
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation and properties of a thin membrane based on sodium alginate grafting acrylonitrile†
Lijun Yang a,
Jing Guo*ab,
Jing Wua,
Yunming Yanga,
Sen Zhang*a,
Jingxing Songa,
Qingda Ana and
Yumei Gonga
a,
Jing Guo*ab,
Jing Wua,
Yunming Yanga,
Sen Zhang*a,
Jingxing Songa,
Qingda Ana and
Yumei Gonga
aDalian Ploytechnic University, Liaoning 116034, P. R. China. E-mail: guojng8161@163.com; 20430155@qq.com
bLiaoning Engineering Technology Research Center of Function Fiber and its Composites, Dalian Ploytechnic University, Dalian 116034, P. R. China
First published on 30th October 2017
Abstract
Sodium alginate (SA) was modified for better water resistance with potassium persulfate (as the initiator) and acrylonitrile (as the monomer) via free radical graft copolymerization, and the corresponding sodium alginate-acrylonitrile graft copolymer film (SA-g-AN) was prepared. The modified SA film was characterized by measuring contact angle and water solubility and with DSC. The copolymerization mechanism and the microstructure of the graft copolymer were studied by FT-IR, 1H NMR, TEM and SEM. The correlated properties of the SA-g-AN film materials were measured with AFM, TG, XRD and DMA. The results indicated that the pyran ring of SA was open during the reaction, the morphology of the product exhibited a solid spherical cluster, and the density of the graft copolymer decreased as the content of acrylonitrile increased. The water resistance was improved after SA was modified by acrylonitrile. Both its crystallinity and thermal stability improved by introducing the acrylonitrile unit. The storage and loss modulus were in between those of SA and PAN. The SA-g-AN materials had a better film formation property and an excellent adsorbability towards heavy metal ions. These results have proven that this is a better development prospect on the field of sewage treatment.
1. Introduction
Currently, physical and chemical approaches are the main methods to modify polymer materials. Moreover, chemical modification plays an irreplaceable role in controlling the structure of a molecular chain.1,2 Sodium alginate is extracted from marine algae as well as naturally biodegradable polymer materials with excellent properties of non-toxicity, water solubility and oxygen permeability,3–6 which makes it have extensive applications in food, medicine and chemical industries, etc.7–10 Sodium alginate possesses excellent properties for film and fiber formation11,12 and can chelate with divalent metal ions to form an eggshell structure,13–16 which has been used to prepare film materials, hydrogel microspheres or the as-spun fibers for extensive applications in wound dressing,17–20 tissue engineering,21–23 and medicine controlled-release, etc.24,25 There is a lot of hydroxyl and carboxyl groups in alginate molecular, which leads to an excessive swelling and a sharp decline in mechanical properties, which in turn causes the loss of usability of alginate film, hydrogel or fiber materials in the existence of water, particularly in saline solution.26,27 It limits the application of alginate products to some extent. Thus, how to solve the problem of poor water resistance of alginate has attracted more and more attention. Much effort has been made by researchers to modify the water resistance or the hydrophobicity of SA at the molecular level.28,29 Hubert et al. used C12 to modify the hydrophobicity, and the resulting alginates have a good hydrophobicity after the modification.30 Nyström et al. also obtained a good hydrophobic modified alginate material by using β-cyclodextrin, and the obtained product showed great potential to be applied in drug-release application.31 Galant et al. reported hydrophobically modified alginate (Alg–CONH–C8) by coupling n-octylamine to the backbone carboxylic acid groups using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC-HCl).32 V. G. Babak et al. used dodecyl chains to modify sodium alginate and found drastically different hydrophobic behaviors with different long alkyl chains.33The reaction to produce acrylonitrile graft sodium alginate copolymer was also studied by Samir et al. long time ago, with Fenton's reagent and ceric ammonium nitrate as an initiator. The optimized reaction conditions of grafting were determined by varying the concentrations of the reactants.34,35 N. Radhakrishnan et al. also studied the graft copolymerization of acrylonitrile with sodium alginate and obtained significant copolymerization results.36 However, both Samir and Radhakrishnan only analyzed the effects of reaction conditions, such as the reaction time, temperature, pH, and monomer and initiator concentrations, on grafting copolymerization and did not perform detailed research on the applications and properties of the SA-g-AN copolymer as the membrane materials. Trivedi et al.37 prepared the acrylonitrile-sodium salt copolymer by using ceric as the initiator. The structure and morphology of the copolymer were characterized by SEM. A spherical particle morphology was observed.
In this work, we proposed to modify SA using acrylonitrile as a modification monomer, aimed to introduce the hydrophobic polyacrylonitrile side group into the SA molecules via graft copolymerization to improve the water resistance of SA, and then solved the problem of poor water resistance of SA during the application as functional materials. Potassium persulfate (as the initiator) and acrylonitrile (as the monomer) were employed to synthesize sodium alginate-acrylonitrile graft copolymer (SA-g-AN) via free radical graft copolymerization, and the chemical structure and corresponding polymerization mechanism were analyzed by FT-IR and 1H NMR. The SA-g-AN film materials were prepared by a casting process, and the comprehensive properties of the films were studied by AFM, TG, XRD, DMA, DSC, contact angle and aqueous solubility tests. The structural properties of SA, PAN and SA-g-AN films were comparatively investigated.
The experimental results that the target product SA-g-AN was successfully synthesized via free radical polymerization, but the grafting mechanism was not that hydrogen atom was transferred to form active sites and then drove acrylonitrile toward free radical polymerization.
The introduction of polyacrylonitrile unit could improve the water resistance of sodium alginate effectively. In the microstructure, the as-prepared SA-g-AN graft copolymer materials had the characteristics of looseness and light weight with solid microspheres. The product remained stable in both water and DMF without dissolution, which further proved the success of the graft copolymerization. The SA-g-AN graft copolymer had better film-formation characteristics, and the thermal stability and water resistance were both improved as the content of acrylonitrile increased, but the surface flatness of the films was decreased. The dynamic mechanical analysis indicated that the introduction of polyacrylonitrile could reduce the glass transition temperature (Tg) and the storage and loss moduli. The research results proved that the modification of water resistance by introducing acrylonitrile was effective and the as-prepared graft copolymer had a promising prospect in the application as film materials.
2. Experimental
2.1 Materials
Sodium alginate (SA) was of food grade with a molecular weight of 7 × 106 and viscosity of 700 cps, (Qingdao Bright Moon Seaweed Group Co., Ltd, China.). Acrylonitrile (AN), purified by two times distillation. Potassium peroxydisulfate, N,N-dimethylformamide (DMF) and dimethylsulfoxide (DMSO) were of analytical grade and purchased from Sinopharm Chemical Reagent Co., Ltd, China.2.2 Synthesis of SA-g-AN and thin films fabrication
SA and AN were added to a three-neck flask with various mass ratios of 1/8, 1/11 and 1/14. The mixture was allowed to react in a water bath at 70 °C for 1.5 h with a rotation speed of 450 rpm. The corresponding mechanism of the graft copolymerization is presented in Fig. 1. The raw product was washed twice with DMF to remove the polyacrylonitrile homopolymer, and then the residue was filtered. After that, the product was washed twice with deionized water to remove unreacted SA, and the SA-g-AN copolymer was obtained by vacuum filtration. SA-g-AN was dissolved in DMSO to prepare 2 wt% diluted solution for film casting, and the SA-g-AN film was obtained after drying at 70 °C. | ||
| Fig. 1 The reaction mechanism of the SA-g-AN graft copolymerization. | ||
2.3 Characterization
The chemical structure of SA-g-AN materials was determined by a Spectrum-One B Fourier-transformed infrared spectroscope (with KBr discs) with a scan range of 4500–500 cm−1 and a resolution of 4 cm−1. The 1H NMR spectra of the SA-g-AN copolymer materials were recorded on a Bruker ADVANCE III nuclear magnetic resonance spectrometer with a frequency range of 6–430 MHz and DMSO as a solvent. The differential scanning calorimetry analysis for the SA-g-AN graft copolymer was performed on an 822e differential scanning calorimeter analyzer (DSC, Mettler Toledo) under the N2 atmosphere in a temperature range of 25–300 °C and a heating rate of 10 °C min−1. Their micromorphology was observed on a JSM-7800F field emission scanning electron microscopy (FE-SEM, JEOL) system. The aggregation state of the SA-g-AN graft copolymer was measured on a JEM-2100 (UHR) transmission electron microscope with a point resolution of 0.19 nm and a line resolution of 0.14 nm (TEM, JEOL). The morphology of SA-g-AN was observed by a Nanoscope IV atomic force microscope. The water resistance of the SA-g-AN films was evaluated by an XG-CANC1 optical contact angle measuring device (Shanghai Xuanyi Chuangxi Industrial Equipment). The wide-angle X-ray diffraction patterns of the SA-g-AN films were collected using a diffractometer-6000 X-ray diffractometer with a Cu radiation, a tube voltage of 40 KV, a tube current of 2.5–80 mA, and a 2θ range of 5–75° and a scanning rate of 4° min−1. The thermal stability of the fibers was measured on a TGA-Q50 thermal gravimetric analyzer under N2 atmosphere in a temperature range of 0–700 °C, a heating rate of 20 °C min−1 and a gas flow of 40 ml min−1. The response of molecular motion of SA-g-AN films to an external temperature variation was determined by a Q800 dynamic mechanical thermal analyzer (DMA, TA) in a temperature range of 30–160 °C and a heating rate of 3 °C min−1. The dimension of the sample was 0.5 cm × 3 cm × 0.1 cm. The density test was performed on a DX-300DT density tester, from Shenzhen Qun Long Instrument Equipment Co., Ltd. using ethanol as a test medium at room temperature. The copper ion adsorption experiment was carried out in the CuSO4 aqueous solution and the concentration was 1.4257 mg L−1. The adsorption rate (AR) was calculated using the following equation:
 | (2.1) |
3. Results and discussion
3.1 Microstructure and water resistance performance
The microstructure of SA-g-AN graft copolymer and the water-soluble test results are presented in Fig. 2. It can be seen that rod-like SA exhibits characteristics of looseness and light weight after graft copolymerization. In order to investigate the characteristics of its microstructure, the micromorphology is observed by SEM and TEM. As can be observed in the SEM image, the products from the graft copolymerization are spherical particles and the diameter distribution of these microspheres is uniform with a diameter of about 200 nm. There exists an obvious two-phase structure in the microsphere with algae in the disperse phase and PAN in the continuous phase. The TEM image shows that the microsphere is solid. The density of SA-g-AN was tested and the results are summarized in Table 1. It is found that the density of SA-g-AN (ρ = 1.09g cm−3, w/w = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11) is lower than that of SA (ρ = 1.72 g cm−3), and the reduction trend in density intends to enlarge with increasing content of AN. The analysis reveals that the characteristics of looseness and light weight might result from the stronger short-range repellent effect between the rigid ring structure of SA and the PAN side groups, which forces the macromolecular distance to become wider leading to density reduction and light weight. However, SA and PAN are incompatible, which results in the gap between two structures and two phases to reduce the density. During the solubility experiment, the AN modified SA material exhibits no solubility in water or DMF and the copolymer does not dissolve in water after 60 h, which indicates a successful graft copolymerization. It also proves that the introduction of the PAN unit significantly improves the poor water resistance of SA.
:11) is lower than that of SA (ρ = 1.72 g cm−3), and the reduction trend in density intends to enlarge with increasing content of AN. The analysis reveals that the characteristics of looseness and light weight might result from the stronger short-range repellent effect between the rigid ring structure of SA and the PAN side groups, which forces the macromolecular distance to become wider leading to density reduction and light weight. However, SA and PAN are incompatible, which results in the gap between two structures and two phases to reduce the density. During the solubility experiment, the AN modified SA material exhibits no solubility in water or DMF and the copolymer does not dissolve in water after 60 h, which indicates a successful graft copolymerization. It also proves that the introduction of the PAN unit significantly improves the poor water resistance of SA.
 | ||
| Fig. 2 Microstructure SEM image (a), TEM image (b), water-resistance test (c and d) in which the SA-g-AN copolymer was immersed in different times in water (c1–c5). Herein, the ratio of SA/AN is 1/11 in all samples and the water-resistance test sample is after filtration at room temperature. | ||
| Sample | SA/AN mass ratio | Density [g cm−3] | Error limitation |
|---|---|---|---|
| SA | 1/0 | 1.72 | ±0.03 |
| SA-g-AN | 1/8 | 1.43 | ±0.01 |
| SA-g-AN | 1/11 | 1.25 | ±0.02 |
| SA-g-AN | 1/14 | 1.19 | ±0.02 |
3.2 Structure characterization and reaction mechanism of SA-g-AN
The FT-IR and 1H-NMR spectra of SA and the SA-g-AN graft copolymer are exhibited in Fig. 3(a and b). In the FT-IR spectrum of SA, the peak at 1605 cm−1 is attributed to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, the peak at 1413 cm−1 is caused by the in-plane bending vibration of C–H of the molecular skeleton, the peak at 1093 cm−1 is assigned to the stretching vibration of C–O and C–C from the pyran ring, and the peak at 1033 cm−1 is attributed to the stretching vibration of C–OH. In the FT-IR spectrum of SA-g-AN, the absorbance peak of –OH at 3345 cm−1 obviously decreases, the vibration peak of –CN appears at 2245 cm−1, and the characteristic peak of the saturated C–H emerges at 2927 and 1450 cm−1. The observation proves that the PAN unit has been introduced into the backbone of SA successfully. As seen in Fig. 3(c and d), the active proton peak of the pyran ring of SA at 3.8–4.5 ppm disappears in the 1H-NMR spectrum of SA-g-AN, and a new absorbance peak of the saturated proton appears at 1.5–3.5 ppm. The proportion of the area of the peaks at 2.05 ppm and 3.15 ppm is close to 1:2, which is attributed to the proton of methylene and methine groups in the AN side groups. The peak at 7.96 ppm is attributed to the proton of the carbonyl group. In the FT-IR derivative spectrum of SA-g-AN, the characteristic peaks at 1413 cm−1, 1033 cm−1 and 1092 cm−1 in SA disappear, while the characteristic peak of the carbonyl group appears at 1740 cm−1. It reveals that the pyran ring is open under the effect of K2S2O8 and the aldehyde group is generated, which forms an active point at a ring-opening location, and the graft copolymerization with the AN unit occurs following the specific route as shown in Fig. 1.
O bond, the peak at 1413 cm−1 is caused by the in-plane bending vibration of C–H of the molecular skeleton, the peak at 1093 cm−1 is assigned to the stretching vibration of C–O and C–C from the pyran ring, and the peak at 1033 cm−1 is attributed to the stretching vibration of C–OH. In the FT-IR spectrum of SA-g-AN, the absorbance peak of –OH at 3345 cm−1 obviously decreases, the vibration peak of –CN appears at 2245 cm−1, and the characteristic peak of the saturated C–H emerges at 2927 and 1450 cm−1. The observation proves that the PAN unit has been introduced into the backbone of SA successfully. As seen in Fig. 3(c and d), the active proton peak of the pyran ring of SA at 3.8–4.5 ppm disappears in the 1H-NMR spectrum of SA-g-AN, and a new absorbance peak of the saturated proton appears at 1.5–3.5 ppm. The proportion of the area of the peaks at 2.05 ppm and 3.15 ppm is close to 1:2, which is attributed to the proton of methylene and methine groups in the AN side groups. The peak at 7.96 ppm is attributed to the proton of the carbonyl group. In the FT-IR derivative spectrum of SA-g-AN, the characteristic peaks at 1413 cm−1, 1033 cm−1 and 1092 cm−1 in SA disappear, while the characteristic peak of the carbonyl group appears at 1740 cm−1. It reveals that the pyran ring is open under the effect of K2S2O8 and the aldehyde group is generated, which forms an active point at a ring-opening location, and the graft copolymerization with the AN unit occurs following the specific route as shown in Fig. 1.
 | ||
| Fig. 3 The chemical structure of the SA-g-AN graft copolymer: (a) infrared spectra of SA and SA-g-AN, (b) infrared derivative spectra of SA and SA-g-AN, (c) 1H-NMR of SA, and (d) H-NMR of SA-g-AN. All samples with SA/AN = 1/11(W/W), the infrared spectra of SA-g-AN and SA were obtained by KBr pellet technique. Hydrogen nuclear magnetic resonance spectra were recorded with the SA-g-AN solution dissolved in DMSO. | ||
3.3 Properties of SA-g-AN thin films
The XRD pattern of SA-g-AN is presented in Fig. 4(a), in which the diffraction curve of SA exhibits a large dispersing diffraction peak, indicating that most of the inner parts of the material are in the amorphous state. Compared with SA, SA-g-AN has a stronger crystallinity property. There appears a larger crystalline peak at 16.8°, which is attributed to the (110) lattice plane. Thus, it can be concluded that the degree of crystallinity of the graft copolymer is contributed by the AN unit. Polyacrylonitrile polymer with a better crystallinity property and the introduction of a certain amount of acrylonitrile units can form an independent motion unit in the copolymer molecules, arranging orderly to enter the lattice and end the crystallization. In order to investigate the effect of the introduction of the AN unit on the thermal stability of the graft copolymer, DSC tests for various graft polymers were performed. The corresponding curves are presented in Fig. 4(b). In the DSC curve of SA-g-AN, an exothermic peak of bound water at about 100 °C disappears, and an exothermic peak from the decomposition emerges at about 255 °C, indicating that the introduction of AN to seaweed macromolecules reduces the moisture absorption significantly. It also proves, from the thermodynamic aspect, the successful modification of SA changing its water resistance by introducing AN, which is consistent with the experimental result from the water-solubility test.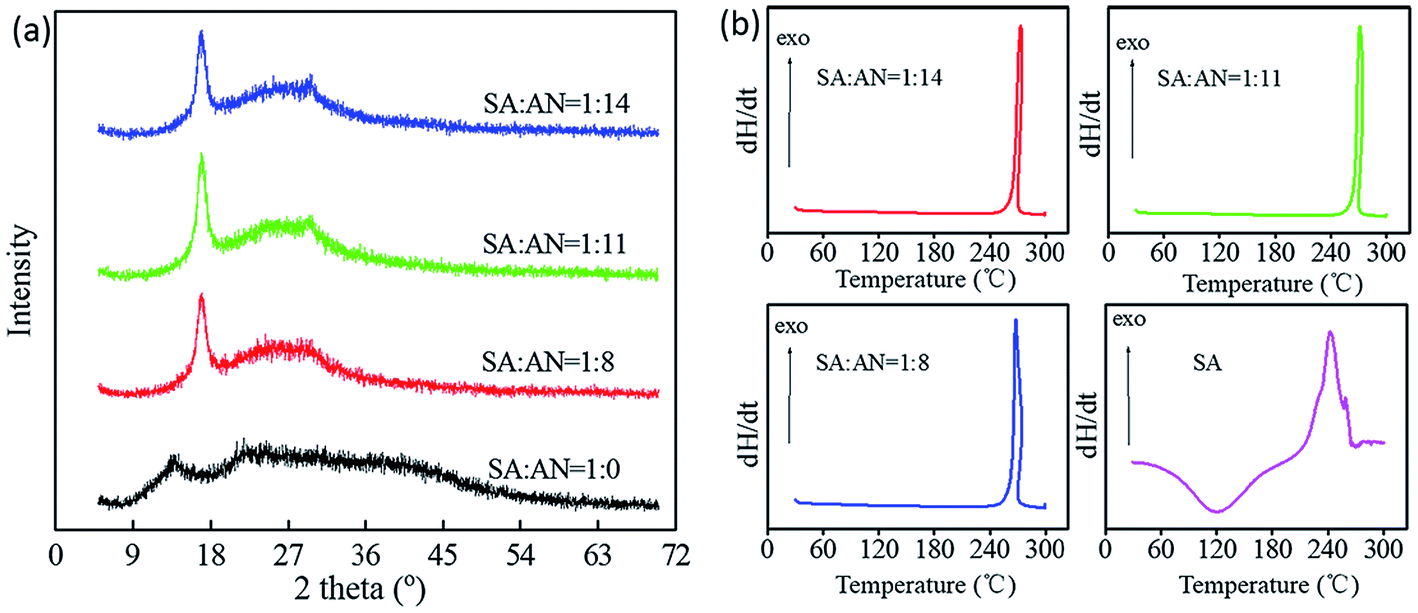 | ||
| Fig. 4 XRD curves (a) and DSC curves (b) of SA-g-AN. SA and AN raw ratio as followed 1/8, 1/11, and 1/14 (W/W). All samples are dried for 12 hours at 70 °C to remove the moisture. | ||
SA macromolecule is a linear macromolecular polysaccharide extracted from algae. The introduction of the PAN side group into the backbone makes the macromolecular interaction become more intensive, and this type of interaction affects the motion patterns of the intramolecular segment, the side groups and other motion units to some extents. In order to clearly describe the response of SA-g-AN film against an external stress during the application, the dynamic mechanical test is performed, and the results are taken in comparison with those of SA and the PAN film. The corresponding curves of loss modulus, storage modulus and tanδ versus temperature are presented in Fig. 5(a–c).
 | ||
| Fig. 5 (a) Loss modulus, (b) storage modulus, (c) tanδ, and (d) TG curves of SA-g-AN. SA and AN raw ratio is 1/11 (W/W), the size of the membrane sample is 0.5 cm × 3 cm × 0.1 cm, test frequency of 1 Hz and the heating rate of 3°C min−1. The TG samples are dried for 12 hours at 70 °C. | ||
As exhibited in Fig. 5(a and b), at the same temperature, the loss and storage modulus of SA-g-AN are lower than those of SA and higher than those of PAN. SA has a rigid pyran ring structure and contains a large amount of polar hydroxyl and carboxyl groups, which lead to a much higher intermolecular interaction. Thus, the rigidity and viscosity are higher. In case of PAN, there exist strong polar cyano groups possessing a larger dipole moment, which forces them to attract as well as repulse. Thus, the intermolecular interaction is relatively weaker and the loss and storage modulus are lower. After the PAN side group is introduced in the SA backbone, the original pyran ring structure is partly destroyed, which results in a decrement in molecular regularity, a reduction in rigidity, weakening in intermolecular interaction and decrease in the loss and storage moduli. It can be seen in Fig. 5(c) that the Tg of the SA-g-AN copolymer is 92 °C, which is obviously lower than that of the SA film (Tg = 108 °C) but higher than that of the PAN film (Tg = 80 °C). It indicates that the introduction of AN strengthens the motion ability of the segment, and it might be because the intermolecular distance of algae increases after introducing long side groups, the intermolecular interaction is weakened, and the molecular motion ability is enhanced.
The TG curves of the SA-g-AN copolymer are shown in Fig. 5(d). The first stage of thermal weight loss for SA appears at about 90 °C, which is attributed to the loss of free water in SA. The thermal weight loss rate reaches up to the maximum value at about 230 °C, and this decomposition temperature corresponds to the degradation of the molecular skeleton and the glycosidic bond of SA. Compared with that observed from the TG curves of SA, the thermal stability of SA-g-AN is significantly improved, and the maximal decomposition rate appears at about 250 °C. Moreover, the degradation peak at about 80 °C disappears because the introduction of PAN side group screens the heat of SA in the backbone, which improves the thermal stability. Besides, it is easy for SA-g-AN to undergo cyclization and carbonization under heat, and such reactions improve the thermal stability and heat insulation performance to achieve a great improvement in the thermal stability of the whole materials.
The results from the contact angle test and AFM images of the SA-g-AN graft copolymer with various ratios are presented in Fig. 6. It can be observed that all of the samples have excellent film formation properties. As shown in the AFM images, the surface of SA film is smooth and flat, the corresponding root-mean-square of the surface roughness is 0.487 nm, and the fluctuation of the surface of SA-g-AN graft copolymer becomes larger as the content of acrylonitrile increases. The root-mean-square of the surface roughness increases and the film surface becomes rougher. When SA/AN = 1/14, the root-mean-square of surface roughness reaches 4.12 nm. In the contact angle test of SA and SA-g-AN, the hydrophilicity of SA is very large, and it results in swelling and even dissolution during the experiment. As the content of acrylonitrile increases, the surface of the copolymer film material becomes rougher, the contact angle gets larger, and the hydrophobicity of the SA-g-AN film increases, which would be originated by introducing the hydrophobic PAN unit leading to the reduction in the interfacial energy of the material surface against aqueous molecules.
 | ||
| Fig. 6 AFM images and contact angle test of the SA-g-AN film with various contents of acrylonitrile: (a) SA/AN = 1/0; (b) SA/AN = 1/8; (c) SA/AN = 1/11 and (d) SA/AN = 1/14. | ||
The SA-g-AN films have an extensive application in the field of sewage treatment of heavy metal ions due to the water resistance improvement of SA. It has many hydroxyl and carboxyl groups, which can be chelated with divalent and trivalent metal ions. Furthermore, acrylonitrile side chains in the copolymer not only improve the water resistance but also enhance the adsorption capacity of heavy metal ions after alkali treatment. During the treatment in sodium hydroxide solution, cyano groups transform to amide and carboxyl groups. The transformation mechanism is shown in Fig. 7(a). The heavy metal ion adsorption performance of the SA-g-AN films with different ratios is demonstrated in Fig. 7(b). The SA-g-AN film has a good resistance to water after the modification. It cannot be dissolved in the solution with heavy metal ions. During the adsorption process, the Cu2+ in the solution can be removed effectively. With the increase in copolymerization (i.e. the SA/AN ratio), the Cu2+ adsorption rate increases. When the SA/AN ratio is 1:14, the Cu2+ adsorption rate (AR) reaches 80.3%. The experimental data prove that the SA-g-AN film may have a broad application prospect on the field of sewage treatment.
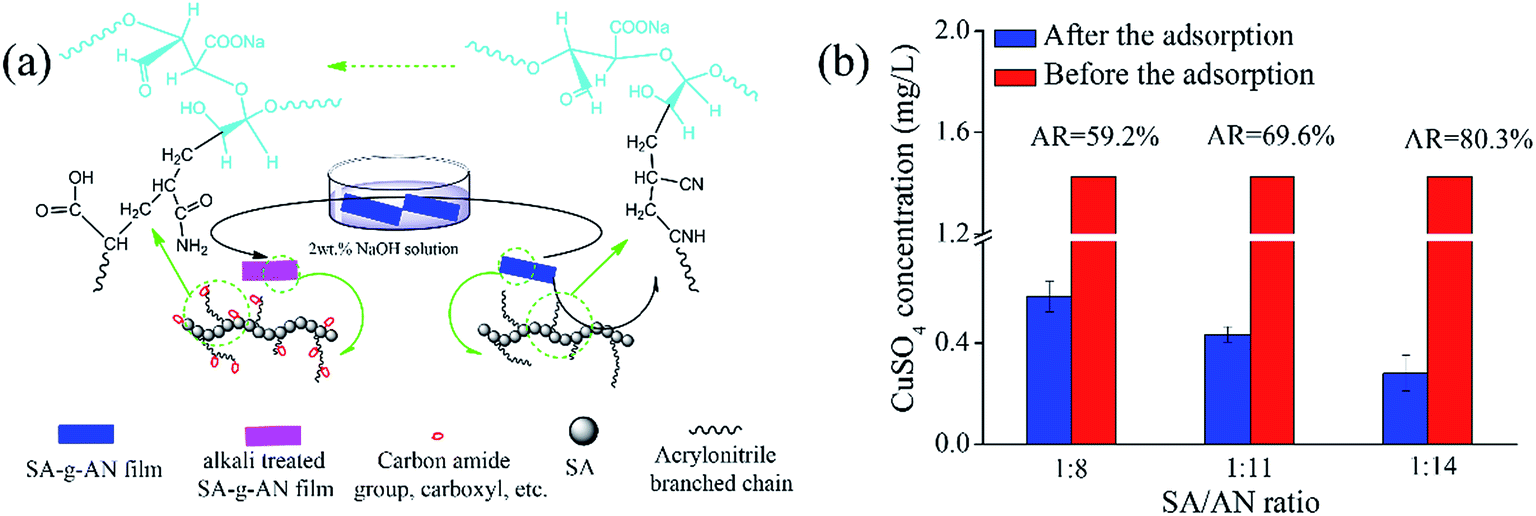 | ||
| Fig. 7 (a) Molecular structure evolution mechanism of the SA-g-AN film after alkali treatment and (b) the heavy metal ion adsorption performance of the SA-g-AN films with different ratios. SA/AN ratios are 1/8, 1/11 and 1/14. The SA-g-AN film mass/CuSO4 solution mass = 1:120 (w/w) and the alkali treatment time is 20 min at 25 °C. | ||
4. Conclusions
In this study, acrylonitrile was applied to modify the water resistance property of SA, and the SA-g-AN graft copolymer material with an improved water resistance was prepared successfully. It was discovered that SA underwent ring-opening reaction in the presence of potassium persulfate, and it reacted with acrylonitrile to form a graft copolymer. The SA-g-AN copolymer exhibited the characteristics of looseness and light weight, and it had a solid spherical morphology. The modified SA-g-AN graft copolymer materials had a better film formation property, and the roughness of the film surface increased with increasing content of acrylonitrile, as observed by AFM. The as-prepared film material has a certain crystallinity. The thermal stability and hydrophobicity were improved as the content of acrylonitrile increased. The DMA analysis indicated that the graft copolymer has a better dynamic mechanical property, in which the storage and loss modulus were in between those of the PAN and SA films. This research has provided an effective methodology to modify the water resistance of sodium alginate.Conflicts of interest
We declare that we do not have any commercial or associative interest that represents a conflict of interest in connection with the work submitted.Acknowledgements
Financial support for this research is provided by the National Science Foundation of China (No. 51773024) and (No. 51373027), the National Science Foundation of Liaoning (2015020221), National Natural Science Foundation of China (No.51403028), and Surface Project of Liaoning Science and Technology Department (No.20170540077).Notes and references
- S. S. N. J. Edgar, Biomacromolecules, 2011, 12, 4095–4103 CrossRef PubMed.
- S. Liu, Y. Li and L. Li, Carbohydr. Polym., 2017, 160, 62–70 CrossRef CAS PubMed.
- P. Gurikov, S. Raman, D. Weinrich, M. Fricke and I. Smirnova, RSC Adv., 2015, 11, 7812–7818 RSC.
- A. Abbaszaddrafi and M. Mahkam, RSC Adv., 2015, 4, 2378–2382 Search PubMed.
- M. Kamila, B. Richard, B. Mischa and H. Johannes, Macromolecules, 2014, 47, 771–776 CrossRef.
- H. Hecht and S. Srebnik, Biomacromolecules, 2016, 17, 2160–2167 CrossRef CAS PubMed.
- A. Sood, V. Arora, J. Shah, R. Kotnala and T. Jain, Mater. Sci. Eng., C, 2017, 80, 274–281 CrossRef CAS PubMed.
- C. Feng, R. Song, S. Guohui, K. Ming, B. Zixian, L. Yang, C. Xiaojie, C. Dongsu, H. Park and C. Xiguang, Biomacromolecules, 2014, 15, 985–996 CrossRef CAS PubMed.
- L. Jian Wei, Z. Yong Liang, G. Zhao Xia, H. Ping and Y. Jian, Polymer, 2006, 47, 8026–8031 CrossRef.
- B. Muhammad, I. Munawar and H. Hongbo, Biochem. Eng. J., 2016, 109, 153–161 CrossRef.
- S. Shilpa, S. Pallab, C. Arun and G. Siddhartha, RSC Adv., 2012, 2, 5837–5843 RSC.
- Y. Lijun, G. Jing, Z. Sen and G. Yumei, Int. J. Biol. Macromol., 2017, 99, 166–172 CrossRef PubMed.
- D. Wei, Z. Jianfei, Z. Yunhang, W. Yanan and S. Bi, Carbohydr. Polym., 2017, 157, 1650–1656 CrossRef PubMed.
- R. Ahmad, R. Kumar and S. Haseeb, Arabian J. Chem., 2012, 5, 353–359 CrossRef CAS.
- A. Blandino, M. Macías and D. Cantero, J. Biosci. Bioeng., 1999, 6, 686–689 CrossRef.
- Q. Yimin, H. Huiqun and L. Aixiang, J. Appl. Polym. Sci., 2006, 101, 4216–4221 CrossRef.
- N. Khuathan and T. Pongjanyakul, Int. J. Pharm., 2014, 460, 63–72 CrossRef CAS PubMed.
- F. Agnely, I. Iliopoulos and R. Zana, Langmuir, 2000, 25, 9921–9927 CrossRef.
- J. Kimb, J. KilParka and J. Hoon Kimb, Int. J. Pharm., 2008, 359, 79–86 CrossRef PubMed.
- K. Shalumon, K. Anulekha, S. Nai and K. Chennazhi, Int. J. Biol. Macromol., 2011, 49, 247–254 CrossRef CAS PubMed.
- Y. Jen Ming, J. Hao Yang, T. Shu Chun and C. Ding, Mater. Sci. Eng., C, 2016, 66, 170–177 CrossRef PubMed.
- L. Honglin, Z. Guifu, X. Guangyao and L. Chunzhi, Mater. Technol., 2017, 32, 78–84 CrossRef.
- J. Venkatesan, I. Bhatnagar and P. Manivasagan, Int. J. Biol. Macromol., 2015, 72, 269–281 CrossRef CAS PubMed.
- L. Wanga, R. Sheltona and P. Cooper, Biomaterials, 2003, 24, 3475–3481 CrossRef.
- C. Valeria Liew, L. Wah Chan, A. Ling Ching and P. Wan Sia Heng, Int. J. Pharm., 2006, 309, 25–37 CrossRef PubMed.
- H. Chang, H. Liang and F. Qu, J. Membr. Sci., 2016, 499, 429–441 CrossRef CAS.
- A. Charfi, H. Jang and J. Kim, Bioresour. Technol., 2017, 240, 106–114 CrossRef CAS PubMed.
- A. Matin, H. Shafi, M. Wang, Z. Khan, K. Gleason and F. Rahman, Desalination, 2016, 379, 108–117 CrossRef CAS.
- G. Shi, Y. Che, Y. Zhou, X. Bai and C. Ni, J. Mater. Sci., 2015, 50, 7835–7841 CrossRef CAS.
- A. Sinquin, P. Huber and E. Dellacherie, Langmuir, 1993, 9, 3334–3337 CrossRef CAS.
- V. Burckbuchler, A. L. Kjøniksen, C. Galant, R. Lund, C. Amiel, K. D. Knudsen and B. Nyström, Biomacromolecules, 2006, 7, 1871–1878 CrossRef CAS PubMed.
- G. Celine, A. Kjoniksen, G. Nguyen, D. Knudsen and B. Nystrom, J. Phys. Chem. B, 2006, 110, 190–195 CrossRef PubMed.
- G. Babak, A. Skotnikova, G. Lukina, S. Pelletier, P. Hubert and E. Dellacherie, J. Colloid Interface Sci., 2000, 225, 505–510 CrossRef PubMed.
- S. Samir, P. Chanedrakant and C. Harikrishna, High Perform. Polym., 1992, 4, 151–159 CrossRef.
- S. Samir, P. Chanedrakant and C. Harikrishna, J. Appl. Polym. Sci., 2010, 51, 1421–1426 Search PubMed.
- N. Radhakrishnan, Y. Lakshminarayana, S. Uma Devi and K. Srinivasan, J. Macromol. Sci., Part A: Pure Appl.Chem., 1994, 31, 581–591 CrossRef.
- J. H. Trivedia, K. Kaliab, N. K. Patela and H. C. Trivedi, Carbohydr. Polym., 2005, 60, 117–125 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08532j |
| This journal is © The Royal Society of Chemistry 2017 |