
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biological and chemical guided isolation of 3,4-secograyanane diterpenoids from the roots of Pieris formosa†
Chang-Shan Niu,
Yong Li,
Yun-Bao Liu,
Shuang-Gang Ma,
Fei Liu,
Li Li,
Song Xu,
Xiao-Jing Wang,
Sheng Liu,
Ru-Bing Wang,
Jing Qu* and
Shi-Shan Yu *
*
State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing 100050, China. E-mail: yushishan@imm.ac.cn; qujing@imm.ac.cn
First published on 12th September 2017
Abstract
Seventeen new 3,4-secograyanoids (1–17), together with seven known compounds (18–24), were isolated from the roots of Pieris formosa. Their structures with absolute configurations were characterized by a series of spectroscopic methods and X-ray diffraction. Compounds 1, 2, 4–8, 10–13, and 16–24 exhibited significant analgesic activity at 5.0 mg kg−1 (ip) compared to vehicle-injected mice (p < 0.05). In particular, compounds 16 and 17 showed highly potent activities with inhibition rates of 63.5% and 69.9%, respectively.
Introduction
Grayanoids, a special type of diterpenoid, exist exclusively in the Ericaceae family with diverse structures and wide bioactivities.1 To date, almost two hundred grayanoids with eleven carbon skeletons, including grayanane,2,3 1,5-secograyanane,4 3,4-secograyanane,5 9,10-secograyanane,6 1,10:2,3-disecograyanane,7 kalmane,8 1,5-secokalmane,9 leucothane,10 micranthane,11 mollane,12 and rhodomollane,13 have been reported. Notably, 3,4-secograyanane is a rare type of grayanoid, and only seventeen 3,4-secograyanoids have been isolated and characterized. Several grayanoids display analgesic and insect antifeedant activities.Pieris formosa (Wall) D. Don, a traditional Chinese medicine, has been used to treat tinea and scabies.14 In previous phytochemical investigations of P. formosa, fifteen grayananes and three diterpenes with an unusual carbon skeleton were isolated and characterized, and most compounds were found to exhibited an analgesic effect.15,16 In our on-going efforts, biological and chemical guided fractionation led to the isolation of seventeen new 3,4-secograyananes (1–17), together with seven known 3,4-secograyanoids (18–24) (see Fig. 1). Herein, we report their isolation, structural elucidation, and bioactivity. The main text of the article should appear here with headings as appropriate.
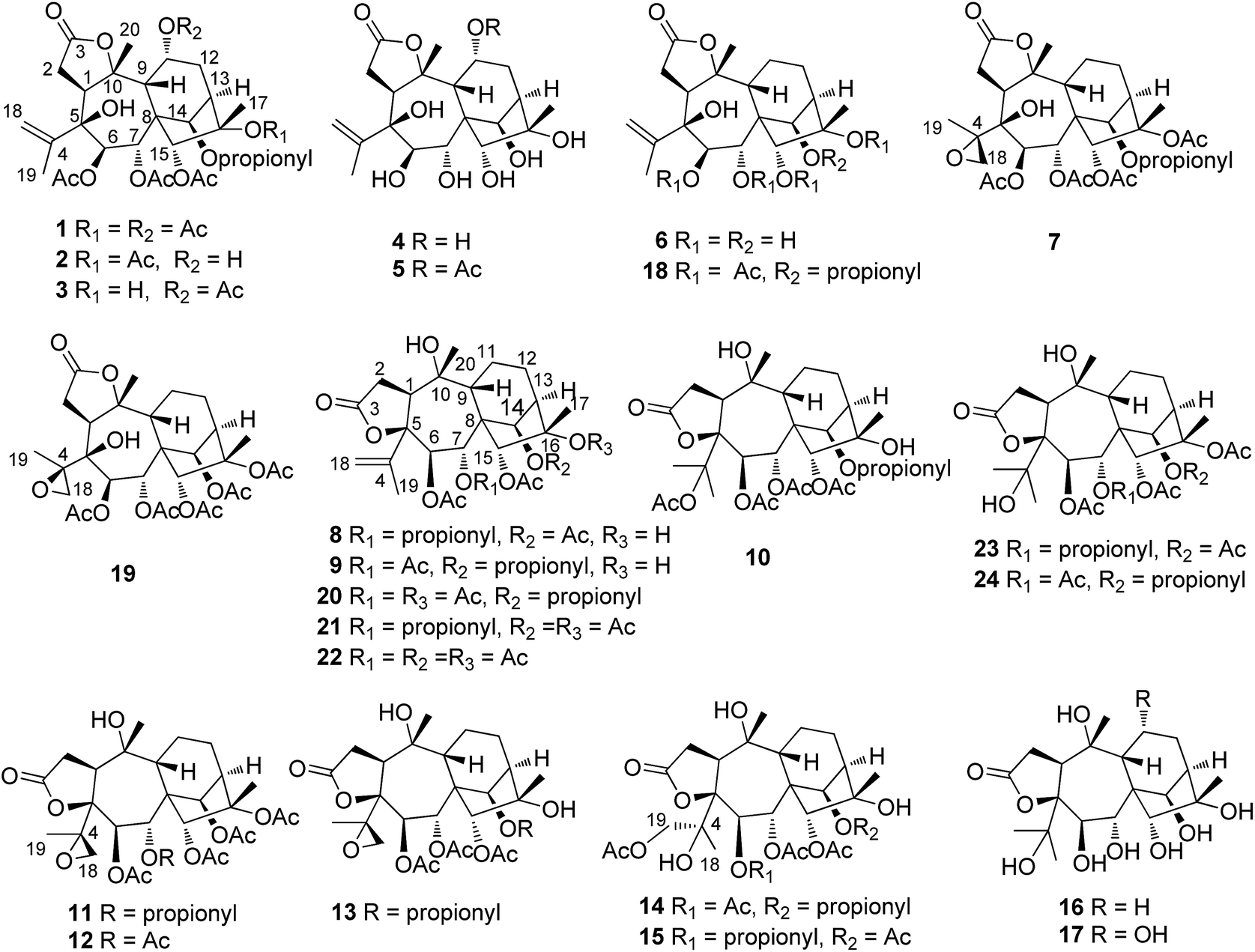 | ||
| Fig. 1 Chemical structures of the isolated compounds 1–24. | ||
Results and discussion
Biological and chemical guided fractionation and isolation of the roots of P. formosa
The fractionation and isolation procedures were guided by acetic acid-induced writhing test and online HPLC/UV/ESIMS2 analyses. The roots of P. formosa were extracted with 95% EtOH and the evaporation of the solvent under reduced pressure subsequently yielded a black residue. The residue was mixed with diatomite (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) and Soxhlet was successively extracted thereafter using a gradient of petroleum ether, CH2Cl2, EtOAc and CH3OH. Acetic acid-induced writhing test showed analgesic activity for the CH2Cl2 portion (D) and the EtOAc portion (E), with 20% and 25% writhe inhibitions (100 mg kg−1, op), respectively. Subsequently, the CH2Cl2 portion (D) and the EtOAc portion (E) were applied to a macroporous resin D101 column and eluted with a gradient of 30% EtOH–H2O, 50% EtOH–H2O and 70% EtOH–H2O, to yield three fractions, respectively (D1–D3, and E1–E3). Fractions D1, D2, E1, and E2 showed analgesic activity with 20%, 33%, 73%, and 41% writhe inhibition (100 mg kg−1, op), respectively. Fractions D1 and D2 were separated through a polyamide column and eluted with H2O–EtOH (100:0, 30:70, 50:50, 70:30, v/v), to yield eight fractions (D1a–D1d and D2a–D2d). Fractions E1 and E2 were separated through a polyamide column and eluted with a gradient of H2O–EtOH (100:0, 30:70, 60:40, v/v), to yield six fractions (E1a–E1c and E2a–E2c). Online HPLC/UV/ESIMS2 analyses showed that there were many constituents that exhibited a maximum absorption band at λ 210–220 nm in the UV spectra and [M − 60]+, [M − 2 × 60]+, and [M − 3 × 60]+ fragment ions in the positive MS2 mode in fractions D1a, D2a, and D2b (see Fig. S1–S3†), characteristic of the highly acylated 3,4-secograyanane diterpenoids.5 Similarly, there were many constituents that exhibited a maximum absorption band at λ 210–220 nm in the UV spectra and [M − 18]+, [M − 2 × 18]+, and [M − 3 × 18]+ fragment ions in the positive MS2 mode in fractions E1a, E1b, and E2a (see Fig. S4–S6†), characteristic of the highly hydroxylated grayanane diterpenoids. Finally, seventeen new 3,4-secograyanoids (1–17), together with seven known 3,4-secograyanoids (18–24), were isolated from the fractions D1a, D2a, D2b, E1a, E1b, and E2a by a combination of different chromatographic methods. Acetic acid-induced writhing test showed that compounds 1, 2, 4–8, 10–13, and 16–24 exhibited significant analgesic activity at 5.0 mg kg−1 (ip).
:2) and Soxhlet was successively extracted thereafter using a gradient of petroleum ether, CH2Cl2, EtOAc and CH3OH. Acetic acid-induced writhing test showed analgesic activity for the CH2Cl2 portion (D) and the EtOAc portion (E), with 20% and 25% writhe inhibitions (100 mg kg−1, op), respectively. Subsequently, the CH2Cl2 portion (D) and the EtOAc portion (E) were applied to a macroporous resin D101 column and eluted with a gradient of 30% EtOH–H2O, 50% EtOH–H2O and 70% EtOH–H2O, to yield three fractions, respectively (D1–D3, and E1–E3). Fractions D1, D2, E1, and E2 showed analgesic activity with 20%, 33%, 73%, and 41% writhe inhibition (100 mg kg−1, op), respectively. Fractions D1 and D2 were separated through a polyamide column and eluted with H2O–EtOH (100:0, 30:70, 50:50, 70:30, v/v), to yield eight fractions (D1a–D1d and D2a–D2d). Fractions E1 and E2 were separated through a polyamide column and eluted with a gradient of H2O–EtOH (100:0, 30:70, 60:40, v/v), to yield six fractions (E1a–E1c and E2a–E2c). Online HPLC/UV/ESIMS2 analyses showed that there were many constituents that exhibited a maximum absorption band at λ 210–220 nm in the UV spectra and [M − 60]+, [M − 2 × 60]+, and [M − 3 × 60]+ fragment ions in the positive MS2 mode in fractions D1a, D2a, and D2b (see Fig. S1–S3†), characteristic of the highly acylated 3,4-secograyanane diterpenoids.5 Similarly, there were many constituents that exhibited a maximum absorption band at λ 210–220 nm in the UV spectra and [M − 18]+, [M − 2 × 18]+, and [M − 3 × 18]+ fragment ions in the positive MS2 mode in fractions E1a, E1b, and E2a (see Fig. S4–S6†), characteristic of the highly hydroxylated grayanane diterpenoids. Finally, seventeen new 3,4-secograyanoids (1–17), together with seven known 3,4-secograyanoids (18–24), were isolated from the fractions D1a, D2a, D2b, E1a, E1b, and E2a by a combination of different chromatographic methods. Acetic acid-induced writhing test showed that compounds 1, 2, 4–8, 10–13, and 16–24 exhibited significant analgesic activity at 5.0 mg kg−1 (ip).
Characterization of compounds 1–17
Pieristoxinlactone A (1) was obtained as a colorless crystal, and its molecular formula was determined to be C33H44O15 based on the HRESIMS [M + Na]+ ion peak m/z 703.2568 (calcd 703.2572), indicating twelve indices of hydrogen deficiency. Its 1H NMR spectrum (see Table 1) showed one triplet methyl (δH 1.35, t, J = 7.0 Hz) and eight singlet methyls (δH 2.25, 2.24, 2.19, 2.18, 2.12, 2.00, 1.73, 1.70). In the low-field region, a pair of AB doublets (δH 6.41, 5.64, d, J = 10.0 Hz) and an exchangeable hydroxy proton (δH 7.26) were also observed. The 13C NMR (see Table 2) and DEPT spectra displayed 33 carbon resonances, ascribed to nine methyls, four methylenes (one olefinic at δC 113.9), eight methines (five oxygenated at δC 92.2, 75.6, 74.0, 68.6, 67.5), and twelve quaternary carbons (seven ester carbonyls at δC 174.8, 174.1, 171.9, 170.7, 170.3, 169.1, 168.9; one olefinic at δC 147.9; three oxygenated at δC 88.3, 88.2, 76.3). The 1H–1H COSY (see Fig. 3) and HSQC spectra revealed the presence of four spin systems: C(1)H–C(2)H2, C(6)H–C(7)H, C(9)H–C(11)H–C(12)H2–C(13)H–C(14)H, and a propionyl unit. These structural features suggested that 1 was a highly acylated 3,4-secograyanane. Further HSQC and HMBC experiments (see Fig. 3) enabled the full assignments of the 1H and 13C NMR spectra of 1. Compound 1 showed the same planar structure as secorhodomollolide D5. NOESY correlations (see Fig. 3) from H-14 to H-1 and H-12a and from H-6 to H-1, H-14, and H3-19 revealed that these protons were α-oriented. In contrast, the NOESY correlations from H-7 to H-9 and H3-20, from H-15 to H-9 and H-12b, from H3-17 to H-12b, suggested that these protons were β-oriented. Furthermore, the small coupling constant between H-9 and H-11 (3JH9,H11 = 5.0 Hz) in the 1H NMR spectrum, with the aid of NOESY correlations from H-11 to H-9 and H-12b, indicated that H-11 was β-orientated. To further corroborate the structural assignment and establish the absolute configuration, a single X-ray diffraction [Cu Kα radiation; Flack parameter: 0.00(8)] of 1 (see Fig. 2; CCDC: 1548366†) was successfully carried out, allowing an explicit assignment of the absolute configuration of 1 as 1S, 5S, 6R, 7R, 8S, 9R, 10R, 11R, 13R, 14R, 15R, 16S. Thus, the structure of 1 was defined, and it was named pieristoxinlactone A.| No. | 1a | 2a | 3b | 4a | 5b |
|---|---|---|---|---|---|
| a Recorded at 500 MHz.b Recorded at 600 MHz. | |||||
| 1 | 3.74, dd (13.5, 8.0) | 4.45, dd (13.5, 7.0) | 3.65, dd (10.2, 6.5) | 4.49, dd (13.5, 7.0) | 3.52, dd (13.2, 7.2) |
| 2 | a 3.12, dd (17.5, 13.5), b 2.51, dd (17.5, 8.0) | a 3.13, dd (17.5, 13.5), b 2.36, dd (17.5, 7.0) | a 3.12, dd (18.6, 10.2), b 2.50, overlap | a 3.16, dd (17.0, 13.5), b 2.41, dd (17.0, 7.0) | a 3.12, dd (17.4, 13.2), b 2.47, dd (17.4, 7.2) |
| 6 | 5.64, d (10.0) | 6.03, d (9.0) | 5.74, overlap | 4.88, d (9.5) | 4.78, d (9.0) |
| 7 | 6.41, d (10.0) | 6.84, d (9.0) | 6.63, d (10.8) | 5.52, d (9.5) | 5.45, d (9.0) |
| 9 | 3.58, d (5.0) | 2.48, overlap | 3.21, brs | 1.98, d (2.5) | 2.40, brs |
| 11 | 5.70, t (5.0) | 4.71, dd (7.0, 5.0) | 5.76, overlap | 4.70, d (5.5, 2.5) | 5.71, dd (4.8, 3.6) |
| 12 | a 2.46, brd (11.0), b 2.15, m | a 2.40, overlap, b 2.11, dd (16.0, 5.5) | a 2.45, overlap, b 2.18, overlap | a 2.15, dd (15.5, 9.5), b 2.03, dd (15.5, 5.5) | a 2.22, dd (16.8, 9.6), b 2.11, dd (16.8, 5.4) |
| 13 | 3.62, d (10.0) | 3.45, d (9.5) | 3.48, overlap | 2.59, d (9.5) | 2.52, d (9.6) |
| 14 | 7.04, brs | 7.40, brs | 6.92, brs | 6.25, brs | 5.57, brs |
| 15 | 5.36, s | 5.66, s | 5.45, s | 4.40, s | 4.39, s |
| 17 | 1.73, s | 1.94, s | 1.57, s | 1.56, s | 1.53, s |
| 18 | a 5.68, brs, b 5.12, brs | a 5.64, brs, b 5.05, brs | a 5.68, brs, b 5.14, brs | a 5.56, brs, b 5.04, brs | a 5.58, brs, b 5.12, brs |
| 19 | 2.00, s | 1.83, s | 1.96, s | 2.07, s | 2.08, s |
| 20 | 2.12, s | 2.17, s | 2.10, s | 1.92, s | 1.88, s |
| 6-OAc | 1.70, s | 1.75, s | 1.75, s | ||
| 7-OAc | 2.25, s | 2.15, s | 2.20, s | ||
| 11-OAc | 2.19, s | 2.15, s | 2.04, s | ||
| 14-OPr | a 2.78, m, b 2.41, overlap, 1.35, t (7.0) | a 2.60, m, b 2.47, overlap, 1.30, t (7.5) | a 2.67, m, b 2.57, m, 1.27, t (7.0) | ||
| 15-OAc | 2.24, s | 2.23, s | 2.24, s | ||
| 16-OAc | 2.18, s | 2.06, s | |||
| 5-OH | 7.26, s | 7.18, s | 6.30, s | ||
| 11-OH | 7.61, d (3.5) | ||||
| No. | 1a | 2a | 3b | 4a | 5b | 6b | 7a | 8b | 9b | 10b | 11a | 12b | 13b | 14b | 15b |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Recorded at 125 MHz.b Recorded at 150 MHz. | |||||||||||||||
| 1 | 42.6 | 42.7 | 43.3 | 42.4 | 43.7 | 42.9 | 41.9 | 54.7 | 54.7 | 51.5 | 52.6 | 52.5 | 52.7 | 49.6 | 49.6 |
| 2 | 30.5 | 31.0 | 30.7 | 31.5 | 31.2 | 32.3 | 31.3 | 32.3 | 32.3 | 33.6 | 32.7 | 32.7 | 32.7 | 33.7 | 33.8 |
| 3 | 174.1 | 174.8 | 174.0 | 175.4 | 174.5 | 174.9 | 174.5 | 174.6 | 174.7 | 175.0 | 174.4 | 174.3 | 174.3 | 175.4 | 175.4 |
| 4 | 147.9 | 148.5 | 147.6 | 151.4 | 150.3 | 150.2 | 60.2 | 147.9 | 147.8 | 89.7 | 62.0 | 62.0 | 62.0 | 76.9 | 76.9 |
| 5 | 76.3 | 76.6 | 76.4 | 77.0 | 76.7 | 76.0 | 73.0 | 90.8 | 90.8 | 92.4 | 87.8 | 87.7 | 87.6 | 92.3 | 92.3 |
| 6 | 74.0 | 74.1 | 73.9 | 73.4 | 73.8 | 75.2 | 74.0 | 70.0 | 70.0 | 68.9 | 69.8 | 69.8 | 69.9 | 70.1 | 69.9 |
| 7 | 67.5 | 66.7 | 67.4 | 72.5 | 72.4 | 74.4 | 67.9 | 68.5 | 68.8 | 68.5 | 68.4 | 68.6 | 69.1 | 68.7 | 68.7 |
| 8 | 53.3 | 53.1 | 53.4 | 54.4 | 53.9 | 53.8 | 52.9 | 56.1 | 56.1 | 56.0 | 55.7 | 55.7 | 56.1 | 55.9 | 55.9 |
| 9 | 60.8 | 57.6 | 58.8 | 57.0 | 57.0 | 53.6 | 56.9 | 47.6 | 47.5 | 47.4 | 47.3 | 47.2 | 46.8 | 48.1 | 48.1 |
| 10 | 88.3 | 89.2 | 88.1 | 89.8 | 88.6 | 90.2 | 89.3 | 76.9 | 76.9 | 76.8 | 76.4 | 76.4 | 76.5 | 76.4 | 76.5 |
| 11 | 68.6 | 62.8 | 68.4 | 63.2 | 68.1 | 19.0 | 18.8 | 20.5 | 20.5 | 20.6 | 20.6 | 20.6 | 21.0 | 20.3 | 20.3 |
| 12 | 30.6 | 35.1 | 31.4 | 35.8 | 31.6 | 23.7 | 22.6 | 25.4 | 25.4 | 25.3 | 25.5 | 25.4 | 25.5 | 25.1 | 25.1 |
| 13 | 41.5 | 44.9 | 47.8 | 50.9 | 49.7 | 50.4 | 42.5 | 50.4 | 50.6 | 50.4 | 45.6 | 45.7 | 51.3 | 50.2 | 49.6 |
| 14 | 75.6 | 78.1 | 77.8 | 79.6 | 79.3 | 80.4 | 76.9 | 80.1 | 80.1 | 80.3 | 79.1 | 79.1 | 80.1 | 80.0 | 80.2 |
| 15 | 92.2 | 88.9 | 90.6 | 91.2 | 92.0 | 90.6 | 91.1 | 86.9 | 86.7 | 86.7 | 86.5 | 86.3 | 86.2 | 87.3 | 87.3 |
| 16 | 88.2 | 89.9 | 81.1 | 82.3 | 81.8 | 81.5 | 88.4 | 80.0 | 80.2 | 80.0 | 88.2 | 88.2 | 80.2 | 80.1 | 80.0 |
| 17 | 18.9 | 22.7 | 23.8 | 26.3 | 25.5 | 24.1 | 19.1 | 21.9 | 21.7 | 21.8 | 19.0 | 19.0 | 21.9 | 21.6 | 21.7 |
| 18 | 113.9 | 113.0 | 113.9 | 111.7 | 112.3 | 112.3 | 49.5 | 112.1 | 112.0 | 22.8 | 52.7 | 52.7 | 52.7 | 23.3 | 23.3 |
| 19 | 19.2 | 18.8 | 19.1 | 19.7 | 20.0 | 19.9 | 18.3 | 19.2 | 19.1 | 20.8 | 17.6 | 17.5 | 17.5 | 68.4 | 68.4 |
| 20 | 30.2 | 27.4 | 28.8 | 26.9 | 27.8 | 27.5 | 29.6 | 33.3 | 33.4 | 34.0 | 33.1 | 33.0 | 33.1 | 33.7 | 33.7 |
| 4-OAc | 169.0, 22.5 | ||||||||||||||
| 6-OAc/OPr | 169.1, 19.9 | 169.1, 20.0 | 169.2, 19.9 | 169.6, 20.4 | 169.2, 20.6 | 169.2, 20.5 | 169.2, 21.1 | 169.2, 20.6 | 169.2, 20.5 | 169.3, 20.9 | 168.8, 21.3 | 172.2, 27.9, 9.0 | |||
| 7-OAc/OPr | 170.7, 21.5 | 170.1, 21.6 | 170.2, 21.5 | 170.5, 21.6 | 172.6, 28.1, 9.1 | 169.5, 21.4 | 169.4, 21.6 | 173.0, 28.4, 9.4 | 169.8, 21.5 | 169.6, 21.5 | 169.5, 21.6 | 169.4, 21.6 | |||
| 11-OAc | 168.9, 21.3 | 169.0, 21.3 | 169.2, 21.6 | ||||||||||||
| 14-OAc/OPr | 174.8, 28.3, 9.2 | 173.6, 28.5, 9.5 | 173.2, 28.4, 9.1 | 174.8, 28.4, 9.3 | 170.0, 21.7 | 173.1, 28.5, 9.1 | 173.1, 28.6, 9.1 | 170.7, 22.0 | 170.6, 21.9 | 173.1, 28.4, 9.1 | 173.2, 28.6, 9.1 | 170.0, 22.1 | |||
| 15-OAc | 171.9, 20.8 | 171.0, 21.0 | 171.5, 20.9 | 171.7, 20.8 | 171.9, 21.0 | 171.8, 20.9 | 171.7, 20.9 | 171.9, 21.0 | 171.8, 20.9 | 171.4, 20.5 | 171.9, 20.9 | 171.9, 20.9 | |||
| 16-OAc | 170.3, 22.9 | 169.8, 22.5 | 170.3, 22.9 | 170.1, 22.8 | 170.0, 22.7 | ||||||||||
| 19-OAc | 170.9, 20.8 | 170.9, 20.8 | |||||||||||||
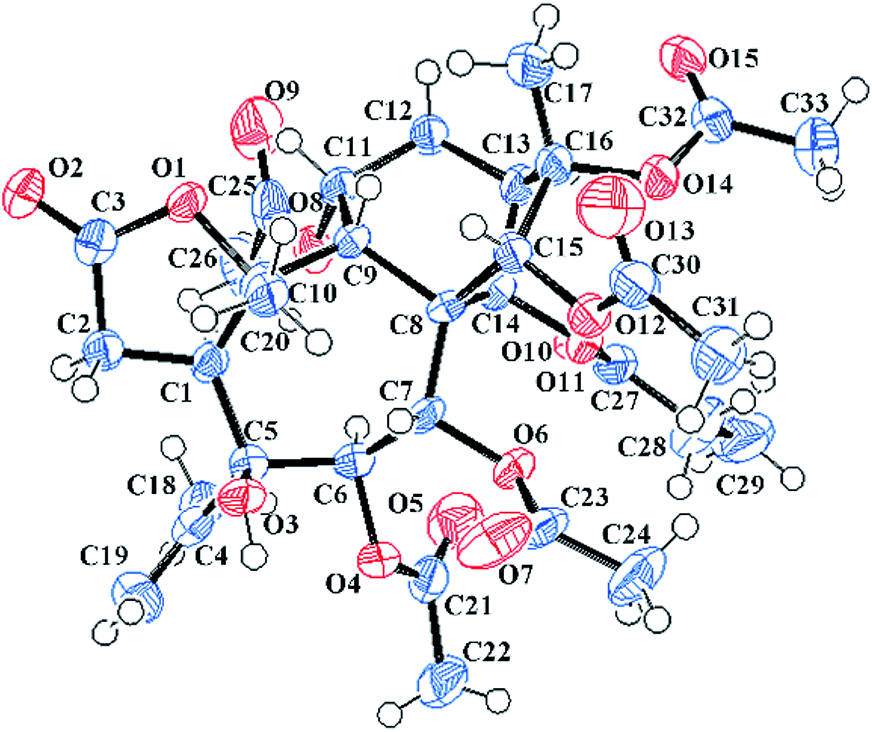 | ||
| Fig. 2 X-ray crystal structure of 1. | ||
Pieristoxinlactone B (2) was assigned the molecular formula C31H42O14 based on the HRESIMS ion at m/z 661.2477 [M + Na]+, indicative of eleven degrees of unsaturation. The 1H and 13C NMR data (see Tables 1 and 2) of 2 closely resembled those of 1, with the only difference between the two compounds was a hydroxy group replacing the acetoxyl group at C-11 of 2. The observation of upfield shift of H-11 (ΔδH 1.69) and C-11 (ΔδC 5.8) of 2 compared to those of 1 and the key HMBC correlation from 11-OH to C-11 confirmed the above deduction. The NOESY correlations from H-14 to H-1, H-6, and H-12a, and from H-1 to H3-19 and 11-OH, suggested these protons are α-oriented. The NOESY correlations from H-9 to H-7, H-11, H-15, and H3-17, and from H3-20 to H-7 and 5-OH indicated that these protons are β-oriented. Thus, the structure of compound 2 was determined and it was given the trivial name pieristoxinlactone B.
Pieristoxinlactone C (3) exhibited the molecular formula of C31H42O14, similar to 2, as deduced from the HRESIMS ion (m/z 661.2467 [M + Na]+). Analysis of the NMR data (see Tables 1 and 2) clearly indicated that 3 is closely related to 1. The major differences are that the signals due to an acetyl group was absent, and the resonances for C-13, C-15, C-16, and C-17 were shifted by ΔδC + 6.3, −1.6, −7.1, and +4.9 ppm, respectively, compared to those of 1. This finding indicated that the acyloxy group at C-16 in 1 was replaced by a hydroxy group in 3. The NOESY correlations from H-14 to H-1, H-6, from H-6 to H3-19 and 11-OH, suggested that these protons are α-orientated. The NOESY correlations from H-9 to H-15 and H3-17, and from H3-20 to H-7 indicated that these protons were β-orientated. Furthermore, NOESY correlations from H-9 to H-11, together with the small coupling constant of H-9 (brs), indicated that H-11 was also β-orientated. Thus, the structure of 3 was established as assigned, and it was named pieristoxinlactone C.
Pieristoxinlactone D (4) had the molecular formula C20H30O9 based on its HRESIMS ion at m/z 415.1984 [M + H]+, indicative of six degrees of unsaturation. The 1H NMR (see Table 1), together with the HSQC showed three tertiary methyls (δH 2.07, 1.92, 1.56), a terminal double bond (δH 5.56, 5.04, brs) and a pair of AB doublets (δH 5.52, 4.88, d, J = 9.5 Hz) in the low-field region. The 13C NMR (see Table 2) and DEPT spectra displayed 20 carbon resonances (one olefinic quaternary carbon resonance should appear at δC 151.4) ascribed to three methyls, three methylenes (one olefinic at δC 111.7), eight methines (five oxygenated at δC 91.2, 79.6, 73.4, 72.5, 63.2), and six quaternary carbons (one ester carbonyl at δC 175.4; one olefinic at δC 151.4; three oxygenated at δC 89.8, 82.3, 77.0). The 1H–1H COSY (see Fig. 3) and HSQC spectra established the presence of three spin systems: C(1)H–C(2)H2, C(6)H–C(7)H and C(9)H–C(11)H–C(12)H2–C(13)H–C(14)H. Analysis of the 1H and 13C NMR data of 4 showed that its structure was closely related to 1 except for the absent of six acyl groups. A more detailed analysis of the 2D NMR data (see Fig. 3) revealed that 4 is based on the same carbon skeleton as 1. The NOESY correlations (see Fig. 3) from H-14 to H-1, H-6, and H-12a and from H-6 to H-19 established these protons were spatially and were α-orientated. On the other hand, the NOESY correlations from H-7 to H-9 and H3-20, from H-9 to H-15 and H3-17, and from H-12b to H-11 and H3-17 suggested the β-orientation of H-7, H-9, H-11, H-12b, H-15, and H3-17. The small coupling constant between H-11 and H-12b (3JH11,H12b = 5.5 Hz) in the 1H NMR spectrum also proved that H-11 was β-orientated. Therefore, the structure of 4 was assigned as shown, and it was named pieristoxinlactone D.
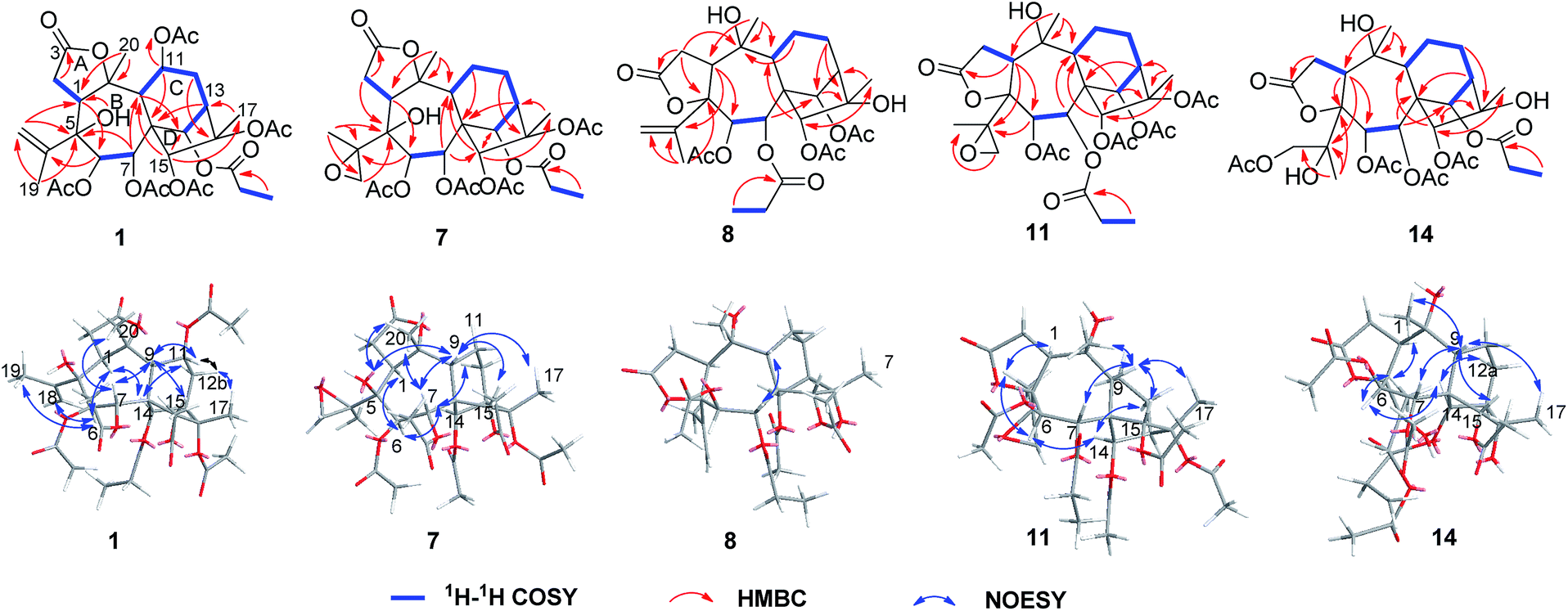 | ||
| Fig. 3 Key 1H–1H COSY, HMBC and NOESY correlations of compounds 1, 7, 8, 11, and 14. | ||
Pieristoxinlactone E (5) possessed a molecular formula of C22H32O10 as assigned by the HRESIMS data (m/z 457.2075 [M + H]+). Comparison of the NMR data of 5 (see Tables 1 and 2) with those of 4 revealed that they are structural analogues. The only difference between the two compounds was the presence of an acetoxyl group at C-11 of 5 instead of a hydroxy group in 4, which was further confirmed by the HMBC correlation from H-11 to the acetate carbonyl carbon (δC 169.2). Therefore, the structure of 5 was assigned as shown, and it was named pieristoxinlactone E.
The molecular formula of 6 was assigned as C20H30O8, by the HRESIMS ion at m/z 421.1831 [M + Na]+ and 13C NMR data. Analysis of the NMR data (see Tables 2 and 3) indicated that the structure of 6 closely resembled that of 4 with the only difference was the absence of the hydroxy group at C-11 in 6, which was confirmed by the spin system C(9)H–C(11)H2–C(12)H2–C(13)H–C(14)H verified by 1H–1H COSY and HSQC spectra. Further 2D NMR experiments enabled the full assignments of the 1H and 13C NMR data of 6. Therefore, 6 was assigned as shown, and was named pieristoxinlactone F.
| No. | 6b | 7a | 8b | 9b | 10b |
|---|---|---|---|---|---|
| a Recorded at 500 MHz.b Recorded at 600 MHz. | |||||
| 1 | 3.20, dd (13.2, 6.6) | 3.43, dd (13.0, 7.0) | 3.09, t (10.2) | 3.09, t (10.2) | 3.62, dd (12.0, 7.8) |
| 2 | a 3.13, dd (16.2, 13.2), b 2.55, dd (16.2, 6.6) | a 3.55, overlap, b 3.11, dd (17.5, 7.0) | a 2.94, overlap, b 2.91, overlap | a 2.94, overlap, b 2.91, overlap | a 3.11, dd (19.2, 12.0), b 2.94, dd (19.2, 7.8) |
| 6 | 4.65, d (8.4) | 5.60, d (10.0) | 6.59, d (9.0) | 6.60, d (9.0) | 6.78, d (10.2) |
| 7 | 5.27, d (8.4) | 5.35, d (10.0) | 5.78, d (9.0) | 5.77, d (9.0) | 5.77, d (10.2) |
| 9 | 2.37, t (6.0) | 3.23, dd (11.5, 7.0) | 2.71, brd (7.2) | 2.66, brd (7.8) | 2.66, brd (7.8) |
| 11 | a 1.63, overlap, b 1.61, overlap | a 1.78, m, b 1.71, overlap | a 2.12, m, b 1.78, m | a 2.12, m, b 1.77, m | a 2.13, overlap, b 1.81, overlap |
| 12 | a 1.93, dd (15.0, 7.8), b 1.57, overlap | a 2.06, overlap, b 1.57, m | a 2.15, m, b 1.67, m | a 2.15, m, b 1.67, m | a 2.16, overlap, b 1.65, overlap |
| 13 | 2.41, brd (7.8) | 3.53, brd (7.0) | 2.49, brs | 2.47, brs | 2.49, overlap |
| 14 | 4.93, brs | 6.15, brs | 6.94, brs | 6.99, brs | 6.86, brs |
| 15 | 4.26, s | 5.35, s | 5.44, s | 5.46, s | 5.48, s |
| 17 | 1.53, s | 1.73, s | 1.45, s | 1.46, s | 1.46, s |
| 18 | a 5.54, s, b 5.08, s | a 3.29, d (5.5), b 2.48, d (5.5) | a 5.42, s, b 4.89, s | a 5.41, s, b 4.88, s | 1.91, s |
| 19 | 1.99, s | 1.61, s | 1.99, s | 1.99, s | 2.01, s |
| 20 | 1.84, s | 2.05, s | 1.47, s | 1.47, s | 1.57, s |
| 4-OAc | 1.90, s | ||||
| 6-OAc | 2.01, s | 2.02, s | 2.03, s | 2.15, s | |
| 7-OAc/OPr | 2.29, s | a 2.58, m, b 2.47, m, 1.23, t (6.0) | 2.22, s | 2.27, s | |
| 14-OAc/OPr | a 2.79, m, b 2.47, overlap, 1.36, t (7.5) | 2.23, s | a 2.58, m, b 2.51, m, 1.26, t (6.0) | a 2.55, m, b 2.50, overlap, 1.27, t (7.8) | |
| 15-OAc | 2.24, s | 2.10, s | 2.13, s | 2.14, s | |
| 16-OAc | 2.17, s | ||||
| 5-OH | 5.65, s | ||||
| 10-OH | 7.29, s | 7.30, s | 7.32, s | ||
| 16-OH | 3.64, s | 3.94, s | |||
The HRESIMS ion at m/z 661.2478 [M + Na]+ and the 13C NMR data of 7 suggested a molecular formula of C31H42O14. The 1H and 13C NMR data of 7 (see Tables 2 and 3) closely resembled those of pierisoid A17 except for the location of the propionyloxyl. Key HMBC correlations (see Fig. 3) from H-7 to one acetyl carbonyl (δC 170.5) and from H-14 to the propionyl carbonyl (δC 174.8) suggested that one acetyloxyl and the propionyloxyl were connected to C-7 and C-14, respectively. Further HSQC and HMBC experiments (see Fig. 3) enabled the full assignments of the 1H and 13C NMR spectra of 7 to be carried out. The NOESY correlations (see Fig. 3) from H-14 to H-1, H-6, and H-12a established that these protons were located on the same face of the ring, while the NOESY correlations (see Fig. 3) from H-7 to H-9 and H3-20, from H-9 to H-15 and H3-17, and from 5-OH to H-9 and H3-20 suggested these protons were on the other face of the ring. However, the chiral quaternary C-4 was in the side chain and its relative configuration couldn't be assigned. Subsequently, an X-ray single-crystal structure of 7 (see Fig. 4; CCDC: 1548369†), obtained using Cu Kα radiation [Flack parameter: 0.04(7)], not only permitted the unambiguous assignment of the propionyloxyl but also defined its absolute configuration as 1S, 4R, 5S, 6R, 7R, 8S, 9R, 10R, 13R, 14R, 15R, 16S. Thus, the structure of 7, named pieristoxinlactone G, was finally determined.
 | ||
| Fig. 4 X-ray crystal structure of 7. | ||
The molecular formula C29H40O12 of pieristoxinlactone H (8) was assigned according to the HRESIMS ion at m/z 603.2398 [M + Na]+. The 1H and 13C NMR data of 8 (see Tables 2 and 3) revealed a close resemblance to those of secorhodomollolide A,5 and the only difference between the two compounds involved a hydroxy group at C-16 of 8 replacing an acetoxy group of secorhodomollolide A. Compared to secorhodomollolide A, the 1H NMR resonances for H-13 and H3-17 of 8 were shifted by ΔδH −1.15 and −0.25, while the 13C NMR resonances for C-13, C-16, and C-17 of 8 were shifted by ΔδC +5.5, −8.1, +3.0 ppm, respectively, suggesting the presence of a hydroxy group at C-16 in 8. The NOESY correlations (see Fig. 3) from H-9 to H-7 and H-15, from H-15 to H-11b and H3-17, from H3-17 to H-12b suggested the β-orientation of these protons. The large coupling constant between H-6 and H-7 (3JH6,H7 = 9.0 Hz) in the 1H NMR spectrum indicated that H-6 and H-7 were in the trans configuration. Therefore, H-6 was α-orientated. Moreover, the NOESY correlations from H-14 to H-12a, from H3-19 to H-1 and H-6, and from H-1 to 10-OH established these protons to be α-orientated. Thus, the structure of pieristoxinlactone H (8) was assigned as drawn.
Compound 9 had the same molecular formula C29H40O12 as 8, as assigned by the HRESIMS (m/z 603.2417 [M + Na]+) and 13C NMR data. Its NMR data were highly similar to those of 8 except for the slight variations in the chemical shifts of H-14 (ΔδH +0.05), C-7 (ΔδC +0.3), and the propionyl carbonyl (ΔδC +0.5), suggesting that the propionyloxyl group may connect to C-14 in 9. The key HMBC correlations from H-7 to one acetyl carbonyl (δC 169.5) and from H-14 to the propionyl carbonyl (δC 173.1) confirmed the above assignment. The relative configuration of 9 was assigned as the same as 8 by their similar NMR and NOESY data. Therefore, the structure of 9 was assigned, and it was named pieristoxinlactone I.
The molecular formula C31H44O14 of 10 was identical to 24, as determined by 13C NMR data and the HRESIMS ion at m/z 663.2618 [M + Na]+. The general features of the NMR data of 10 (see Tables 2 and 3) resembled those of 24, except for the substituents at C-4 and C-16. The observation of the upfield shift of H-13 (ΔδH 1.11) and H3-17 (ΔδH −1.11), and downfield shift of H3-18 (ΔδH 0.24) and H3-19 (ΔδH 0.36) in the 1H NMR spectrum of 10, as well as the downfield shift of C-4 (ΔδC 13.8), C-13 (ΔδC 5.3), and C-17 (ΔδC 2.9) and an upfield shift of C-4 (ΔδC 8.2) in the 13C NMR spectrum of 10 compared with those of 24 suggested that an acetyloxyl group and a hydroxy group were connected to C-4 and C-16, respectively. The HMBC cross-peaks from H-1 and H3-19 to C-4 (δC 89.7) and from H-14, H-13 and H3-17 to C-16 (δC 80.0) reconfirmed the above assignment. Compound 10, pieristoxinlactone J, was thereby structurally characterized.
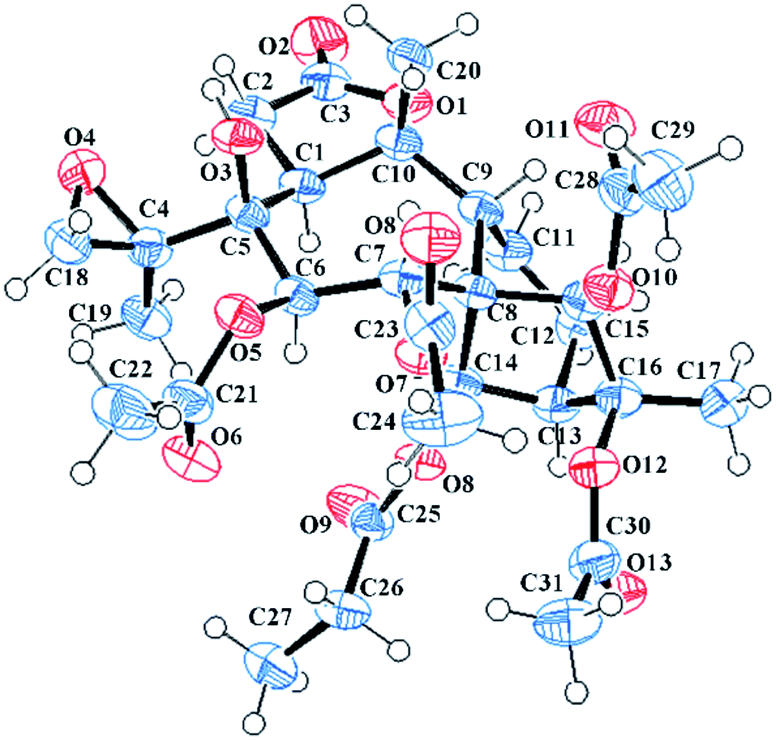
The molecular formula C31H42O14 was assigned to 11 with eleven indices of hydrogen deficiency by the HRESIMS ion at m/z 661.2469 [M + Na]+ and 13C NMR data. The NMR data of 11 (see Tables 2 and 4) showed many similarities to those of 23, revealing that these two compounds are homologues, except for the presence of a 4,18-oxirane group in 11. Compared to 23, the C-4 carbon resonance was shielded (ΔδC 13.9) and the C-18 carbon resonance was deshielded (ΔδC 26.9), suggesting that an oxirane group was present between C-4 and C-18. Moreover, the key HMBC correlations (see Fig. 3) from H3-19 to C-4 (δC 62.0) and C-18 (δC 52.7) further confirmed this deduction. The NOESY correlations (see Fig. 3) from H-14 to H-6 and H-12a and from H3-19 to H-1 and H-6 suggested that these protons are spatially close and α-orientated. In contrast, the NOESY correlations from H-9 to H-7, H-15, H3-17, and H3-20 demonstrated the β-orientation of H-7, H-9, H-15, H3-17, and H3-20. However, the relative configuration of C-4 still remains uncertain because C-4 was on the side chain. Finally, an X-ray diffraction study using the anomalous dispersion radiation of Cu Kα [Flack parameter: −0.07(15)] of 11 (see Fig. 5; CCDC: 1548367†) defined its absolute configuration as 1S, 4R, 5S, 6R, 7R, 8S, 9R, 10R, 13R, 14R, 15R, 16S. Thus, the structure of 11 was determined, and it was named pieristoxinlactone K.
| No. | 11a | 12b | 13b | 14b | 15b |
|---|---|---|---|---|---|
| a Recorded at 500 MHz.b Recorded at 600 MHz. | |||||
| 1 | 3.42, brd (11.5) | 3.41, dd (11.4, 7.8) | 3.41, dd (11.4, 8.4) | 4.09, t (10.2) | 4.09, t (10.2) |
| 2 | a 3.00, dd (19.0, 11.5), b 2.81, dd (19.0, 8.0) | a 2.99, dd (18.6, 11.4), b 2.79, dd (18.6, 7.8) | a 3.01, dd (18.6, 11.4), b 2.80, dd (18.6, 8.4) | a 3.03, m, b 2.98, m | a 3.03, m, b 2.98, m |
| 6 | 6.55, d (9.5) | 6.55, d (9.6) | 6.60, d (9.0) | 6.62, d (8.4) | 6.68, d (9.0) |
| 7 | 5.80, d (9.5) | 5.76, d (9.6) | 5.84, d (9.0) | 5.73, d (8.4) | 5.73, d (9.0) |
| 9 | 2.64, brd (8.0) | 2.61, d (7.8) | 2.52, overlap | 2.74, brd (8.4) | 2.74, brd (7.8) |
| 11 | a 2.09, overlap, b 1.78, m | a 2.11, m, b 1.75, m | a 2.08, m, b 1.79, m | a 2.12, overlap, b 1.79, m | a 2.11, overlap, b 1.79, m |
| 12 | a 2.18, overlap, b 1.66, overlap | a 2.17, m, b 1.65, m | a 2.14, m, b 1.65, m | a 2.18, overlap, b 1.67, m | a 2.17, overlap, b 1.67, m |
| 13 | 3.61, t (4.0) | 3.58, t (3.6) | 2.46, t (3.0) | 2.44, m | 2.48, overlap |
| 14 | 7.04, brs | 7.03, brs | 7.14, brs | 6.78, brs | 6.75, brs |
| 15 | 5.53, s | 5.52, s | 5.51, s | 5.45, s | 5.45, s |
| 17 | 1.71, s | 1.71, s | 1.47, s | 1.46, s | 1.47, s |
| 18 | a 3.35, d (5.5), b 2.59, d (5.5) | a 3.34, d (5.4), b 2.59, d (5.4) | a 3.35, d (4.8), b 2.60, d (4.8) | 1.74, s | 1.75, s |
| 19 | 1.62, s | 1.61, s | 1.61, s | a 4.76, d (11.4), b 4.70, d (11.4) | a 4.78, d (11.4), b 4.69, d (11.4) |
| 20 | 1.49, s | 1.49, s | 1.50, s | 1.47, s | 1.47, s |
| 6-OAc | 2.14, s | 2.14, s | 2.13, s | 2.17, s | a 2.52, m, b 2.45, overlap, 1.16, t (7.2) |
| 7-OAc/-OPr | a 2.68, m, b 2.56, overlap, 1.34, t (7.5) | 2.29, s | 2.25, s | 2.26, s | 2.24, s |
| 14-OAc/-OPr | 2.24, s | 2.22, s | a 2.65, m, b 2.53, overlap, 1.28, t (7.2) | a 2.60, m, b 2.50, m, 1.26, t (7.2) | 2.23, s |
| 15-OAc | 2.13, s | 2.13, s | 2.12, s | 2.14, s | 2.14, s |
| 16-OAc | 2.15, s | 2.15, s | |||
| 19-OAc | 2.06, s | 2.05, s | |||
| 10-OH | 7.55, s | 7.51, s | 7.50, s | 7.33, s | 7.32, s |
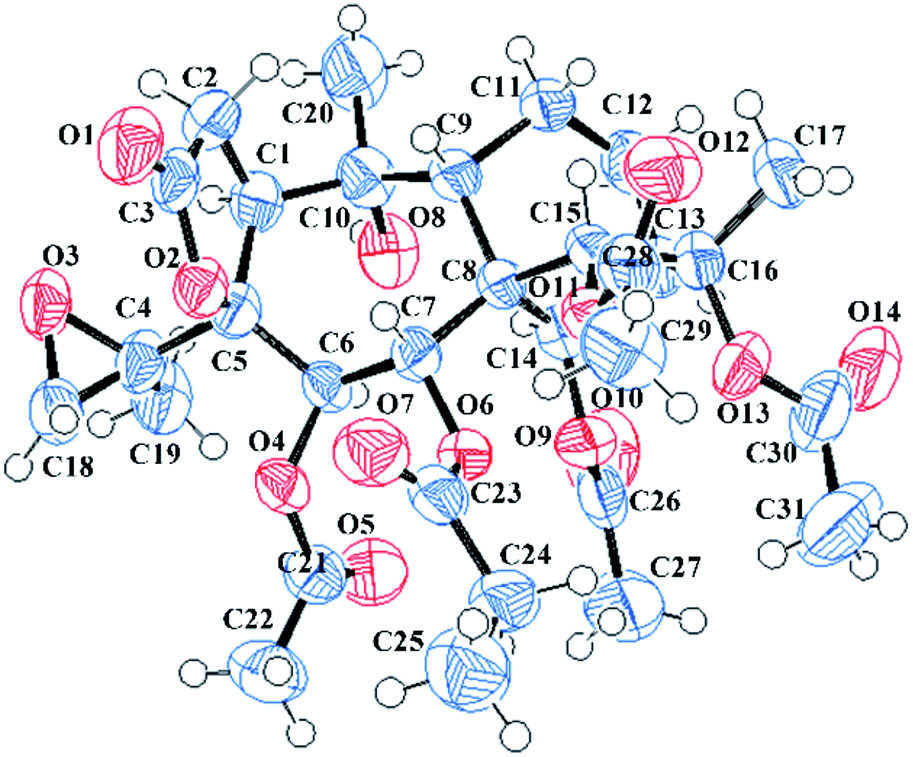 | ||
| Fig. 5 X-ray crystal structures of 11. | ||
The molecular formula of 12, C30H40O14, was determined by the HRESIMS ion at m/z 647.2307 [M + Na]+. A careful analysis of the NMR data of 12 revealed 12 to be based on a 3,4-secograyanane skeleton in a similar manner to 11, except for an acetoxy group substituent at C-7 in 12 instead of a propionyloxy group in 11. The key HMBC cross-peak from H-7 to an acetyl carbonyl carbon (δC 169.8) confirmed the assignment. The structure of compound 12, pieristoxinlactone L, was thus defined as depicted.
Compound 13 had a molecular formula of C29H40O13, as assigned by HRESIMS (m/z 619.2355 [M + Na]+). The 1H and 13C NMR spectra of compound 13 (see Tables 2 and 4) were similar to those of 11, suggesting that these two compounds were homologues. Compound 13 differs from 11 by substitutions at C-7, C-14, and C-16. The HMBC correlations from H-7 to one acetyl carbonyl (δC 169.6) and from H-14 to the propionyl carbonyl (δC 173.1) suggested that C-7 and C-14 were connected to an acetoxyl and propionyloxyl, respectively. In addition, compared to 11, the resonances of H-13 and H3-17 were shifted by ΔδH −1.15 and −0.24, and the resonances of C-13, C-16, and C-17 were shifted by ΔδC +5.7, −8.0, and +2.9 ppm, respectively, suggesting a hydroxy group substituent at C-16 in 13. Thus, the structure of 13 was eventually determined, and it was named pieristoxinlactone M.
Compound 14 exhibited a molecular formula of C31H44O15 based on the HRESIMS ion at m/z 679.2572 [M + Na]+ and with the aid of 13C NMR data. The 1H and 13C NMR spectra of 14 (see Tables 2 and 4) were similar to those of 10, differing only in the presence of a hydroxy group and an oxygenated methylene group (δC 68.4) and the absence of a methyl group in 14. Compared to 10, the C-4 carbon resonance of 14 was shielded (ΔδC 12.8), revealing that the hydroxy group substituent was at C-4. Moreover, the HMBC correlations (see Fig. 3) from H2-19 to one acetyl carbonyl (δC 170.9) and from H3-18 to C-4 (δC 76.9) and C-19 (δC 68.4) indicated that the hydroxy group and one acetyl group were placed at C-4 and C-19, respectively. The NOESY correlations (see Fig. 3) from H-14 to H-12a and H-6 and from H3-18 to H-1 and H-6 established the α-orientations of H-1, H-6, H-12a, H-14, and H3-18, while correlations from H-9 to H-7, H-15, H3-17, and H3-20 confirmed the β-orientations of these protons. The relative configuration of C-4 remains uncertain because C-4 is located on the side chain. Finally, the X-ray diffraction [Cu Kα; Flack parameter: 0.04(16)] of 14 (see Fig. 6; CCDC: 1548368†) was successfully performed, which not only confirmed the assigned structure but also established the absolute configuration as 1S, 4S, 5S, 6R, 7R, 8S, 9R, 10R, 13R, 14R, 15R, 16S. Thus, the structure of pieristoxinlactone N (14) was characterized as shown.
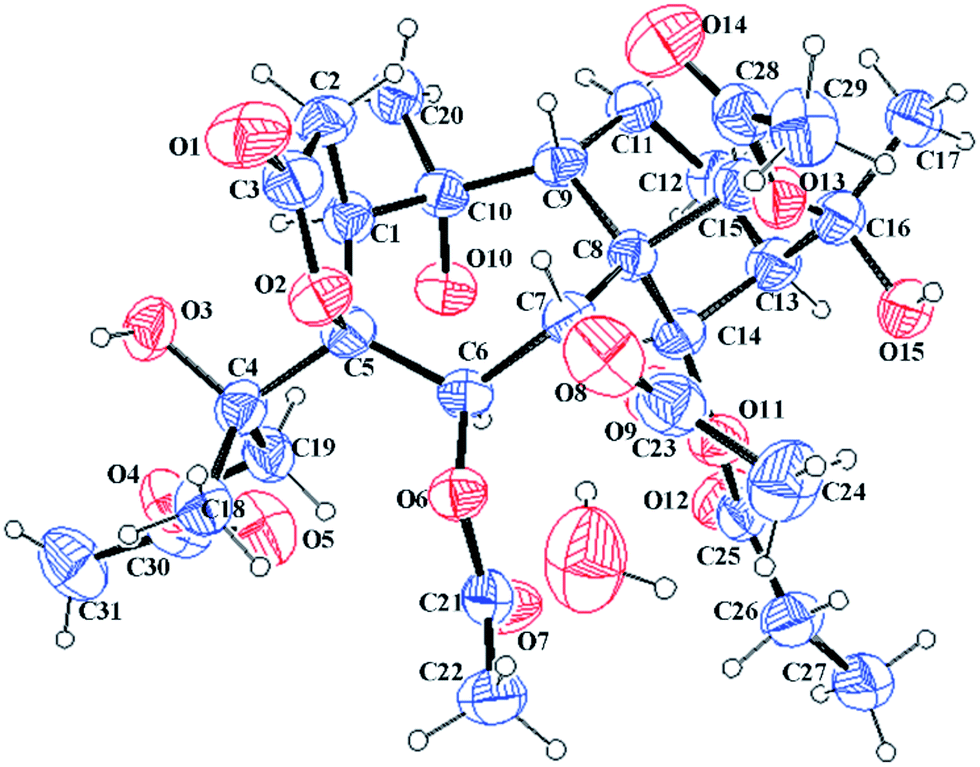 | ||
| Fig. 6 X-ray crystal structures of 14. | ||
The molecular formula of 15 was deduced to be C31H44O15, identical to that of 14, as indicated by the HRESIMS ion at m/z 679.2572 [M + Na]+. NMR data analysis showed that its structure was closely related to that of 14, differing only in the location of the propionyloxyl group (at C-6 in 15; at C-14 in 14). The key HMBC correlations from H-6 to the propionyl carbonyl (δC 172.2) and from H-14 to one acetyl carbonyl (δC 170.0) confirmed that the propionyloxyl group was connected to C-6. Thus, the structure of compound 15 was determined as drawn, and it was named pieristoxinlactone O.
The HRESIMS ion peak at m/z 417.2123 [M + H]+ and the 13C NMR data suggested a molecular formula of C20H32O9 for compound 16. Apart from the signals for the five acyl groups, the 1H and 13C NMR data of 16 (see Table 5) and 10 were similar, suggesting that these two compounds were homologues. Further 1H–1H COSY, HSQC, and HMBC experiments allowed for the full assignments of the 1H and 13C NMR spectra of 16. Compound 16 shared the same carbon skeleton as 10. The NOESY correlations from H-14 to H-12a, H3-19 and from H-1 to H-2a, H-6, and H3-19 demonstrated that the α-orientation of these protons, while the NOESY correlations from H-9 to H-7 and H-15, H3-20 to H-2b, and from H-15 to H3-17 confirmed the β-orientation of these protons. Compound 16, named pieristoxinlactone P, was thereby structurally characterized.
| No. | 16 | 17 | ||
|---|---|---|---|---|
| δH (J in Hz) | δC | δH (J in Hz) | δC | |
| 1 | 3.80, dd (13.2, 5.4) | 51.5 | 3.71, dd (11.4, 10.8) | 51.1 |
| 2 | a 3.10, dd (18.0, 13.2), b 2.72, dd (18.0, 5.4) | 35.0 | a 3.04, dd (18.0, 10.8), b 2.98, dd (18.0, 11.4) | 34.7 |
| 3 | 176.6 | 176.2 | ||
| 4 | 76.7 | 75.6 | ||
| 5 | 95.2 | 95.1 | ||
| 6 | 5.60, brs | 70.4 | 5.61, d (7.2) | 71.6 |
| 7 | 4.65, brs | 73.6 | 5.08, d (7.2) | 70.9 |
| 8 | 57.6 | 57.6 | ||
| 9 | 2.02, overlap | 47.2 | 2.41, d (4.2) | 54.9 |
| 10 | 77.6 | 78.2 | ||
| 11 | a 1.96, overlap, b 1.56, m | 21.0 | 4.54, t (4.8) | 68.1 |
| 12 | a 1.91, overlap, b 1.60, m | 26.0 | a 2.39, overlap, b 2.02, dd (15.6, 4.2) | 34.7 |
| 13 | 2.42, t (3.0) | 51.2 | 2.58, brd (10.2) | 46.8 |
| 14 | 5.73, brs | 81.1 | 5.89, brs | 77.8 |
| 15 | 4.20, s | 88.2 | 4.04, s | 90.7 |
| 16 | 81.2 | 82.2 | ||
| 17 | 1.50, s | 22.5 | 1.61, s | 23.3 |
| 18 | 2.05, s | 28.1 | 1.86, s | 27.9 |
| 19 | 1.82, s | 25.9 | 1.76, s | 26.8 |
| 20 | 1.47, s | 34.7 | 1.67, s | 33.6 |
| 11-OH | 6.53, s | |||
Compound 17 possessed the molecular formula of C20H32O10, as assigned by the HRESIMS ion at m/z 433.2065 [M + H]+ and 13C NMR data. The NMR data of 17 resembled those of 16, except for the only difference of the presence of an oxygenated methine carbon and the absence of a methylene carbon in 17. The spin system C(9)H–C(11)H–C(12)H2–C(13)H–C(14)H established by 1H–1H COSY and HSQC spectra, suggested that a hydroxy group was connected to C-11 (δC 68.1). The α-orientation of 11-OH was confirmed by the NOESY correlations from H-11 to H-9 and H3-20. The structure of pieristoxinlactone Q (17) was thus defined as shown.
The seven known diterpenoids, namely, secorhodomollolide C (18),5 pierisoid B (19),17 secorhodomollolide B (20),5 secorhodomollolide A (21),5 pierisformotoxin G (22),18 pierisformotoxin B (23),19 and pierisformotoxin A (24),19 were identified by the comparison of the experimental and previously reported spectroscopic data.
The antinociceptive activities of compounds 1, 2, 4–8, 10–13, and 16–24 were evaluated using an acetic acid-induced writhing test. As shown in Fig. 7a, all evaluated compounds exhibited significant analgesic activity at 5.0 mg kg−1 (ip) compared to vehicle-injected mice (p < 0.05). In particular, compounds 16 and 17 showed highly potent activities, with writhe inhibition rates of 63.5% and 69.9%, respectively. To further understand the action of the antinociceptive activities of compounds 16 and 17, three doses (5.0, 1.0, and 0.1 mg kg−1) were employed in an acetic acid-induced writhing test. The results revealed that 16 and 17 were able to reduce acetic acid-induced writhes in a dose-dependent manner (Fig. 7b), with ID50 values of 1.24 and 0.72 mg kg−1, respectively.
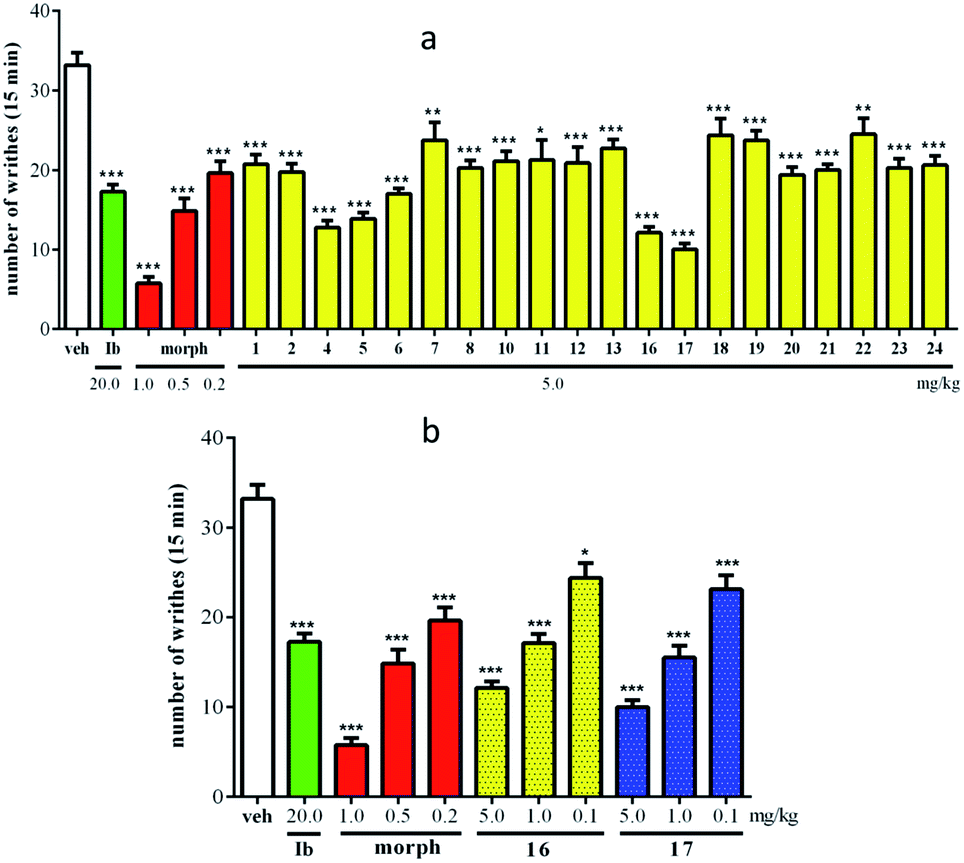 | ||
| Fig. 7 (a) Antinociceptive activities of the purified compounds from the roots of P. formosa in an acetic acid-induced writhing test. (b) Antinociceptive activities of various concentrations of 16 and 17 in an acetic acid-induced writhing test. Ibuprofen (Ib) and morphine (morph) were used as control drugs. Each bar and vertical line represents the mean ± SEM of the values obtained from 8 mice. *p < 0.05, **p < 0.01, ***p < 0.001, significant differences between the test compound or control drug groups and vehicle group (veh) (one-way ANOVA followed by the Bonferroni test). | ||
Experimental
General experimental procedures
Optical rotations were measured on an AUTOPOL V polarimeter. IR spectra were recorded using a Nicolet 5700 FT-IR microscope (FT-IR microscope transmission). 1D and 2D NMR spectra were obtained using an INOVA-500 or a Bruker-600 spectrometer. Online HPLC/UV/ESIMS2 analyses were recorded on an Agilent 1100 Series liquid chromatography system chromatograph. HRESIMS data were recorded on an Agilent 6520 Accurate-Mass Q-TOF LC/MS spectrometer. Preparative HPLC was performed using a Shimadzu LC-6AD instrument equipped with RID-20A and SPD-20A detectors, using a YMC-Pack RP-C18 column (250 × 10 mm, 5 μm) or a COSMOSIL packed column (250 × 10 mm, 5 μm). Column chromatography was performed using polyamide (30–60 mesh, Changzhou Changfeng Chemical Factory, China), macroporous resin D101 (Qingdao Marine Chemical Factory, China), MCI (Mitsubishi Chemical Corporation), and ODS (50 μm, Merck, Germany). TLC was carried out with glass precoated Si gel GF254 plates (Qingdao Marine Chemical Factory, China). Spots were visualized by spraying with 10% H2SO4 in EtOH–H2O (95:5, v/v) followed by heating.
Plant material
Roots of Pieris formosa (120 kg) were collected in Chuxiong Yi Autonomous Prefecture in Yunnan Province, People's Republic of China, in June 2014 and were identified by Prof. Lin Ma (Department of Natural Products Chemistry, Institute of Materia Medica, Chinese Academy of Medical Sciences). A voucher specimen (ID-s-2747) was deposited in the herbarium at the Department of Medicinal Plants, Institute of Materia Medica, Chinese Academy of Medical Sciences.Extraction and isolation
The isolation procedures were guided by acetic acid-induced writhing test and online HPLC/UV/ESIMS2 analyses as described above. The roots of Pieris formosa (120 kg) were extracted with 95% EtOH (150 l × 3 h × 3). The evaporation of the solvent under reduced pressure yielded a black residue (5.0 kg). The residue was mixed with diatomite (1:2) and then successively Soxhlet extracted using a gradient of petroleum ether, CH2Cl2, EtOAc and CH3OH. The CH2Cl2 portion (D) and the EtOAc portion (E) were then applied to a macroporous resin D101 column (10 kg) and eluted with a gradient of 30% EtOH–H2O, 50% EtOH–H2O and 70% EtOH–H2O, to yield six fractions (D1–D3, and E1–E3). Fractions D1 (60 g) and D2 (57 g) were separated through a polyamide column and eluted with H2O–EtOH (100:0, 30:70, 50:50, 70:30, v/v), to yield eight fractions (D1a–D1d and D2a–D2d). Fractions E1 (20 g) and E2 (35 g) were separated through a polyamide column and eluted with a gradient of H2O–EtOH (100:0, 30:70, 60:40, v/v), to yield six fractions (E1a–E1c and E2a–E2c). Fraction D1a (4 g) was separated via an MCI column and eluted with a gradient of H2O–MeOH (1:9–9:1, v/v), to obtain four subfractions D1a1–D1a4. Subfraction D1a1 (0.9 g) was purified by semipreparative HPLC with MeCN–H2O (23:77, v/v, 4.5 ml min−1), to yield 13 (4.5 mg, tR = 37.5 min), 14 (1.0 mg, tR = 34.5 min) and 15 (0.6 mg, tR = 32.5 min). Fraction D2a (4 g) was separated through a MCI column and eluted with a gradient of H2O–MeOH (1:9–9:1, v/v), to obtain four subfractions D2a1–D2a4. Subfraction D2a1 (1.5 g) was purified further using semipreparative HPLC with MeCN–H2O (30:70, v/v, 4.0 ml min−1), to yield 1 (26.0 mg, tR = 70.0 min), 18 (11.0 mg, tR = 72.5 min), 20 (83.0 mg, tR = 57.5 min), and 21 (180.0 mg, tR = 55.5 min). Subfraction D2a2 (0.5 g) was purified further using semipreparative HPLC with MeCN–H2O (22.5:77.5, v/v, 4.0 ml min−1), to yield 3 (1.2 mg, tR = 75.0 min), 8 (2.5 mg, tR = 79.0 min), 10 (3.5 mg, tR = 80.0 min), and 19 (14.0 mg, tR = 91.0 min). Subfraction D2a3 (1.3 g) was purified further using semipreparative HPLC with MeCN–H2O (24:76, v/v, 4.0 ml min−1), to yield 11 (36.0 mg, tR = 103.0 min), 12 (2.5 mg, tR = 70.0 min), 23 (16.0 mg, tR = 61.0 min), and 24 (71.0 mg, tR = 67.0 min). Fraction D2b (8 g) was separated through an ODS column and eluted with 10% CH3OH–H2O, 30% CH3OH–H2O, 50% CH3OH–H2O, and 70% CH3OH–H2O, successively, to yield four fractions (D2b1–D2b4). Subfraction D2b3 (1.5 g) was purified by reversed-phase semipreparative HPLC with MeCN–H2O (23:77, v/v, 4.0 ml min−1), to yield 2 (5.7 mg, tR = 115.5 min), 7 (8.0 mg, tR = 151.5 min), 9 (1.0 mg, tR = 104.5 min), and 22 (7.6 mg, tR = 114.5 min). Fraction E1a (3 g) was chromatographed on a YMC RP-C18 column (17% CH3CN–H2O, v/v, 5 ml min−1) to yield seven fractions (E1a1–E1a7). Subfraction E1a2 (0.6 g) was purified further using semipreparative HPLC with MeCN–H2O (8:92, v/v, 4.5 ml min−1), to yield 4 (6.5 mg, tR = 21.5 min) and 6 (5.0 mg, tR = 38.0 min). Fraction E1b (5 g) was separated through an ODS column and eluted with 10% CH3OH–H2O, 30% CH3OH–H2O, 50% CH3OH–H2O, and 70% CH3OH–H2O, successively, to yield four fractions (E1b1–E1b4). Subfraction E1b1 (2 g) was chromatographed on a YMC RP-C18 column (14% CH3CN–H2O, v/v, 5 ml min−1) to yield four fractions (E1b1a–E1b1d). Subfraction E1b1a (0.3 g) was purified by semipreparative HPLC with MeCN–H2O (12:88, v/v, 5.0 ml min−1), to yield 5 (5.0 mg, tR = 25.5 min). Fraction E2a (15 g) was separated via an MCI column and eluted with a gradient of H2O–MeOH (1:9–9:1, v/v), to obtain five subfractions E2a1–E2a5. Subfraction E2a1 (1 g) was purified further using a semipreparative HPLC with MeCN–H2O (6:94, v/v, 5.0 ml min−1), to yield 16 (6.0 mg, tR = 32.0 min) and 17 (6.0 mg, tR = 18.5 min).
X-ray crystallographic analysis
The crystals of compounds 1, 7, 11, and 14 were obtained from recrystallization in a mixture of solvents (MeOH/H2O, 10:1) at room temperature. The X-ray crystallography studies of the four compounds were conducted according to the usual procedure (for details, see Tables S1–S4 in the ESI†). The crystallographic data for compounds 1, 7, 11, and 14 reported in this paper have been deposited at the Cambridge Crystallographic Data Centre (deposition No. CCDC 1548366, 1548369, 1548367, and 1548368† for 1, 7, 11, and 14, respectively).
Animals
All animal care and experimental procedures were in accordance with the current guidelines of the National Institutes of Health (NIH). Animal experiments were approved by the Ethics Committee of Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College.Acetic acid-induced writhing test
Groups of 8 Kunming mice were used as control and test mice. The mice were given an intraperitoneal injection of 1% v/v acetic acid solution 15 minutes after the administration of the compounds (blank control received the vehicle, 0.9% NaCl, 5 ml kg−1, ip; positive control received the morphine, 0.5 mg kg−1, ip). The mice were placed individually in glass boxes. The numbers of writhes produced in these mice were counted for 20 minutes. For scoring purposes, a writhe was indicated by a stretching of the abdomen with a simultaneous stretching of at least one hind limb.Conclusions
In summary, herein we have reported seventeen new 3,4-secograyanoids (1–17), together with seven known compounds (18–24), which were isolated from the roots of Pieris formosa. Their structures with absolute configurations were characterized by a series of spectroscopic methods and X-ray diffraction. Compounds 1, 2, 4–8, 10–13, and 16–24 exhibited significant analgesic activity at 5.0 mg kg−1 (ip) compared to vehicle-injected mice (p < 0.05). In particular, compounds 16 and 17 showed highly potent activities with writhe inhibition rates of 63.5% and 69.9%, respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81673314) and CAMS Innovation Fund for Medical Sciences (No. 2016-I2M-1-010). The authors are grateful to the Department of Instrumental Analysis at our institute for the spectroscopic measurements.References
- Y. Li, Y. B. Liu and S. S. Yu, Phytochem. Rev., 2013, 12, 305–325 CrossRef CAS.
- W. H. Tallent, M. L. Riethof and E. C. Horning, J. Am. Chem. Soc., 1954, 79, 4548–4554 CrossRef.
- P. Narayanan, M. Rohrl, K. Zechmeister and W. Hoppe, Tetrahedron Lett., 1970, 11, 3943–3944 CrossRef.
- S. Fushiya, H. Hikino and T. Takemoto, Tetrahedron Lett., 1974, 15, 183–186 CrossRef.
- S. J. Wang, S. Lin, C. G. Zhu, Y. C. Yang, S. Li, J. J. Zhang, X. G. Chen and J. G. Shi, Org. Lett., 2010, 12, 1560–1563 CrossRef CAS PubMed.
- Z. Y. Wu, H. Z. Li and W. G. Wang, Phytochem. Lett., 2012, 5, 87–90 CrossRef CAS.
- Y. Li, Y. B. Liu, J. J. Zhang, Y. H. Li, J. D. Jiang, S. S. Yu, S. G. Ma, J. Qu and H. N. Lv, Org. Lett., 2013, 15, 3074–3077 CrossRef CAS PubMed.
- J. W. Burke, R. W. Doskotch, C. Z. Ni and J. Clardy, J. Am. Chem. Soc., 1989, 111, 5831–5833 CrossRef CAS.
- S. Z. Zhou, S. Yao, C. P. Tang, C. Q. Ke, L. Li, G. Lin and Y. Ye, J. Nat. Prod., 2014, 77, 1185–1192 CrossRef CAS PubMed.
- A. Furusaki, N. Hamanaka and H. Miyakoshi, Chem. Lett., 1972, 1, 783–786 CrossRef.
- M. K. Zhang, Y. Zhu, G. Q. Zhan, P. H. Shu, R. J. Sa, L. Lei, M. Xiang, Y. B. Xue, Z. W. Luo, Q. Wan, G. M. Yao and Y. H. Zhang, Org. Lett., 2013, 15, 3094–3097 CrossRef CAS PubMed.
- Y. Li, Y. B. Liu, Y. L. Liu, C. Wang, L. Q. Wu, L. Li, S. G. Ma, J. Qu and S. S. Yu, Org. Lett., 2014, 16, 4320–4323 CrossRef CAS PubMed.
- Y. Li, Y. B. Liu, H. M. Yan, Y. L. Liu, Y. H. Li, H. N. Lv, S. G. Ma, J. Qu and S. S. Yu, Sci. Rep., 2016, 6, 36572 CrossRef PubMed.
- J. S. Chen and S. Zheng, Chinese Poisonous Plants; Science, Beijing, 1987, pp. 226–227 Search PubMed.
- C. S. Niu, Y. Li, Y. B. Liu, S. G. Ma, F. Liu, L. Li, S. Xu, X. J. Wang, R. B. Wang, J. Qu and S. S. Yu, Org. Lett., 2017, 19, 906–909 CrossRef CAS PubMed.
- C. S. Niu, Y. Li, Y. B. Liu, S. G. Ma, L. Li, J. Qu and S. S. Yu, Tetrahedron, 2016, 72, 44–49 CrossRef CAS.
- C. H. Li, X. M. Niu and Q. Luo, Org. Lett., 2010, 12, 2426–2429 CrossRef CAS PubMed.
- Z. Y. Wu, Y. D. Li and G. S. Wu, Chem. Pharm. Bull., 2011, 59, 492–495 CrossRef CAS PubMed.
- W. G. Wang, Z. Y. Wu, R. Chen, H. Z. Li, H. M. Li, Y. D. Li, R. T. Li and H. R. Luo, Chem. Biodiversity, 2013, 10, 1061–1071 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed 1D and 2D NMR, HRESIMS, IR, and X-ray crystallography data. CCDC 1548366–1548369. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra08635k |
| This journal is © The Royal Society of Chemistry 2017 |