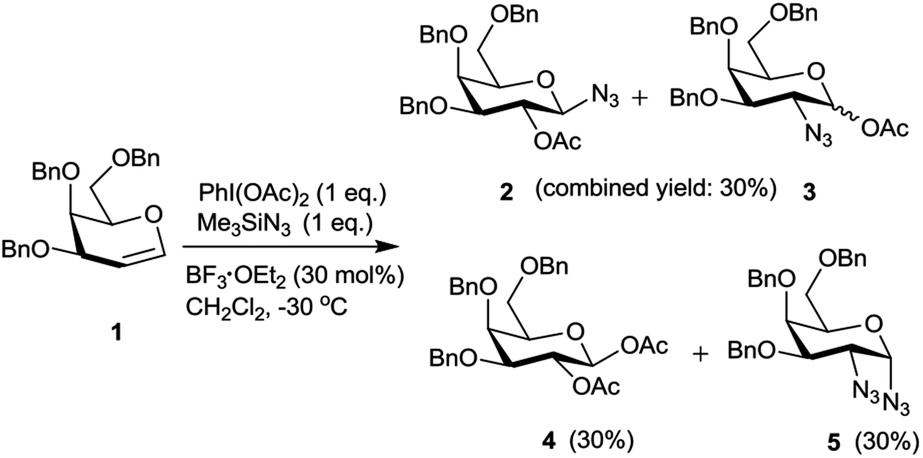 | ||
| Scheme 1 Exploratory reaction of galactal derivative 1 with PIDA–Me3SiN3. | ||
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | N3-Source | Equiv. | Solvent | Temp (°C) | Time (h) | Yield (%) |
| a Reaction conditions: glycals (0.36 mmol), TMSN3, PhI(OCOCF3)2 (0.36 mmol), TMSOTf (0.10 mmol), CH2Cl2, −30 °C, N2 atmosphere, isolated yields after purification by silica gel column chromatography.b Yield based on recovered starting material. | |||||||
| 1 | None | NaN3 | 2 | CH3CN | rt | 24 | No reaction |
| 2 | None | TMSN3 | 2 | CH2Cl2 | 0 | 24 | No reaction |
| 3 | BF3·OEt2 | TMSN3 | 2 | CH2Cl2 | −30 | 0.5 | 38b |
| 4 | BF3·OEt2 | TMSN3 | 3 | CH2Cl2 | −30 | 0.5 | 44b |
| 5 | BF3·OEt2 | Bu4NN3 | 3 | CH2Cl2 | −30 | 0.5 | 37b |
| 6 | TMSOTf | Bu4NN3 | 3 | CH2Cl2 | −30 | 0.5 | 33b |
| 7 | TMSOTf | TMSN3 | 3 | CH2Cl2 | −30 | 0.5 | 61 |
 | ||
| Scheme 2 Percentage yield and α/β ratio of the 1,2-diazides. | ||
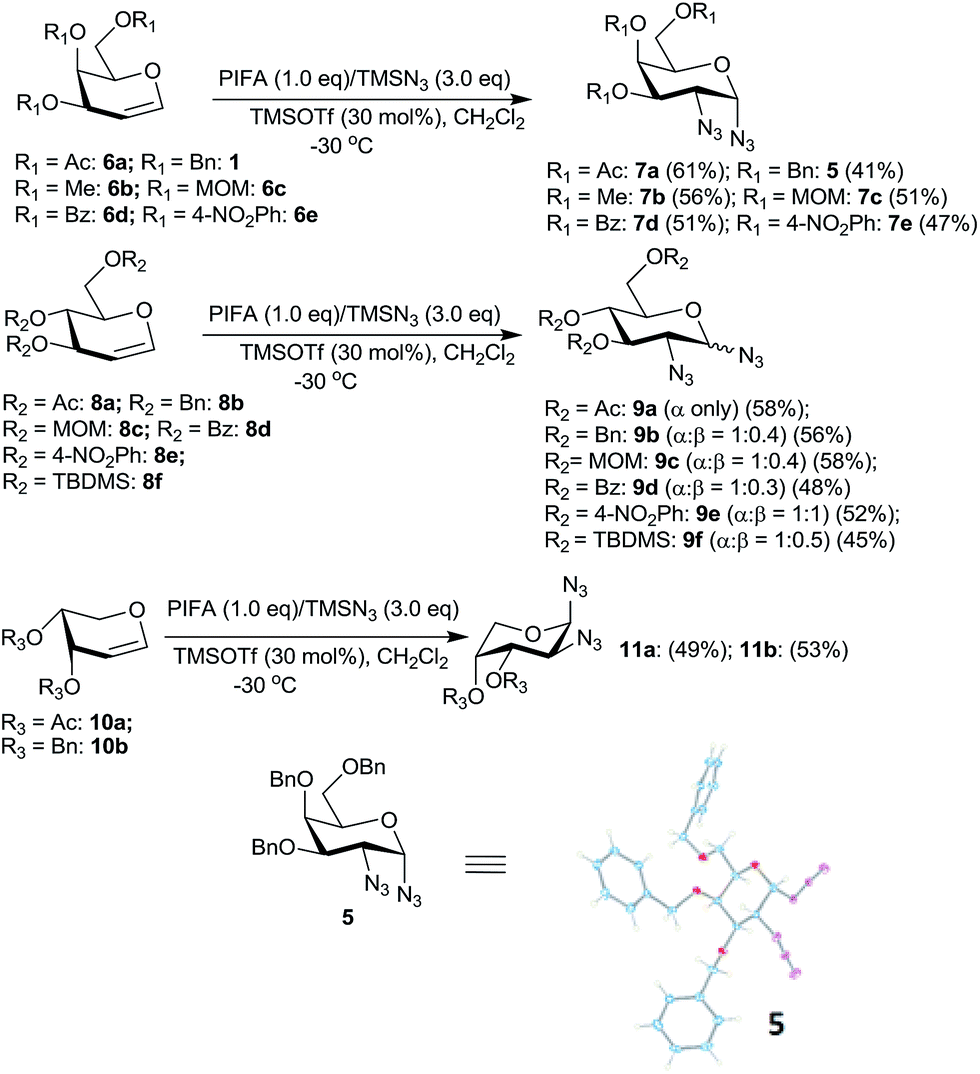
It was generally found that while the galactal derivatives led exclusively to cis-1,2-diazides, the glucal derivatives gave a mixture of α- and β-anomers in varied ratios with the α-anomer being major in most of the cases. The structures and the α/β ratios were established based on COSY, NOE, homonuclear decoupling and DEPT experiments.16a Thus, for example, in the 1H NMR spectrum of compound 7a the anomeric proton (H-1) appeared as a doublet at δ 5.48 with J = 4.12 Hz. The homonuclear decoupling of H-1 led the proton H-2 to appear as a doublet, from a doublet of doublet, with J = 11.00 Hz indicating that H-2 and H-3 are trans-diaxial. Further, in the NOE experiments no enhancement was observed for protons H-3 and H-5 when H-1 was irradiated and vice versa indicating that H-1 is equatorially oriented. In addition, the structure of the galactal derivative 5 was proved by single crystal X-ray analysis.16b Likewise, the spectral data of 1,2-diazide 9a, derived from glucal 8a, also showed that the azido moieties are α-oriented.16a Thus, in NOE experiments, irradiation of proton H-4 at δ 5.00 led to the enhancement of signal for H-2 at δ 3.61 and vice versa indicating that H-2 is axially oriented. On the other hand, no enhancement was observed for H-3 and H-5 protons when H-1 was irradiated at δ 5.44 suggesting that H-1 is equatorially oriented.
Interestingly, the arabinal derivatives 10a and 10b gave the corresponding 1,2-diazides 11a and 11b having 1C4 conformations which was confirmed by spectral studies.16a In case of 11a, irradiation of H-1 at δ 5.43 in the homonuclear decoupling experiment led the doublet of doublet for H-2 to appear as a doublet with J = 10.6 Hz indicating that H-2 and H-3 are diaxially oriented. Likewise, irradiation of H-4 at δ 5.27 caused the doublet of doublet for H-3 at δ 5.13 to appear as a doublet with J = 10.6 Hz. Further, in NOE experiments irradiation of H-3 did not lead to the enhancement of the signal for H-1, and also irradiation of H-2 did not show the enhancement of H-4. These data confirm that H-2 and H-3 are diaxially oriented and that 11a possesses 1C4 conformation. In a similar fashion, the structure of 11b was established.
It is important to note that in the absence of an acid catalyst no reaction was found to take place between a glycal derivative and PIFA–Me3SiN3 reagent combination indicating that the azido radical, if formed,15 does not add on to the electron rich glycal double bond. Therefore we presume that in the present case, the reaction does not proceed via radical pathway,17a instead it proceeds via ionic pathway similar to the ones proposed by Moriarty,17b and Kirschning.17c Thus, in situ generated PhI(N3)2 upon π-interaction (complex A in Scheme 3) with the double bond from the α-side (in case of a galactal and a glucal derivative) leads to intermediate B and transfer of the azide moiety to C-2 occurs in an SNi fashion from the α-side only. Following this (or simultaneously) the second azide preferentially attacks at the anomeric carbon of the galactal derivative from the α-side (path a) due to steric hindrance caused by the substituents at C-3, C-4 and C-5. In case of glucal derivatives, however, some product was also formed due to β-attack of the azide moiety via path b since the glucal derivatives show less steric bias from the β-side as compared to galactal derivatives.
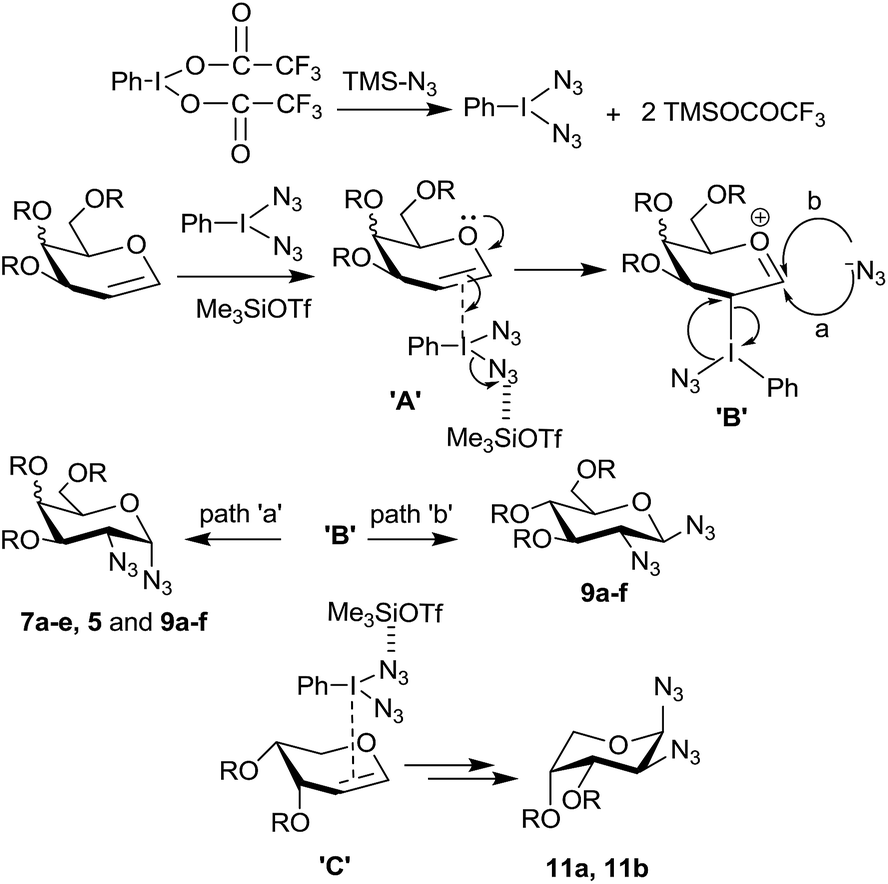 | ||
| Scheme 3 Proposed mechanism for the formation of 1,2-diazides. | ||
Further, in case of arabinal derivatives it appears that the double bond preferentially forms π-complex from the less hindered β-side (complex C), followed by azide ion attachments at C-2 and C-1 from the opposite face to the two –OR groups, in a similar manner as happens in galactal cases, eventually leading to the observed 1,2-diazides 11a and 11b (Scheme 3) which assume 1C4 conformations. It is clear that the success of the 1,2-diazide formation is mainly due to the low nucleophilicity of the trifluoroacetate ion compared to the azide ion.
In order to demonstrate the utility of these 1,2-diazides, we have utilized the click chemistry to prepare two bis-triazoles, one from a non-sugar and the other from a sugar based alkyne. Thus, 1,2-diazide 5 was reacted with 2 eq. of phenylacetylene 12 to form the bis-triazole 13 (Scheme 4) in 80% yield using standard conditions.3d Likewise, a sugar derived alkyne18 14 led to the corresponding bis-triazole 15, a pseudotrisaccharide,3 in 75% yield. The structures of these triazoles were confirmed from their spectral data.16a
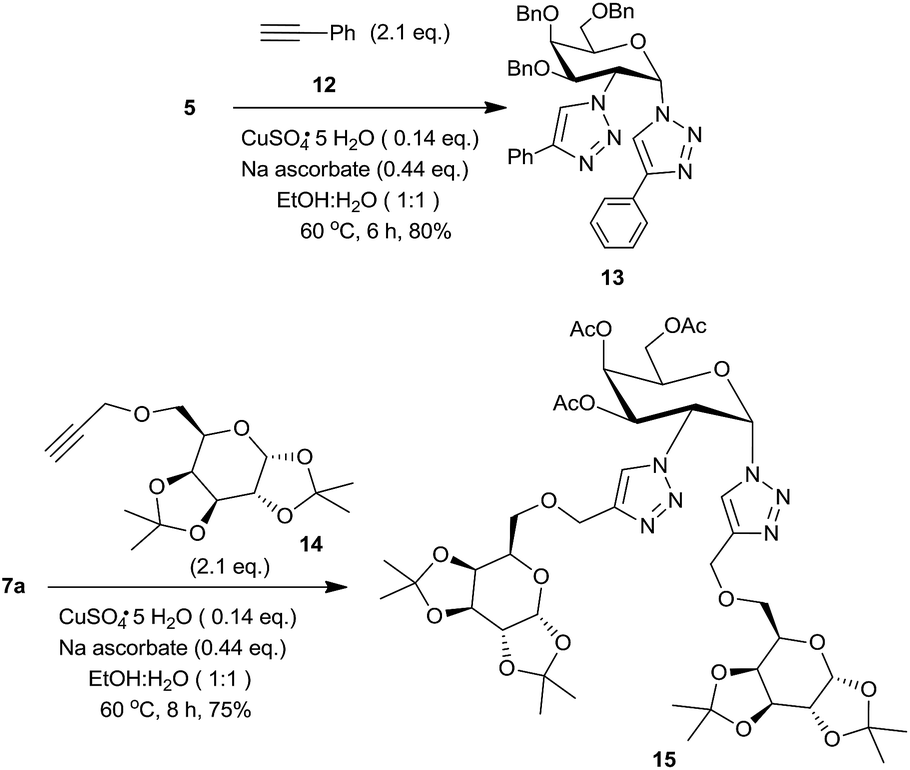 | ||
| Scheme 4 Conversion of 1,2-diazides into bis-triazole 13 and pseudotri-saccharide 15. | ||
The importance of N-glycopeptides is apparent from the introduction part (vide supra) and a number of synthetic approaches have been reported to procure them, and newer synthetic routes are still being developed. In view of this, we demonstrate the utility of 1,2-diazides 7a and 9a to form the corresponding 2-azido-N-glycopeptides 18–23 (Scheme 5). Thus, selective reduction of the anomeric azide of 7a and 9a was carried out with ammonium tetrathiomolybdate [(NH4)2MoS42−]19 to give the corresponding amino compounds 16 and 17 and, without isolating them, the crude amines were coupled with a number of amino acids and small peptides, mediated by 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)20a to lead to the corresponding 2-azido-N-glycopeptides 18–23. The stereochemistry of the 2-azido-N-glycopeptides was confirmed to be 1,2-trans based on COSY, HETCOR and homonuclear decoupling experiments16a of 18. Thus, selective decoupling of C1NH at δ 6.05 led the anomeric proton to appear as a doublet with coupling constant J = 9.96 Hz indicating that both H-1 and H-2 are axial. The facile mutarotation of free anomeric amines has been reported in many cases4b,20b,c and their functionalization mainly leads to the β-anomers. It is therefore not surprising that in the present case, reduction of α-azides to the corresponding free amines exclusively lead to β-glycopeptides via mutarotation. The azido moiety at C-2 of the azido-glycopeptide 19 was readily reduced21 with Zn/AcOH/Ac2O to form the 2-amino-N-glycopeptide 24 in 85% yield, and thus the present method forms an alternate route to 2-amino-N-glycopeptides.
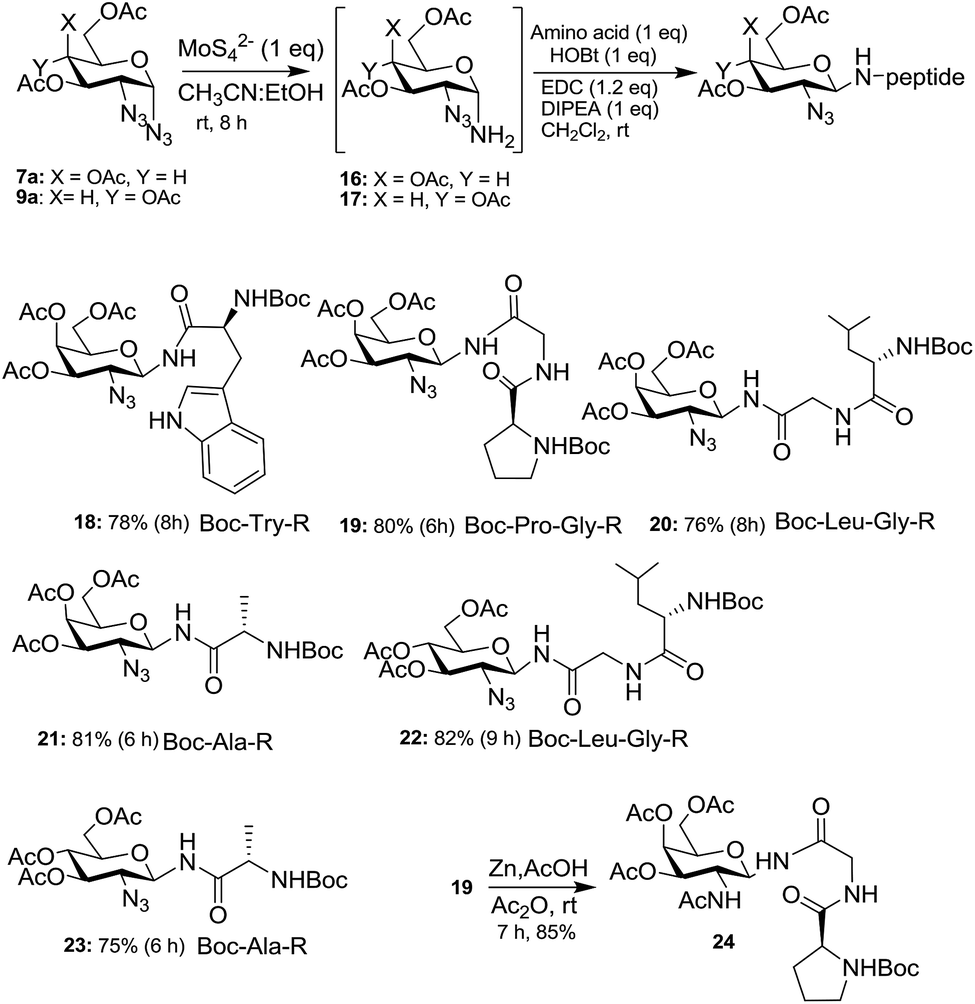 | ||
| Scheme 5 Selective reduction of 1,2-diazides and synthesis of β-N-glycopeptides. | ||
Having explored the conversion of glycals to 1,2-diazides using PIFA–Me3SiN3 reagent system, we studied their reactivity with PIDA–Me3SiN3 under analogous conditions. As discussed above, the galactal derivative 1 (Scheme 1) upon reaction with PIDA and Me3SiN3 (both 1 eq.) gave a mixture of four products 2–5 and thus it was not a synthetically useful reaction. We therefore examined the reaction conditions to incorporate both, the acetate (more nucleophilic compared to trifluoroacetate ion) as well as the azide moieties onto glycals. Our aim was to procure synthetically useful 1-azido-2-acetoxy sugars and/or 2-azido-1-acetoxy sugars, and to avoid the formation of 1,2-diacetates and 1,2-diazides. After exploring various combinations of the azide source and Lewis acids (Table 2), it became clear that a combination of 3 eq. of Me3SiN3 and 1 eq. of PIDA along with 30 mol% of TMSOTf at −30 °C was optimum to form vicinal azidoacetates. Thus, galactal derivative 1 led to the formation of an inseparable mixture of two products, 1-azido-2-acetoxy sugar 2 and 2-azido-1-acetoxy sugar 3 in 61% combined yield (Scheme 6). The two compounds were formed in 1:2 ratio which was ascertained after selectively reducing the mixture of 2 and 3 with ammonium tetrathiomolybdate (vide supra) followed by acetylation that gave chromatographically separable 25 and unreacted 3.22a Further, 2-azido-1-acetoxy sugar 3 was found to be a mixture of two anomers (α/β = 63/37). The glucal derivative 8b, on the other hand, gave a chromatographically separable mixture of 2616a and 2722a (α/β anomers = 60/40) in 1:2 ratio. Compound 26 was again reduced and acetylated to form 2822b which was spectroscopically characterized. Interestingly, the arabinal derivative 10b (ref. 23) led to a single product 29 in 56% yield whose structure was established based on spectral data and also by single crystal X-ray analysis.16b
| Entry | Catalyst | N3-Source | Equiv. | Solvent | Temp (°C) | Time (h) | Yield (%) | ||
|---|---|---|---|---|---|---|---|---|---|
| (2 + 3) | 4 | 5 | |||||||
| a Reaction conditions: glycals (0.36 mmol), TMSN3 (1.10 mmol), PhI(OAc)2 (0.36 mmol), TMSOTf (0.10 mmol), CH2Cl2, −30 °C, N2 atmosphere, isolated yields after purification by silica gel column chromatography. | |||||||||
| 1 | BF3·OEt2 | TMSN3 | 1 | CH2Cl2 | −30 | 1 | 30% | 30% | 21% |
| 2 | BF3·OEt2 | TMSN3 | 2 | CH2Cl2 | −30 | 1 | 20% | 20% | 40% |
| 3 | BF3·OEt2 | TMSN3 | 3 | CH2Cl2 | −30 | 0.5 | 52% | — | 12% |
| 4 | BF3·OEt2 | Bu4NN3 | 3 | CH2Cl2 | −30 | 0.5 | 46% | — | 17% |
| 5 | TMSOTf | Bu4NN3 | 3 | CH2Cl2 | −30 | 0.5 | 38% | — | 30% |
| 6 | TMSOTf | TMSN3 | 3 | CH2Cl2 | −30 | 0.5 | 61% | — | — |
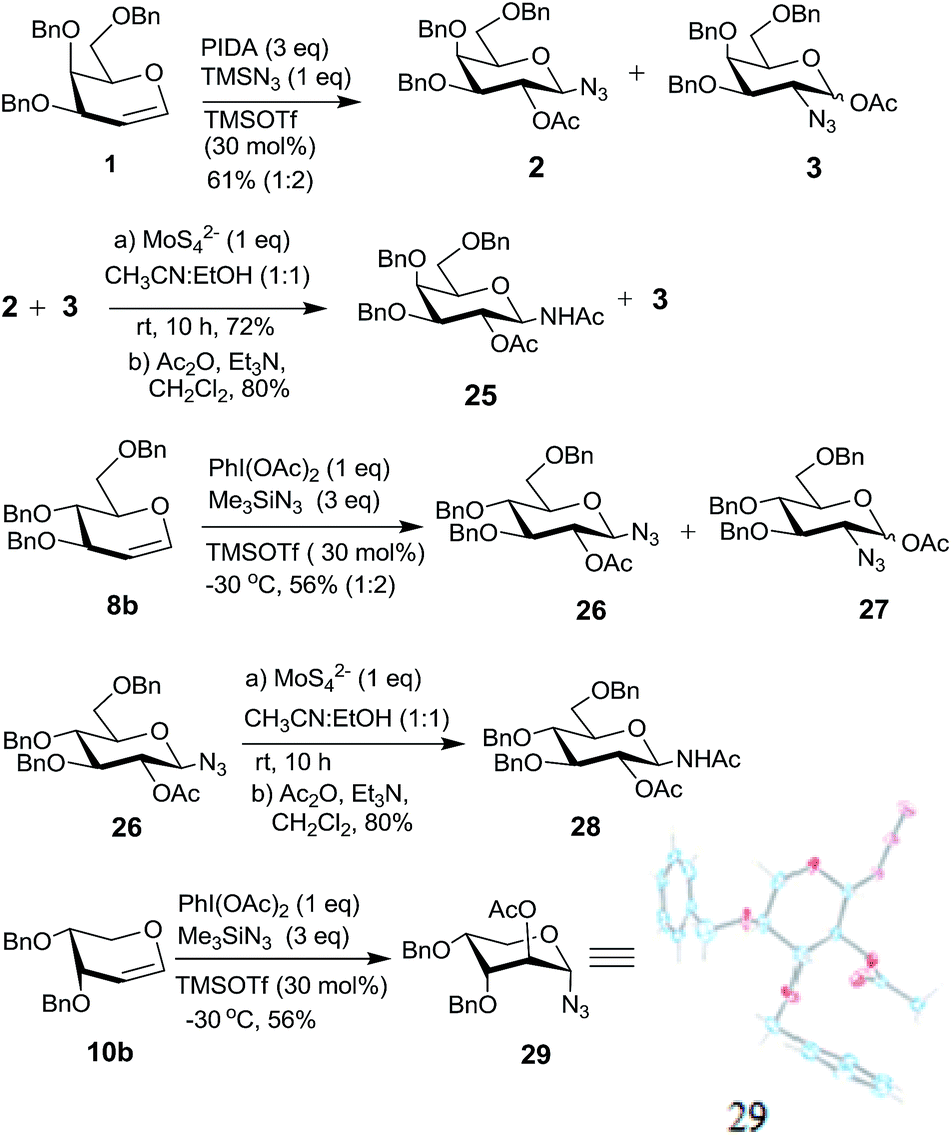 | ||
| Scheme 6 Reaction of glycal derivatives 1, 8b and 29 with PIDA–Me3SiN3. | ||
Based on these product distributions in the reactions with glycal derivatives 1 and 8b with PIDA–Me3SiN3 reagent system we propose a tentative mechanism as shown in Scheme 7. Accordingly, we expect PIDA to react with excess of Me3SiN3 (3 eq.) resulting into species X and TMSOAc with the equilibrium being more favorable on the right side. Reaction of glycals 1 and 8b with the species X in presence of TMSOTf should lead to a π-complex A which should generate another molecule of TMSOAc on the way to intermediate ‘B’. Thus, chances of TMSOAc acting as a preferred nucleophile to form products 3 and 27 appear to be high compared to TMSN3. Subsequently, the intermediate B will undergo attack of the acetate ion either from α- or β-side (paths a and b) at the anomeric carbon accompanied by an SNi type reaction with the azide ion at C-2 to give compounds 3 and 27 with the loss of PhI. Likewise, formation of 2 and 26 can be rationalized by invoking intermediates C (a π-complex) and D in the reaction of glycals with PIDA. The π-complex C will expel a molecule of TMSOAc only upon the formation of the intermediate D. Thus at any given time, during this pathway, more amount of Me3SiN3 is available to attack at intermediate D which leads to the observed products 2 and 26.
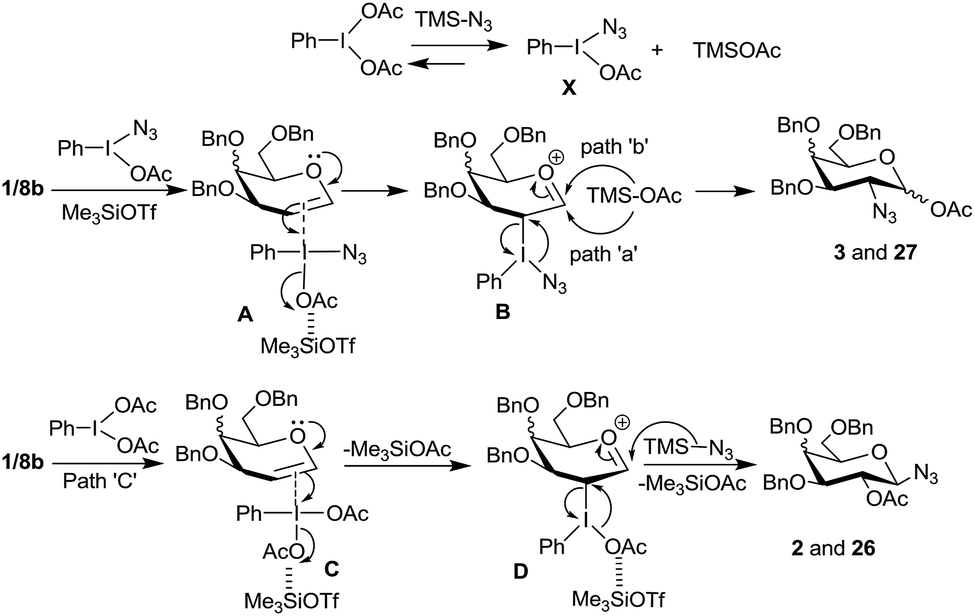 | ||
| Scheme 7 Proposed mechanisms for the reaction of glycals with PIDA and Me3SiN3. | ||
In case of arabinal derivative 10b, a complex similar to ‘C’ should form from the β-face to avoid the steric repulsions from the two –OBn groups. This should be followed by azide ion attack in an intermolecular fashion from the axial orientation at the anomeric carbon. Subsequent acetate ion attack at C-2 occurs, in an intramolecular fashion, again from the axial side leading to the observed product 29. It is however not clear at this moment why the product 29 prefers to have 4C1 conformation as against compounds 11a and 11b which prefer 1C4 conformations.
We further checked the reactivity of 2,4,6-tri-O-acetylated glycals 6a and 8a towards the PIDA–Me3SiN3 reagent system. The reaction was extremely sluggish in presence of 30 mol% of TMSOTf even at room temperature for prolonged reaction times. Gradual increase in the amount of TMSOTf ultimately led to the use of 1 eq. of it for complete consumption of the starting material at −30 °C. However, the galactal derivative 6a, interestingly, led to 2-hydroxy-1-azido sugar 30 as a product in 68% yield (Scheme 8) whose structure was established based on spectral data including COSY, NOE, homonuclear decoupling and DEPT experiments.16a Thus, in NOE experiments, irradiation of H-3 at δ 4.89, led to the enhancement of the signals for protons H-1 and H-5 at δ 4.19 and δ 3.96 respectively. Also, when H-1 at δ 4.19 was irradiated, the signals for H-3 and H-5 were enhanced suggesting that H-1 is axially oriented. In the homonuclear decoupling experiment of 30 when H-4 at δ 5.36 was irradiated, the proton H-3 appeared as a doublet with J = 3.68 Hz suggesting that H-2 is equatorially oriented. These data support the structure assigned to 30. Compound 30 was also derivatized to 31 and 32 and their structures confirmed based on spectral analysis. On the other hand, the glucal derivative 8a led to the Ferrier reaction to form 3324 in 76% yield as an anomeric mixture (α:β = 60:40).
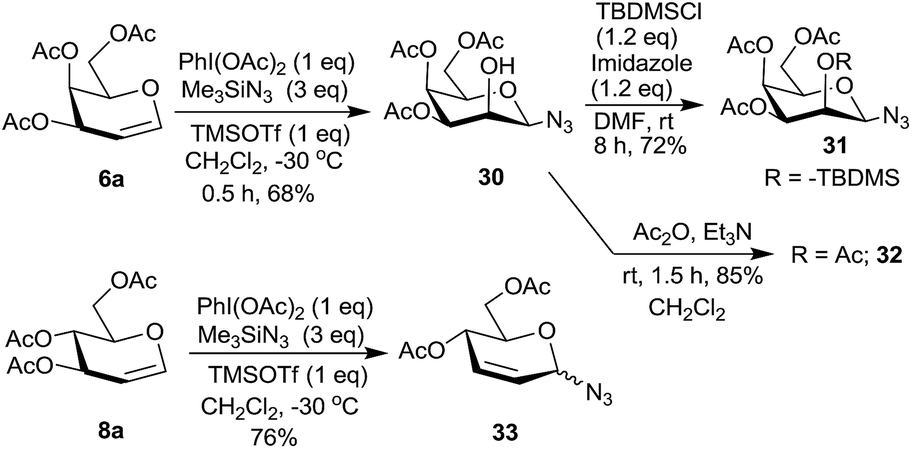 | ||
| Scheme 8 Reaction of glycal derivatives 6a and 8a with PIDA–Me3SiN3. | ||
Mechanistically, in case of 2,4,6-tri-O-acetyl galactal 6a, the intermediate F (Scheme 9), resulting from the π-complex E, should allow azide ion attack from the β-side leading to the intermediate G. It is possible that intermediate G then undergoes intramolecular participation by C4–OAc group to form intermediate I which then hydrolytically decomposes to give 30. Since the glucal derivative 8a does not give similar reaction, instead undergoes the Ferrier reaction, intramolecular participation of C4–OAc group in 6a becomes crucial.
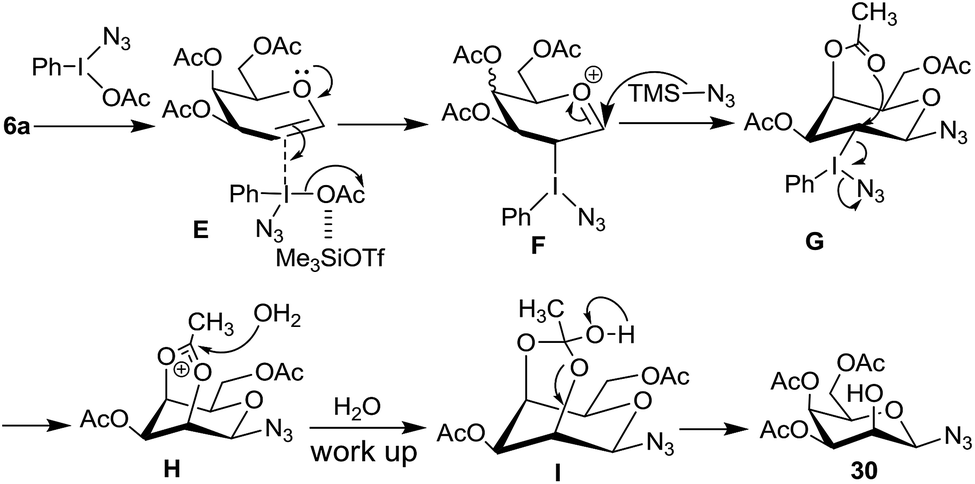 | ||
| Scheme 9 Proposed mechanisms for the reaction of glycal 6 with PIDA and Me3SiN3. | ||
In view of the importance of 2-amino-O-glycosides, 1-acetoxy-2-azido galactose derivative 3 was hydrolyzed25 with benzyl amine to form 2-azido-3,4,6-tri-O-benzyl-galactopyranose 34 (Scheme 10). The corresponding trichloroacetimidate 35, a glyosyl donor, was readily prepared by following a literature procedure26 and was reacted with 36,27 an orthogonal glycosyl acceptor having a thioglycoside linkage, followed by the click reaction with sugar derived alkyne 14 to form a pseudotrisaccharide 38.16a
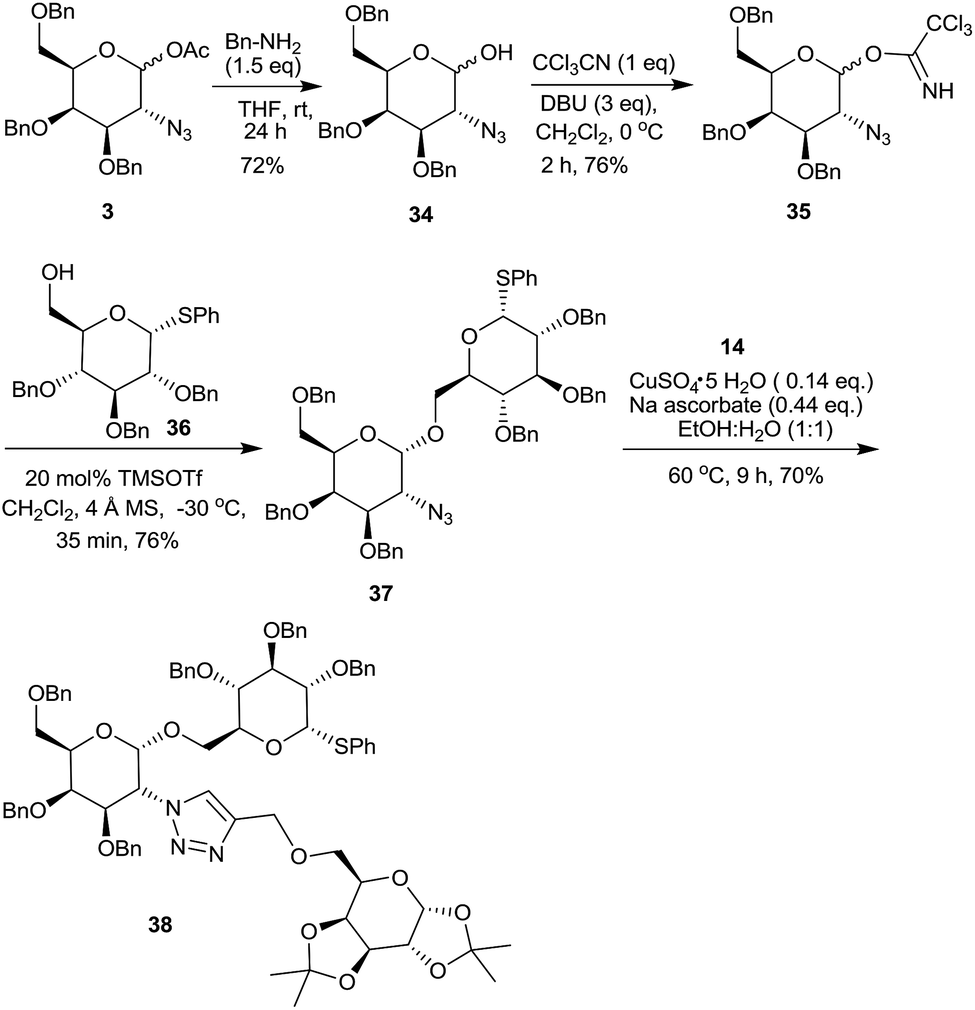 | ||
| Scheme 10 Synthesis of a pseudotrisaccharide 38. | ||
Further, we also show the importance of 1-azido-2-acetoxy sugar derivative 29 in the synthesis of a piperidine triol 44, that belongs to a class of potential glycosidase inhibitors9h,i (Scheme 11). Thus, the acetate moiety of azidoacetate 29 was deprotected and converted to the MOM derivative 40 via 39 under standard reaction conditions. Subsequently, the anomeric azide group was reduced with LiAlH4 followed by protection of the resulting amine as N-nosyl group to give 41 which was cyclized to the piperidine derivative 42 under the Mitsunobu conditions. Deprotections of the nosyl, benzyl and the MOM groups led to the formation of the piperidine 44, an iminosugar as a potential glycosidase inhibitor.28
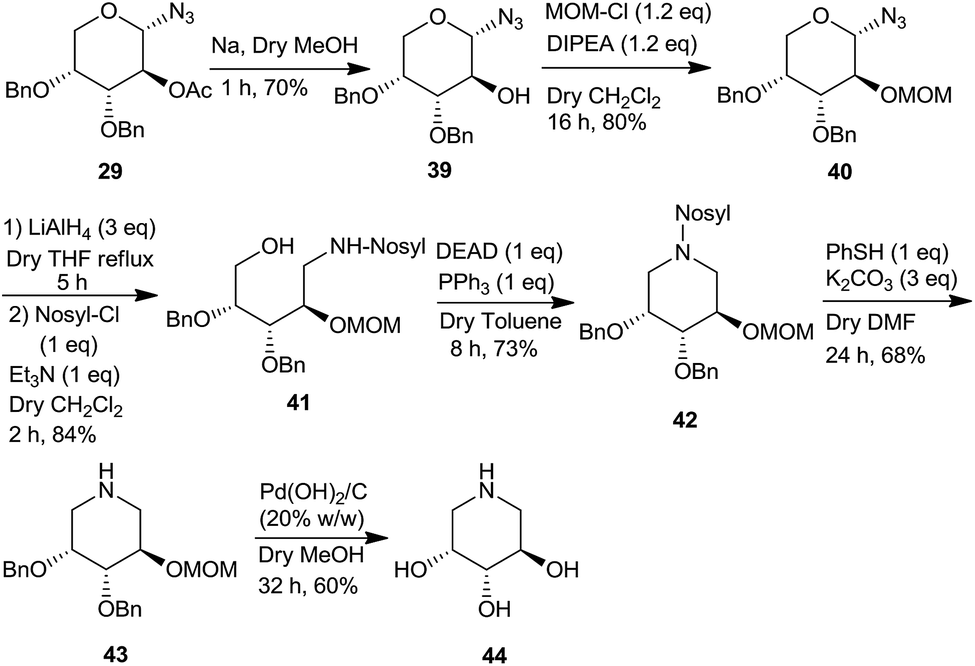 | ||
| Scheme 11 Synthesis of a piperidine derivative 44 from the azidoacetate 29. | ||
Conclusions
In conclusion, we have shown that the reagent system PIFA–Me3SiN3 in presence of TMSOTf as a catalyst conveniently converts glycals into the corresponding 1,2-diazido derivatives. These vicinal azido derivatives are converted to 2-azido-N-glycopeptides, which are convenient precursors of 2-amino-N-glycopeptides, and also into a pseudotrisaccharide 15. Likewise, glycals are converted into a mixture of 1-azido-2-acetoxy sugars and 2-azido-1-acetoxy sugars using the reagent system PIDA–Me3SiN3 in the presence of TMSOTf as a catalyst. The –OAc group of 2-azido-1-acetates can be hydrolyzed and converted into a trichloroacetimidate group for effecting glycosylations. One of the azidoacetates 3 was converted into a pseudotrisaccharide 38 after glycosylation followed by click chemistry. Further, 1-azido-2-acetoxy sugar derivative 29, derived from the arabinal derivative 10b, was eventually converted into a piperidine triol 44, a potential glycosidase inhibitor. Tentative mechanisms have been proposed to account for the formation of different azido sugars.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Department of Science and Technology, New Delhi, India, for a J. C. Bose National Fellowship (No. SR/S2/JCB-26/2010) to Y. D. V. A. C. thanks the Council of Scientific and Industrial Research, New Delhi for a Senior Research Fellowship.Notes and references
- (a) S. Mirabella, F. Cardona and A. Goti, Org. Biomol. Chem., 2016, 14, 5186–5204 RSC and references cited therein; (b) J. Zeng, G. Sun, W. Yao, Y. Zhu, R. Wang, L. Cai, K. Liu, Q. Zhang, X.-W. Liu and Q. Wa, Angew. Chem., Int. Ed., 2017, 56, 5227–5231 CrossRef CAS PubMed.
- (a) C. Pöhner, V. Ullmann, R. Hilpert, E. Samain and C. Unverzagt, Tetrahedron Lett., 2014, 55, 2197–2200 CrossRef; (b) J. Kerékgyértó, L. Kalmár, Z. Szurmai, O. Hegyi and G. K. Tóth, Int. J. Pept. Res. Ther., 2012, 18, 1–5 CrossRef; (c) T. Buskas, S. Ingale and G.-J. Boons, Glycobiology, 2006, 16, 113R–136R CrossRef CAS PubMed; (d) H. Herzner, T. Reipen, M. Schultz and H. Kunz, Chem. Rev., 2000, 100, 4495–4537 CrossRef CAS PubMed; (e) G. K. Rawal, A. Kumar, U. Tawar and Y. D. Vankar, Org. Lett., 2007, 9, 5171–5174 CrossRef CAS PubMed; (f) R. Lahiri, S. Dharuman and Y. D. Vankar, Chimia, 2012, 66, 905–912 CrossRef CAS PubMed; (g) V. Ullmann, M. Rädisch, I. Boos, J. Freund, C. Pöhner, S. Schwarzinger and C. Unverzagt, Angew. Chem., Int. Ed., 2012, 51, 11566–11570 CrossRef CAS PubMed; (h) C. M. Kaneshiro and K. Michael, Angew. Chem., Int. Ed., 2006, 45, 1007–1081 CrossRef PubMed; (i) R. Joseph, F. B. Dyer and P. Garner, Org. Lett., 2013, 15, 732–735 CrossRef CAS PubMed.
- (a) L. Marmuse, S. A. Nepogodiev and R. A. Field, Org. Biomol. Chem., 2005, 3, 2225–2227 RSC; (b) V. K. Tiwari, B. B. Mishra, K. B. Mishra, N. Mishra, A. S. Singh and X. Chen, Chem. Rev., 2016, 116, 3086–3240 CrossRef CAS PubMed and references cited therein; (c) S. Chandrasekaran and R. Ramapanicker, Chem. Rec., 2017, 17, 63–70 CrossRef CAS PubMed; (d) S. Tejera, R. L. Dorta and J. T. Vázquez, Tetrahedron: Asymmetry, 2016, 27, 896–909 CrossRef CAS.
- (a) Y. S. Reddy, A. P. John Pal, P. Gupta, A. A. Ansari and Y. D. Vankar, J. Org. Chem., 2011, 76, 5972–5984 CrossRef CAS PubMed; (b) B. G. Reddy, K. P. Madhusudanan and Y. D. Vankar, J. Org. Chem., 2004, 69, 2630–2633 CrossRef CAS PubMed; (c) G. K. Rawal, S. Rani, K. P. Madhusudanan and Y. D. Vankar, Synthesis, 2007, 294–298 CAS; (d) T. Cui, R. Smith and X. Zhu, Carbohydr. Res., 2015, 416, 14–20 CrossRef CAS PubMed; (e) M. L. Lepage, A. Bodlenner and P. Compain, Eur. J. Org. Chem., 2013, 1963–1972 CrossRef CAS; (f) Z. Györgydeák, L. Szilágyi and H. Paulsen, J. Carbohydr. Chem., 1993, 12, 139–163 CrossRef; (g) G. Pastuch-Gawołek, K. Malarz, A. Mrozek-Wilczkiewicz, M. Musioł, M. Serda, B. Czaplinska and R. Musiol, Eur. J. Med. Chem., 2016, 112, 130–144 CrossRef PubMed.
- (a) R. Ahuja, N. K. Singhal, B. Ramanujam, M. Ravikumar and C. P. Rao, J. Org. Chem., 2007, 72, 3430–3442 CrossRef CAS PubMed; (b) E. Honda and D. Y. Gin, J. Am. Chem. Soc., 2002, 124, 7343–7352 CrossRef CAS PubMed.
- (a) R. U. Lemieux and R. M. Ratcliffe, Can. J. Chem., 1979, 57, 1244–1251 CrossRef CAS; (b) R. R. Schmidt and Y. D. Vankar, Acc. Chem. Res., 2008, 41, 1059–1073 CrossRef CAS PubMed and references cited therein; (c) R. Bodner, B. K. Marcellino, A. Severino, A. L. Smenton and C. M. Rojas, J. Org. Chem., 2005, 70, 3988–3996 CrossRef CAS PubMed; (d) J. Liu and D. Y. Gin, J. Am. Chem. Soc., 2002, 124, 9789–9797 CrossRef CAS PubMed; (e) J. Liu, V. D. Bussolo and D. Y. Gin, Tetrahedron Lett., 2003, 44, 4015–4018 CrossRef CAS; (f) D. A. Griffth and S. J. Danishefsky, J. Am. Chem. Soc., 1990, 112, 5811–5819 CrossRef; (g) B.-Y. Lee, P. H. Seeberger and D. V. Silva, Chem. Commun., 2016, 52, 1586–1589 RSC; (h) L. Corcilius, G. Santhakumar, R. S. Stone, C. J. Capicciotti, S. Joseph, J. M. Matthews, R. N. Ben and R. J. Payne, Bioorg. Med. Chem., 2013, 21, 3569–3581 CrossRef CAS PubMed.
- (a) B. Premdjee, A. L. Adams and D. Macmillan, Bioorg. Med. Chem. Lett., 2011, 21, 4973–4975 CrossRef CAS PubMed; (b) G. Arsequell and G. Valencia, Tetrahedron: Asymmetry, 1999, 10, 3045–3094 CrossRef CAS; (c) C. Brocke and H. Kunz, Bioorg. Med. Chem., 2002, 10, 3085–3112 CrossRef CAS PubMed.
- (a) P. K. Kancharla, Y. S. Reddy, S. Dharuman and Y. D. Vankar, J. Org. Chem., 2011, 76, 5832–5837 CrossRef CAS PubMed; (b) J. Thiem and T. Wiemann, Angew. Chem., Int. Ed. Engl., 1990, 29, 80–82 CrossRef; (c) Y.-A. Yuan, D.-F. Lu, Y.-R. Chen and H. Xu, Angew. Chem., Int. Ed., 2016, 55, 534–538 CrossRef CAS PubMed; (d) R. Joseph, F. B. Dyer and P. Garner, Org. Lett., 2013, 15, 732–735 CrossRef CAS PubMed; (e) V. Kumar and N. G. Ramesh, Chem. Commun., 2006, 4952–4954 RSC; (f) V. Kumar and N. G. Ramesh, Org. Biomol. Chem., 2007, 5, 3847–3858 RSC; (g) S. Xiang, J. Ma, B. K. Gorityala and X.-W. Liu, Carbohydr. Res., 2011, 346, 2957–2959 CrossRef CAS PubMed; (h) Y. V. Mironov, A. A. Sherman and N. E. Nifantiev, Tetrahedron Lett., 2004, 45, 9107–9110 CrossRef CAS.
- (a) A. Palanivel, S. Dharuman and Y. D. Vankar, Tetrahedron: Asymmetry, 2016, 27, 1088–1100 CrossRef CAS; (b) A. Mallick, N. Kumari, R. Roy, A. Palanivel and Y. D. Vankar, Eur. J. Org. Chem., 2014, 5557–5563 CrossRef CAS; (c) S. Dharuman and Y. D. Vankar, Org. Lett., 2014, 16, 1172–1175 CrossRef CAS PubMed; (d) A. A. Ansari, P. Rajasekaran, M. M. Khan and Y. D. Vankar, J. Org. Chem., 2014, 79, 1690–1699 CrossRef CAS PubMed; (e) A. A. Ansari and Y. D. Vankar, RSC Adv., 2014, 4, 12555–12567 RSC; (f) A. A. Ansari and Y. D. Vankar, J. Org. Chem., 2013, 78, 9383–9395 CrossRef CAS PubMed; (g) S. Dharuman, P. Gupta, P. K. Kancharla and Y. D. Vankar, J. Org. Chem., 2013, 78, 8442–8450 CrossRef CAS PubMed; (h) Y. S. Reddy, P. K. Kancharla, R. Roy and Y. D. Vankar, Org. Biomol. Chem., 2012, 10, 2760–2773 RSC; (i) P. Gupta and Y. D. Vankar, Eur. J. Org. Chem., 2009, 1925–1933 CrossRef.
- (a) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435 CrossRef CAS PubMed; (b) V. V. Zhdankin, ARKIVOC, 2009, i, 1–62 Search PubMed.
- (a) A. Kirschning, C. Plumeier and L. Rose, Chem. Commun., 1998, 33–34 RSC; (b) P. Pandit, K. S. Gayen, S. Khamarui, N. Chatterjee and D. K. Maiti, Chem. Commun., 2011, 47, 6933–6935 RSC; (c) M. Islam, N. D. Tirukoti, S. Nandi and S. Hotha, J. Org. Chem., 2014, 79, 4470–4476 CrossRef CAS PubMed; (d) T. R. Reddy, D. S. Rao, K. Babachary and S. Kashyap, Eur. J. Org. Chem., 2016, 291–301 CrossRef CAS.
- L. Shi, Y.-J. Kim and D. Y. Gin, J. Am. Chem. Soc., 2001, 123, 6939–6940 CrossRef CAS PubMed.
- (a) K. Wu, Y. Liang and N. Jiao, Molecules, 2016, 21, 1–21 Search PubMed; (b) B. B. Snider and H. Lin, Synth. Commun., 1998, 28, 1913–1922 CrossRef CAS; (c) M.-Z. Lu, C.-Q. Wang and T.-P. Loh, Org. Lett., 2015, 17, 6110–6113 CrossRef CAS PubMed; (d) D. A. Kamble, P. U. Karabal, P. V. Chouthaiwale and A. Sudalai, Tetrahedron Lett., 2012, 53, 4195–4198 CrossRef CAS; (e) G. Fumagalli, P. T. G. Rabet, S. Boyd and M. F. Greaney, Angew. Chem., Int. Ed., 2015, 54, 11481–11484 CrossRef CAS PubMed.
- (a) Y. V. Mironov, A. A. Sherman and N. E. Nifantiev, Tetrahedron Lett., 2004, 45, 9107–9110 CrossRef CAS; (b) Y.-A. Yuan, D.-F. Lu, Y.-R. Chen and H. Xu, Angew. Chem., Int. Ed., 2016, 55, 534–538 CrossRef CAS PubMed.
- (a) Y. Kita, H. Tohma, K. Hatanaka, T. Takada, S. Fujita, S. Mitoh, H. Sakurai and S. Oka, J. Am. Chem. Soc., 1994, 116, 3684–3691 CrossRef CAS; (b) P. Li, J. Zhao, C. Xia and F. Li, Org. Chem. Front., 2015, 2, 1313–1317 RSC; (c) Y. Kita, H. Tohma, T. Takada, S. Mitoh, S. Fujita and M. Gyoten, Synlett, 1994, 427–428 CrossRef CAS; (d) K. Matcha and A. P. Antonchick, Angew. Chem., Int. Ed., 2013, 52, 2082–2086 CrossRef CAS PubMed; (e) R. Narayan and A. P. Antonchick, Chem.–Eur. J., 2014, 20, 4568–4572 CrossRef CAS PubMed; (f) K. Matcha, R. Narayan and A. P. Antonchick, Angew. Chem., Int. Ed., 2013, 52, 7985–7989 CrossRef CAS PubMed.
- (a) See the ESI† for spectral details; (b) CCDC 1554252 for compound 5 and CCDC 1554251 for compound 29.
- (a) For the formation of PhI(N3)2 from PhIO–Me3SiN3 and its utility in presence of TEMPO via radical pathway see: (i) P. Magnus, M. B. Roe and C. Hulme, J. Chem. Soc., Chem. Commun., 1995, 263–265; (ii) P. Magnus, J. Lacour, P. A. Evans, M. B. Roe and C. Hulme, J. Am. Chem. Soc., 1996, 118, 3406–3418; (b) R. M. Moriarty and J. S. Khosrowshahi, Tetrahedron Lett., 1986, 27, 2809–2812 CrossRef CAS; (c) A. Kirschning, Eur. J. Org. Chem., 1998, 2267–2274 CrossRef CAS.
- A. Mishra and V. K. Tiwari, J. Org. Chem., 2015, 80, 4869–4881 CrossRef CAS PubMed.
- P. R. Sridhar, K. R. Prabhu and S. Chandrasekaran, J. Org. Chem., 2003, 78, 5261–5264 CrossRef PubMed.
- (a) J. L. Torres, I. Haro, E. Bardaji, G. Valencia, J. M. Garcia-Anton and F. Reig, Tetrahedron, 1988, 44, 6131–6136 CrossRef CAS; (b) A. Bianchi and A. Bernardi, J. Org. Chem., 2006, 71, 4565–4577 CrossRef CAS PubMed; (c) A. D. Dorsey, J. E. Barbarow and D. Trauner, Org. Lett., 2003, 5, 3237–3239 CrossRef CAS PubMed.
- R. J. Payne, S. Ficht, S. Tang, A. Brik, Y.-Y. Yang, D. A. Case and C.-H. Wong, J. Am. Chem. Soc., 2007, 129, 13527–13536 CrossRef CAS PubMed.
- (a) N. V. Bovin, S. E. Zurabvan and A. Y. Khorlin, Carbohydr. Res., 1981, 98, 25 CrossRef CAS; (b) A. A. Pavia, S. N. Ung-Chhun and J. L. Durand, J. Org. Chem., 1981, 46, 3158–3160 CrossRef CAS.
- K. Matsuo and M. Shindo, Tetrahedron, 2011, 67, 971–975 CrossRef CAS.
- S. Chandrasekhar, C. R. Reddy and G. Chandrashekar, Tetrahedron Lett., 2004, 45, 6481–6484 CrossRef CAS.
- S. Kopitzki and J. Thiem, Eur. J. Org. Chem., 2013, 4008–4016 CrossRef CAS.
- (a) R. R. Schmidt and J. Michel, Angew. Chem., Int. Ed., 1980, 19, 731–732 CrossRef; (b) J. Kalikanda and Z. Li, J. Org. Chem., 2011, 76, 5207–5218 CrossRef CAS PubMed.
- L. X. Wang, N. Sakairi and H. Kuzuhara, J. Chem. Soc., Perkin Trans. 1, 1990, 1677–1682 RSC.
- X. Garrabou, J. A. Castillo, C. G. Hélaine, T. Parella, J. Joglar, M. Lemaire and P. Clapés, Angew. Chem., Int. Ed., 2009, 48, 5521–5525 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: CCDC 1554251 and 1554252. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra08637g |
| ‡ M.Sc. research project participant (2016). |
| This journal is © The Royal Society of Chemistry 2017 |