
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in hybrid measurement methods based on atomic force microscopy and surface sensitive measurement techniques
Stephan Handschuh-Wang
a,
Tao Wang
b and
Xuechang Zhou
 *a
*a
aCollege of Chemistry and Environmental Engineering, Shenzhen University, Shenzhen 518060, P. R. China. E-mail: xczhou@szu.edu.cn; Fax: +86-755-26536141; Tel: +86-755-26536627
bFunctional Thin Films Research Center, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, P. R. China
First published on 9th October 2017
Abstract
AFM was proposed in the 1990s as a tool to determine the topography at a sub-micrometer level. Since then AFM proved to be a valuable tool in material science and physical chemistry. Soon after successful application of AFM, it was employed for biological applications. The success of AFM can be ascribed to the high sensitivity, versatility, and resolution (1000 times higher resolution compared to optical microscopy). However, it intrinsically lacks amongst others chemical sensitivity. These limitations of AFM can be overcome by coupling it to orthogonal (complementary) optical (i.e. microscopy techniques) or non-optical techniques (i.e. Kelvin method). Such combinations are capable to garner information on morphological and mechanical properties of a sample together with fluorescence, spectroscopic (IR/Raman/fluorescence), electric, temperature or conductivity. Recently, advances in microscopy and super-resolution techniques enable the collection of even more than 2 measurands. Furthermore, new combinations, such as the photothermal-induced resonance (PTIR), a combination of IR spectroscopy and AFM, and scanning near-field ellipsometry microscopy (SNEM), a combination of ellipsometry and AFM, are actively researched, widening the adoptability of orthogonal combinations relying on AFM, while researchers devote tremendous effort to enhance established hybrid methods, such as tip-enhanced Raman spectroscopy (TERS). Here, we introduce hybrid measurement techniques, relying on AFM and complementary techniques. The main focus of these hybrid techniques is on the combination with optical techniques as there has been much progress in the past few years and we can contribute our expertise in this field. For each combination, the working principle is explained briefly and applications of such combinations are pointed out. Finally, a short comprehension is given with basic information on resolution and field of application of all major combinations. We envision that this review is helpful to those confronted with combined measurements for the first time, for experienced researchers, needing quick access to recent literature as it may convey new ideas for problems in analysis, as well as for researchers, who develop novel combined methods.
 Stephan Handschuh-Wang | Stephan Handschuh-Wang obtained his Diploma degree in physical chemistry from the University of Technology Dortmund in 2010. He pursued his PhD with Professor Holger Schönherr at the University of Siegen on the study of soft matter interfaces and obtained his PhD in 2016. Dr Handschuh-Wang is currently a postdoctoral fellow of Prof. Xuechang Zhou's group at Shenzhen University. His previous and current research interests are focused on phenomena at the interface of preferably soft matter, in particular, the characterization and application of novel functional interfaces. |
 Tao Wang | Tao Wang obtained her bachelor and Master degree in materials science and engineering from the Dalian University of Technology in 2011. She pursued her PhD with Professor Xin Jiang at the University of Siegen on the study of surface science and technology and obtained her PhD in 2015. Dr Tao Wang is currently a postdoctoral fellow of Prof. Yongbing Tang's group at Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences. Her previous and current research interests are focused on surface coating, surface functionalization and interactions of molecules in applications of mechanical engineering, materials science, and biomedicine. |
 Xuechang Zhou | Prof. Xuechang Zhou is currently an Associate Professor of College of Chemistry and Environmental Engineering at Shenzhen University. Prof. Zhou received his B.S in polymer chemistry (Advisor: Prof. Guangzhao Zhang) at the University of Science and Technology of China in 2005, and Mphil and PhD degrees in Department of Chemistry (Prof. Bo Zheng's group) at the Chinese University of Hong Kong in 2007 and 2010, respectively. From 2010 to 2014, he worked in Prof. Zijian Zheng's group as a Postdoctoral at the Hong Kong Polytechnic University focusing on the development of flexible and wearable electronics, and surface patterning technique. He joined the College of Chemistry and Environmental Engineering at the Shenzhen University as an Associate Professor in 2014. His research interests included flexible and stretchable electronics, sensors, surface analysis and environmental engineering. |
1. Introduction
Advances in science are closely related to advances in the design and construction of analysis tools. Here, confocal laser scanning microscopy should be mentioned with its unprecedentedly high lateral resolution and optical slicing ability. A second measurement technique, which made further advances possible, was atomic force microscopy. The atomic force microscope (AFM) rapidly emerged as an invaluable tool for direct measurement of topography and intermolecular forces with atomic resolution for a broad spectrum of applications, such as electronics,1–3 semiconductors,4 materials and manufacturing,5 polymers,6 biology, and biomaterials.7 An AFM is able to measure a sample at ambient conditions8 and even in liquid9–11 or buffer solutions,12 which is in contrast to scanning tunneling microscopy (STM), scanning electron microscopy (SEM) and transmission electron microscopy (TEM). This feature renders AFM especially useful for in vitro measurements of biological samples.13 Furthermore, AFM can employ several operation modes, which yields additional capabilities and makes it a powerful tool for several applications. These operation modes are for example contact mode, intermittent mode (topography), lateral force microscopy (friction) and noncontact mode, force modulation (elasticity) and phase imaging (dissipation). Nanoindentation can be utilized to determine in situ the mechanical properties of specific points and areas of a sample without change of the cantilever, tip, measurement equipment, and sample.14–16 A widespread technique is force modulation microscopy (FMM), which is utilized to characterize mechanical properties of polymers or composite material to detect inhomogeneities or contaminations.17,18 In biological regime, AFM can be utilized to determine biomechanical properties, changes in mechanical properties of cells, membranes and other biomaterials.19,20 AFM can be utilized to determine the adhesion of the tip and the surface. By functionalizing the tip/tip-apex with different probes, such as nanoparticles21 or molecules,22,23 interaction forces between two surfaces, a surface and a biological molecule or two biological molecules (single molecule force spectroscopy (SMFS)) can be determined.24,25 AFM is utilized in a wide range of disciplines in natural sciences, including solid state physics (e.g. identification of atoms at surfaces, sensor of electric charge and dynamic lateral friction microscopy),26 semiconductor science (e.g. topography, conductivity and current sensing),27 molecular engineering (e.g. nano-manipulation),28 polymer chemistry (e.g. surface structure, chemical sensing and viscoelasticity)29,30 surface chemistry (e.g. roughness, height, friction and surface rheology),31 molecular biology (e.g. DNA and chromosome studies and monitoring biological processes),32,33 cell biology (cell imaging & mechanical properties)34 and medicine (e.g. virology and tissue/organ imaging).35,36 Besides, new operation modes for the AFM are actively developed, such as scanning probe lithography37–40 and non-contact mode, and nowadays multiparametric AFM imaging is possible,41 further diversifying the field of application.However, AFM as a stand-alone technique retains a few limitations. Identification of the surface termination or chemical composition of a sample is beyond its reach, as AFM is an indirect measurement technique it intrinsically lacks chemical information.42 Furthermore, AFM resembles a technique relying on the analysis of a surface by a probing stick (cantilever), without seeing the morphology directly. This often results in challenging and time-consuming search for a specific region of interest (ROI), similar to finding the light-switch in a dark room. Hence, many novel AFMs are equipped with a camera, facilitating laser alignment and ROI identification. Yet, the camera is typically only capable of ascertaining a rough knowledge of the surface (mm range), whereas the AFM measures in μm and nm range. Therefore, combining AFM with optical microscopy, confocal laser scanning microscopy or super-resolution techniques provide an advantage over stand-alone AFM techniques.
A combination of AFM and an orthogonal optical measurement technique, such as confocal laser scanning microscopy (CLSM),43,44 Raman (tip-enhanced Raman spectroscopy (TERS))45 or infrared (photothermal-induced resonance (PTIR)),46 or non-optical techniques, like scanning thermal force microscopy,47 makes it possible to detect chemical information,48 identify the chemical composition49 or detect the thermal conductivity50 at high (lateral) resolution, which also offers new opportunities to gather multidimensional data from a given system.51,52 In combinations of an optical technique with AFM, both techniques retain their individual capabilities, which means the resolution and the information garnered by a method stays the same (obviously, the case for CLSM) or is in some cases improved (for tip-enhanced methods, such as TERS and PTIR). Furthermore, new capabilities arising from the versatility of the AFM can expand the capability of a combined setup. For example, an AFM-CLSM combination can make use of fluorescence modulation effects, which arise from the AFM tip in the vicinity of fluorescent molecules. These effects can either be enhancement53,54 or quenching55,56 of fluorescence, leading to a change in the excited state lifetime57 and can be utilized to enhance the spatial resolution of the CLSM/FLIM (fluorescence lifetime imaging microscopy).58
Tremendous effort is concentrated on the implementation of non-optical methods, such as Kelvin probe59 or thermal microscopy50 into the AFM. These combined methods allow concomitant measurement of topography with a localized surface potential,60 thermal information61 or thermal conductivity,50 which is important for material science and engineering, for example, mapping of semiconductors and their work functions.62
This review covers the combination of optical and non-optical surface sensitive techniques with the AFM. An overview of the hybrid methods discussed in this article is given in Fig. 1. For comparison, firstly, super-resolution techniques are introduced, which are capable of increasing the resolution of optical images, with its advantages and drawbacks. Subsequently, the main focus is on combinations of optical methods, especially fluorescence microscopy, and CLSM, as with these examples the alignment, an important issue for hybrid methods, can be introduced easily. Afterwards, combinatory methods based on AFM-CLSM are introduced, such as the combination of AFM with FLIM and stimulated emission depletion microscopy (STED), which are viable and recently developed candidates for predominantly addressing biological samples. Important representatives beside fluorescence-based methods are introduced hereafter, for example combination of AFM with Raman, tip-enhanced Raman scattering (TERS), and infrared spectroscopy, photothermal-induced resonance (PTIR). Thereafter, combinations of AFM with non-optical methods are introduced, such as the combination of AFM with Kelvin's method (a method for determining the contact potential difference), Kelvin probe force microscopy (KPFM), and with thermal conductivity measurement, scanning thermal microscopy (SThM). Examples out of scientific publications illustrate the applicability of these orthogonal measurement techniques and their scope. Finally, the advantages and disadvantages of these techniques are evaluated and the recent progress of hybrid methods relying on AFM is assessed.
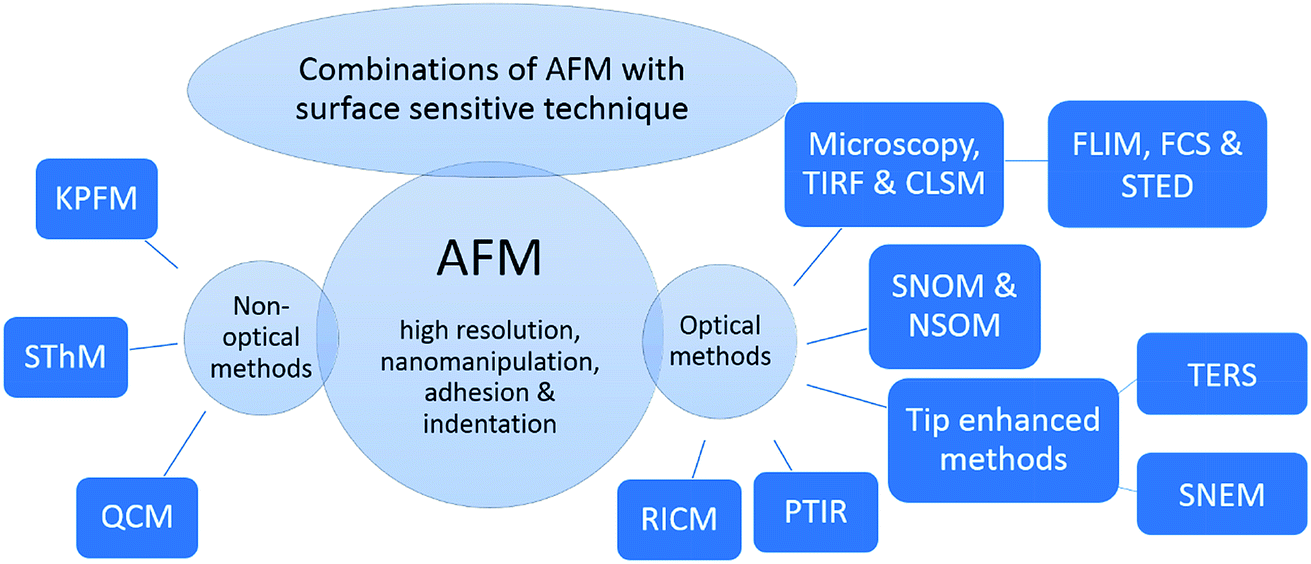 | ||
| Fig. 1 Overview of hybrid measurement techniques based on a surface sensitive technique and AFM. The abbreviations in this figure denote for the following: KPFM – Kelvin probe force microscopy, SThM – scanning thermal microscopy, QCM – quartz crystal microbalance, TIRF – total internal reflection fluorescence, CLSM – confocal laser scanning microscopy, FLIM – fluorescence lifetime imaging microscopy, FCS – fluorescence correlation spectroscopy, STED – stimulated emission depletion, SNOM – scanning near-field optical microscopy, NSOM – near-field scanning optical microscopy, TERS – tip-enhanced Raman scattering, SNEM – scanning near-field ellipsometry microscopy, PTIR – photothermal-induced resonance and RICM – reflection interference contrast microscopy. | ||
As this review article focuses on the recent advances of hybrid methods and novel hybrid methods since 2009, this article cannot cover the basic principle of the AFM and its different measurement modes. The interested reader is referred to the well-established literature regarding AFM, for example the books by Eaton, West: Atomic force microscopy63 for a more theoretic overview and Schönherr, Vancso: Scanning Force Microscopy of Polymers64 for a more practical introduction and the recent inspiring reviews by Zhong and Yan65 (AFM for nanomaterial research), Allison et al.66 (AFM for biological samples) and Dufrêne et al.67 (imaging modes, AFM for molecular and cell biology).
2. Combination of AFM with optical surface sensitive methods
Well-established methods in biology and nanotechnology, i.e. for cell-imaging or imaging of nanoparticles, are fluorescence microscopy and confocal fluorescence microscopy or confocal laser scanning microscopy (CLSM).68,69 CLSM was first patented in 1957 by Marvin Minsky.70 Its impact for research applications arises from the drastic improvement of optical resolutions to an unprecedented level of up to 180 nm lateral (x−, y) and up to 500 nm axial (z) resolution due to the spatial filtering technique.71,72 This technique allows optical slicing of the sample and construction of a three-dimensional image without collection of out of focus light.73 This feature allows to image for example cells, tissues, 3D scaffolds/sponges, which showed promise in copious application, including microfluidics and flexible electronics,74–77 or nanostructures and subsequently illustrate them 3-dimensional.78–80 Nowadays, the research is shifting towards nanotechnology (for example nanocontainers) and fundamental biology of cells, which are in a size-range of tens to few hundred nm.81 Here, optical microscopy methods lack sufficient resolution due to the diffraction limit described by Abbe.82 Several attempts to overcome the diffraction limit were successful. These methods are introduced briefly in the following paragraph.2.1 Super-resolution techniques
The general name of approaches overcoming the resolution limit is super-resolution fluorescence microscopy. As an emerging method, the super-resolution reversible saturable optical linear (fluorescence) transitions (RESOLFT) microscopy has to be mentioned,83,84 as it is suitable for in vivo studies.85 STED (stimulated emission depletion) and ground state depletion (GSD) microscopy rely on the working principle of RESOLFT.86 As STED is a well-established method, since Stefan Hell received the Nobel Prize in Chemistry in 2014 for his outstanding work in super-resolved microscopy,87 this technique is shortly introduced here. However, other – not less intriguing –super-resolution techniques include PALM88 (Photoactivated Location Microscopy), FIONA89 (Fluorescence Imaging with One Nanometer Accuracy), a method for accurately determining the position of a fluorophore by ascertaining the center of its emission pattern,89 and STORM90 (Stochastic Optical Reconstruction Microscopy), a stochastic approach based on FIONA and relying on the transfer of the majority of fluorophores to a non-fluorescent dark state (see also the topical review of Hell et al.85). In STED, two well-aligned lasers are used. The first laser possesses the typical Gaussian beam profile and is used to excite the fluorophores in the sample, whereas the second laser possesses a donut-shaped beam profile and is used to de-excite all but the fluorophores in the center of the beam. Afterwards, the fluorescence emission of molecules in this small region (smaller than diffraction limit) is detected. As STED is a scanning technique, the image is generated by moving the beams over the sample while recording the fluorescence intensity. A lateral resolution of down to 2.4 nm was reported recently.91 Typically, the resolution is in the order of tens of nanometers.92 The resolution can be adjusted by the intensity of the depletion pulse, the lateral STED-enhanced resolution/PSF (point spread function) is given by eqn (1).93 The axial resolution, however, remains for a typical experiment rather low (approx. 40–500 nm).94
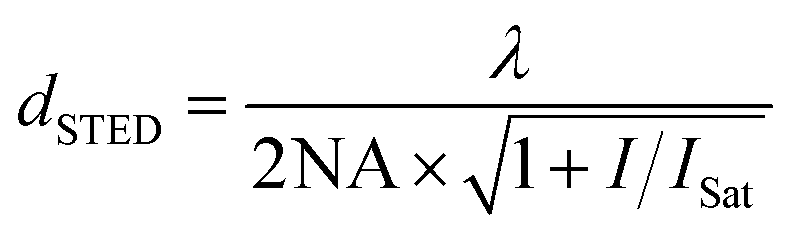 | (1) |
The drawbacks of STED are, that (a) only a small variety of fluorophores may be used for STED due to their stability against photo-bleaching97 and (b) short fluorescence lifetimes i.e. of samples with a high concentration of reporter dye (self-quenching) cannot be determined with a high accuracy due to the STED-induced decay.98 Also the analysis of the amplitudes will yield in falsified results as the decay is not used between 0 and 2 ns after the excitation pulse. Here, the normal fluorescence lifetime imaging microscopy has an advantage over the STED/FLIM as the instrument response function (IRF) is well defined and for fitting the excited state lifetimes the whole decay may be used, which yields in high accuracy of short lifetimes and a quantitative analysis of different populations. Therefore, other techniques and combinations of measurement techniques are developed or are used.
2.2 Coupling far-field optical microscopy and its variations with the AFM
Fig. 2 shows different acquisition modes. The two major methods of data acquisition of a combined measurement are either concomitant (Fig. 2a) or successive (Fig. 2b).58,109 Some measurements or analysis are even made comparative, meaning AFM and optical measurements may not be at the same position of the sample (not aligned). A researcher should decide carefully if it is favorable to measure simultaneously or execute the measurements successively. A combined measurement may save time and an interaction of the tip with the sample may be observed due to tip-enhanced fluorescence or tip-induced quenching.57,110 This may be desired to determine the location of a dye molecule on a surface or in a polymer matrix. On the other hand, the determination of fluorescence properties of fluorophores and the topography measurement have to be conducted successively to extirpate undesired changes in the fluorescence properties of the fluorophore.
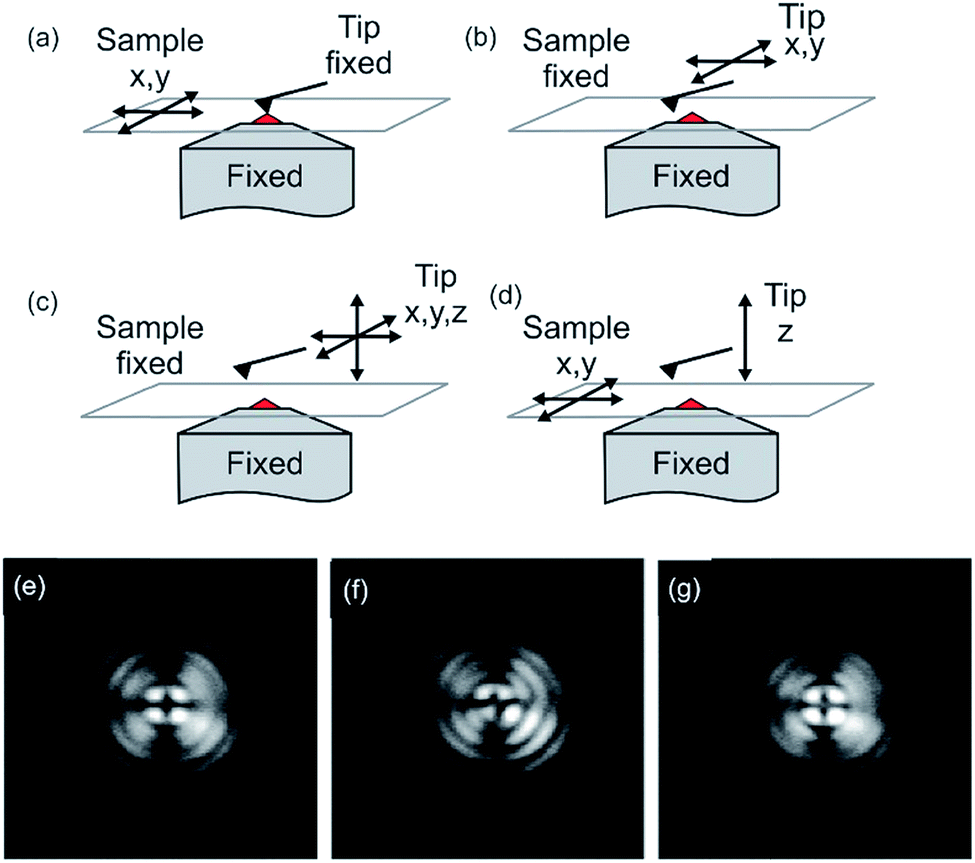 | ||
| Fig. 2 Modes of operation of the atomic force fluorescence microscope (AFFM): (a) simultaneous imaging by scanning the sample between a fixed tip and a fixed focus; (b) AFM imaging by lateral tip scanning with a specific ROI in the excitation volume; (c) force-extension imaging with the tip moving in three dimensions; (d) force–extension imaging with the tip ramping perpendicular to the surface and the sample moving in lateral directions. Reprinted with permission from ref. 44 Copyright (2005) John Wiley and Sons. (e–f) Beam diagnostic camera images of back-reflected light from the sample surface with the AFM tip retracted (e), with AFM tip visible as a distortion (f) and after alignment (g). For all images, the focus had been moved towards the AFM tip to show the distortion of the pattern more clearly. Reprinted with permission from the authors and ref. 110 Copyright (2010) Society of Photo-Optical Instrumentation Engineers. | ||
To combine far-field optical microscopy techniques, such as fluorescence microscopy, confocal microscopy and FLIM or other methods, such as total internal reflection fluorescence (TIRF), fluorescence correlation spectroscopy (FCS) and super-resolution techniques, with the AFM a specific setup is needed, which has to be aligned perfectly (up to a few nanometers). The combined AFM-optical microscopy technique consists out of a sample scanning AFM and an inverted microscope (e.g. CLSM). The microscope can be equipped with different excitation wavelengths. For optical detection with high resolution, an (oil) immersion objective with a high numeric aperture (1.45 or even higher) can be utilized. The link between the two measurement systems is the AFM sample stage, which is mounted directly on the inverted microscope chassis replacing the original sample stage. The sample stage has to be mounted carefully onto the inverted microscope, especially, taking into account the necessary space needed for the objective of the microscope, as well as its operating distance. It might be necessary to insert spacers. This setup allows measuring concomitant or successively optical microscopy and AFM.
For alignment of the orthogonal methods, first, the cantilever tip is adjusted to the cross hairs in the eyepiece of the microscope by manual positioning of the sample stage. Now, the cantilever tip is brought into contact with the surface and the microscope is adjusted to measure backscattering light. Keep in mind to reduce the excitation intensity to protect the detector. Acquired images of the backscattering of the cantilever indicate the position of the tip. The tip apex is typically visible in the backscattering images as a bright spot.58 Alternatively, the scattering of the incident light, which can be observed by a CCD camera (beam diagnostic camera, see Fig. 2e–g), can be utilized for fine adjustment, if the tip apex is not visible in the backscattering image.109 The quality of alignment is in this process clearly visible in the distortion of the reflected light originating from the retracted tip (Fig. 2f). Rather a strong distortion of the symmetry is observed for a misaligned setup, while high symmetry of the backscattered light is obtained for a well-aligned (AFM tip in the center of distortion pattern) setup, as shown in Fig. 2g.
A problem for combined AFM and microscopy measurements in the past was the presence of background noise in microscopy images, which is related to the laser utilized for the AFM optical lever.122 This is bypassed either by measuring consecutively or by measuring concomitant while using a high-quality filter.122 While utilizing filters, a potentially useful part of the spectrum cannot be analyzed due to the blocking of these wavelengths. This is especially a problem for microscopic systems, utilizing the emitted fluorescence light for generation of spectra. By employing a laser with a wavelength of approx. 1050 nm for the AFM, the background in microscopy images as well as problems with photobleaching or unspecific excitation of the sample can be avoided.123 Kassies et al. designed such a combination of an AFM with a confocal microscope (atomic force fluorescence microscope (AFFM)), where the AFM laser was shifted to a wavelength of 1050 nm. They constructed an AFFM with the capability of measuring simultaneously topography and fluorescence images, and the capability to measure fluorescence spectra at specific points of interest. They showed that they can measure on membrane fragments from Rhodobacter sphaeroides, containing both LH1 and LH2 (LH = light-harvesting pigment–protein complex).123 Furthermore, they demonstrated that measurements in a combined optical and force extension/nanomanipulation mode is possible with their setup.123
As shown previously, the exact alignment may be difficult and time-consuming. Another problem during a combined experiment is the drift in an AFM during measurement,121 which necessitates keeping an eye on the alignment of the measurement equipment. Timmel et al. utilized an alignment correction method, which uses the cantilever tip as a reference landmark.121 At first, they validated their method with beads in a complex artificial sample and subsequently, the method was utilized to maintain alignment during measurement of membrane structures in fixed and living human fibroblasts (biological sample). They showed that the fluorescence signals and the topographic structures were well-aligned and that nanoscale membrane structures could be related to the fluorescence spot.121
Lately, Staunton et al. presented a method for quantitative mechanical characterization of soft, heterogeneous samples in 3D via a combination of AFM, CLSM and finite element analysis (FEA). They showed the capability of their combined technique for the quantification of the elastic properties of cells (here metastatic breast adenocarcinoma) in an extracellular matrix (here collagen hydrogels). With this method, it is possible to decouple the response of a cell from its soft surrounding (extracellular matrix, ECM). By this means, although the response of the indentation experiment is based on both, the cell and the soft surrounding, the elastic properties of the cell and the ECM can be determined quantitatively.124
The fluorescence lifetime (decay time, eqn (7)), τF, can be defined by use of a simple kinetic scheme (first-order rate constants), where all radiative (fluorescence, eqn (3)) and all nonradiative pathways (inter system crossing (ISC), internal conversion (IC), dissociation) have to be considered.
| M + hν → 1M* × Ia (absorption) | (2) |
| 1M* → M + hν × kR (fluorescence) | (3) |
| 1M* → 3M* × kISC (intersystem crossing) | (4) |
| 1M* → M × kIC (internal conversion) | (5) |
| 1M* → products × kD (dissociation, bleaching) | (6) |
| τF = (kR + kIC + kISC + kD)−1 | (7) |
There are several techniques to determine the decay times of fluorescence. The most common technique is TCSPC. It relies on the accurate time measurement of single photon events (e.g. fluorescence). The laser pulse serves as a reference for the start time, whereas the stop time is determined by the fluorescence photon. These photon detection events are binned and converted into a histogram. For this histogram, a small time bin down to 1 ps is used. The photon events are collected by adding +1 in the time bin proportional to the start-stop time. After many cycles, the distribution of such detection times builds up.126–128 Afterwards, the data can be analyzed by eqn (8) (exponential decay).
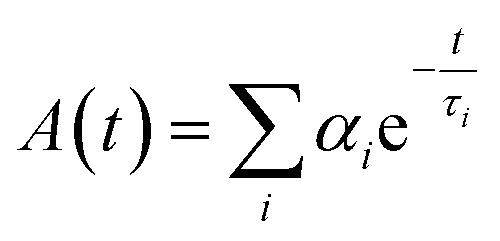 | (8) |
The fluorescence lifetime values can yield information about the molecular microenvironment of a fluorescent molecule, i.e. viscosity,129,130 pH,131 temperature,132 polarity133,134 and solvation.135 Furthermore, the excited state lifetime of a fluorophore can be altered by ionic strength,136 oxygen concentration,137 binding to macromolecules, changes in protein orientation, change in protein folding,138 a critical binding reaction139 and the proximity of molecules that can deplete the excited state by resonance energy transfer (RET).138,140 Therefore, fluorescence lifetimes can be utilized as indicators of these parameters. There are various applications utilizing fluorescence lifetime, i.e. in calcium and other chemical detection,141 clinical detection142 and determination of transport processes (diffusion).143,144 Although FLIM is typically featured in biological studies, it also finds application in polymer science and surface analysis.109,145 A combination of FLIM with AFM potentially yields information of biological processes occurring inside cells, which would not be possible without FLIM.
Recently, a few articles concerning themselves with this kind of combination, which belong to an a-SNOM approach, were published. An advantage, which was exploited in these articles, was the higher optical resolution due to short ranged tip-induced quenching and inherent fluorescence lifetime decline. Schulz et al. dried a diluted sample of ATTO 647N on a clean glass slide. The density of the dye molecules was in the order of 1 molecule per μm2. During the measurement in the combined mode, the diffraction limited spots of the fluorophores show typical single molecule features like blinking and beaching. All fluorescence spots possessed a dark area in their center, which were typically in the order of 20–40 nm FWHM. The dark areas were attributed to energy transfer to the AFM tip.110 Here, the excited state lifetime changed by advancing the tip towards the organic dye. A drop of excited state lifetime with decreasing distance was observed. Thus, this setup possesses the ability to measure with high resolution without the need of tedious and sophisticated super-resolution equipment, such as specialized dyes, optics, and lasers. A few years later, single digit nanometer resolution via the same approach was shown (see Fig. 3). Concomitantly, the fluorescence lifetime information yielded knowledge about the orientation of the dye molecules. Schulz et al. found that the molecular transition dipole moments lie parallel to the sample surface.58 AFM (intermittent mode) and FLIM were also used by Yoskovitz et al.57 CdSe quantum dots coated with multiple shells (7.3 nm diameter) were placed onto a cleaned glass surface. AFM was measured in tapping mode isochronal with fluorescence intensity and the time-correlated single photon counting data. The obtained data showed a fluorescence enhancement for silicon tips at approximately 100 nm laterally away from the fluorophore. This data is in agreement with the literature, where the fluorescence enhancement is assigned to a field enhancement at the apex of the tip.146,147 Furthermore they showed that excited state lifetime yields in an enhancement of the signal-to-noise ratio, and also significantly improved resolution.57
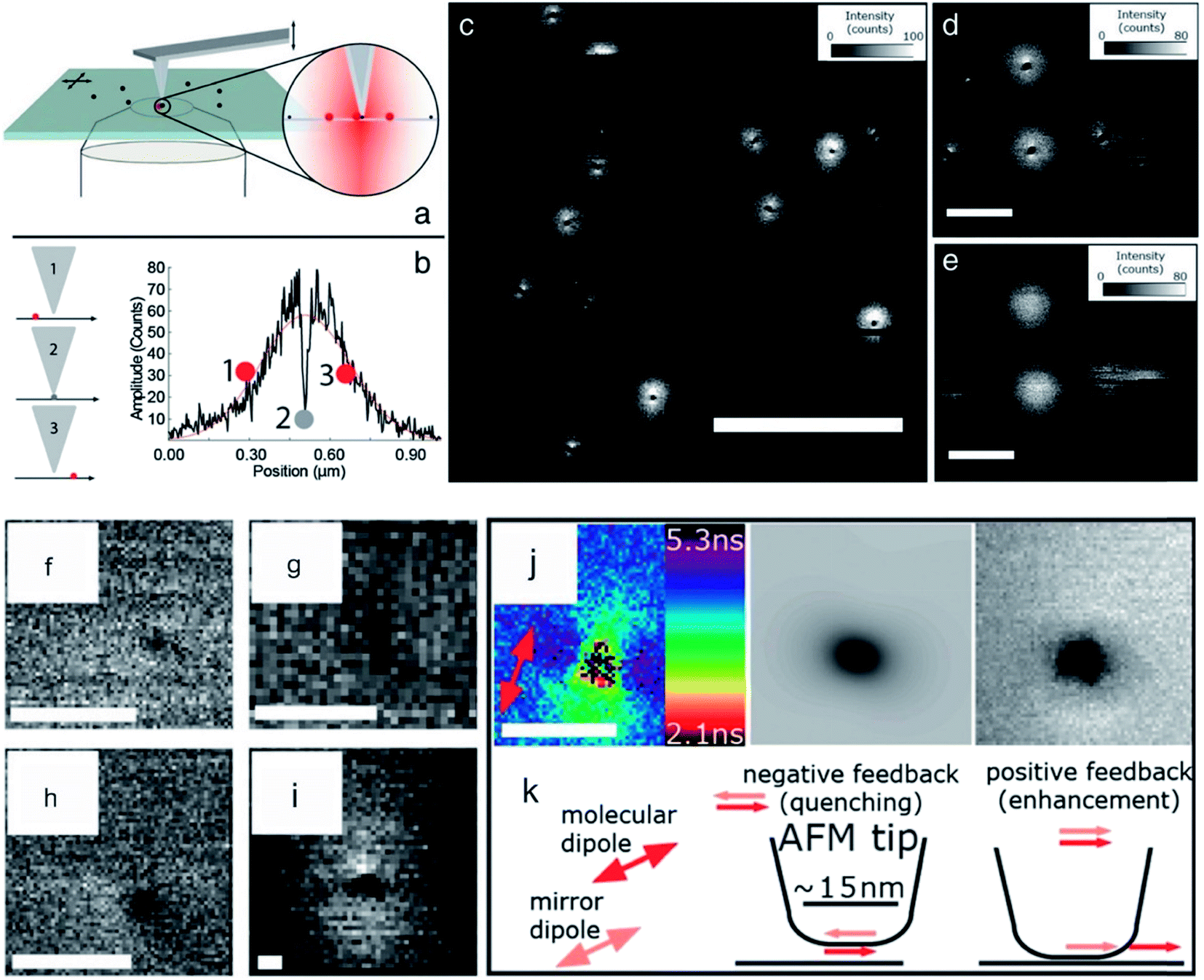 | ||
| Fig. 3 (a) Scheme of hybrid AFM-FLIM setup. (b) Scheme of tip-induced quenching and course of intensity versus separation distance between tip and fluorophore. (c) Fluorescence intensity data of a sample containing single dye molecules (ATTO 655) with the AFM tip approached to the surface. (d) Magnification of (c). (e) Same ROI as in (d) is imaged with retracted tip. The scale bar for (c) is 2 μm and for (d) and (e) 500 nm. For (f), (g), (h) the FWHMs are 6.8 nm, 7.5 nm, and 5.9 nm, respectively. (i) Triangle shape due to a blunt tip. (j) Fluorescence lifetime distribution, the calculated fluorescence intensity and the measured fluorescence intensity of a quenching spot with a FWHM of 16 nm is shown. (k) Scheme of the physical mechanism leading to quenching. Reprinted from open-access article ref. 58 under the terms of the Creative Commons Attribution License. | ||
Lately, a combination of AFM-FLIM in a far field approach was utilized to study surface nanobubbles.109 Surface nanobubbles are nanocavities filled with gas (air), which nucleate at the interface between solids and liquids (typically water). For this approach water was stained with Rhodamine 6G (Rh6G). Rh6G appeared to be enriched at the liquid–gas interface, which developed during nanobubbles nucleation, as shown in Fig. 4b. The surface nanobubbles appeared brighter in the confocal fluorescence microscopy images than the interphase (glass–water) without nanobubbles (compare Fig. 4d and g). The fluorescence intensity image (bright nanobubbles) and the topography image of the nanobubbles were in good agreement. Here, the excited state lifetime analysis served as proof of a water–air interface. TCSPC decays measured at the location of a nanobubble yielded a tri-exponential decay, whereas TCSPC decays on the substrate–solid interface (without nanobubbles) yielded a bi-exponential decay (compare Fig. 4e and h). The third excited state lifetime, which was approx. 0.6 ns, was attributed to the liquid–air interface, as this lifetime is not present in a sample without nanobubbles.109 Prior to this, the existence of this air/water interface was discussed in the academic community as these nanobubbles would be unstable due to high inner pressure (curvature).148
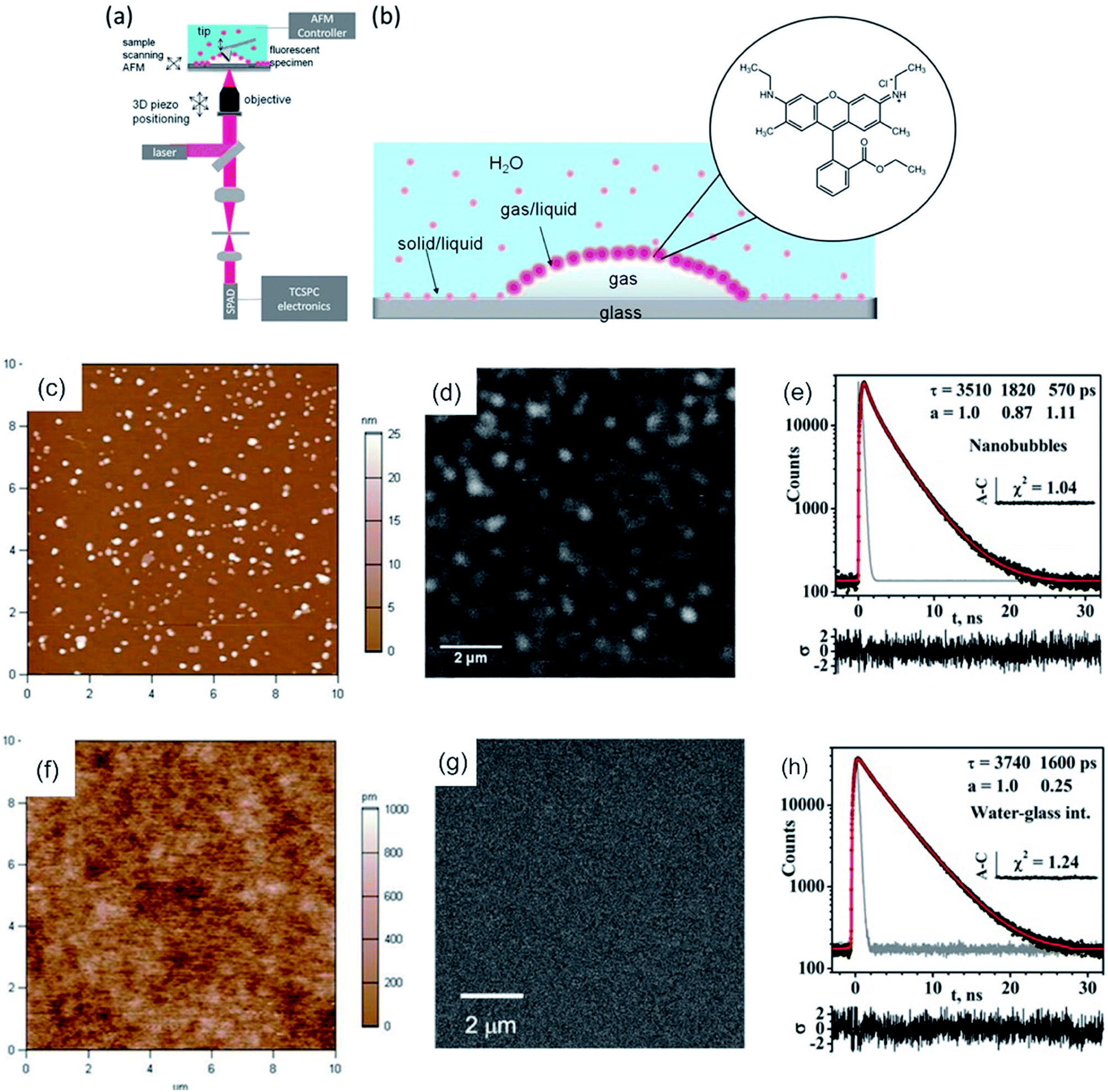 | ||
| Fig. 4 (a) Scheme of the hybrid AFM-FLIM microscope. (b) Scheme of the partitioning of the fluorophore Rh6Gat the interface and in water. Nanobubbles nucleated by an ethanol–water exchange on the piranha-cleaned glass in an aqueous 850 nMRh6G solution: (c) AFM height image; (d) confocal fluorescence microscopy intensity image in the same ROI as in (c). (e) TCSPC fluorescence decay curve of Rh6G obtained from TTTR data of panel (d). (f) AFM height image and (g) confocal fluorescence microscopy intensity images of UV-ozone cleaned glass after the ethanol–water exchange in aqueous Rh6G solution (850 nM). (h) TCSPC decay curve of Rh6G obtained from TTTR data in panel (g). Reprinted with permission from .109 Copyright (2016) American Chemical Society. | ||
He et al. employed AFM correlated with single-molecule fluorescence intensity/lifetime imaging microscopy (their acronym: AFM-SMFLIM) to investigate interfacial electron-transfer processes of a mixed ((m-ZnTCPP/TiO2 nanoparticles) + m-ZnTCPP)/cover glass sample. The correlated information of fluorescence blinking of each individual dye molecule and its fluorescence lifetime along with the surrounding topography explained the coupling strength inhomogeneity of each dye molecule with TiO2 nanoparticles (NPs) and its effect on electron transfer intermittency and thus, yielded molecular level understanding of the interfacial electron transfer reactivity.149
A combination of these two techniques was utilized typically in the analysis of liquid films and membranes.150,151 The first study utilizing AFM-FCS was done by Burns et al. in 2005. It was utilized to analyze the relative partitioning and the mobility of labeled probes in gel and disordered liquid phase of bilayers consisting of 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) lipid domains in 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC).152 For their analysis, at first, sites with different domains were mapped by combined AFM and CLSM measurement, allowing the correlation of topography (AFM) with probe partitioning between gel and liquid domains (CLSM). The FCS analysis showed that two components are present in the DOPC fluid phase (fast and slow), which was ascribed to membrane heterogeneities.150 Subsequently, in the group of Schwille, the AFM-FCS technique was utilized to characterize dynamic and structural organization of phase-separated membranes161 and to show coexistence of the liquid disordered and liquid ordered phase for ceramides, which forms gel-like domains.153 The formation of these ceramide-rich domains was found to be dependent on the acyl chain length of the ceramide.151
Many publications rely on the superior lateral and axial resolution of AFM over TIRF and the single photon accuracy of TIRF.168,171 These qualities were employed in the study of myosin labeled with tetramethylrhodamine-5-maleimide.168 Myosin self-assembled to asymmetric filaments. The cross-correlation of AFM and TIRF (height/volume vs. intensity) yielded structural information of the assembly. The intensity versus height ratio implies that myosin heads form a shell around the filament axis, which is consistent with the heads exposed to the outside.168 Although TIRF is the technique with lower resolution, it was capable to detect the association of β-amyloidDOPC/DPPC lipid bilayers earlier than AFM.171
AFM is often exploited for its force measurement capability and its ability to manipulate microscopic objects (micromanipulation) or even manipulation on an atomic scale (nanomanipulation).169,172–174 Such an application is the single cell manipulation via AFM to study the integrin Mac-1(cell)-fibrinogen interactions in comparison to the intermolecular fibrinogen interactions.172 Force transmission, number, and arrangement of focal adhesion points of the cells can be analyzed by exerting a force on the apical membrane of cells, which adhere to a transparent and thin surface.173 Recently, TIRF was utilized to stimulate a single T cell by light (via photoactivable Rac protein) while simultaneously the mechanical response of the cell was measured via AFM.115 This method depicts an inverted approach of the AFM-TIRF setup, as typically the AFM is utilized as a nanomanipulator. Single cells could be seeded with fluorescent beads (fluorescent nanoparticles), which could be imaged subsequently with the TIRF setup.175 An interesting approach to monitoring the genome release of adenovirus capsids was shown by Ortega-Esteban et al. In their research, similar to CLSM and FLIM essays for release studies, the rupture and release of fluorophores was observed by TIRF while the virus membrane was weakened and finally ruptured by the AFM (active trigger, see Fig. 5).174 They could show that the loss of the initial penton in frame 7 leads to a marginal incline in fluorescence but not to a reduction in height, whereas the rapid decrease of height after frame 17 signals the start of the complete disassembly, accompanied by a strong increase in fluorescence intensity.174 By advancing towards fluorescent nanoparticles/molecules, fluorescence modulation techniques similar to CLSM can be utilized. Eckel et al. used fluorescent CdSe nanocrystals and gold-modified AFM tips to enhance the emission, the tip-nanocrystal distance for maximum emission was 22 nm, or to quench emission effectively.170 Here, the enhancement was attributed to resonant exciton-plasmon coupling.176 An enhancement of the previously mentioned method could be achieved by Lee et al. as they compensate for the photo brightening effect, which enables quantitative measurements.177
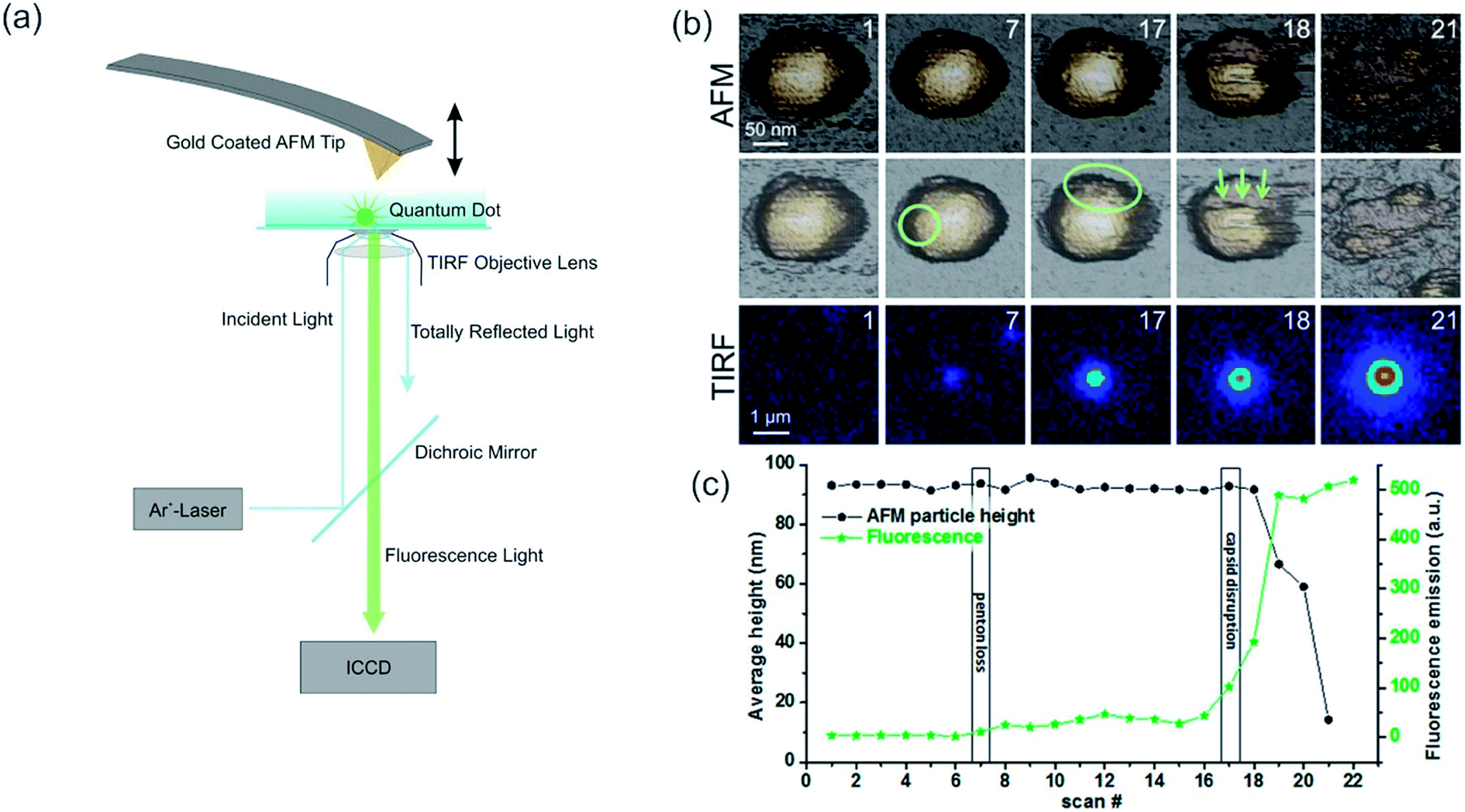 | ||
| Fig. 5 (a) Scheme of a combined AFM-TIRF setup, the incident laser wavelength and the setup for the AFM may vary. Reprinted from open-access article ref. 178 under the terms of the Creative Commons Attribution License. (b, c) Slow unpacking of adenovirus capsids. (b) The first row shows the AFM images of the fatigue experiment of a mature particle obtained in dynamic mode. The second row indicates the loss of the first penton in frame 7 with a circle. The ellipse in frame 17 shows the nucleation point for further disruption. The arrows indicate the direction of further disassembly in this experiment. After each AFM frame, the scan was stopped, the excitation laser was switched on, and 10 fluorescence images were recorded and averaged (third row). For the next AFM frame, the laser was turned off to limit bleaching of the YOYO-1 dye molecules. The time between consecutive images was 2 min. (c) Evolution of the average height (left axis, black) and fluorescence emission (right axis, green), corresponding to the fatigue experiment. Reprinted with permission from ref. 174 Copyright (2015) American Chemical Society. | ||
2.3 AFM coupled to super-resolution techniques
A further improvement of the AFM-CLSM combination was achieved by adopting super-resolution techniques, such as STED,179 FIONA,180 dSTORM181 and PALM.181 Such a combination has several advantages, amongst others, other fluorescent-based techniques can be adapted at least for the STED, such as FCS,182,183 FLIM,184 fluorescence recovery after photobleaching (FRAP),185 multicolor imaging186,187 and steady-state fluorescent probing. Via super-resolution techniques the lateral and axial resolution is enhanced up to 20 nm and 100 nm, respectively, rendering subcellular structures observable in detail. Higher optical resolution was observed for non-commercial setups. Similar to an AFM-CLSM combination, the AFM-super resolution techniques benefit from the fast acquisition times of the optical method, although nowadays special AFM equipment is available with high temporal resolution.188 Therefore, via this combination tracking of several small molecules in close proximity, is possible (super-resolution technique) and simultaneous either nanomanipulation,189,190 determination of change in morphology or viscoelastic properties of a sample.191 Similar to the CLSM, the chemical information/surrounding of a fluorophore can be evaluated, which proves to be difficult via the AFM. However, biomolecules and polymers are invisible to single-molecule localization microscopy (SMLM) as it only detects fluorophores with a specific absorption range and has to be stained. Furthermore, the z-resolution in SMLM methods is still in the order of 100–500 nm, although advances in SMLM for example via a mirror-reflected approach yield in higher axial resolution.192 The successful combination of STED with AFM was first reported in 2012 by Harke et al.191 As the used experimental setup is similar to the setup for a combined AFM-CLSM experiment the representation of the setup was waived. AFM-STED was demonstrated at first for 40 nm fluorescing crimson spheres at dry and wet conditions. Subsequently, microtubules of Cos7 cells labelled with Atto 647 N were imaged and correlated to mechanical variations in the Young's modulus (see also Fig. 6).191 Chacko et al. showed that measurement artifacts of the optical image can be accounted for by AFM and the images can be corrected afterwards, i.e. by calibration via a Labview procedure.193 Furthermore, AFM subdiffraction nanomanipulation of fluorescent crimson microspheres visualizes the high-resolution targeting capability of the system.193 Jianqiang et al. facilitated the combined setup by utilizing a super continuum laser and for the first time concomitant and synchronized optical and topographic images were obtained. They applied their setup for nanobeads with a resolution of 42 nm as well as cells.194 Recently, dSTORM and PALM were combined with the AFM to measure f-actin (dSTORM) and living mammalian cells (PALM). While the AFM images illustrate the dynamics of the cell membrane a PALM series monitors the changes in protein clusters over time.181 Similarly, a combination of AFM with FIONA was introduced by Fronczek et al.195 It was utilized to identify different proteins in multi-protein–DNA complexes with a resolution of up to 8 nm. Nowadays, commercial solutions for STED are available, which can be upgraded with FLIM and an AFM module. Therefore, these systems are expected to receive increased attention, especially in biological research areas. | ||
| Fig. 6 Cos7 cells labeled with Atto 647 N, (a) confocal image, (b) STED image, (c) 3D AFM height image extracted from AFM force curves (45 × 45 pixilated scan), (d) confocal image and (e) STED image (both linear deconvolved) (f) map of Young's modulus calculated from AFM force curves. Scale bars in in (a) and (b): 2 μm, axes bars in (c)–(f): 3 μm. Reprinted from open-access article ref. 191 under the terms of the Creative Commons Attribution License. | ||
2.4 Near-field optical microscopy (SNOM/NSOM)
The idea of SNOM was probably first conceived by Ash et al. in 1972.196 By use of 3 cm microwave radiation, a resolution of λ/60 in one dimension and λ/20 for two-dimensional objects was achieved, but technical difficulties prevented the extension of this technique towards shorter wavelengths. 2007 by Oshikane et al. the lateral and vertical resolution was determined to be 20 and 5–2 nm, respectively.197 SNOM is a nano-optical imaging technique that enables light–matter interaction for the purpose of extracting relevant information about nanoscale objects. There are two inherently different methods in scanning near-field optical microscopy. One can make use of a sub-wavelength aperture from which excitation light emanates or through which the emission from the sample is detected, or both. This is called aperture SNOM. The resolution in aperture SNOM is proportional to the radius of the aperture. However, as the aperture size decreases the amount of light admitted into the aperture decreases as well and signal levels drop. This results in a trade-off between signal levels and spatial resolution leading to a practical resolution limit for aperture SNOM of λ/10.198 The other method is executed by probing highly localized interactions of excitation and emission photons with a sub-wavelength structure. These structures are usually in the range of a few nanometers to a few tens of nanometers (aNSOM or NSOM).199,200 However, the contrast mechanism is not fully understood and images may contain topographical artifacts.201 Apertureless NSOM is typically measured via a bottom-illumination mode,202–206 which yields in advantages in light confinement and background noise reduction. As the probing tip has to be in close proximity to the glass surface, the measurement by aNSOM is restricted to thin samples, which diminishes its applicability. Recently, an aNSOM was developed with the capability to measure via bottom-illumination and top-illumination mode, as well as a measure in water and in air.207 The resolution was found to be independent on the illumination mode, but was lower in water (32 ± 7 nm) than in air (25 ± 6 nm).207 Furthermore, the resolution can be enhanced by amplifying the incident and scattered electric fields at the tip itself.208 These results indicate that aNSOM in top-illumination mode (also called s-SNOM) is able to measure thicker samples, which are not measurable via the bottom-illumination mode. Both, the setup of apertureless and aperture NSOM are shown in Fig. 7, indicating the different approaches to enhanced resolution.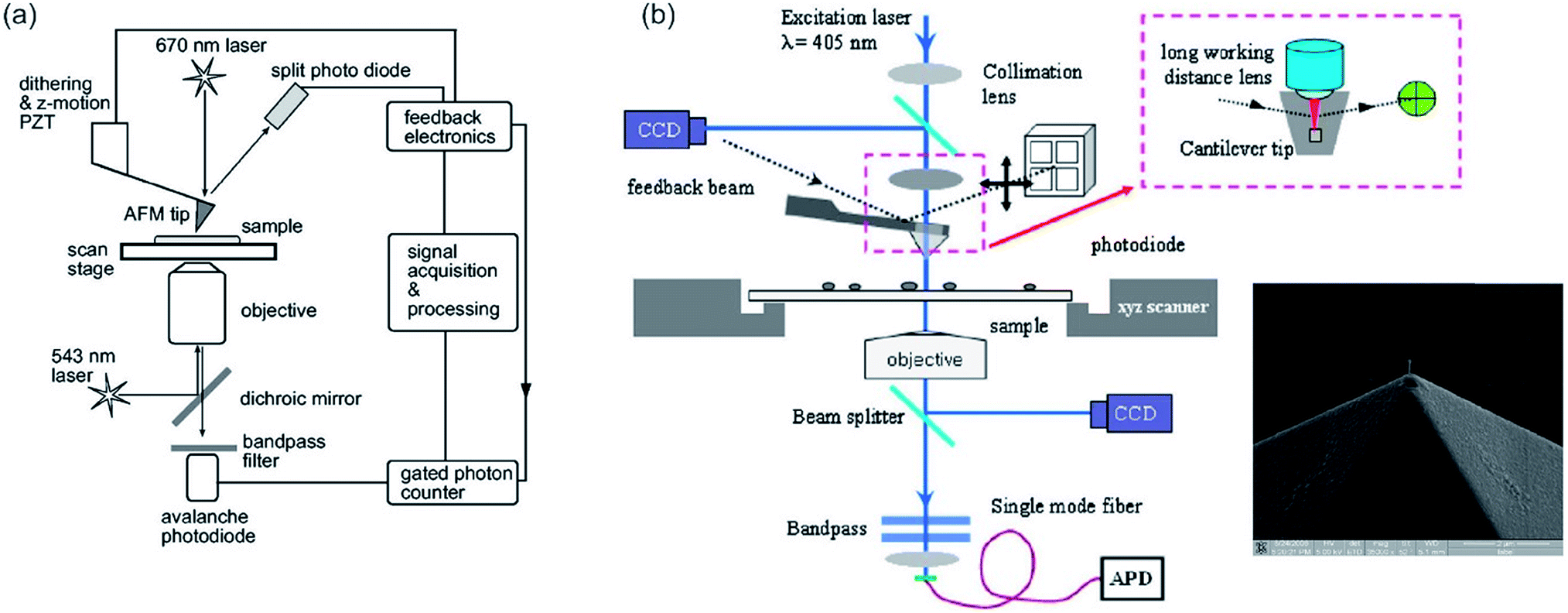 | ||
| Fig. 7 (a) Schematic diagram of an apertureless NSOM. Reprinted with permission from,199 Copyright (2000) AIP Publishing. (b) Aperture SNOM, the inset shows the cantilever tip with its aperture. Reprinted with permission from,209 Copyright (2008) Elsevier. | ||
Nowadays, s-SNOM is coupled to IR to resolve intermolecular interactions with nanometer spatial resolution,210,211 which is yet, similar to PTIR, another method to chemically identify surfaces with high spatial resolution. This field is under vast development and recently numerous research articles were published.23,212 For example, Pollard et al. investigated as a proof of principle the self-organization of amphiphilic block copolymers into hydrophilic and hydrophobic domains. By their approach, they were able to chemically map the surface with high spatial resolution while obtaining exquisite sensitivity, which can be used for the unravelling of intra- and intermolecular interaction (see Fig. 8).211 Still, SNOM methods are burdened with various artifacts, i.e. due to a broken or blunt tip. There is up to now no method to evaluate if an image is without an imaging artifact.213 As SNOM relies on the concomitant measurement of the optical and force microscopy, a fast and simple survey scan similar to typical optical microscopy is not possible anymore. On these grounds and because aperture SNOM is still often utilized (more artifacts, lower resolution compared to other methods) a combination of conventional optical microscopy may be favorable to SNOM and s-SNOM, especially for large three-dimensional samples (optical slicing ability). Applications of NSOM/SNOM and the novel s-SNOM are mostly biological appliances, such as cell visualization and analysis.214 However, also applications in microscopy for materials are well known for example, analysis of semiconducting heterostructures, such as quantum dots, quantum wires and disordered quantum wells, material imaging, e.g. local crystal structures, crystal defects and doping,215 and observation of intra- and intermolecular interaction in both biological and material science fields.211
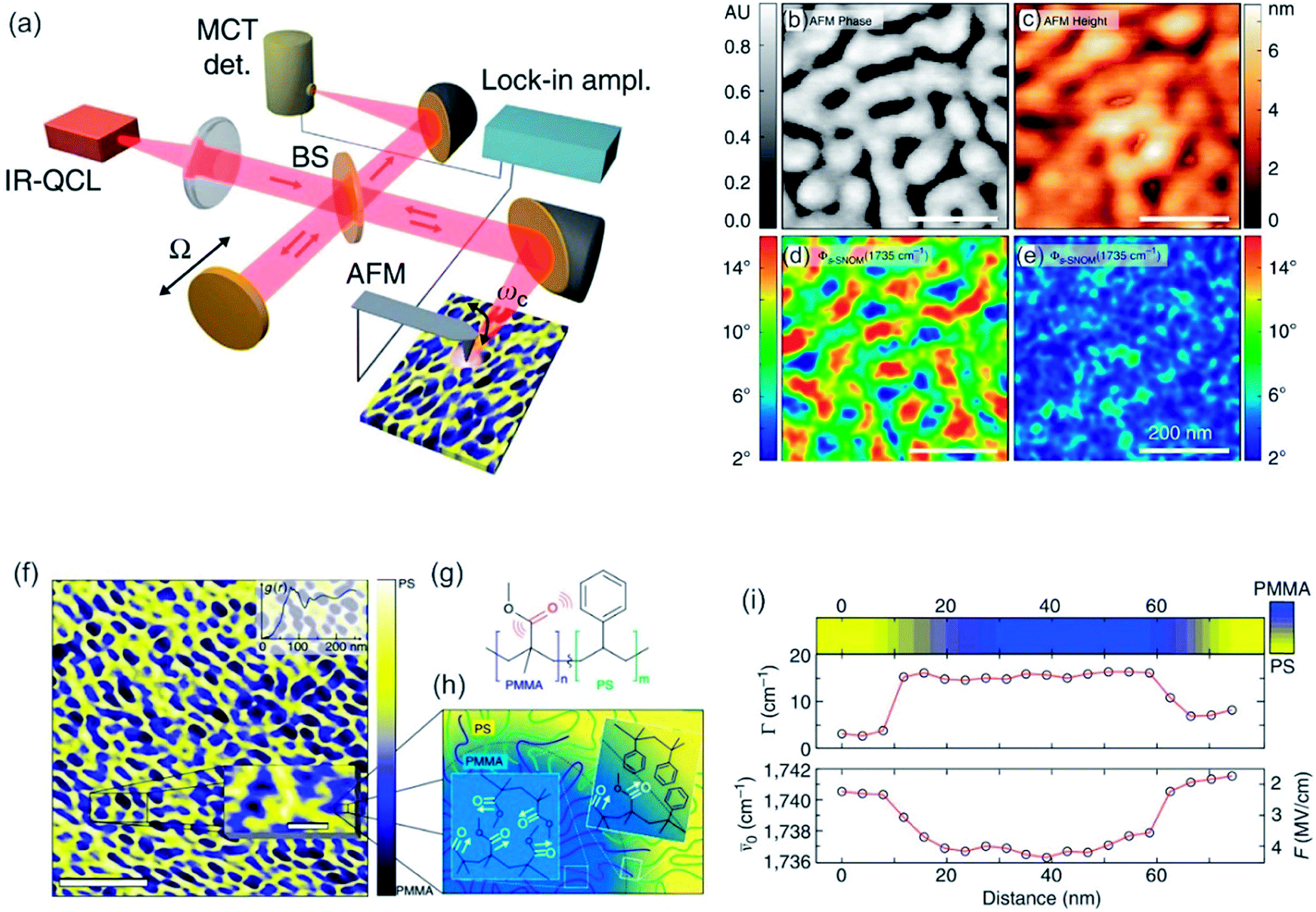 | ||
| Fig. 8 (a) Scheme of interferometric s-SNOM experiment. QLC = quantum cascade laser, MCT IR detector = HgCdTe infrared detector, BS = beam splitter (b–e) sample scans of quasi-lamellar PS-b-PMMA showing AFM tapping phase (b), AFM height (c) and near-field phase (fs-SNOM) (d) and off resonance (e) with the carbonyl mode. Scale bars 200 nm. (f) Nanoscale chemical map of self-phase separated amphiphilic block copolymer PS-b-PMMA derived from carbonyl resonant s-SNOM imaging. Scale bar 750 nm. In the inset a magnification of a small ROI is shown (scale bar 100 nm). (g) The molecular structure of block copolymer. (h) Schematic of domain morphology of PS-b-PMMA. (i) Spectrally resolved line cut showing the local relative concentration of PMMA as measured by the s-SNOM phase (top color map), linewidth (middle), and spectral shift (bottom), with derived local electric field F. Reprinted with permission from ref. 211, Copyright (2014) Macmillan Publishers Ltd., Nature Publishing Group. | ||
2.5 AFM & Raman spectroscopy – tip-enhanced Raman spectroscopy (TERS)
Raman spectroscopy is the complementary technique to IR spectroscopy as most vibrations are either Raman or IR active, and it is also orthogonal to AFM as Raman is able to gather chemical information of a desired sample.216,217 The former refers to the different selection rule compared to IR. Raman occurs mainly for symmetric vibrations when the polarizability changes upon excitation. Raman spectroscopy relies on the inelastic scattering of monochromatic light (nowadays laser), while the molecule (sample) is excited to a virtual energy state. From this state, an inelastically scattered photon emanates, which can possess either higher (anti-Stokes) or lower (Stokes) energy than the incoming photon. The difference in energy of the resulting scattered photon can be related to the energy differences in the rotational and vibrational states. Raman spectroscopy is widely utilized in chemistry and material science,218–221 as chemical bonds, functional groups and even different hybridization states are detectable.222,223 Especially, it is utilized in applications related to analytical chemistry,224 forensic science (analysis of drugs or paints),225,226 geology and mineralogy (e.g. gemstone identification),227 biology (e.g. secondary structure of polypeptides/proteins, phospholipids),228–231 material science232,233 (e.g. semiconductors, carbon materials and polymers) and life science (e.g. pharmaceutical analysis).234,235Tip-enhanced Raman spectroscopy (TERS) combines the chemical sensitivity of surface-enhanced Raman spectroscopy (SERS) with the high spatial resolution of the AFM to concomitant garner chemical and morphological information at the nanometer length-scale. TERS is a near-field microscopy technique in which the Raman signal is enhanced by an apertureless probe. The probe is a sharp metal-coated AFM-tip, which is positioned at the center of a laser focus. The enhancement of the electromagnetic (EM) field and therefore of the Raman signal can be ascribed to a combination of localized surface plasmon (LSP) resonance and lightning rod effect.236 The enhancement effect is confined to the vicinity of the tip-apex. Hence, TERS overcomes the resolution limit conventional Raman spectroscopy is bound to. The resolution of TERS is typically in the order of 20 nm (see Fig. 9),231,237,238 but can be enhanced by specific reflection modes up to 1 nm.239
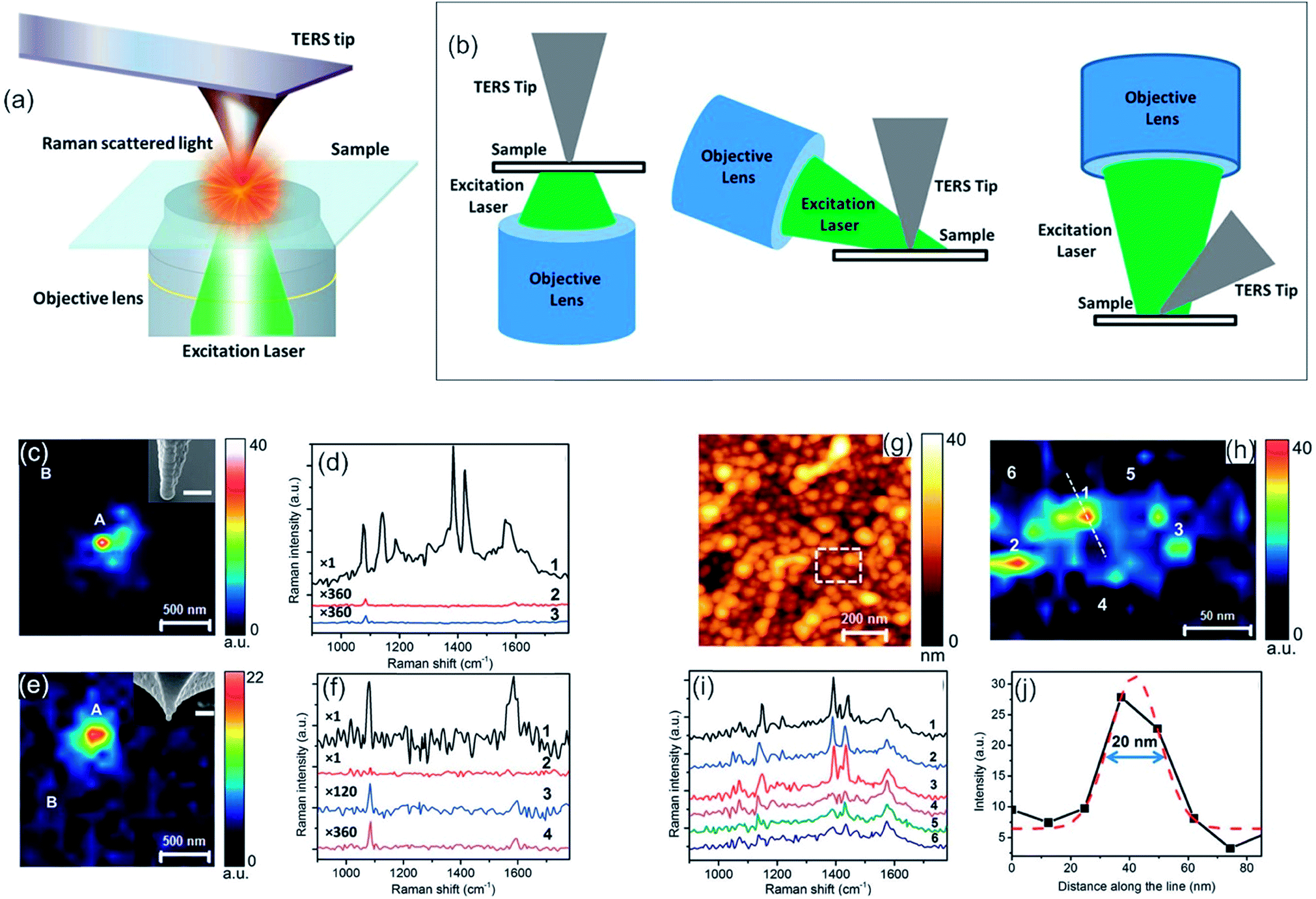 | ||
| Fig. 9 (a) Schematic diagram of a TERS set-up in transmission mode. (b) Schematic diagrams of most commonly used TERS configurations. Bottom illumination (left), side illumination (middle) and top illumination (right). (a, b) Reprinted from open-access article ref. 240 under the terms of the Creative Commons Attribution License. (c) Mapping of photocatalytic reaction occurring at the Ag-coated TERS tip-apex using the 1142 cm−1 peak intensity of DMAB. In the inset, a SEM image of the tip is shown. Scale bar: 100 nm. (d) Comparison of the near-field spectrum from position A (1) in (c) with the tip in contact with the pMA/polymethylmethacrylat (PMMA) surface, its far-field spectrum (3) and far-field spectrum from position B (2) with the tip in contact with the surface. (e) Raman mapping of the alumina-protected Ag-coated TERS tip-apex using the 1086 cm−1 peak intensity of pMA. SEM image of the tip is shown in the inset. Scale bar: 100 nm. (f) Comparison of near-field and far-field spectra of locations A and B from (e). (1) Near-field pos. A (2–4) far-field spectra. (g) AFM topography image of a glass substrate covered with Ag nanoparticles. (h) TERS map from the dashed rectangle marked in (g), showing the variation of the 1142 cm−1 Raman peak intensity of DMAB. (i) Near-field spectra from the positions marked in (h). (j) Intensity profile along the dotted line marked in (h), showing the spatial resolution of the TERS map. Gaussian fit to the intensity profile is shown by the dashed red curve. (c–j) Reprinted with permission from ref. 238, Copyright (2015) the Royal Society of Chemistry. | ||
TERS has been utilized in a broad range of applications in biology, such as pathogens,241 cell membranes242 and peptides,243 in materials science,244 such as polymer blends, and chemical engineering, i.e. in catalysis238,245,246 for detection of intermediates or as a trigger for chemical reactions.247 For example, Kumar et al. investigated in situ photocatalytic oxidation of (p-mercaptoaniline) pMA to (p,p′-dimercaptoazobenzene) DMAB on gold nanoparticles with TERS (see Fig. 10c–f). Compared to far-field Raman spectroscopy, the integration time could be reduced while the peaks are more pronounced due to the enhancement effect in TERS, as shown in Fig. 10d and f.238 Due to the broad spectrum of TERS and its applications a thorough illustration of TERS would fill another review by its own. Therefore, we want to refer the interested reader to the reviews of Kumar et al. and Jiang et al., which summarize recent advances in TERS together with its advantages and weaknesses.240,248 TERS itself is developed further, even though it is already utilized for many applications. Important issues, which are encountered, are low fabrication reproducibility of the AFM-tip, which becomes manifest in differing Raman enhancement factor or induced plasmonic dipole factors from tip to tip,249 short plasmonic lifetimes, which are assigned to metal delamination (Au-tips) or chemical alteration (Ag-tips)250,251 and low signal-to-noise ratio due to far-field scattering/emission. Extensive research was performed on solving these issues. Improved fabrication reproducibility of the tip was achieved by utilizing an increased amount of wet-chemically synthesized silver nanowires (AgNWs), which are bound to the apex of the tip. This leads to improved electric contact, mechanical strength, and reproducibility,252 which can be exploited by the commercialization of such modified tips. However, TERS probes relying on Ag-tips were found to lose the plasmonic enhancement of the tip during the first 4 hours of exposure to the environment (oxygen/humidity). It was shown that the lifetime of a TERS-tip based on Ag can be maintained up to 5 months by utilizing an environment with sub-ppm oxygen and moisture concentration (typically in a glovebox). It was also mentioned that Ag based tips can be protected for storage and measurement either by the inert atmosphere with previous mentioned low oxygen content and low humidity or by covering the tip with a thin (≤3 nm) layer of alumina.253,254 Furthermore, enhancement of TERS can be achieved by change of the illumination geometry, resulting in an even improved TERS signal,45 which was observed for Raman spectra of malachite green isothiocyanate and thiophenol in water and in air (see Fig. 10a–f), and tip modification, which renders the combined setup sensitive to pH change.255
 | ||
| Fig. 10 (a) Experimental setup for solid/liquid TERS. (b) Scheme of the TERS setup (side illumination) (c) tip focusing process imaged in air (top) and water (bottom) (d) STM images of malachite green isothiocyanate (MGITC)/Au(111) in air (top) and in water (bottom). (e) Comparison of Raman spectra of MGITC Au(111) obtained via TER resonant (TERR) in air (e-a), far-field Raman in air (e-b), TERR in MilliQ water (e-c), far-field Raman in water (e-d) and signal of tip (e–e). (f) Comparison of Raman spectra of PhS/Au(111) obtained via TER resonant (TERR) in air (f-a), far-field Raman in air (f-b), TERR in MilliQ water (f-c), far-field Raman in water (f-d). Reprinted from open-access article ref. 45 under the terms of the Creative Commons Attribution License (CC-BY). | ||
2.6 AFM & IR-spectroscopy – photothermal-induced resonance (PTIR) and infrared photoinduced force microscopy (IR-PiF)
IR spectroscopy is one of the oldest and widespread analysis techniques for the analysis of solid samples. However, IR spectroscopy can be also utilized for liquids,256 soft matter257 and gases258 to identify chemical species. It is based on the absorption of light from the spectrum of a light source. Nowadays, IR is measured via the Fourier-transform technique also known as Fourier-transform infrared spectroscopy (FTIR),259 which enables fast and highly accurate measurement of whole spectra. Molecule vibrations, which change the dipole moment of the molecule, are IR active and may absorb light of a specific wavelength. This absorption of a specific wavelength can successively be translated into specific bonds of a molecule or related to small molecules themselves. Recently, the FTIR technique was enhanced by coupling it to an optical microscope.260 This results in FTIR microscopy with a lateral resolution of down to 10 μm (compare diffraction limit). In an actual experiment, this value is with a scan size of 20 × 20 μm bigger, as it is difficult to obtain a good signal to noise ratio and enough IR radiation.261 The most important operation modes of FTIR are transmission, diffuse reflectance infrared Fourier transform (DRIFT) and attenuated total reflectance (ATR).262 The transmission mode can be utilized for gases, liquids, and solids, whereas the ATR method can only be utilized for liquids and solids. For a thorough explanation of FTIR and IR, its mathematics and its operation modes please refer to standard textbooks, e.g. “Fourier Transform Infrared (FTIR) Spectroscopy”.263IR and FTIR spectroscopy are widely utilized in biology/biophysics (e.g. protein conformational changes),264,265 environmental science (e.g. gas analysis),266–268 chemistry (e.g. identification of molecules or polymers and chemical reactions in the gas phase),269 surface analysis270–274 (e.g. surface contamination, thin film analysis and analysis of surface termination) and polymer analysis275–277 (e.g. degree of crystallinity).
A combination of AFM with infrared spectroscopy or typically Fourier transform infrared spectroscopy is called AFM-IR, but often it is abbreviated PTIR (photothermal-induced resonance) due to its working principle. Dazzi et al. developed the concept of PTIR in 2010.278 PTIR relies on the excitation of molecules (vibration) by a tunable pulsed IR laser. The infrared light emitted from the pulsed IR laser is absorbed by the molecules leading to a quick thermal expansion. This expansion induces an oscillation of the AFM cantilever, which is detected via the deflection of the detection laser beam. By utilizing a Fourier transformation, frequencies and amplitudes can be acquired. Here, the obtained amplitudes are related to the strength of the cantilever oscillation. Studies utilizing PTIR have analyzed the chemical information of biological samples, such as proteins,279,280 lipids and lipid vesicles,281,282 cells,283,284 bacteria,48,279,285 polymer samples and thin films,49,286–288 medical devices,289 drugs,290,291 nanostructures,292–294 metal–organic frameworks (MOFs),295 organo-halide perovskites,296,297 and nano-patterned metals298 at a resolution up to 50 nm. Here the improved resolution of the PTIR serves to analyze nanoscale regions of relatively thin samples. The amplitude of the PTIR signal (S, see eqn (9)) is dependent on the sample thickness (z), absorbed energy per unit area (Uabs), the thermal conductivity of the sample (η) and thermal expansion of the sample αexp (see eqn (9)). The PTIR signal was found to increase linearly with thickness of the sample up to a thickness of 1 μm. Therefore, a sample for PTIR needs to possess a significant thickness of 4–15 nm.46,289,299
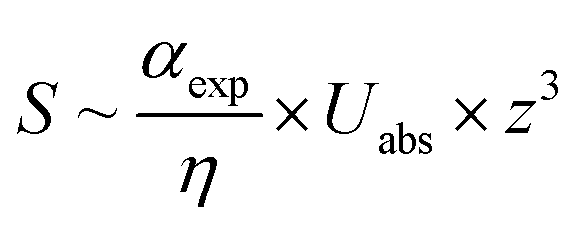 | (9) |
The PTIR method was enhanced by utilizing resonance enhancement in 2014.299 The improved method relies on the Q-fold amplification of the signal, by matching the laser repetition rate of a quantum cascade laser to the resonance frequency of the cantilever oscillation. The oscillating cantilever senses the sample expansion and the absorbed energy is transferred to the cantilever yielding in an amplification factor Q of approx. 100. This method is visualized in Fig. 11. Lu et al. utilized this specialized setup with a p-polarized pulsed mid-infrared laser to induce photo expansion of polyethylene glycol methyl ether thiol (PEG) islands adsorbed on a gold modified mica surface. By this means, the Q-factor was enhanced to around 93 and thus, the sensitivity was estimated to 30 molecules of PEG with a thickness of 2 nm. Via this method, a resolution of up to 25 nm was achieved (Fig. 11i) while even monolayers can be analyzed.299 Researchers have shown that polymers and self-assembled monolayers (SAMs) can be investigated via resonance enhanced-atomic force microscopy-infrared spectroscopy (RE-AFM-IR).299,300 Recently, the coupling of mode synthesizing atomic force microscopy (MS-AFM)301 with PTIR was proposed,281 vesicles inside of bacteria could be detected and analyzed in situ.281 This method can potentially be generalized to nanocontainers inside soft matter.
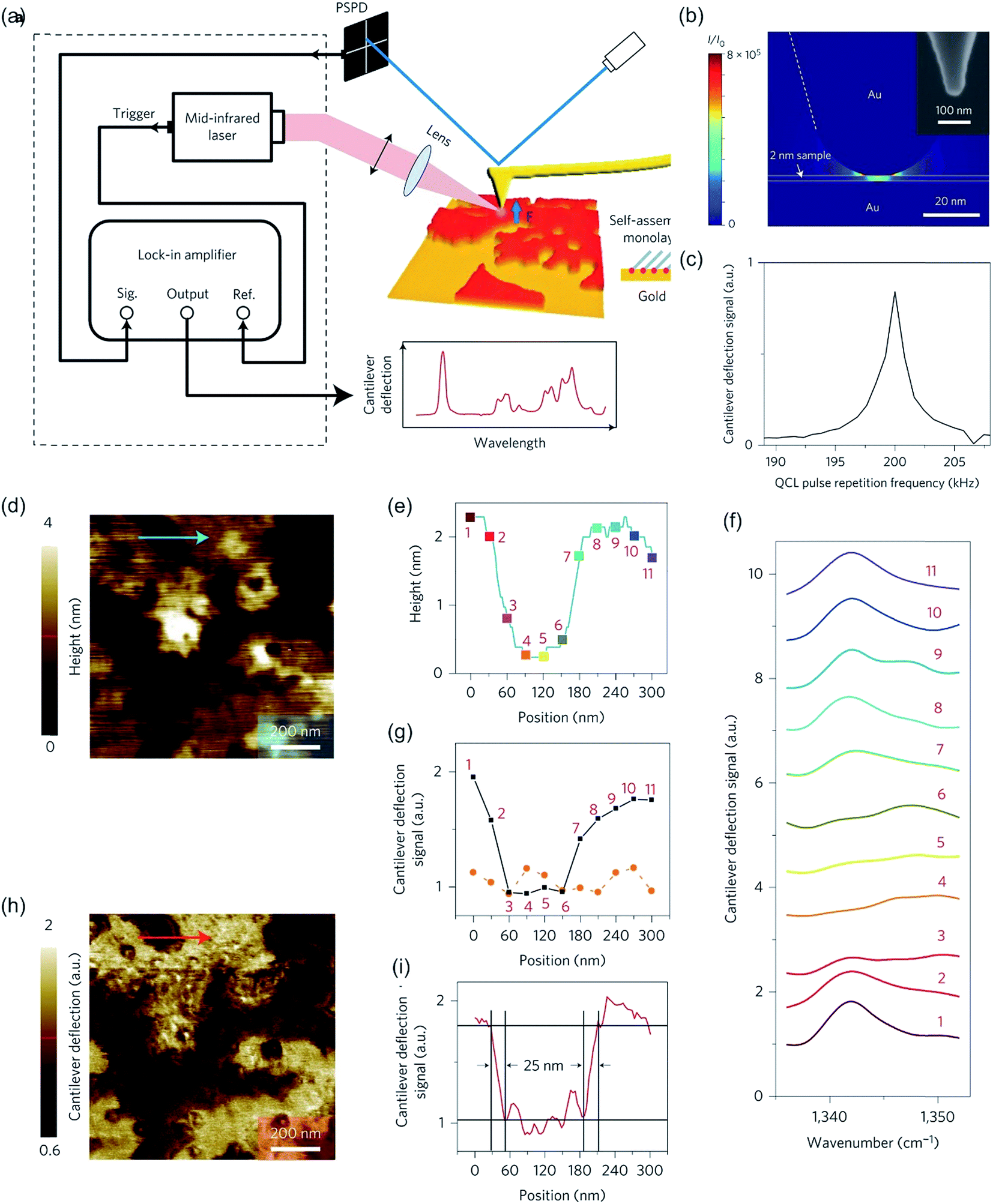 | ||
| Fig. 11 (a). Schematic of the experimental set-up of PTIR. (b) 3D simulation of the tip-enhancement of the light intensity (I/I0). For simulation, a molecular monolayer on gold with a thickness of 2 nm, a mid-infrared light incident angle of 75° to the surface normal and a geometry similar to the AFM tip was modeled with a radius of curvature of 25 nm and a half-cone angle of 17°. A SEM micrograph of the AFM tip is shown in the inset. (c) Experimental dependence of the lock-in output on the repetition frequency of quantum cascade laser pulses. (d) AFM topography image of PEG monolayer islands acquired in contact mode. (e) Line height profile along the blue arrow is shown in (d). (f) PTIR spectra taken at the positions indicated in (e). (g) Comparison of cantilever deflection signal between incident laser wavelength of 1342 cm−1 (PEG absorption line, black squares) and 1552 cm−1, no PEG absorption (orange circles). Data points were extracted from spectra in (f). (h) Mid-infrared mapping of monolayer islands at 1342 cm−1. Peg rich regions are bright, whereas at dark regions PEG is absent. (i) Signal along the line scan shown with a red arrow in (h), indicating a spatial resolution of 25 nm. Reprinted with permission from ref. 299. Copyright (2014) Macmillan Publishers Ltd. (Nature publishing group). | ||
Nowadays, PTIR is superseded by infrared photoinduced force microscopy (IR-PiFM) and recently, by peak force infrared microscopy (PFIR),302 both methods are related to PTIR. However, IR-PiFM is different from other spectroscopically sensitive force-detection technique as it can be operated in non-contact mode, generating high-resolution measurements in the sub-10 nm range303 while it can be conducted at ambient conditions without harmful contact between tip and sample, as shown in Fig. 12a.304,305 For example, Nowak et al. utilized this technique for chemical imaging of self-assembled block copolymer polystyrene-b-epoxydicyclopentadiene methacrylate (PS-b-PEDCPMA) patterns. Specifically, an incident mid-IR laser was employed to pulse at fm = f1 − f0, where f0 and f1 are the first and second mechanical eigenmode resonances of the cantilever. The topography of the sample was recorded by the AFM feedback system at f1, and the PiFM was concurrently recorded at f0 by the feedback laser position-sensitive detector and lock-in electronics. The sample was raster-scanned under the tip to generate the image. The incident light was polarized along the tip axis to maximize the signal coupling of the dipole–dipole force along the vertical direction of the cantilever vibration. High spatial resolution and chemical specificity were obtained by resolving and identifying each domain with ∼40 nm pitch (see Fig. 12b–i).304
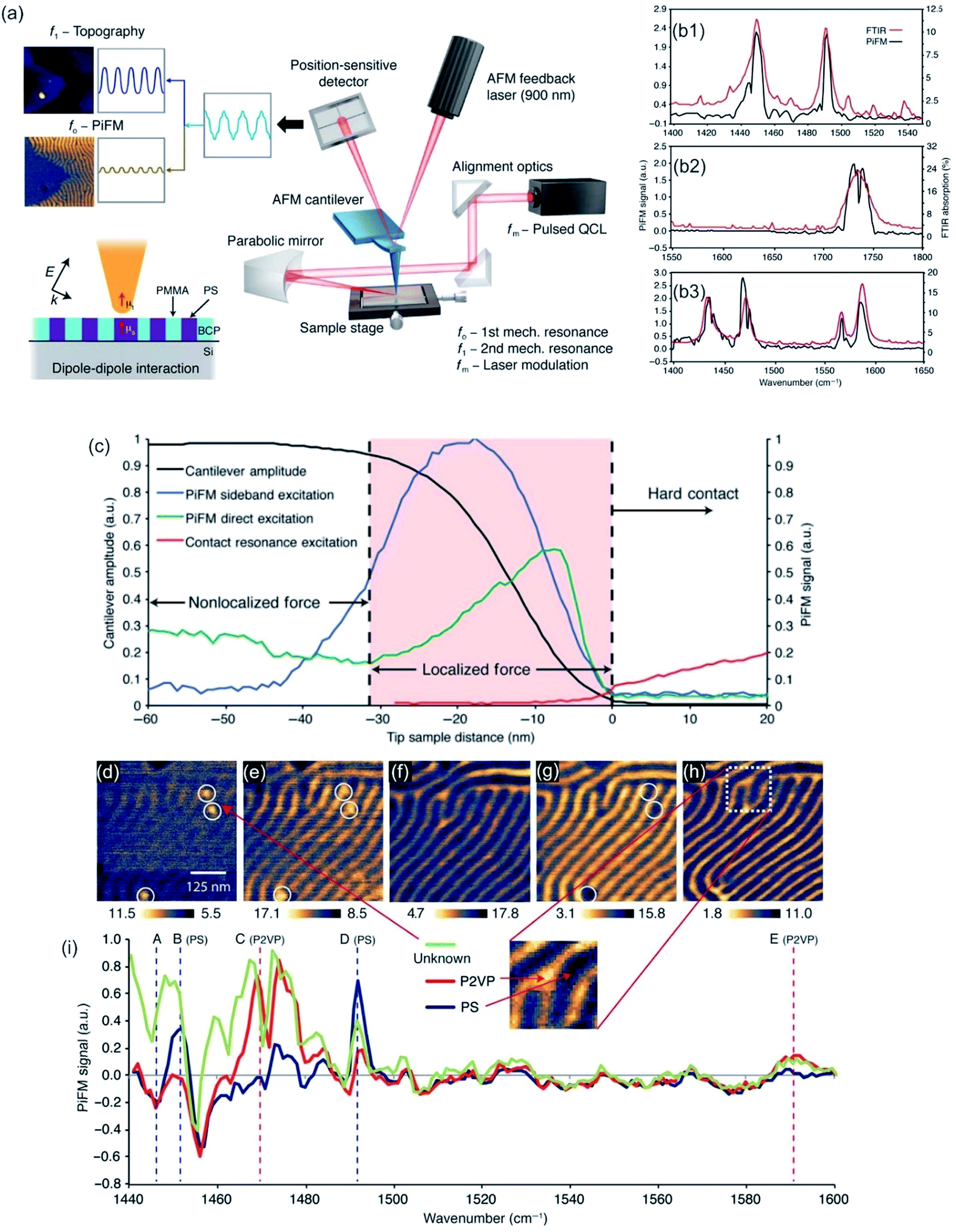 | ||
| Fig. 12 (a) Schematic of IR-PiFM experiment. (b) Comparison of PiFM and FTIR spectra of homopolymers polystyrene (PS, b1), polymethylmethacrylate (PMMA, b2), and poly(2-vinylpyridine) (P2VP, b3). (c) The PiF signal and cantilever amplitude as a function of tip-sample distance on 10 nm-thick PS-b-PEDCPMA brush prepared on Si and excited with 1733 cm−1. Fingerprint region of PS-b-P2VP. (d to h) PiFM imaging at (d) 1447 cm−1, (e) 1452 cm−1, (f) 1469 cm−1, (g) 1492 cm−1, and (h) 1589 cm−1. (i) Spectra were taken from point locations shown in (h), which were normalized against a Si background. Reprinted from open-access article ref. 304 under the terms of the Creative Commons Attribution License. | ||
Similarly, PFIR is capable of chemical imaging, a collection of infrared spectra, and mechanical mapping at a spatial resolution of 10 nm. An advantage of this technique is the possibility to gather on top of chemical and topographical data also mechanical data, such as adhesion or moduli.302 Wang et al. showed its appliance, for example for the nanoscale phase separation in block copolymers.302
2.7 AFM & ellipsometry - scanning near-field ellipsometry microscopy (SNEM)
Ellipsometry detects the changes of polarization of incident light while it is reflected from a surface. The polarization change is measured via the ellipsometry angles. These angles are related to parameters, such as thickness and the refractive index of the sample.306 The method is utilized to characterize material properties, such as composition,307 thickness (depth),308 crystalline nature,309 doping concentration,310 electrical properties311 and other material properties. AFM and ellipsometry were first combined in 2001, a scheme of such a combination is shown in Fig. 13a, to characterize concomitant optical (local refractive index) and topographical properties of thin polymer films.312 Via exploiting the electric field enhancement at a gold-coated AFM tip the signal to noise ratio (see Fig. 13b and c), as well as the resolution of the ellipsometry images, was enhanced by Tranchida et al., which was ascribed to lightning-rod effect and localized surface plasmons (see also TERS enhancement).313 The field enhancement was shown to be strongly distance dependent, denoting a significant effect at distances lower than 100 nm. They were able to visualize concomitant with the AFM and the ellipsometry nanoparticles embedded in poly(methyl methacrylate) and microphases in microphase-separated block copolymer films (vide Fig. 13c). For the enhancement effect, a tip with a gold surface is needed. Gold garners atmospheric contaminants and therefore a steady degradation in performance is observed in SNEM as well as TERS. Cumurcu et al. proposed a protective layer of ethanethiol (self-assembled monolayer), which yields consistent and reproducible results for scanning near-field ellipsometry microscopy for at least 5 days (storage at ambient conditions).314 The combined SNEM method was advanced by Cumurcu et al. to a concomitant tapping mode SNEM measurement, which was applied for microphase-separated morphology of polystyrol-block-poly(2-vinylpyridine) (PS-b-P2VP) block copolymer thin films, reducing the inflicted damage originating from contact of the tip with the surface.315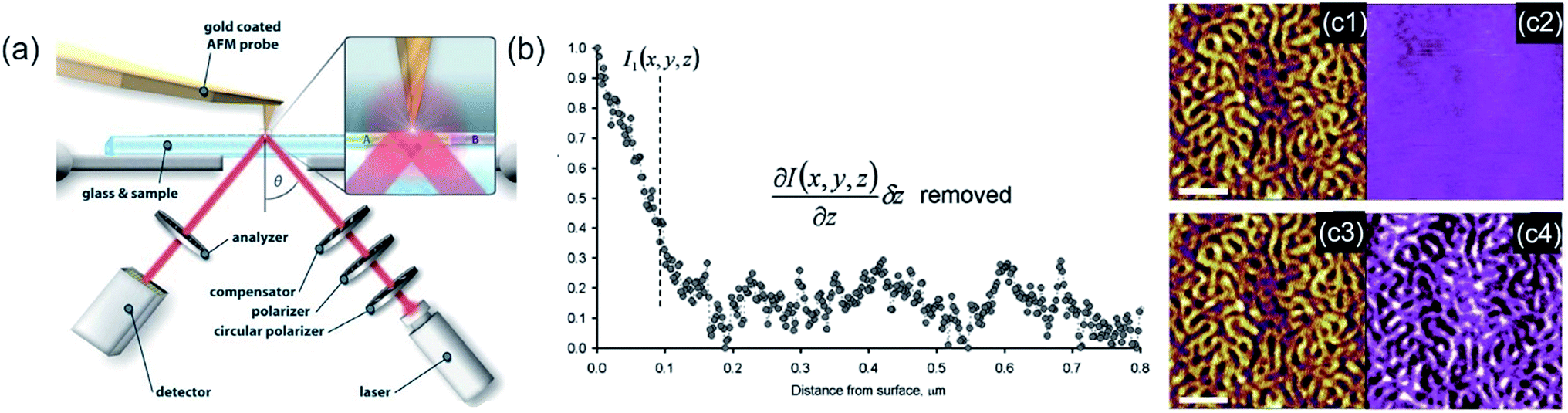 | ||
| Fig. 13 (a) Scheme of the experimental setup of a SNEM experiment. The angle of incidence is fixed to around θ = 40°. Reprinted with permission from,315 Copyright (2013) Elsevier. (b) Intensity vs. distance curve, obtained with a gold coated AFM tip during acquisition of a force–displacement curve on glass, corrected for the effect of background and interference. (c) SNEM data of micro-phase separated PS2092-b-PtBA1055 block copolymer film on glass: (c1, c3) AFM height image (the scale bar represents 500 nm), (c2) optical image acquired without AFM tip in proximity of sample (the z-scale covers 0–12 mV) and the corresponding tip-enhanced SNEM optical image (c4). (b, c) Reprinted with permission from ref. 313 Copyright (2011) the Royal Society of Chemistry. | ||
2.8 A combination of AFM with reflection interference contrast microscopy (RICM)
The method RICM was invented in the 1960s to study the interaction of cells with a glass substrate.316 It is an interferometric technique that allows the determination of the vertical distance and of the contact area between the interfaces.317 RICM is a label-free method (similar to IR, Raman, and ellipsometry) and can be implemented into a standard inverted microscope with little investment. A RICM setup comprises out of a monochromatic light source, an inverted microscope equipped with an anti-flex objective and a CCD camera. The light beam passes an aperture diaphragm (AD) and a field diaphragm (FD), which is positioned with respect to the sample and objective to obtain a Köhler illumination. This illumination method generates an extremely even illumination. Subsequently, the light is linear polarized, reflected by a semi-reflecting mirror and passed through a quarter wave plate, which renders the light circularly polarized. The light is transmitted and reflected by the glass substrate and the sample (i.e. a cell). The reflected light of different distances from the glass surface (i.e. membrane a few nanometers away from the glass surface) interfere with polarized light reflected directly at the glass surface, which leads to constructive or destructive interference. The interference pattern is subsequently recorded as a two-dimensional matrix of intensities known as Newton rings. Out of these rings, the distance between two surfaces can be calculated by eqn (10).
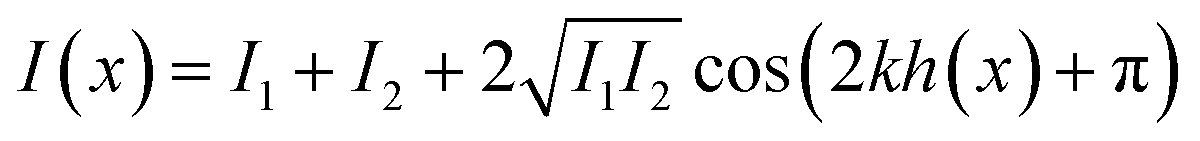 | (10) |
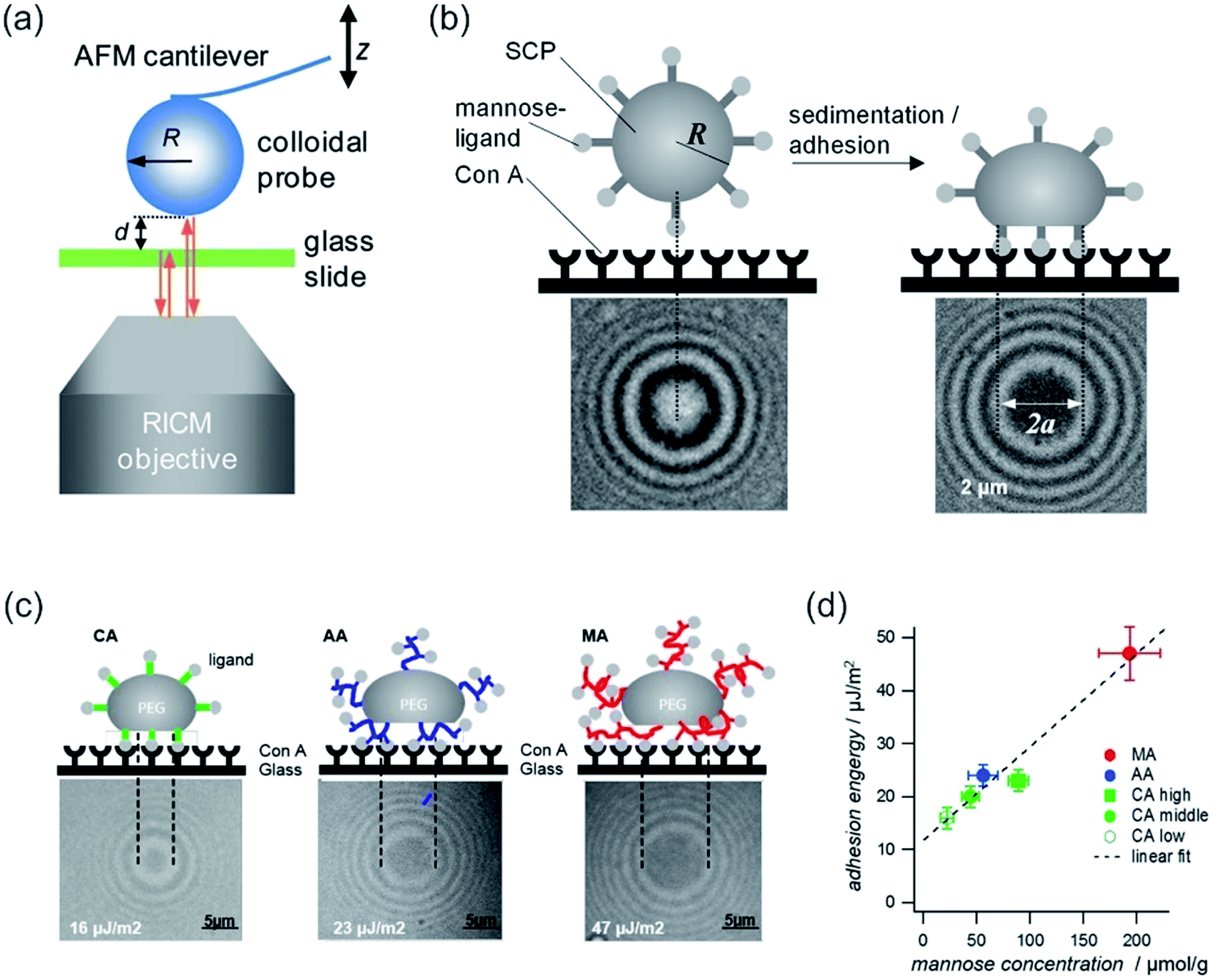 | ||
| Fig. 14 (a) Combined colloidal probe AFM/RICM setup. Reprinted with permission from ref. 320. Copyright (2012) American Chemical Society. (b) SCP adhesion measurement sketch (top): a mannose-functionalized PEG-SCP sediments onto a Concanavalin A (ConA) receptor functionalized surface (left). Binding of mannose units to ConA (right). Central circular interference minimum (bottom, denoted by 2a). (c) Scheme of SCP deformation of polyethylenglycol–crotonic acid (PEG–CA) particles, PEG–MA (MA = methacrylic acid) and PEG–AA (AA = acrylic acid). Below, the corresponding interference patterns are presented. (d) Plot of the adhesion energy vs. mannose concentrations for SCPs with grafting type from (c) and PEG–CA SCPs with varying density of grafting units. Reprinted from open-access article ref. 321 under the terms of the Creative Commons Attribution License. | ||
In a combination with AFM, RICM can be a powerful tool, as it can assess the contact area of an AFM-tip with the surface/polymer membrane. Therefore, it is a pivotal technique to study inter-surface interactions. The significance of the combination was boosted by advent of soft colloidal probes in AFM measurements,318 as by RICM the actual contact area is accessible,319 which further improved the sensitivity, for example for adhesion measurements.
Typically, the position of an interface can be detected easy and precise by an AFM. Hyaluronan brushes and other soft materials, i.e. hydrogels, possess in their swollen state a soft matter/water interface, which is difficult to locate by AFM. At this interface, an analysis of mechanical parameters (elasticity or viscosity) is difficult. Attili et al. utilized a combination of colloidal probe AFM and RICM to determine forces and concomitant the distances between a probe and an interface.320 The compressive mechanics of films of the polysaccharide hyaluronan that was end-grafted to a supported lipid bilayer via a biotin anchor was investigated. The compressive response of the polymer brushes was in good agreement with a theoretical model of polymer brushes. Furthermore, via RICM linear instrumental drifts of the AFM can be corrected, as well as cantilever sensitivity was obtained without additional reference measurements.320
Instead of compression, adhesion of a soft colloidal probe (SCP) was investigated by Schmidt et al.321 The SCP was covered with a mannose ligand, which binds under contact to a surface covered with Concanavalin A (ConA) receptors (see Fig. 14b and c). The interaction energy between the SCP and the protein surface could be evaluated as the surface area is accessible via RICM by analysis of the extent of the central circular interference minimum. A linear relationship between the mannose density and adhesion energy was found (vide Fig. 14d), excluding affinity differences between mannose units. Therefore, multivalent binding types, such as chelate and subsite, were ruled out.321
2.9 AFM coupled to surface plasmon resonance (SPR)
With surface plasmon resonance (SPR) adsorption of molecules onto metal interfaces can be determined.322 SPR is a label-free method,323 possesses a high sensitivity,324 is able to monitor changes in adsorption in real-time,324 consumes low sample volumes325 and is a quantitative measurement. Therefore, determination of quantitative kinetic rate constants is feasible (i.e. adsorption or desorption rate constants).326,327 In SPR the Kretschmann configuration328 is often utilized, which is shown in Fig. 15a. The Kretschmann configuration comprises of a thin metal film (silver or gold) at the interface of two dielectric media. A laser beam is focused onto a prism with refractive index (n1). The light is, dependent on the angle of incident, either reflected or adsorbed (resonance angle). The resonance angle changes upon the refractive index of the solution of interest, for example by adsorption. For the measurement, the solution of interest needs to possess a lower refractive index (n2). The surface plasmon resonance angle shift, which is originating from the change in refractive index, is utilized to determine the quantitative adsorption of molecules. SPR and AFM were already combined in 1995 by Chen et al. and Shakesheff et al.329,330 As the two systems are geometric complementary (AFM measured from the top of the sample, whereas SPR measures from bottom) a combination of these two methods is fairly easy. The time resolution of the SPR together with the high sensitivity can substitute in a combined AFM-SPR measurement the lack of time resolution (for most AFM equipment) of the AFM. Early studies have analyzed the degradation of biodegradable polymer films on silver. The combination can make use of synergy of both capabilities, as AFM can determine structural changes of the polymer films during the degradation, whereas SPR determines the thinning of the polymer film, as shown in Fig. 15b and c.329 For conjugated polymer electropolymerization, a combined in situ AFM-SPR-electrochemical analysis yields in surface morphological properties and domain growth of the film by AFM, whereas via SPR & electrochemical analysis the electrochemical behavior depending on the polymer is accessible.331 Similarly, both techniques have been employed for analysis of mucoadhesion of pectins332 and for real-time measurements of changes in the mammalian membrane mimics structure induced by antimicrobial peptides.333 For the latter, a detailed model for antimicrobial peptide action as a function of the degree of bilayer disruption was proposed by a comparative AFM, SPR, and polarization interferometry (DPI) analysis.333 | ||
| Fig. 15 (a) Instrumental configuration for AFM + SPR. Reprinted with permission from ref. 52 Copyright (2009) Royal Society of Chemistry. (b) Time dependence of SPR angle shift during degradation of a poly(orthoester) (POE) film exposed to flowing (1 ml s−1) acidic solution (pH 3.5). The arrows indicate the acquisition of corresponding scanning force microscopy (SFM) scans (c) at the time t = 0 min (c-a), t = 15 min (c-b), t = 29 min (c-c), t = 39 min (c-d), t = 44 min (c-e), and t = 49 min (c-f). (b, c) Reprinted with permission from ref. 334 Copyright (1996) AIP Publishing LLC. | ||
3. Combination of AFM with non-optical surface sensitive methods
3.1 Combination of AFM with Kelvin method – Kelvin probe force microscopy (KPFM)
By rendering the AFM tip and the sample conductive, the work function or surface potential can be obtained with a high lateral resolution, which can be lower than 10 nm.335 The method is commonly denoted as Kelvin probe force microscopy (KPFM) and was first introduced in 1991 by Nonnenmacher et al.336 KPFM measures the contact potential difference (CPD) of a conductive AFM tip and a conductive sample. CPD is defined by the work functions of the tip (ΦTip) and the sample (ΦSample) and the elementary charge (e).
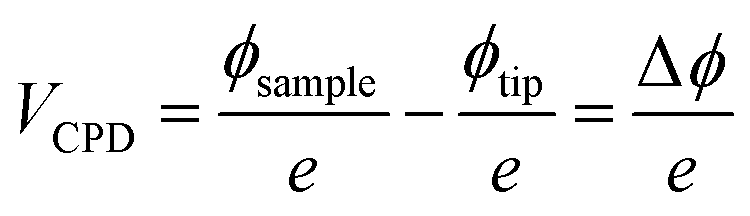 | (11) |
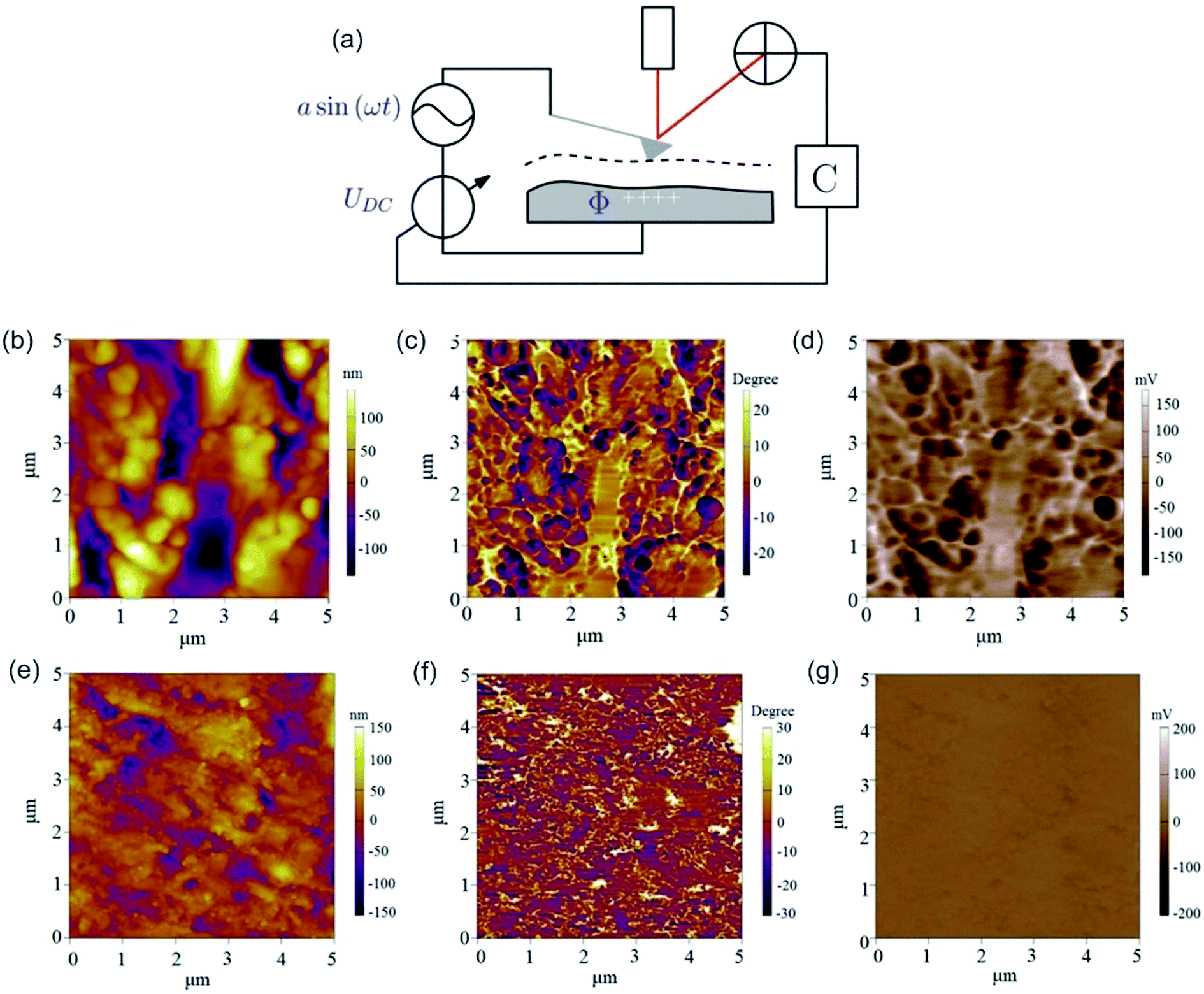
By applying both, an AC (VAC also UAC) and a DC (VDC also UDC) voltage to the AFM tip, the work function of the sample is assessed. Here, the DC voltage repeals the CPD difference between tip and sample surface, which is oscillated due to the applied AC voltage. The cantilever oscillates at a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) sin(ωt) (sinusoidal excitation signal). a denotes for the amplitude and ω for the frequency of the sinusoidal electrical signal. The feedback controller C compensates for the surface potential Φ indicated by + + + + by adjusting a DC (bias) voltage UDC, as shown in Fig. 16a.337 As a consequence of these applied signals, the resulting force has three contributions, two oscillatory terms ω and 2ω and a bias, which contributes to the topographical signal. The contact potential difference can be obtained by a lock-in technique by tuning the bias voltage UDC to a value, which nulls the oscillating force signal (ω). The contact potential difference is this needed bias voltage.
sin(ωt) (sinusoidal excitation signal). a denotes for the amplitude and ω for the frequency of the sinusoidal electrical signal. The feedback controller C compensates for the surface potential Φ indicated by + + + + by adjusting a DC (bias) voltage UDC, as shown in Fig. 16a.337 As a consequence of these applied signals, the resulting force has three contributions, two oscillatory terms ω and 2ω and a bias, which contributes to the topographical signal. The contact potential difference can be obtained by a lock-in technique by tuning the bias voltage UDC to a value, which nulls the oscillating force signal (ω). The contact potential difference is this needed bias voltage.
 | ||
| Fig. 16 (a) Schematic illustration of the Kelvin probe force microscopy setup. Reprinted from open-access article ref. 337 under the terms of the Creative Commons Attribution License. (b–d) AFM and KPFM of Al-6061 cv 3%. (b) AFM height, (c) AFM phase and (d) KPFM potential mapping of Al-6061 covetic 3% sample. V can be computed by inverting and scaling the signal by one half (V = −2b) (e-g) AFM and KPFM of pure Al-6061. (e) AFM height (f) AFM phase and (g) KPFM potential mapping of a pure Al-6061 sample of the same region. Reprinted with permission from ref. 338 Copyright (2016) Elsevier. | ||
The surface potential can be determined by at least 4 methods. Here, the KPFM is the most versatile and possesses distinct advantages over standard Kelvin probe (KP), photoemission spectroscopy (PES) and scanning electron microscopy (SEM). The measurement principle of KP and KPFM is similar. The energy resolution of KP method is slightly better compared to KPFM, but KP method relies on averaging over the whole sample, indicating no or low lateral resolution is obtainable. Via PES both, lateral resolution of approximately 100 nm and 20 meV energy resolution can be obtained.339 However, KPFM possesses enhanced lateral and energy resolution. However, for a sample surface containing adsorbents, KPFM cannot distinguish between surface band bending and surface dipoles created by adsorbents from the sample surface.340 PES yields in the spectral distribution of the surface potential, which can be utilized in a comparative approach to determine band bending and surface dipole contributions, independently.340 A qualitative measure of the surface potential can be obtained with a lateral resolution of approximately 70 nm by SEM via electron beam induced current.341 KPFM has an energy resolution of 5–20 meV and a lateral resolution of 10 nm or better,335 depending on the utilized measurement mode.
Typically, a KPFM experiment is conducted in either an amplitude modulation mode (AM) or frequency modulation mode (FM). Here, FM-KPFM is considered the method with higher lateral resolution (up to sub-nanometer), whereas AM-KPFM possesses a higher energy resolution of up to 5 meV. The enhanced VCPD precision can be related to a higher signal-to-noise ratio originating from the resonance peak of the cantilever, whereas in FM mode the VCPD is determined by FM demodulator, which increases the noise in a measurement.342
Ongoing research is devoted to enhancing the time resolution, because the scan speed of FM mode is slow343 and AM mode measurements are burdened with artifacts, such as the stray capacitance effect344 and interfering signals, leading to topographical coupling.345,346 Several approaches try to couple the lateral resolution and reproducibility of FM mode KPFM with enhanced time resolution similar to AM mode KPFM, such as time-resolved electrostatic force microscopy, pump-probe KPFM,347 general acquisition mode,348 open loop (OL) KPFM349 and Heterodyne (H) KPFM.350 For example, H-KPFM improved the scan rate while maintaining the spatial resolution and voltage sensitivity comparable to FM-KPFM.59 Furthermore, it is not susceptible to artifacts such as stray capacitance (AM mode), topographical oscillation (FM mode) and AC inductive coupling (AM mode) artifacts.59,346 However, artifacts originating from collisions with the surface persist.
KPFM is applied for characterization of metal351 and semiconductor surfaces,352 organic films353 and biological membranes.354 Surface potential mapping is utilized for distinguishing carbon from the alloying elements, such as Al, Zn and Mg, as the surface potential difference is relatively small for carbon (similar work function as the tip (Pt)), as is illustrated in Fig. 16b–e.338 In contrast, the potential difference between the metals and tip are high.338 For example, Jaim et al. utilized KPFM to analyze the difference between Al-6061 cv 3% and Al-6061 and substantiated the presence and the ribbon networks of carbon embedded in the Al-matrix of Al-6061 cv 3%. They observed a correlation of the dark regions in the AFM phase image (Fig. 16b) and KPFM image (Fig. 16c), which was ascribed to carbon as it possesses a lower work function compared to the Al-matrix surface potential. Consequently, Al-6061 does not show this correlation between AFM phase and KPFM image, which appears to possess a rather homogeneous CPD. Another common area of application of KPFM is the analysis of semiconductors.352,355,356 Here, the KPFM method is sensitive enough to determine the dopant concentration in the range of 1014 cm−3 to 1019 cm−3.356 However, the contact potential difference is altered by surface defects,357 exploitable for detecting of defects.358 By illumination with a sub-bandgap laser, band bending is reduced and higher contrast in CPD in conjunction with reduced influence on surface defect states is achieved.356 KPFM can be applied for Langmuir–Blodgett films of organic molecules on water (subphase) to assess the packing of the organic molecules at varying surface pressure by means of the CPD.353 Furthermore, KPFM can be utilized to ascertain charge transfer from Pt adsorbates to TiO2 surface359 and analysis of charge separation for the benchmark of organic solar cells, which for example consist of poly(3-hexylthiophene) (P3HT)/fullerene (C60) acid-methyl ester (PCBM).155 An interesting application, which also shows the high lateral resolution and sensitivity, is the analysis of quantum size effects (QSE) proposed by Späth et al. In contrast to other techniques, KPFM was able to directly address the single digit nanometer height of single Pb islands on Si while simultaneously assessing the local work function (see Fig. 17).360 Yamagishi et al. utilized KPFM to visualize trapped charges in organic thin-film transistor (OTFT) channels.361 KPFM was utilized in the biological area to examine the effects of surface potential on microbial adhesion on various metals. The surface potential of the microbial was dependent on adsorbents and was linked with changes in cellular metabolism and motility.60
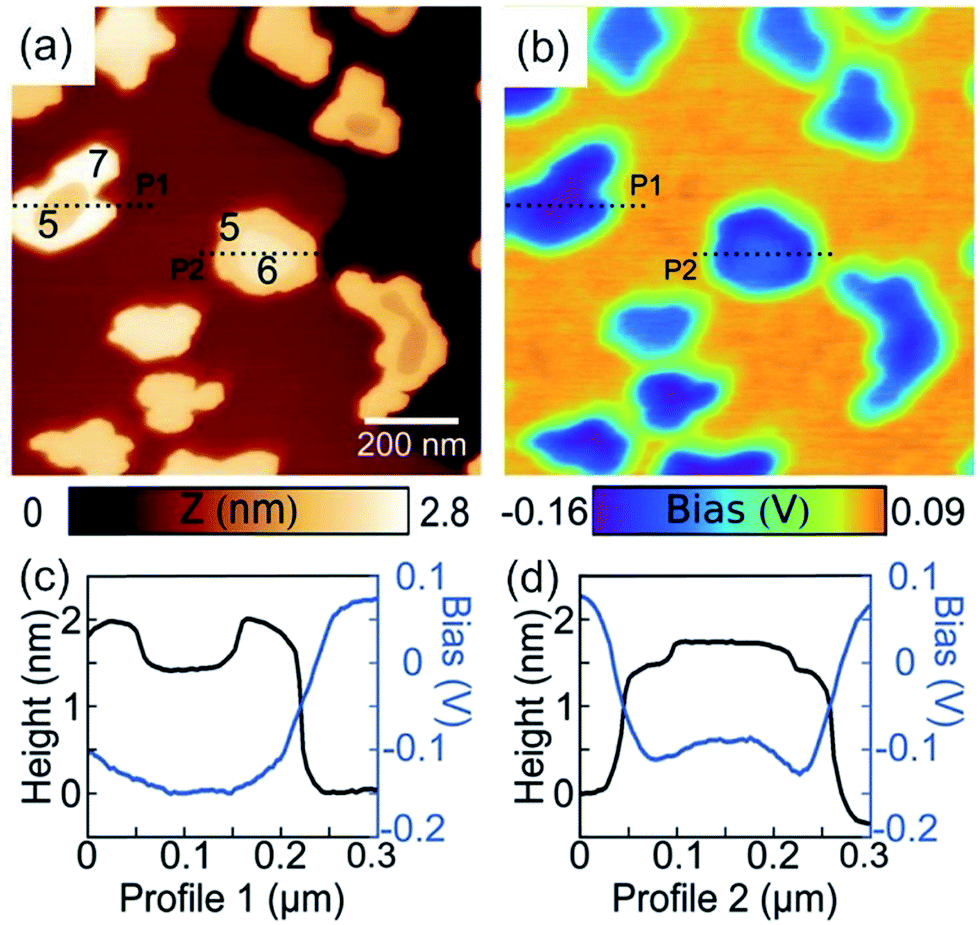 | ||
| Fig. 17 SFM topographic (a) and simultaneously obtained Kelvin images (b) of ultra-thin Pb islands on Si(111). The local height in atomic layers measured from the wetting layer is indicated by the numbers in the images. (c, d) Profiles over Pb islands along the dashed lines marked in (a, b). Reprinted with permission from ref. 360 Copyright (2017) Royal Society of Chemistry. | ||
3.2 AFM combined with quartz crystal microbalance (QCM)
QCM is a method to detect and quantify change of the mass by a shift in the frequency of vibration of a piezoelectric device.362 The frequency shift (ΔF, eqn (12)) of the resonator is dependent on the fundamental frequency of quartz resonator (F0), change of mass on the surface of quartz resonator (Δm) and the vibrating surface area (s).
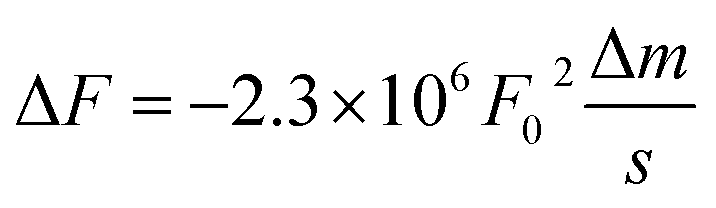 | (12) |
In addition to the detection of mass change, it may provide information about the conformation of molecules adsorbed to the surface by damping of the oscillation (amplitude). The latter information is often hard to come by, as changes in shape cannot directly be related to a QCM response. On the other hand, it is feasible to analyze changes in shape by AFM. Therefore, it is conducive for analysis of QCM data. For a combined measurement, the oscillation amplitude has to be lower than the desired resolution of the AFM experiment. The first combination was reported in 1998 by Iwata et al. and was used to evaluate the morphology and mass change at the liquid/solid interface during the electrodeposition of Ag on a Pt thin film.363 For electrochemistry purposes, a specialized three-electrode cell was utilized, comprising of an Au coated quartz crystal working electrode (WE), Ag wire reference electrode (RE), and Zn foil counter electrode (CE) (see Fig. 18a). Such a setup was successful in performing in situ electrochemistry, as shown in Fig. 18b–d.363–365 For example, Smith et al. were able to confirm that the growth mechanism of electrodeposited Zn resembles a 3D progressive model.365 Thus, this hybrid method imparts a realistic prospect of understanding of electrochemical nucleation and growth, which may be transferable to other issues outside electrochemistry. Most recently, Kelvin probe microscopy was annexed to an AFM-QCM combination to improve the versatility.366 This approach was utilized to measure the response of tin oxide-based sensors and determine during adsorption of analyte in situ the work function of the sensor nanomaterial (i.e. SnO2, Pd-doped SnO2, and bromocresol purple-doped HfO2).366
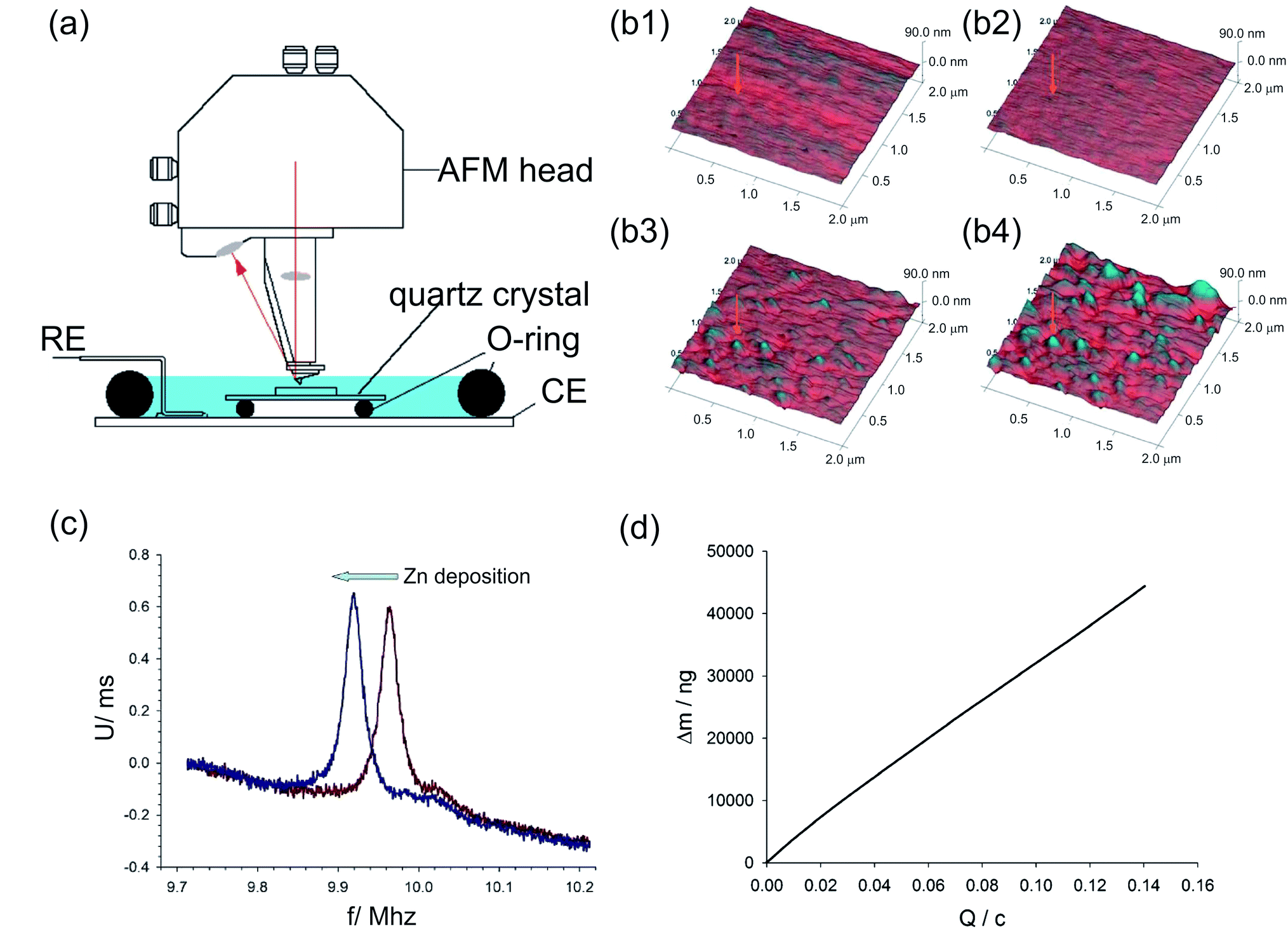 | ||
| Fig. 18 (a) Schematic of a custom-built AFM-QCM hybrid especially designed for electrochemistry, also called electrochemical QCM. (b) Tapping mode, AFM images recorded in liquid at a concentration of 0.3 M ZnCl2 in ethaline of an Au coated resonating quartz crystal under electrochemical control (−1.1 V versus Ag wire). Height images show Zn deposition at times (b1)t = 0 s (bare Au), (b2)t = 120 s, (b3)t = 240 s and (b4)t = 360 s. (c) Acoustic impedance plots recorded for the quartz crystal before (red trace) and after (blue trace) Zn deposition. Raw data with no baseline correction. (d) Deposited mass, Δm, versus passed charge (Q), acquired simultaneously with AFM images during deposition. Reprinted with permission from ref. 365 Copyright (2009) American Chemical Society. | ||
3.3 AFM combined with thermal imaging – scanning thermal microscopy (SThM)
Scanning thermal microscopy (SThM) maps the local temperature and thermal conductivity of interfaces. SThM allows thermal measurements at the nano-scale. Besides temperature and energy transport, processes involving the exchange of energy and entropy with the surroundings are feasible, such as thermal properties of materials,367 thermal conductivity,50 heat capacity, glass transition temperature,368 latent heat, enthalpy, etc.In 1986, shortly after the scanning tunneling microscope was invented, Williams and Wickramasinghe reported a nanoscale measurement exploiting thermal phenomena.369 Several scanning thermal microscopes were established, which are mainly based on atomic force microscopy. Here, the combined system with the AFM diversifies the applicability, for example by performing measurements as a function of the tip-sample force and distance370,371 or by varying sensor types and geometries. The working principle of SThM is dependent on the utilized thermal method. The methods can be classified according to the temperature-dependent mechanism into either (a) thermovoltage,372–374 (b) change in electrical resistance,375–377 (c) fluorescence378 or (d) thermal expansion.379,380 However, these methods are still under rapid development and other techniques have been proposed, for example a method based on simultaneous registration of the static and the dynamic electrical resistance of the probe driven by the sum of DC and AC currents, which is suitable for thin films deposited on thick substrates.381
(a) Thermovoltage based measurements can be conducted either in contact or in non-contact mode. Crucial, but also limiting its applicability is the need for an electrically conducting surface of the sample. The working principle of thermovoltage methods can be tunneling thermometry382 or the point-contact thermocouple method.383 As probes in thermovoltage-based methods, thermocouples (see Fig. 19c)374,384 and Schottky diode are utilized.385,386 Accessible via these methods are the temperature and the thermal conductivity387,388 at a minuscule surface area. The lateral resolution utilizing a thermovoltage method is in the nm range and both the lateral resolution and probe thermal time constant of an experiment was substantial enhanced by miniaturization of the cantilever, the tip and the junction at the tip.374,384 Thermovoltage methods are per se not quantitative due to the dependence between temperature rise measured locally on the size of the heated area (heat transfer also via gas), as shown in Fig. 19b. Vacuum conditions (below 0.1 Pa) with an active thermal feedback scheme allows maintaining the tip temperature equal to the sample surface temperature.389 Nevertheless, null-point SThM (NP-SThM)390 and the previously mentioned ultra-high vacuum method circumvent this limitation, and outstanding thermal (∼15 mK) and spatial (∼10 nm) resolutions were obtained (see also Fig. 19d–f).61 Thermal conductivity is probed via the two omega (2ω) method.387,388 To this end, an AC current at a frequency ω is applied to the thermoelectric probe. The current passing from the SThM probe to the sample surface via the thermocouple junction generates heat at a frequency of 2ω, which leads to an oscillation of the junction temperature at 2ω and the amplitude of the 2ω signal from the thermocouple junction is monitored.387 Recently, a 2ω method was proposed utilizing a micro-thermocouple probe placed on a Quartz Tuning Fork (QTF), which circumvents the imprecise surface detection by optical deflection methods.391 Nevertheless, some artifacts persist during the measurement, for example Kim et al. observed a different temperature profile during the measurement of a heated Pt line in slow and fast scan mode, which was assigned to differences in contact area between tip and sample, resulting in a change in thermal resistance between the tip and the sample, as shown in Fig. 19e and f.61
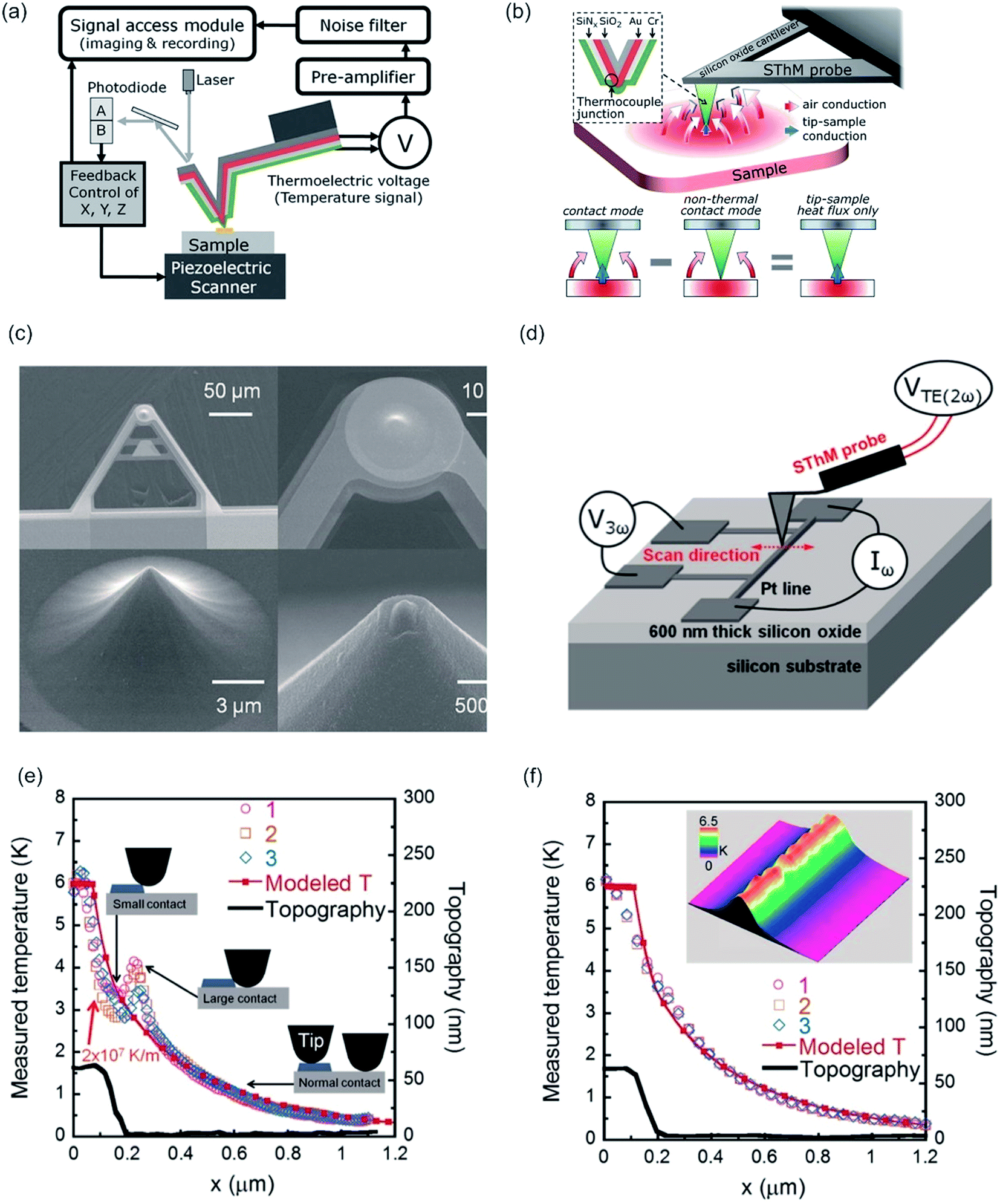 | ||
| Fig. 19 (a) Experimental setup of SThM and (b) principle of quantitative thermal profiling. The SThM probe with a nano thermocouple junction integrated at the end of the tip, as shown in the inset, is mounted on an AFM. Below a scheme illustrating both contact mode and nonthermal contact mode is shown. (a, b) Reprinted with permission from ref. 390 Copyright (2011) American Chemical Society. (c) SEM micrographs of the SThM probe with an integrated AuCr thermocouple. The AuCr thermocouple junction is at the apex of the tip (d ∼ 100 nm) (d) setup used to demonstrate quantitative thermal measurements. The temperature and topography of a heated Pt line are concomitantly mapped by scanning in a direction perpendicular to the Pt line. (e) Temperature profile obtained during slow scan, along with the modeled temperature profiles and the topographical profile of the Pt line. The inset depicts the contact conditions between the probe and the Pt line during the scan. (f) Temperature profile and topography obtained during fast scan. The inset depicts a four-dimensional map of the temperature field (color) and topography. The scan size was 200 × 200 nm2. (c–f) Reprinted with permission from ref. 61 Copyright (2012) American Chemical Society. | ||
(b) The change in electrical resistance can be utilized to determine the temperature at a small region on a sample. A heater with frequency ω heats the sample at 2ω frequency, similar to the 2ω method. The temperature oscillation at a frequency of 2ω results in an oscillation of the resistance of the second probe, which is thermal sensor, at 2ω.
(c) The fluorescence intensity depends strongly on the temperature, as the emitted intensity Iem is proportional to the population of excited states. By monitoring the fluorescence intensity at a given frequency, the temperature change can be derived from the change in fluorescence intensity. To enhance this approach, Aigouy et al. proposed gluing a fluorescent particle to the tip apex, which is afterward utilized for the local temperature measurement.378
(d) Thermal expansion (thermoacoustic effect) can be utilized to measure the increase of temperature as thermal expansion coefficient is usually in a range close to 10−5 m K−1 and the dilation can be measured accurately via AFM. The sample can be either heated by Joule heating392 or optically,393 similar to AFM-IR.289
Several approaches to enhance the lateral and thermal resolution have been investigated. In the beginning, in SThM a Wollaston wire probe with a temperature coefficient a = 0.00166 K−1 and time response of approximately 200 ms (in air) was utilized, which limits the lateral resolution of an experiment.394,395 Smaller metallic probes attached to cantilevers with a low spring constant (0.35 N m−1) and a tip radius of around 50 nm improved the lateral resolution.396,397 Furthermore, this probe enhanced the time response to a few tens of ms while maintaining a similar temperature coefficient as the Wollaston wire.398 Recently, high thermal conductivity nanowires (NW) attached to a tip apex were proposed to address the low efficiency of thermal coupling in SThM approach, while maintaining the lateral resolution.399 It has to be mentioned that the tip-sample heat transfer is crucial for the lateral resolution. Menges et al. developed a method, probing a time-dependent and a time-averaged heat flux signal between a self-heated scanning probe sensor and a temperature-modulated sample, permitting the elimination of tip-sample contact-related artifacts, a major step of SThM towards nanoscale thermometry.400
As stated previously, SThM can measure temperature (passive mode), thermal conductivity (active mode) and local phase–change transition (active mode) of a sample. The measurement of temperature is exploited for point studies and mappings of the amplitude of the steady periodic temperature field. It was utilized for the characterization of the temperature profile measurements of a PN thermoelectric couple401 and hot-spots in integrated circuits.402 Similar to KPFM, SThM in thermal mode appears useful for failure localization and analysis of integrated circuits.403,404 By the aforementioned smaller probes, heat dissipation, thermal conductivity or transport pathways in multiwall carbon nanotubes (MWCNs), single wall carbon nanotubes (SWCNs), graphene oxide (see Fig. 20a and b)405 and various other nanocomponents was determined.406 Furthermore, Joule self-heated nanomaterials, such as graphene nanoribbons (GNRs),407 silicon nanowire, nanowire diode408 and nanoscale metal interconnect structures (see Fig. 20c–h),400 and photoinduced heating409 were analyzed via this method. The thermal conductivity of materials was probed at the level of minuscule subsurface volumes for bulk ZnO and simple binary intermetallic clathrates (Ba8Si46), porous bulks,410–413 mesoporous bulks414 and thin films of submicrometric thickness.415 Recently, SThM was utilized to determine the penetration depth of an adhesive in wood (subsurface imaging).47 Via the measurement of the heat conductivity, the phase transition between martensite-austenite of NiTi microstructures was accessible.416 Furthermore, the glass transition Tg and melting temperature Tm of polymers or polymer blends at submicron scale is accessible (method is also named nano-TA).368
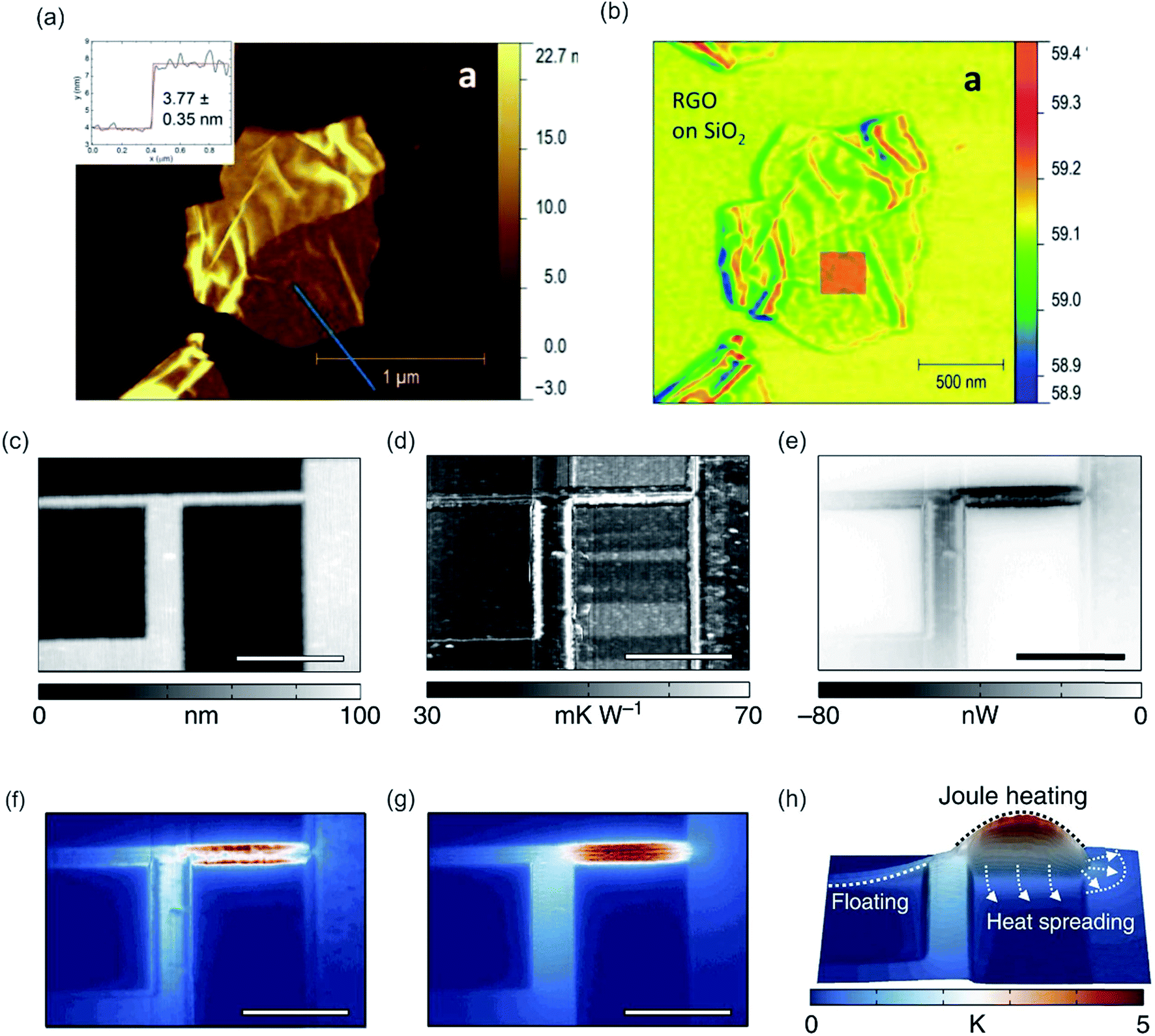 | ||
| Fig. 20 Topography (a) and SThM (b) maps of RGO nanoplates deposited on a Si/SiO2. Reprinted with permission from ref. 405 Copyright (2016) Elsevier. A bipolar voltage bias is applied to the electrical interconnect shown in (c) as height image (scale bar, 1 mm). Joule heating of the structure is characterized by simultaneously mapping local variations in (d) the tip-sample thermal resistance and (e) the tip-sample heat flux. (f) Artefacts arising from constant, position-independent (x, y) tip-sample thermal resistance (color bar see h) (g) calculated true sample temperature incline by combining position-dependent thermal resistance variations with the local heat flux maps. (h) Relative temperature increase encoded in both height and color, as well as heat spreading into the SiNx/Si substrate and the electrically floating interconnect segment. Reprinted from open-access article ref. 400 under the terms of the Creative Commons Attribution License. | ||
4. Conclusion and future aspects
The combination of several orthogonal techniques is a fertile research field. Especially, hybrid analysis tools comprising out of two complementary techniques, which can compensate for their intrinsic weaknesses, are employed and refined while novel approaches are investigated in recent years. All these attempts to design hybrid analysis tools with nanometer resolution are driven by the search for advanced means to analyze and characterize new nanomaterials with high sensitivity and spatial resolution as well as to analyze processes occurring at the nanoscale, be it transport processes in biological samples or electronic processes at metal interfaces. Therefore, a strong emphasis was on combinations of optical methods, which are impeded by its low resolution, and AFM. Here, tremendous effort has been devoted to the improvement of resolution, signal-to-noise ratio and alignment/drift of the AFM. Furthermore, novel methods, such as SNEM and PTIR were actively developed, which yielded in the development of IR-PiFM, a noninvasive method of combined AFM and IR measurement with an unprecedentedly high optical resolution when compared with other IR measurement techniques. Still, some issues remain unsolved, for instance, the lifetime of the tip-apex in SNEM, PTIR and TERS is short, which results in a loss of signal after a few hours. Nonetheless, these tip-enhanced methods are invaluable nowadays as they provide chemical fingerprints or chemical structures at sub-diffraction resolution. Combinations out of AFM and an optical microscopy can be advanced by adding further measurement techniques, such as fluorescence spectroscopy, fluorescence anisotropy and fluorescence lifetime. Although commercial solutions, at least for FLIM in combination with AFM are present, their alignment and computerized control still lack. These combined methods based on optical measurement techniques were mainly but not exclusively applied to biological samples (e.g. lipids, proteins, cells, viruses, particles, or biofilms), although these techniques also possess applications in material science and surface science (see surface nanobubbles).In contrast, the non-optical techniques were predominantly utilized in applications related to material science and engineering (e.g. semiconductors and nanomaterials). Surface analysis techniques, relying on non-optical techniques, have emerged and effort has been devoted to these surface analysis techniques (KPFM and SThM) to improve their lateral resolution as well as their time resolution and precision. Still, resolution of these techniques is lower than for AFM on its own. Here, a variation of tip/tip-apex geometry and tip material has shown to enhance these parameters. Further advancement would benefit material science and engineering, as these methods are utilized to examine the properties of semiconductors for computers, which are presently undergoing miniaturization.
Some techniques, like QCM, SPR, FCS and RICM, are rarely combined with the AFM, which can be related to different acquisition times, issues with alignment and/or lack of data interpretation. However, these hybrid methods potentially give insight into processes appearing at interfaces at a molecular level. For example, despite the scarce application of RICM in literature, it has the potential to become a vital technique for determination of the actual contact area, which is important for adhesion measurements of soft colloidal probes.
In spite of the fact that most of the combined techniques are still under development and improvement, and equipment optimization is pending, hybrid techniques are highly important in the areas of biology, surface science (physical chemistry) and material science. Subsequently, Table 1 shows a comprehensive list of all major combined techniques mentioned in this review, together with their typical resolution, the field of application, advantages, and drawbacks, serving as a decision guidance for the interested reader. We anticipate that this review will provide a comprehensive overview of the hybrid measurement techniques based on the combination of surface sensitive techniques with the atomic force microscope. Besides, it will impart knowledge of the field of application of a specific combination in conjunction with its intrinsic advantages and limitations and thus serve as a viable tool for deciding on the most beneficial measurement method.
| Resolution | Samples | Measurand | Advantages | disadvantages | |
|---|---|---|---|---|---|
| a For thermovoltage measurements.b Resolution of the complementary technique, resolution of AFM remains sub nm (lateral) and sub Angström (axial).c Tip enhancement effects may apply, pre. = predominant, fl. = fluorescence, ident. = identification NC = noncontact. | |||||
| SNOM, s-SNOM | Lateral ≈ 30 nm, axial ≈ 10 nm | Membranes, carbon nanotubes, polymers, biological samples | Refractive index, reflectivity, transparency, polarization, fluorescence… | High resolution, high sensitivity (single molecule) | Low working distance, scan time, analysis difficult, artifacts |
| AFM-fl. microscopy | Lateral ≈ see CLSMb,c, axial ≈ 2000 nmb,c | Pre. biological | Intensity, location, movement | Very fast acquisition time | Low axial resolution, label required |
| AFM-TIRF | Lateral ≈ see CLSMb,c, axial ≈ 20–100 nmb,c | Pre. biological | Intensity, location, movement | Very fast acquisition time, high sensitivity, selective excitation at interface | Limited to the interface (around 100 nm), label required |
| AFM-CLSM | Lateral ≈ 200 nmb,c, axial ≈ 500 nmb,c | Pre. biological | Intensity, location, movement | Fast acquisition, optical slicing, high sensitivity | Low axial resolution, label required |
| AFM-FCS | See AFM-CLSM, volume ≈ 1 fL | Pre. membranes | Concentration, size diffusion coefficient | Correlate diffusion with topography, high sensitivity | Drift in AFM, artifacts, label required |
| AFM-FLIM | See AFM-CLSMb,c | Pre. biological, surface analysis | Intensity, fl. lifetime, movement/diffusion | Modulation effects, fast, multidimensional | Proximity of tip can alter lifetimes, artifacts label required |
| AFM-STED | Lateral ≈ 20 nmb, axial ≈100 nmb | Pre. biological | Intensity, movement/diffusion | High resolution, high sensitivity | Bleaching, specific dyes, special label required |
| PTIR/RE-AFM-IR | Lateral 25 nm/10 nm | Biological, thin films | IR spectrum | Chemical ident. and fingerprint, label-free | Tip lifetime limited, contact mode required |
| IR-PiFM, PFIR | See PTIR | Biological, thin films, monolayers, polymers | IR spectrum | Chemical ident. and fingerprint, NC mode | Tip lifetime limited |
| TERS | Lateral 1–20 nm | Biological, thin films, polymers | Raman spectrum | Chemical ident. and fingerprint, label-free | Tip lifetime limited |
| KPFM | Better than 10 nm, 5–20 meV | Semiconductor, organic films, metals | Contact potential difference | High resolution, defects detectable, label-free | Artifacts |
| SNEM | Lateral ≤ λ/10 | Semiconductor, biology, polymers | Complex refractive index, thickness | Composition accessible, label-free | Tip lifetime limited |
| AFM-RICM | Lateral nanometer, axial nanometer | Soft interfaces, soft probes | Contact surface area, distance | Fast and real time, label-free | Limited to distance and contact area |
| AFM-QCM | Dependent on the size of QCM crystalb | Adsorption of particles, polymers, proteins… | Frequency shift, adsorbed mass | High accuracy, real time, label-free | Analysis difficult, scarce |
| AFM-SPR | Lateral 600 nmb | Adsorption/binding of molecules/proteins | Refractive index, adsorbed mass | Fast and real time, label-free | Thin metal surface needed |
| SThM (also nano-TA) | Lateral up to 10 nm, heating ∼100 nm, up to 15 mK | Semiconductors, polymers, metals, nanocomposite | Temperature, thermal conductivity, phase transitions | Probing into the sample, fast local heating, label-free | Conductivity needed, several heat transport pathwaysa |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Natural Science Foundation of China (21674064 and 21404072) for financial support of this work. We thank Lili Wang for fruitful discussions.References
- P. Knittel, B. Mizaikoff and C. Kranz, Anal. Chem., 2016, 88, 6174–6178 CrossRef CAS PubMed.
- J. M. Mativetsky, Y.-L. Loo and P. Samori, J. Mater. Chem. C, 2014, 2, 3118–3128 RSC.
- S. Liang, Y. Li, T. Zhou, J. Yang, X. Zhou, T. Zhu, J. Huang, J. Zhu, D. Zhu, Y. Liu, C. He, J. Zhang and X. Zhou, Adv. Sci., 2017, 4, 1600313 CrossRef PubMed.
- I. S. Yahia, V. Ganesh, M. Shkir, S. AlFaify, H. Y. Zahran, H. Algarni, M. M. Abutalib, A. Al-Ghamdi, A. M. El-Naggar and A. M. AlBassam, Phys. B, 2016, 496, 9–14 CrossRef CAS.
- G. R. Luis and L. Jian, J. Phys.: Condens. Matter, 2009, 21, 483001 CrossRef PubMed.
- M. D'Acunto, F. Dinelli and P. Pingue, Beilstein J. Nanotechnol., 2015, 6, 2278–2289 CrossRef PubMed.
- J. A. Barreto, W. O'Malley, M. Kubeil, B. Graham, H. Stephan and L. Spiccia, Adv. Mater., 2011, 23, H18–H40 CrossRef CAS PubMed.
- Q. Huang, H. Wu, P. Cai, J. B. Fein and W. Chen, Sci. Rep., 2015, 5, 16857 CrossRef CAS PubMed.
- B. Song, K. Chen, M. Schmittel and H. Schönherr, Langmuir, 2016, 32, 11172–11178 CrossRef CAS PubMed.
- H. Beyer, T. Wagner and A. Stemmer, Beilstein J. Nanotechnol., 2016, 7, 432–438 CrossRef CAS PubMed.
- B. Song, Y. Zhou and H. Schönherr, Langmuir, 2016, 32, 11179–11187 CrossRef CAS PubMed.
- C. Formosa and E. Dague, in Advanced Microscopy in Mycology, ed. S. T. E. Dahms and J. K. Czymmek, Springer International Publishing, Cham, 2015, pp. 125–141, DOI:10.1007/978-3-319-22437-4_7.
- M. Gaczynska, P. Karpowicz, C. E. Stuart, M. G. Norton, J. H. Teckman, E. Marszal and P. A. Osmulski, PLoS One, 2016, 11, 19 Search PubMed.
- R. Gahlin and S. Jacobson, Wear, 1998, 222, 93–102 CrossRef CAS.
- K. Miyahara, N. Nagashima, T. Ohmura and S. Matsuoka, Nanostruct. Mater., 1999, 12, 1049–1052 CrossRef.
- S. Sundararajan and B. Bhushan, J. Mater. Res., 2001, 16, 437–445 CrossRef CAS.
- S. Amelio, A. V. Goldade, U. Rabe, V. Scherer, B. Bhushan and W. Arnold, Thin Solid Films, 2001, 392, 75–84 CrossRef CAS.
- I. A. Morozov, Macromolecules, 2016, 49, 5985–5992 CrossRef CAS.
- S. Z. Lian, R. Y. Shi, X. Huang, X. L. Hu, B. Song, Y. S. Bai, B. Yang, J. Y. Dong, Z. J. Du, Y. Y. Zhang, J. M. Jia, N. Ma, G. Guo and M. Y. Wang, Oncol. Rep., 2016, 36, 984–990 CrossRef CAS PubMed.
- Z. S. Fang, C. R. Jiang, Y. Feng, R. X. Chen, X. Y. Lin, Z. Q. Zhang, L. H. Han, X. D. Chen, H. Y. Li, Y. B. Guo and W. Y. Jiang, Biochim. Biophys. Acta, Mol. Cell Res., 2016, 1863, 2245–2254 CrossRef CAS PubMed.
- J. Li, C. Zhang, P. Cheng, X. Chen, W. Wang and J. Luo, Langmuir, 2016, 32, 5593–5599 CrossRef CAS PubMed.
- S. Senapati and S. Lindsay, Acc. Chem. Res., 2016, 49, 503–510 CrossRef CAS PubMed.
- G. Zito, G. Rusciano, A. Vecchione, G. Pesce, R. Di Girolamo, A. Malafronte and A. Sasso, Sci. Rep., 2016, 6, 16 CrossRef PubMed.
- X. L. Kou, W. Zhang and W. K. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 21055–21062 CAS.
- X. Y. Hu, H. Z. Lei, X. Q. Zhang and Y. Zhang, Microsc. Res. Tech., 2016, 79, 721–726 CrossRef CAS PubMed.
- J. Onoda, K. Niki and Y. Sugimoto, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 155309 CrossRef.
- R. D. Whaley, B. Gopalan, M. Dagenais, R. D. Gomez, F. G. Johnson, S. Agarwala, O. King and D. R. Stone, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.--Process., Meas., Phenom., 1998, 16, 1007–1011 CrossRef CAS.
- B. Schuler, S. Fatayer, F. Mohn, N. Moll, N. Pavliček, G. Meyer, D. Peña and L. Gross, Nat. Chem., 2016, 8, 220–224 CrossRef CAS PubMed.
- Q. Chen, H. Schonherr and G. J. Vancso, Soft Matter, 2009, 5, 4944–4950 RSC.
- D. Hoffman, I. Miskioglu, K. E. Aifantis and J. Drelich, J. Adhes. Sci. Technol., 2012, 26, 1201–1220 CAS.
- C. Kumari, K. Bhat and R. Bansal, J. Conservative Dent., 2016, 19, 56–62 CrossRef CAS PubMed.
- T. Hayashida and K. Umemura, Colloids Surf., B, 2016, 143, 526–531 CrossRef CAS PubMed.
- V. Horňáková, J. Přibyl and P. Skládal, Monatsh. Chem., 2016, 147, 865–871 CrossRef.
- A. Wang, K. Vijayraghavan, O. Solgaard and M. J. Butte, ACS Nano, 2016, 10, 257–264 CrossRef CAS PubMed.
- M. Marchetti, G. J. L. Wuite and W. H. Roos, Curr. Opin. Virol., 2016, 18, 82–88 CrossRef CAS PubMed.
- E. Kämmer, I. Götz, T. Bocklitz, S. Stöckel, A. Dellith, D. Cialla-May, K. Weber, R. Zell, J. Dellith, V. Deckert and J. Popp, Anal. Bioanal. Chem., 2016, 408, 4035–4041 CrossRef PubMed.
- C.-C. Wu, D. N. Reinhoudt, C. Otto, V. Subramaniam and A. H. Velders, Small, 2011, 7, 989–1002 CrossRef CAS PubMed.
- T. Gan, B. Wu, X. Zhou and G. Zhang, RSC Adv., 2016, 6, 50331–50335 RSC.
- C. Chen, X. Zhou, Z. Xie, T. Gao and Z. Zheng, Small, 2015, 11, 613–621 CrossRef CAS PubMed.
- T. Gan, X. Zhou, C. Ma, X. Liu, Z. Xie, G. Zhang and Z. Zheng, Small, 2013, 9, 2850 CrossRef.
- D. Alsteens, D. J. Müller and Y. F. Dufrêne, Acc. Chem. Res., 2017, 50, 924–931 CrossRef CAS PubMed.
- A. F. Eftaiha, S. M. K. Brunet and M. F. Paige, Current Microscopy Contributions to Advances in Science and Technology, Formatex, 2012, p. 1438 Search PubMed.
- D. H. Hu, M. Micic, N. Klymyshyn, Y. D. Suh and H. P. Lu, J. Lumin., 2004, 107, 4–12 CrossRef CAS.
- R. Kassies, K. O. V. d. Werf, A. Lenferink, C. N. Hunter, J. D. Olsen, V. Subramaniam and C. Otto, J. Microsc., 2005, 217, 109–116 CrossRef CAS PubMed.
- N. M. Sabanes, L. M. A. Driessen and K. F. Domke, Anal. Chem., 2016, 88, 7108–7114 CrossRef PubMed.
- B. Lahiri, G. Holland and A. Centrone, Small, 2013, 9, 439–445 CrossRef CAS PubMed.
- D. L. Xu, Y. Zhang, H. D. Zhou, Y. J. Meng and S. Q. Wang, Holzforschung, 2016, 70, 323–330 CrossRef CAS.
- L. Baldassarre, V. Giliberti, A. Rosa, M. Ortolani, A. Bonamore, P. Baiocco, K. Kjoller, P. Calvani and A. Nucara, Nanotechnology, 2016, 27, 075101 CrossRef CAS PubMed.
- S. Ghosh, H. Remita, L. Ramos, A. Dazzi, A. Deniset-Besseau, P. Beaunier, F. Goubard, P.-H. Aubert, F. Brisset and S. Remita, New J. Chem., 2014, 38, 1106–1115 RSC.
- G. Hwang and O. Kwon, Nanoscale, 2016, 8, 5280–5290 RSC.
- T. Tai, O. Karácsony, V. Bocharova, G. J. Van Berkel and V. Kertesz, Anal. Chem., 2016, 88, 2864–2870 CrossRef CAS PubMed.
- S. Moreno Flores and J. L. Toca-Herrera, Nanoscale, 2009, 1, 40–49 RSC.
- J. M. Gerton, L. A. Wade, G. A. Lessard, Z. Ma and S. R. Quake, Phys. Rev. Lett., 2004, 93, 180801 CrossRef PubMed.
- V. Walhorn, O. Schulz, C. Pelargus, D. Anselmetti and R. Ros, Proc. SPIE, 2007, 6444, 644406 CrossRef.
- Y. Ebenstein, T. Mokari and U. Banin, J. Phys. Chem. B, 2004, 108, 93–99 CrossRef CAS.
- Z. Y. Ma, J. M. Gerton, L. A. Wade and S. R. Quake, Phys. Rev. Lett., 2006, 97, 260801–260804 CrossRef PubMed.
- E. Yoskovitz, D. Oron, I. Shweky and U. Banin, J. Phys. Chem. C, 2008, 112, 16306–16311 CAS.
- O. Schulz, Z. Zhao, A. Ward, M. König, F. Koberling, Y. Liu, J. Enderlein, H. Yan and R. Ros, Opt. Nanoscopy, 2013, 2, 1 CrossRef.
- J. L. Garrett and J. N. Munday, Nanotechnology, 2016, 27, 245705 CrossRef PubMed.
- E. Birkenhauer and S. Neethirajan, J. Visualized Exp., 2014, 93, e52327 Search PubMed.
- K. Kim, W. Jeong, W. Lee and P. Reddy, ACS Nano, 2012, 6, 4248–4257 CrossRef CAS PubMed.
- S. Sadewasser, T. Glatzel, M. Rusu, A. Jäger-Waldau and M. C. Lux-Steiner, Appl. Phys. Lett., 2002, 80, 2979–2981 CrossRef CAS.
- P. Eaton and P. West, Atomic force microscopy, Oxford University Press Inc., New York, 2010 Search PubMed.
- H. Schönherr and G. J. Vancso, Scanning Force Microscopy of Polymers, Springer-Verlag, Berlin-Heidelberg, 2010 Search PubMed.
- J. Zhong and J. Yan, RSC Adv., 2016, 6, 1103–1121 RSC.
- D. P. Allison, N. P. Mortensen, C. J. Sullivan and M. J. Doktycz, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2010, 2, 618–634 CrossRef PubMed.
- Y. F. Dufrêne, T. Ando, R. Garcia, D. Alsteens, D. Martinez-Martin, A. Engel, C. Gerber and D. J. Muller, Nat. Nanotechnol., 2017, 12, 295–307 CrossRef PubMed.
- W. B. Amos and J. G. White, Biol. Cell, 2003, 95, 335–342 CrossRef CAS PubMed.
- A. Ostrowski, D. Nordmeyer, A. Boreham, C. Holzhausen, L. Mundhenk, C. Graf, M. C. Meinke, A. Vogt, S. Hadam, J. Lademann, E. Rühl, U. Alexiev and A. D. Gruber, Beilstein J. Nanotechnol., 2015, 6, 263–280 CrossRef PubMed.
- M. Minsky, Microscopy Apparatus, US Pat., 3.013.467, 1957.
- L. Schermelleh, R. Heintzmann and H. Leonhardt, J. Cell Biol., 2010, 190, 165–175 CrossRef CAS PubMed.
- K. D. Kihm, Near-Field Characterization of Micro/Nano-Scaled Fluid Flows, Springer, Berlin-Heidelberg, 2011 Search PubMed.
- M. Grossi, M. Morgunova, S. Cheung, D. Scholz, E. Conroy, M. Terrile, A. Panarella, J. C. Simpson, W. M. Gallagher and D. F. O'Shea, Nat. Commun., 2016, 7, 10855 CrossRef CAS PubMed.
- S. Liang, Y. Li, J. Yang, J. Zhang, C. He, Y. Liu and X. Zhou, Adv. Mater. Technol., 2016, 1, 1600117 CrossRef.
- S. Liang, Y. Li, Y. Chen, J. Yang, T. Zhu, D. Zhu, C. He, Y. Liu, S. Handschuh-Wang and X. Zhou, J. Mater. Chem. C, 2017, 5, 1586–1590 RSC.
- T. Zhou, J. Yang, D. Zhu, J. Zheng, S. Handschuh-Wang, X. Zhou, J. Zhang, Y. Liu, Z. Liu, C. He and X. Zhou, Adv. Sci., 2017, 4, 1700028 CrossRef PubMed.
- D. Zhu, S. Handschuh-Wang and X. Zhou, J. Mater. Chem. A, 2017, 5, 16467–16497 CAS.
- M. Bohrer, M. Schweitzer, R. Nirnberger and B. Weinberger, Proc. SPIE, 2015, 9636, 963609 CrossRef.
- P. Ye, H. Yu and M. Houshmandi, BMC Oral Health, 2016, 16, 1–9 CrossRef PubMed.
- S. Bosi, R. Rauti, J. Laishram, A. Turco, D. Lonardoni, T. Nieus, M. Prato, D. Scaini and L. Ballerini, Sci. Rep., 2015, 5, 9562 CrossRef CAS PubMed.
- M. Maglione and S. J. Sigrist, Nat. Neurosci., 2013, 16, 790–797 CrossRef CAS PubMed.
- E. Abbe, Archiv für Mikroskopische Anatomie, 1873, 9, 413–420 CrossRef.
- S. Schnorrenberg, T. Grotjohann, G. Vorbruggen, A. Herzig, S. W. Hell and S. Jakobs, eLife, 2016, 5, e15567 Search PubMed.
- P. Hoyer, G. de Medeiros, B. Balazs, N. Norlin, C. Besir, J. Hanne, H. G. Krausslich, J. Engelhardt, S. J. Sahl, S. W. Hell and L. Hufnagel, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3442–3446 CrossRef CAS PubMed.
- S. W. Hell, S. J. Sahl, M. Bates, X. W. Zhuang, R. Heintzmann, M. J. Booth, J. Bewersdorf, G. Shtengel, H. Hess, P. Tinnefeld, A. Honigmann, S. Jakobs, I. Testa, L. Cognet, B. Lounis, H. Ewers, S. J. Davis, C. Eggeling, D. Klenerman, K. I. Willig, G. Vicidomini, M. Castello, A. Diaspro and T. Cordes, J. Phys. D: Appl. Phys., 2015, 48, 443001 CrossRef.
- S. W. Hell, Science, 2007, 316, 1153–1158 CrossRef CAS PubMed.
- S. W. Hell and J. Wichmann, Opt. Lett., 1994, 19, 780–782 CrossRef CAS PubMed.
- M. J. Broadhead, M. H. Horrocks, F. Zhu, L. Muresan, R. Benavides-Piccione, J. DeFelipe, D. Fricker, M. V. Kopanitsa, R. R. Duncan, D. Klenerman, N. H. Komiyama, S. F. Lee and S. G. N. Grant, Sci. Rep., 2016, 6, 24626 CrossRef CAS PubMed.
- A. Yildiz and P. R. Selvin, Acc. Chem. Res., 2005, 38, 574–582 CrossRef CAS PubMed.
- D. Kim, T. J. Deerinck, Y. M. Sigal, H. P. Babcock, M. H. Ellisman and X. Zhuang, PLoS One, 2015, 10, e0124581 Search PubMed.
- D. Wildanger, B. R. Patton, H. Schill, L. Marseglia, J. P. Hadden, S. Knauer, A. Schönle, J. G. Rarity, J. L. O'Brien, S. W. Hell and J. M. Smith, Adv. Mater., 2012, 24, 309–313 CrossRef PubMed.
- C. Eggeling, K. I. Willig and F. J. Barrantes, J. Neurochem., 2013, 126, 203–212 CrossRef CAS PubMed.
- B. Harke, J. Keller and S. W. Hell, Opt. Express, 2008, 16, 4154–4162 Search PubMed.
- M. Dyba and S. W. Hell, Phys. Rev. Lett., 2002, 88, 163901 CrossRef PubMed.
- J. Bückers, D. Wildanger, G. Vicidomini, L. Kastrup and S. W. Hell, Opt. Express, 2011, 19, 3130–3143 CrossRef PubMed.
- E. Auksorius, B. R. Boruah, C. Dunsby, P. M. P. Lanigan, G. Kennedy, M. A. A. Neil and P. M. W. French, Opt. Lett., 2008, 33, 113–115 CrossRef PubMed.
- M. Fernandez-Suarez and A. Y. Ting, Nat. Rev. Mol. Cell Biol., 2008, 9, 929–943 CrossRef CAS PubMed.
- L. Po-Yen, L. Yi-Cheng, C. Chia-Seng and K. Fu-Jen, Jpn. J. Appl. Phys., 2013, 52, 028004 CrossRef.
- C. S. Kim, X. Li, Y. Jiang, B. Yan, G. Y. Tonga, M. Ray, D. J. Solfiell and V. M. Rotello, MethodsX, 2015, 2, 306–315 CrossRef PubMed.
- K. M. Dean and A. E. Palmer, Nat. Chem. Biol., 2014, 10, 512–523 CrossRef CAS PubMed.
- A. Ettinger and T. Wittmann, in Methods in Cell Biology, ed. C. W. Jennifer and W. Torsten, Academic Press, 2014, vol. 123, pp. 77–94 Search PubMed.
- P. R. Sarika, N. R. James, P. R. A. Kumar and D. K. Raj, Mater. Sci. Eng., C, 2016, 68, 251–257 CrossRef PubMed.
- N. Singh and S. C. Bhatla, Nitric Oxide Biol. Ch., 2016, 53, 54–64 CrossRef CAS PubMed.
- M. Wada, Plant Sci., 2013, 210, 177–182 CrossRef CAS PubMed.
- Y. C. Yao, H. Xu, C. Liu, Y. Y. Guan, D. Q. Xu, J. Y. Zhang, Y. L. Su, L. L. Zhao and J. B. Luo, RSC Adv., 2016, 6, 9082–9089 RSC.
- M. J. Chen, C. M. Gao, S. Y. Lu, Y. M. Chen and M. Z. Liu, RSC Adv., 2016, 6, 9164–9174 RSC.
- J. S. P. Wong, M. J. Hu, D. Shi, R. K. Y. Li and J. S. S. Wong, Polymer, 2015, 58, 67–75 CrossRef CAS.
- T. Wang, S. Handschuh-Wang, Y. Yang, H. Zhuang, C. Schlemper, D. Wesner, H. Schonherr, W. J. Zhang and X. Jiang, Langmuir, 2014, 30, 1089–1099 CrossRef CAS PubMed.
- N. Hain, D. Wesner, S. I. Druzhinin and H. Schönherr, Langmuir, 2016, 32, 11155–11163 CrossRef CAS PubMed.
- O. Schulz, F. Koberling, D. Walters, M. Koenig, J. Viani and R. Ros, Proc. SPIE, 2010, 7571, 757109 CrossRef.
- C. Neagu, K. O. v. d. Werf, C. A. J. Putman, Y. M. Kraan, B. G. D. Grooth, N. F. v. Hulst and J. Greve, J. Struct. Biol., 1994, 112, 32–40 CrossRef CAS PubMed.
- K. Lieberman, N. Ben-Ami and A. Lewis, Rev. Sci. Instrum., 1996, 67, 3567 CrossRef.
- M. d. Vestergaard, T. Hamada, M. Saito, Y. Yajima, M. Kudou, E. Tamiya and M. Takagi, Biochem. Biophys. Res. Commun., 2008, 377, 725–728 CrossRef CAS PubMed.
- S.-H. Jung, J.-Y. Park, J.-O. Yoo, I. Shin, Y.-M. Kim and K.-S. Ha, Ultramicroscopy, 2009, 109, 1428–1434 CrossRef CAS.
- S. Cazaux, A. Sadoun, M. Biarnes-Pelicot, M. Martinez, S. Obeid, P. Bongrand, L. Limozin and P.-H. Puech, Ultramicroscopy, 2016, 160, 168–181 CrossRef CAS PubMed.
- Y. R. Silberberg, A. E. Pelling, G. E. Yakubov, W. R. Crum, D. J. Hawkes and M. A. Horton, J. Mol. Recognit., 2008, 21, 30–36 CrossRef CAS PubMed.
- E. L. Grzywa, A. C. Lee, G. U. Lee and D. M. Suter, J. Neurobiol., 2006, 66, 1529–1543 CrossRef CAS PubMed.
- S. Kidoaki and T. Matsuda, J. Biomed. Mater. Res., Part A, 2007, 81, 803–810 CrossRef PubMed.
- A. Sarkar, R. B. Robertson and J. M. Fernandez, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12882–12886 CrossRef CAS PubMed.
- J. Gorelik, A. Shevchuk, M. Ramalho, M. Elliott, C. Lei, C. F. Higgins, M. J. Lab, D. Klenerman, N. Krauzewicz and Y. Korchev, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16018–16023 CrossRef CAS PubMed.
- T. Timmel, M. Schuelke and S. Spuler, Microsc. Microanal., 2014, 20, 514–520 CrossRef CAS PubMed.
- A. Noy and T. R. Huser, Rev. Sci. Instrum., 2003, 74, 1217–1221 CrossRef CAS.
- R. Kassies, K. O. v. d. Werft, A. Lenferink, C. N. Hunter, J. D. Olsen, V. Subramaniam and C. Otto, J. Microsc., 2005, 217, 109–116 CrossRef CAS PubMed.
- J. R. Staunton, B. L. Doss, S. Lindsay and R. Ros, Sci. Rep., 2015, 6, 19686 CrossRef PubMed.
- E. Gaviola, Z. Phys., 1925, 35, 748–756 CrossRef.
- S. Cova, M. Bertolaccini and C. Bussolati, Phys. Status Solidi A, 1973, 18, 11–61 CrossRef CAS.
- S. Kinoshita and T. Kushida, Rev. Sci. Instrum., 1981, 52, 572–575 CrossRef CAS.
- R. Schuyler and I. Isenberg, Rev. Sci. Instrum., 1971, 42, 813–817 CrossRef CAS.
- T. Förster and G. Hoffmann, Z. Phys. Chem., 1971, 75, 63–76 CrossRef.
- M. Kaschke, J. Kleinschmidt and B. Wilhelmi, Laser Chem., 1985, 5, 119–132 CrossRef CAS.
- T. Nakabayashi, S. Oshita, R. Sumikawa, F. Sun, M. Kinjo and N. Ohta, J. Photochem. Photobiol., A, 2012, 235, 65–71 CrossRef CAS.
- V. Helenius, R. Monshouwer and R. v. Grondelle, J. Phys. Chem. B, 1997, 101, 10554–10559 CrossRef CAS.
- D. M. Togashi and A. G. Ryder, J. Fluoresc., 2006, 16, 153–160 CrossRef CAS PubMed.
- M. Y. Berezin, H. Lee, W. Akers, G. Nikiforovich and S. Achilefu, Photochem. Photobiol. Sci., 2007, 83, 1371 CAS.
- D. Magde, G. E. Rojas and P. G. Seybold, Photochem. Photobiol., 1999, 70, 737–744 CrossRef CAS.
- A. L. Koner, P. P. Mishra, S. Jha and A. Datta, J. Photochem. Photobiol., A, 2005, 170(1), 21–26 CrossRef CAS.
- D. A. Rusak, W. H. James, M. J. Ferzola and M. J. Stefanski, J. Chem. Educ., 2006, 83, 1857–1859 CrossRef CAS.
- S. Handschuh-Wang, T. Wang, S. I. Druzhinin, D. Wesner, X. Jiang and H. Schönherr, Langmuir, 2017, 33, 802–813 CrossRef CAS PubMed.
- S. Bernacchi and Y. Mély, Nucleic Acids Res., 2001, 29, e62 CrossRef CAS PubMed.
- D. Llères, S. Swift and A. I. Lamond, Curr. Protoc. Cytom., 2007 Search PubMed , Chapter 12, Unit 12.10.
- J. A. Lakowicz, H. Szmacinski, K. Nowaczyk and M. L. Johnson, Cell Calcium, 1992, 13, 131–147 CrossRef CAS PubMed.
- L. Seveus, M. Vaisala and E. Soini, Cytometry, 1992, 13, 329–338 CrossRef CAS PubMed.
- S. Miersch, M. G. Espey, R. Chaube, A. Akarca, R. Tweten, S. Ananvoranich and B. Mutus, J. Biol. Chem., 2008, 283, 18513–18521 CrossRef CAS PubMed.
- S. Wang and Z.-R. Chen, Photochem. Photobiol. Sci., 2017, 16, 155–158 CAS.
- S. Handschuh-Wang, D. Wesner, T. Wang, P. Lu, K.-S. Tücking, S. Haas, S. I. Druzhinin, X. Jiang and H. Schönherr, Macromol. Chem. Phys., 2017, 218, 1600454 CrossRef.
- J. T. Krug, E. J. Sanchez and X. S. Xie, Appl. Phys. Lett., 2005, 86, 233102 CrossRef.
- V. Protasenko, M. Kuno, A. Gallagher and D. Nesbitt, Opt. Commun., 2002, 210, 11–23 CrossRef CAS.
- J. R. T. Seddon, H. J. W. Zandvliet and D. Lohse, Phys. Rev. Lett., 2011, 107, 116101 CrossRef PubMed.
- Y. He, V. Govind Rao, J. Cao and H. P. Lu, J. Phys. Chem. Lett., 2016, 7, 2221–2227 CrossRef CAS PubMed.
- S. Patil, G. Matei, C. A. Grabowski, P. M. Hoffmann and A. Mukhopadhyay, Langmuir, 2007, 23, 4988–4992 CrossRef CAS PubMed.
- S. Chiantia, N. Kahya and P. Schwille, Langmuir, 2007, 23, 7659–7665 CrossRef CAS PubMed.
- A. R. Burns, D. J. Frankel and T. Buranda, Biophys. J., 2005, 89, 1081–1093 CrossRef CAS PubMed.
- S. Chiantia, N. Kahya, J. Ries and P. Schwille, Biophys. J., 2006, 90, 4500–4508 CrossRef CAS PubMed.
- S. Chiantia, J. Ries, G. Chwastek, D. Carrer, Z. Li, R. Bittman and P. Schwille, Biochim. Biophys. Acta, 2008, 1778, 1356–1364 CrossRef CAS PubMed.
- G. A. Rojas, Y. Wu, G. Haugstad and C. D. Frisbie, ACS Appl. Mater. Interfaces, 2016, 8, 5772–5776 CAS.
- M. S. Itano, A. K. Neumann, P. Liu, F. Zhang, E. Gratton, W. J. Parak, N. L. Thompson and K. Jacobson, Biophys. J., 2011, 100, 2662–2670 CrossRef CAS PubMed.
- J. Kuchlyan, N. Kundu, D. Banik, A. Roy and N. Sarkar, Langmuir, 2015, 31, 13793–13801 CrossRef CAS PubMed.
- X. W. Ng, C. Teh, V. Korzh and T. Wohland, Biophys. J., 2016, 111, 418–429 CrossRef CAS PubMed.
- J. Widengren, U. Mets and R. Rigler, J. Phys. Chem. B, 1995, 99, 13368–13379 CrossRef CAS.
- N. Biswas, R. Bhattacharya, A. Saha, N. R. Janab and J. K. Basu, Phys. Chem. Chem. Phys., 2015, 17, 24238–24247 RSC.
- S. Chiantia, J. Ries, N. Kahya and P. Schwille, ChemPhysChem, 2006, 7, 2409 CrossRef CAS PubMed.
- D. R. Stabley, T. Oh, S. M. Simon, A. L. Mattheyses and K. Salaita, Nat. Commun., 2015, 6, 8307 CrossRef CAS PubMed.
- Y. Fu, P. W. Winter, R. Rojas, V. Wang, M. McAuliffe and G. H. Patterson, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4368–4373 CrossRef CAS PubMed.
- C. U. Chan and C.-D. Ohl, Phys. Rev. Lett., 2012, 109, 174501 CrossRef PubMed.
- J. Ki, P. Arumugam and J. M. Song, Anal. Bioanal. Chem., 2016, 408, 3233–3238 CrossRef CAS PubMed.
- S. Block, V. P. Zhdanov and F. Höök, Nano Lett., 2016, 16, 4382–4390 CrossRef CAS PubMed.
- T. Yamada, R. Afrin, H. Arakawa and A. Ikai, FEBS Lett., 2004, 569, 59–64 CrossRef CAS PubMed.
- A. E. X. Brown, A. Hategan, D. Safer, Y. E. Goldman and D. E. Discher, Biophys. J., 2009, 96, 1952–1960 CrossRef CAS PubMed.
- M. S. Z. Kellermayer, A. Karsai, A. Kengyel, A. Nagy, P. Bianco, T. Huber, A. Kulcsar, C. Niedetzky, R. Proksch and L. Grama, Biophys. J., 2006, 91, 2665–2677 CrossRef CAS PubMed.
- R. Eckel, V. Walhorn, C. Pelargus, J. Martini, J. Enderlein, T. Nann, D. Anselmetti and R. Ros, Small, 2007, 3, 44–49 CrossRef CAS PubMed.
- A. Choucair, M. Chakrapani, B. Chakravarthy, L. Katsaras and L. J. Johnston, Biochim. Biophys. Acta, 2007, 1786, 146–154 CrossRef PubMed.
- W. Christenson, I. Yermolenko, B. Plochberger, F. Camacho-Alanis, A. Ros, T. P. Ugarova and R. Ros, Ultramicroscopy, 2014, 136, 211–215 CrossRef CAS PubMed.
- A. B. Mathur, G. A. Truskey and W. M. Reichert, Biophys. J., 2000, 78, 1725–1735 CrossRef CAS PubMed.
- A. Ortega-Esteban, K. Bodensiek, C. San Martín, M. Suomalainen, U. F. Greber, P. J. de Pablo and I. A. T. Schaap, ACS Nano, 2015, 9, 10571–10579 CrossRef CAS PubMed.
- S. Nishida, Y. Funabashi and A. Ikai, Ultramicroscopy, 2002, 91, 269–274 CrossRef CAS PubMed.
- P. Anger, P. Bharadwadj and L. Novotny, Phys. Rev. Lett., 2006, 96, 113002 CrossRef PubMed.
- S. Y. Lee, K. Nakaya, T. Hayashiab and M. Hara, Phys. Chem. Chem. Phys., 2009, 11, 4403–4409 RSC.
- V. Walhorn, J. Paskarbeit, H. G. Frey, A. Harder and D. Anselmetti, Beilstein J. Nanotechnol., 2011, 2, 645–652 CrossRef CAS PubMed.
- D. Milovanovic, M. Platen, M. Junius, U. Diederichsen, I. A. T. Schaap, A. Honigmann, R. Jahn and G. van den Bogaart, J. Biol. Chem., 2016, 291, 7868–7876 CrossRef CAS PubMed.
- M. Capitanio, D. Maggi, F. Vanzi and F. S. Pavone, J. Opt. A: Pure Appl. Opt., 2007, 9, S157–S163 CrossRef CAS.
- P. D. Odermatt, A. Shivanandan, H. Deschout, R. Jankele, A. P. Nievergelt, L. Feletti, M. W. Davidson, A. Radenovic and G. E. Fantner, Nano Lett., 2015, 15, 4896–4904 CrossRef CAS PubMed.
- D. Wustner, M. Modzel, F. W. Lund and M. A. Lomholt, Chem. Phys. Lipids, 2016, 199, 106–135 CrossRef CAS PubMed.
- M. Koenig, P. Reisch, R. Dowler, B. Kraemer, S. Tannert, M. Patting, M. P. Clausen, S. Galiani, C. Eggeling, F. Koberling and R. Erdmann, Proc. SPIE, 2016, 9712, 97120T CrossRef.
- I. Testa, E. D'Este, N. T. Urban, F. Balzarotti and S. W. Hell, Nano Lett., 2015, 15, 103–106 CrossRef CAS PubMed.
- J. Tonnesen, G. Katona, B. Rozsa and U. V. Nagerl, Nat. Neurosci., 2014, 17, 678–685 CrossRef CAS PubMed.
- A. N. Butkevich, G. Y. Mitronova, S. C. Sidenstein, J. L. Klocke, D. Kamin, D. N. H. Meineke, E. D'Este, P. T. Kraemer, J. G. Danzl, V. N. Belov and S. W. Hell, Angew. Chem., Int. Ed., 2016, 55, 3290–3294 CrossRef CAS PubMed.
- S. C. Sidenstein, E. D'Este, M. J. Bohm, J. G. Danzl, V. N. Belov and S. W. Hell, Sci. Rep., 2016, 6, 26725 CrossRef CAS PubMed.
- N. Kodera, D. Yamamoto, R. Ishikawa and T. Ando, Nature, 2010, 468, 72–76 CrossRef CAS PubMed.
- J. C. Cordova, D. K. Das, H. W. Manning and M. J. Lang, Curr. Opin. Struct. Biol., 2014, 28, 142–148 CrossRef CAS PubMed.
- J. V. Chacko, B. Harke, C. Canale and A. Diaspro, J. Biomed. Opt., 2014, 19, 105003 Search PubMed.
- B. Harke, J. V. Chacko, H. Haschke, C. Canale and A. Diaspro, Opt. Nanoscopy, 2012, 1, 3 CrossRef.
- T. Deguchi, S. Koho, T. Nareoja and P. Hanninen, Opt. Rev., 2014, 21, 389–394 CrossRef.
- J. V. Chacko, C. Canale, B. Harke and A. Diaspro, PLoS One, 2013, 8, e66608 CAS.
- J. Q. Yu, J. H. Yuan, X. J. Zhang, J. L. Liu and X. H. Fang, Chin. Sci. Bull., 2013, 58, 4045–4050 CrossRef CAS.
- D. N. Fronczek, C. Quammen, H. Wang, C. Kisker, R. Superfine, R. Taylor, D. A. Erie and I. Tessmer, Ultramicroscopy, 2011, 111, 350–355 CrossRef CAS PubMed.
- E. A. Ash and O. Nichols, Nature, 1972, 237, 510–512 CrossRef CAS PubMed.
- Y. Oshikane, T. Kataoka, M. Okuda, S. Hara, H. Inoue and M. Nakano, Sci. Technol. Adv. Mater., 2007, 8, 181–185 CrossRef CAS.
- F. Keilmann and R. Hillenbrand, Phil. Trans. R. Soc. A, 2004, 362, 787–805 CrossRef CAS PubMed.
- T. J. Yang, G. A. Lessard and S. R. Quake, Appl. Phys. Lett., 2000, 76, 378–380 CrossRef CAS.
- F. Zenhausern, M. P. O'Boyle and H. K. Wickramasinghe, Appl. Phys. Lett., 1994, 65, 1623 CrossRef CAS.
- Y. Martin, F. Zenhausern and H. K. Wickramasinghe, Appl. Phys. Lett., 1996, 68, 2475 CrossRef CAS.
- A. Kramer, W. Trabesinger, B. Hecht and U. P. Wild, Appl. Phys. Lett., 2002, 80, 1652 CrossRef CAS.
- N. Hayazawa, Y. Inouye and S. Kawata, J. Microsc., 1999, 194, 472–476 CrossRef CAS PubMed.
- J. Azoulay, A. Débarre, A. Richard and P. Tchénio, J. Microsc., 1999, 194, 486 CrossRef CAS PubMed.
- A. Hartschuh, E. J. Sánchez, X. S. Xie and L. Novotny, Phys. Rev. Lett., 2003, 90, 095503 CrossRef PubMed.
- T. Ichimura, N. Hayazawa, M. Hashimoto, Y. Inouye and S. Kawata, Phys. Rev. Lett., 2004, 92, 220801 CrossRef PubMed.
- H. G. Frey, J. Paskarbeit and D. Anselmetti, Appl. Phys. Lett., 2009, 94, 241116 CrossRef.
- N. Behr and M. Raschke, J. Phys. Chem. C, 2008, 112, 3766–3773 CAS.
- W.-S. Chang, S. Bauerdick and M. S. Jeong, Ultramicroscopy, 2008, 108, 1070–1075 CrossRef CAS PubMed.
- Z. Shi, X. Hong, H. A. Bechtel, B. Zeng, M. C. Martin, K. Watanabe, T. Taniguchi, Y.-R. Shen and F. Wang, Nat. Photonics, 2015, 9, 515–519 CrossRef CAS.
- B. Pollard, E. A. Muller, K. Hinrichs and M. B. Raschke, Nat. Commun., 2014, 5, 3587 Search PubMed.
- S. A. Donges, O. Khatib, B. T. O'Callahan, J. M. Atkin, J. H. Park, D. Cobden and M. B. Raschke, Nano Lett., 2016, 16, 3029–3035 CrossRef CAS PubMed.
- N. El-Kork, P. Moretti, B. Jacquier, F. Lei and M. Ismail, J. Nanomater., 2016, 2016, 4089260 Search PubMed.
- L. Andolfi, E. Trevisan, B. Troian, S. Prato, R. Boscolo, E. Giolo, S. Luppi, M. Martinelli, G. Ricci and M. Zweyer, J. Nanobiotechnol., 2015, 13, 2 CrossRef PubMed.
- Y. De Wilde and P. A. Lemoine, AIP Conf. Proc., 2007, 931, 43–52 CrossRef CAS.
- R. Gaifulina, A. T. Maher, C. Kendall, J. Nelson, M. Rodriguez-Justo, K. Lau and G. M. Thomas, Int. J. Exp. Pathol., 2016, 97, 337–350 CrossRef CAS PubMed.
- A. K. Meher and Y.-C. Chen, Anal. Chem., 2016, 88, 9151–9157 CrossRef CAS PubMed.
- K. Luo, Y. D. Xiang, H. M. Wang, L. Xiang and Z. H. Luo, J. Mater. Sci. Technol., 2016, 32, 733–737 Search PubMed.
- C. S. Casari, C. S. Giannuzzi and V. Russo, Carbon, 2016, 104, 190–195 CrossRef CAS.
- T. Kavinkumar and S. Manivannan, J. Mater. Sci. Technol., 2016, 32, 626–632 Search PubMed.
- W. Miranda, S. S. Coutinho, M. S. Tavares, E. Moreira and D. L. Azevedo, J. Mol. Struct., 2016, 1122, 299–308 CrossRef CAS.
- P. K. Chu and L. Li, Mater. Chem. Phys., 2006, 96, 253–277 CrossRef CAS.
- E. V. Efremov, F. Ariese and C. Gooijer, Anal. Chim. Acta, 2008, 606, 119–134 CrossRef CAS PubMed.
- S. Bonhommeau, D. Talaga, J. Hunel, C. Cullin and S. Lecomte, Angew. Chem., 2017, 129, 1797–1800 CrossRef.
- P. C. Gutiérrez-Neira, F. Agulló-Rueda, A. Climent-Font and C. Garrido, Vib. Spectrosc., 2013, 69, 13–20 CrossRef.
- S. Boyd, M. F. Bertino and S. J. Seashols, Forensic Sci. Int., 2011, 208, 124–128 CrossRef CAS PubMed.
- G. Barone, D. Bersani, J. Jehlicka, P. P. Lottici, P. Mazzoleni, S. Raneri, P. Vandenabeele, C. Di Giacomo and G. Larina, J. Raman Spectrosc., 2015, 46, 989–995 CrossRef CAS.
- M. N. Kinalwa, E. W. Blanch and A. J. Doig, Anal. Chem., 2010, 82, 6347–6349 CrossRef CAS PubMed.
- S. Signorelli, S. Cannistraro and A. R. Bizzarri, Appl. Spectrosc., 2016, 71, 823–832 CrossRef PubMed.
- H. J. Butler, L. Ashton, B. Bird, G. Cinque, K. Curtis, J. Dorney, K. Esmonde-White, N. J. Fullwood, B. Gardner, P. L. Martin-Hirsch, M. J. Walsh, M. R. McAinsh, N. Stone and F. L. Martin, Nat. Protoc., 2016, 11, 664–687 CrossRef CAS PubMed.
- N. Kumar, M. M. Drozdz, H. Jiang, D. M. Santos and D. J. Vaux, Chem. Commun., 2017, 53, 2451–2454 RSC.
- P. Mukherjee, S. J. Lim, T. P. Wrobel, R. Bhargava and A. M. Smith, J. Am. Chem. Soc., 2016, 138, 10887–10896 CrossRef CAS PubMed.
- T. Iwasaki, T. Zelai, S. Ye, Y. Tsuchiya, H. M. H. Chong and H. Mizuta, Carbon, 2017, 111, 67–73 CrossRef CAS.
- A. Paudel, D. Raijada and J. Rantanen, Adv. Drug Delivery Rev., 2015, 89, 3–20 CrossRef CAS PubMed.
- L. S. Lawson and J. D. Rodriguez, Anal. Chem., 2016, 88, 4706–4713 CrossRef CAS PubMed.
- M. Asghari-Khiavi, B. R. Wood, P. Hojati-Talemi, A. Downes, D. McNaughtona and A. Mechlera, J. Raman Spectrosc., 2012, 43, 173–180 CrossRef CAS.
- W. Su and D. Roy, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom., 2013, 31, 041808 Search PubMed.
- N. Kumar, B. Stephanidis, R. Zenobi, A. J. Wain and D. Roy, Nanoscale, 2015, 7, 7133–7137 RSC.
- R. Zhang, Y. Zhang, Z. C. Dong, S. Jiang, C. Zhang and L. G. Chen, Nature, 2013, 498, 82–86 CrossRef CAS PubMed.
- N. Kumar, S. Mignuzzi, W. Su and D. Roy, EPJ Tech. Instrum., 2015, 2, 9 CrossRef.
- B. R. Wood, E. Bailo, M. A. Khiavi, L. Tilley, S. Deed and T. Deckert-Gaudig, Nano Lett., 2011, 11, 1868–1873 CrossRef CAS PubMed.
- R. Bohme, M. Richter, D. Cialla, P. Rosch, V. Deckert and J. Popp, J. Raman Spectrosc., 2009, 40, 1452–1457 CrossRef.
- T. Deckert-Gaudig, E. Bailo and V. Deckert, Phys. Chem. Chem. Phys., 2009, 11, 7360–7362 RSC.
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51–87 CrossRef CAS.
- E. M. v. S. Lantman, T. Deckert-Gaudig, A. J. G. Mank, V. Deckert and B. M. Weckhuysen, Nat. Nanotechnol., 2012, 7, 583–586 CrossRef PubMed.
- C. E. Harvey and B. M. Weckhuysen, Catal. Lett., 2015, 145, 40–57 CrossRef CAS PubMed.
- T. Hartman, C. S. Wondergem, N. Kumar, A. v. d. Berg and B. M. Weckhuysen, J. Phys. Chem. Lett., 2016, 7, 1570–1584 CrossRef CAS PubMed.
- N. Jiang, D. Kurouski, E. A. Pozzi, N. Chiang, M. C. Hersam and R. P. V. Duyne, Chem. Phys. Lett., 2016, 659, 16–24 CrossRef CAS.
- T. Mino, Y. Saito and P. Verma, ACS Nano, 2014, 8, 10187–10195 CrossRef CAS PubMed.
- L. T. Nieman, G. M. Krampert and R. E. Martinez, Rev. Sci. Instrum., 2001, 72, 1691–1699 CrossRef CAS.
- L. Opilik, Ü. Dogan, J. Szczerbinski and R. Zenobi, Appl. Phys. Lett., 2015, 107, 091109 CrossRef.
- Y. Fujita, R. Chiba, G. Lu, N. N. Horimoto, S. Kajimoto, H. Fukumura and H. Uji-i, Chem. Commun., 2014, 50, 9839–9841 RSC.
- C. A. Barrios, A. V. Malkovskiy, A. M. Kisliuk, A. P. Sokolov and M. D. Foster, J. Phys. Chem. C, 2009, 113, 8158–8161 CAS.
- N. Kumar, S. J. Spencer, D. Imbraguglio, A. M. Rossi, A. J. Wain, B. M. Weckhuysen and D. Roy, Phys. Chem. Chem. Phys., 2016, 18, 13710–13716 RSC.
- P. Pienpinijtham, S. Vantasin, Y. Kitahama, S. Ekgasit and Y. Ozaki, J. Phys. Chem. C, 2016, 120, 14663–14668 CAS.
- N. Biliškov and G. Baranović, J. Mol. Liq., 2009, 144, 155–162 CrossRef.
- S. Nakata, M.-a. Kakimoto and Y. Imai, Polym. J., 1993, 25, 569–576 CrossRef CAS.
- A. Harrer, R. Szedlak, B. Schwarz, H. Moser, T. Zederbauer, D. MacFarland, H. Detz, A. M. Andrews, W. Schrenk, B. Lendl and G. Strasser, Sci. Rep., 2016, 6, 21795 CrossRef CAS PubMed.
- M. F. H. Al-Kadhemy, Z. S. Rasheed and S. R. Salim, J. Radiat. Res. Appl. Sci., 2016, 9, 321–331 CrossRef CAS.
- S. Prati, E. Joseph, G. Sciutto and R. Mazzeo, Acc. Chem. Res., 2010, 43, 792–801 CrossRef CAS PubMed.
- R. Bhargava and I. W. Levin, in Spectrochemical Analysis Using Infrared Multichannel Detectors, Blackwell Publishing Ltd, 2007, pp. 1–24, DOI:10.1002/9780470988541.ch1.
- M. M. Beasley, E. J. Bartelink, L. Taylor and R. M. Miller, J. Archaeol. Sci., 2014, 46, 16–22 CrossRef CAS.
- J. S. Gaffney, N. A. Marley and D. E. Jones, in Characterization of Materials, John Wiley & Sons, Inc., 2002, DOI:10.1002/0471266965.com107.pub2.
- Á. I. López-Lorente and B. Mizaikoff, Anal. Bioanal. Chem., 2016, 408, 2875–2889 CrossRef PubMed.
- N. Brandes, P. B. Welzel, C. Werner and L. W. Kroh, J. Colloid Interface Sci., 2006, 299, 56–69 CrossRef CAS PubMed.
- A. E. Guber and U. Köhler, J. Mol. Struct., 1995, 348, 209–212 CrossRef CAS.
- M. B. Esler, D. W. T. Griffith, S. R. Wilson and L. P. Steele, Anal. Chem., 2000, 72, 206–215 CrossRef CAS PubMed.
- R. C. Pereira, M. C. Arbestain, M. V. Sueiro and J. A. Maciá-Agulló, Soil Res., 2015, 53, 753–762 CrossRef CAS.
- K. Kachel, D. Siche, S. Golka, P. Sennikov and M. Bickermann, Mater. Chem. Phys., 2016, 177, 12–18 CrossRef CAS.
- O. Planinšek, D. Planinšek, A. Zega, M. Breznik and S. Srčič, Int. J. Pharm., 2006, 319, 13–19 CrossRef PubMed.
- A. Francis, R. Detsch and A. R. Boccaccini, Ceram. Int., 2016, 42, 15442–15448 CrossRef CAS.
- W. H. Zheng, J. T. Hu, S. Rappeport, Z. Zheng, Z. X. Wang, Z. S. Han, J. Langer and J. Economy, Microporous Mesoporous Mater., 2016, 234, 146–154 CrossRef CAS.
- T. Wang, S. Handschuh-Wang, P. Qin, Y. Yang, X. Zhou and Y. Tang, J. Colloid Interface Sci., 2017, 499, 102–109 CrossRef CAS PubMed.
- T. Wang, S. Handschuh-Wang, S. Zhang, X. Zhou and Y. Tang, J. Colloid Interface Sci., 2017, 506, 543–552 CrossRef CAS PubMed.
- Y. Kano, K. Ishikura, S. Kawahara and S. Akiyama, Polym. J., 1992, 24, 135–144 CrossRef CAS.
- Y. Fazli, H. Alijani and K. Khezri, Adv. Polym. Technol., 2016, 35, 260–268 CrossRef CAS.
- A. K. Pal and V. Katiyar, Biomacromolecules, 2016, 17, 2603–2618 CrossRef CAS PubMed.
- A. Dazzi, F. Glotin and R. Carminati, J. Appl. Phys., 2010, 107, 124519 CrossRef.
- L. Baldassarre, V. Giliberti, A. Rosa, M. Ortolani, A. Bonamore, P. Baiocco, K. Kjoller, P. Calvani and A. Nucara, Nanotechnology, 2016, 27, 075101 CrossRef CAS PubMed.
- T. Muller, F. S. Ruggeri, A. J. Kulik, U. Shimanovich, T. O. Mason, T. P. J. Knowles and G. Dietler, Lab Chip, 2014, 14, 1315–1319 RSC.
- P. Vitry, R. Rebois, E. Bourillot, A. Deniset-Besseau, M.-J. Virolle, E. Lesniewska and A. Dazzi, Nano Res., 2016, 9, 1674–1681 CrossRef CAS.
- C. Marcott, M. Lo, K. Kjoller, F. Fiat, N. Baghdadli, G. Balooch and G. S. Luengo, Appl. Spectrosc., 2014, 68, 564–569 CrossRef CAS PubMed.
- C. Policar, J. B. Waern, M.-A. Plamont, S. Clède, C. Mayet, R. Prazeres, J.-M. Ortega, A. Vessières and A. Dazzi, Angew. Chem., Int. Ed., 2011, 50, 860–864 CrossRef CAS PubMed.
- E. Kennedy, R. Al-Majmaie, M. Al-Rubeai, D. Zerulla and J. H. Rice, RSC Adv., 2013, 3, 13789–13795 RSC.
- A. Deniset-Besseau, C. B. Prater, M.-J. Virolle and A. Dazzi, J. Phys. Chem. Lett., 2014, 5, 654–658 CrossRef CAS PubMed.
- F. G. Tang, P. T. Bao and Z. H. Su, Anal. Chem., 2016, 88, 4926–4930 CrossRef CAS PubMed.
- M. Kelchtermans, M. Lo, E. Dillon, K. Kjoller and C. Marcott, Vib. Spectrosc., 2016, 82, 10–15 CrossRef CAS.
- A. M. Katzenmeyer, V. Aksyuk and A. Centrone, Anal. Chem., 2013, 85, 1972–1979 CrossRef CAS PubMed.
- A. Dazzi, J. Saunier, K. Kjoller and N. Yagoubi, Int. J. Pharm., 2015, 484, 109–114 CrossRef CAS PubMed.
- B. Van Eerdenbrugh, M. Lo, K. Kjoller, C. Marcott and L. S. Taylor, J. Pharm. Sci., 2012, 101, 2066–2073 CrossRef CAS PubMed.
- A. J. Harrison, E. A. Bilgili, S. P. Beaudoin and L. S. Taylor, Anal. Chem., 2013, 85, 11449–11455 CrossRef CAS PubMed.
- S. Morsch, Y. W. Liu, S. B. Lyon and S. R. Gibbon, ACS Appl. Mater. Interfaces, 2016, 8, 959–966 CAS.
- L. Gong, D. B. Chase, I. Noda, J. L. Liu, D. C. Martin, C. Y. Ni and J. F. Rabolt, Macromolecules, 2015, 48, 6197–6205 CrossRef CAS.
- V. Aksyuk, B. Lahiri, G. Holland and A. Centrone, Nanoscale, 2015, 7, 3634–3644 RSC.
- S. Laokroekkiat, M. Hara, S. Nagano and Y. Nagao, Langmuir, 2016, 32, 6648–6655 CrossRef CAS PubMed.
- J. Chae, Q. F. Dong, J. S. Huang and A. Centrone, Nano Lett., 2015, 15, 8114–8121 CrossRef CAS PubMed.
- Y. B. Yuan, J. Chae, Y. C. Shao, Q. Wang, Z. G. Xiao, A. Centrone and J. S. Huang, Adv. Energy Mater., 2015, 5, 1500615 CrossRef.
- M. K. F. Lo, A. Dazzi, C. A. Marcott, E. Dillon, Q. C. Hu, K. Kjoller, C. B. Prater and S. W. King, ECS J. Solid State Sci. Technol., 2016, 5, P3018–P3024 CrossRef CAS.
- F. Lu, M. Z. Jin and M. A. Belkin, Nat. Photonics, 2014, 8, 307–312 CrossRef CAS.
- F. Lu and M. A. Belkin, Opt. Express, 2011, 19, 19942–19947 CrossRef CAS PubMed.
- P. Vitry, E. Bourillot, C. Plassard, Y. Lacroute, E. Calkins, L. Tetard and E. Lesniewska, Nano Res., 2015, 8, 2199–2205 CrossRef.
- L. Wang, H. Wang, M. Wagner, Y. Yan, D. S. Jakob and X. G. Xu, Sci. Adv., 2017, 3, e1700255 CrossRef PubMed.
- A. Ambrosio, R. C. Devlin, F. Capasso and W. L. Wilson, ACS Photonics, 2017, 4, 846–851 CrossRef CAS.
- D. Nowak, W. Morrison, H. K. Wickramasinghe, J. Jahng, E. Potma, L. Wan, R. Ruiz, T. R. Albrecht, K. Schmidt, J. Frommer, D. P. Sanders and S. Park, Sci. Adv., 2016, 2, e1501571 Search PubMed.
- J. Jahng, D. A. Fishman, S. Park, D. B. Nowak, W. A. Morrison, H. K. Wickramasinghe and E. O. Potma, Acc. Chem. Res., 2015, 48, 2671–2679 CrossRef CAS PubMed.
- J. N. Hilfiker, M. Stadermann, J. Sun, T. Tiwald, J. S. Hale, P. E. Miller and C. Aracne-Ruddle, Appl. Surf. Sci., 2017, 421, 508–512 CrossRef CAS.
- S. A. Kukushkin and A. V. Osipov, Tech. Phys. Lett., 2016, 42, 175–178 CrossRef CAS.
- J. J. Diaz Leon, D. M. Fryauf, R. D. Cormia, M.-X. M. Zhang, K. Samuels, R. S. Williams and N. P. Kobayashi, ACS Appl. Mater. Interfaces, 2016, 8, 22337–22344 CAS.
- S. Wang, J. Rond, C. Steinberg, M. Papenheim and H.-C. Scheer, Appl. Phys. A: Mater. Sci. Process., 2016, 122, 121 CrossRef.
- K. Dorywalski, I. Maciejewski and T. Krzyżyński, Mechatronics, 2016, 37, 33–41 CrossRef.
- C.-H. Zhai, R.-J. Zhang, X. Chen, Y.-X. Zheng, S.-Y. Wang, J. Liu, N. Dai and L.-Y. Chen, Nanoscale Res. Lett., 2016, 11, 407 CrossRef PubMed.
- P. Karageorgiev, H. Orendi, B. Stiller and L. Brehmer, Appl. Phys. Lett., 2001, 79, 1730–1732 CrossRef CAS.
- D. Tranchida, J. Diaz, P. Schon, H. Schonherr and G. J. Vancso, Nanoscale, 2011, 3, 233–239 RSC.
- A. Cumurcu, J. Diaz, I. D. Lindsay, S. de Beer, J. Duvigneau, P. Schon and G. J. Vancso, Ultramicroscopy, 2015, 150, 79–87 CrossRef CAS PubMed.
- A. Cumurcu, J. Duvigneau, I. D. Lindsay, P. M. Schon and G. J. Vancso, Eur. Polym. J., 2013, 49, 1935–1942 CrossRef CAS.
- A. S. G. Curtis, J. Cell Biol., 1964, 20, 199–215 CrossRef CAS PubMed.
- L. Limozin and K. Sengupta, ChemPhysChem, 2009, 10, 2752–2768 CrossRef CAS PubMed.
- J. Erath, S. Schmidt and A. Fery, Soft Matter, 2010, 6, 1432–1437 RSC.
- D. Gingell and I. Todd, Biophys. J., 1979, 26, 507–526 CrossRef CAS PubMed.
- S. Attili and R. P. Richter, Langmuir, 2012, 28, 3206–3216 CrossRef CAS PubMed.
- S. Schmidt, H. Wang, D. Pussak, S. Mosca and L. Hartmann, Beilstein J. Org. Chem., 2015, 11, 720–729 CrossRef CAS PubMed.
- E. Wassel, S. Jiang, Q. Song, S. Vogt, G. Nöll, S. I. Druzhinin and H. Schönherr, Langmuir, 2016, 32, 9360–9370 CrossRef CAS PubMed.
- P. Palladino, A. M. Aura and G. Spoto, Anal. Bioanal. Chem., 2016, 408, 849–854 CrossRef CAS PubMed.
- K. Johansen, H. Arwin, I. Lundström and B. Liedberg, Rev. Sci. Instrum., 2000, 71, 3530–3538 CrossRef CAS.
- A. Zuber, M. Purdey, E. Schartner, C. Forbes, B. van der Hoek, D. Giles, A. Abell, T. Monro and H. Ebendorff-Heidepriem, Sens. Actuators, B, 2016, 227, 117–127 CrossRef CAS.
- V. Singh, Z. Li, X. Zhou, X. Xu, J. Xu, A. Nand, H. Wen, H. Li, J. Zhu and J. Zhang, RSC Adv., 2016, 6, 3213–3218 RSC.
- L. L. Yin, S. P. Wang, X. N. Shan, S. T. Zhang and N. J. Tao, Rev. Sci. Instrum., 2015, 86, 114101 CrossRef CAS PubMed.
- E. Kretschmann, Opt. Commun., 1972, 6, 185–187 CrossRef.
- K. M. Shakesheff, X. Chen, M. C. Davies, A. Domb, C. J. Roberts, S. J. B. Tendler and P. M. Williams, Langmuir, 1995, 11, 3921–3927 CrossRef CAS.
- X. Chen, K. M. Shakesheff, M. C. Davies, J. Heller, C. J. Roberts, J. B. Tendler and P. M. Williams, J. Phys. Chem. B, 1995, 99, 11537–11542 CrossRef CAS.
- A. Baba, W. Knoll and R. Advincula, Rev. Sci. Instrum., 2006, 77, 064101 CrossRef.
- L. Joergensen, B. Klösgen, A. C. Simonsen, J. Borch and E. Hagesaether, Int. J. Pharm., 2011, 411, 162–168 CrossRef CAS PubMed.
- K. Hall, T.-H. Lee, A. I. Mechler, M. J. Swann and M.-I. Aguilar, Sci. Rep., 2014, 4, 5479 CrossRef CAS PubMed.
- X. Chen, M. C. Davies, C. J. Roberts, K. M. Shakesheff, S. J. B. Tendler, P. M. Williams and J. Davies, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.--Process., Meas., Phenom., 1996, 14, 1582–1586 CrossRef CAS.
- S. Kitamura and M. Iwatsuki, Appl. Phys. Lett., 1998, 72, 3154 CrossRef CAS.
- M. Nonnenmacher, M. P. Oboyle and H. K. Wickramasinghe, Appl. Phys. Lett., 1991, 58, 2921 CrossRef.
- D. Kohl, P. Mesquida and G. Schitter, Microelectron. Eng., 2017, 176, 28–32 CrossRef CAS.
- H. M. I. Jaim, R. A. Isaacs, S. N. Rashkeev, M. Kuklja, D. P. Cole, M. C. LeMieux, I. Jasiuk, S. Nilufar and L. G. Salamanca-Riba, Carbon, 2016, 107, 56–66 CrossRef CAS.
- S. Gunther, B. Kaulich, L. Gregoratti and M. Kiskinova, Prog. Surf. Sci., 2002, 70, 187–260 CrossRef CAS.
- H. Luth, Solid Surfaces, Interfaces and Thin Films, Springer, Berlin, 2001 Search PubMed.
- J. C. Gonzalez, K. L. Bunker and P. E. Russell, Appl. Phys. Lett., 2001, 79, 1567 CrossRef CAS.
- T. Glatzel, S. Sadewasser and M. C. Lux-Steiner, Appl. Surf. Sci., 2003, 210, 84–89 CrossRef CAS.
- U. Zerweck, C. Loppacher, T. Otto, S. Grafström and L. M. Eng, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 125424 CrossRef.
- Z. M. Ma, L. Kou, Y. Naitoh, Y. J. Li and Y. Sugawara, Nanotechnology, 2013, 24, 225701 CrossRef PubMed.
- L. Polak, S. d. Man and R. J. Wijngaarden, Rev. Sci. Instrum., 2014, 85, 046111 CrossRef PubMed.
- S. Barbet, M. Popoff, H. Diesinger, D. Deresmes, D. Théron and T. Mélin, J. Appl. Phys., 2014, 115, 144313 CrossRef.
- J. Murawski, T. Graupner, P. Milde, R. Raupach, U. Zerweck-Trogisch and L. M. Eng, J. Appl. Phys., 2015, 118, 154302 Search PubMed.
- L. Collins, A. Belianinov, S. Somnath, N. Balke, S. V. Kalinin and S. Jesse, Sci. Rep., 2016, 6, 30557 CrossRef CAS PubMed.
- R. Borgani, D. Forchheimer, J. Bergqvist, R. A. Thorén, O. Inganäs and D. B. Haviland, Appl. Phys. Lett., 2014, 105, 143113 CrossRef.
- Y. Sugawara, L. Kou, Z. Ma, T. Kamijo, Y. Naitoh and L. Y. Jun, Appl. Phys. Lett., 2012, 100, 223104 CrossRef.
- M. Nonnenmacher, M. O'Boyle and H. K. Wickramasinghe, Ultramicroscopy, 1992, 44, 268–273 CrossRef.
- A. K. Henning, T. Hochwitz, J. Slinkman, J. Never, S. Hoffmann, P. Kaszuba and C. Daghlian, J. Appl. Phys., 1995, 77, 1888 CrossRef CAS.
- N. Reitzel, T. Hassenkam, K. Balashev, T. R. Jensen, P. B. Howes, K. Kjaer, A. Fechtenkötter, N. Tchebotareva, S. Ito, K. Müllen and T. Bjørnholm, Chem.–Eur. J., 2001, 7, 4894–4901 CrossRef CAS.
- H. F. Knapp, P. Mesquida and A. Stemmer, Surf. Interface Anal., 2002, 33, 108–112 CrossRef CAS.
- M. Tosun, L. Chan, M. Amani, T. Roy, G. H. Ahn, P. Taheri, C. Carraro, J. W. Ager, R. Maboudian and A. Javey, ACS Nano, 2016, 10, 6853–6860 CrossRef CAS PubMed.
- U. Gysin, E. Meyer, T. Glatzel, G. Günzburger, H. R. Rossmann, T. A. Jung, S. Reshanov, A. Schöner and H. Bartolf, Microelectron. Eng., 2016, 160, 18–21 CrossRef CAS.
- C. Sommerhalter, T. Matthes, T. Glatzel, A. Jäger-Waldau and M. C. Lux-Steiner, Appl. Phys. Lett., 1999, 75, 286 CrossRef CAS.
- A. Findlay, D. Marinskiy, P. Edelman, M. Wilson, A. Savtchouk and J. Lagowski, ECS J. Solid State Sci. Technol., 2016, 5, P3087–P3095 CrossRef CAS.
- A. Yurtsever, D. Fernandez-Torre, J. Onoda, M. Abe, S. Morita, Y. Sugimoto and R. Perez, Nanoscale, 2017, 9, 5812–5821 RSC.
- T. Spath, M. Popp, C. Perez Leon, M. Marz and R. Hoffmann-Vogel, Nanoscale, 2017, 9, 7868–7874 RSC.
- Y. Yamagishi, K. Kobayashi, K. Noda and H. Yamada, Appl. Phys. Lett., 2016, 108, 093302 CrossRef.
- G. Sauerbrey, Z. Phys. Chem., 1959, 155, 206 CAS.
- F. Iwata, K. Saruta and A. Sasaki, Appl. Phys. A: Mater. Sci. Process., 1998, 66, S463–S466 CrossRef CAS.
- J. M. Friedt, K. H. Choi, F. Frederix and A. Campitelli, J. Electrochem. Soc., 2003, 150, H229–H234 CrossRef CAS.
- E. L. Smith, J. C. Barron, A. P. Abbott and K. S. Ryder, Anal. Chem., 2009, 81, 8466–8471 CrossRef CAS PubMed.
- V. S. Popov, E. N. Subcheva, A. V. Shelaev, R. G. Pavelko, V. G. Sevastyanov and N. T. Kuznetsov, Theor. Found. Chem. Eng., 2014, 48, 518–523 CrossRef CAS.
- P. Rivière, T. Nypelö, O. J. Rojas, A. Klug, N. Mundigler and R. Wimmer, J. Therm. Anal. Calorim., 2017, 127, 2059–2074 CrossRef.
- M. Yavari, S. Maruf, Y. Ding and H. Lin, J. Membr. Sci., 2017, 525, 399–408 CrossRef CAS.
- C. C. Williams and H. K. Wickramasinghe, Appl. Phys. Lett., 1986, 49, 1587–1589 CrossRef.
- L. Cui, W. Jeong, V. Fernández-Hurtado, J. Feist, F. J. García-Vidal, J. C. Cuevas, E. Meyhofer and P. Reddy, Nat. Commun., 2017, 8, 14479 CAS.
- K. Kim, B. Song, V. Fernández-Hurtado, W. Lee, W. Jeong, L. Cui, D. Thompson, J. Feist, M. T. H. Reid, F. J. García-Vidal, J. C. Cuevas, E. Meyhofer and P. Reddy, Nature, 2015, 528, 387–391 CrossRef CAS PubMed.
- A. Majumdar, J. Lai, M. Chandrachood, O. Nakabeppu, Y. Wu and Z. Shi, Rev. Sci. Instrum., 1995, 66, 3584–3592 CrossRef CAS.
- K. Luo, Z. Shi, J. Lai and A. Majumdar, Appl. Phys. Lett., 1996, 68, 325–327 CrossRef CAS.
- G. Mills, H. Zhou, A. Midha, L. Donaldson and J. M. R. Weaver, Appl. Phys. Lett., 1998, 72, 2900–2902 CrossRef CAS.
- I. W. Rangelow, T. Gotszalk, P. Grabiec, K. Edinger and N. Abedinov, Microelectron. Eng., 2001, 57–58, 737–748 CrossRef CAS.
- Y. Zhang, P. S. Dobson and J. M. R. Weaver, Microelectron. Eng., 2011, 88, 2435–2438 CrossRef CAS.
- A. Gaitas and Y. B. Gianchandani, Ultramicroscopy, 2006, 106, 874–880 CrossRef CAS PubMed.
- L. Aigouy, E. Saïdi, L. Lalouat, J. Labéguerie-Egéa, M. Mortier, P. Löw and C. Bergaud, J. Appl. Phys., 2009, 106, 074301 CrossRef.
- O. Nakabeppu, M. Chandrachood, Y. Wu, J. Lai and A. Majumdar, Appl. Phys. Lett., 1995, 66, 694–696 CrossRef CAS.
- S. Gomès, A. Assy and P.-O. Chapuis, Phys. Status Solidi A, 2015, 212, 477–494 CrossRef.
- J. Bodzenta, J. Juszczyk, A. Kazmierczak-Balata, P. Firek, A. Fleming and M. Chirtoc, Int. J. Thermophys., 2016, 37, 17 CrossRef.
- A. Pavlov, Appl. Phys. Lett., 2004, 85, 2095–2097 CrossRef CAS.
- S. Sadat, A. Tan, Y. J. Chua and P. Reddy, Nano Lett., 2010, 10, 2613–2617 CrossRef CAS PubMed.
- A. Kittel, W. Müller-Hirsch, J. Parisi, S.-A. Biehs, D. Reddig and M. Holthaus, Phys. Rev. Lett., 2005, 95, 224301 CrossRef PubMed.
- S. Heisig, H. U. Danzebrink, A. Leyk, W. Mertin, S. Münster and E. Oesterschulze, Ultramicroscopy, 1998, 71, 99–105 CrossRef CAS.
- T. Leinhos, M. Stopka and E. Oesterschulze, Appl. Phys. A: Mater. Sci. Process., 1998, 66, S65–S69 CrossRef CAS.
- H. H. Roh, J. S. Lee, D. L. Kim, J. Park, K. Kim, O. Kwon, S. H. Park, Y. K. Choi and A. Majumdar, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.--Process., Meas., Phenom., 2006, 24, 2398–2404 CrossRef CAS.
- H. H. Roh, J. S. Lee, D. L. Kim, J. Park, K. Kim, O. Kwon, S. H. Park, Y. K. Choi and A. Majumdar, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.--Process., Meas., Phenom., 2006, 24, 2405–2411 CrossRef CAS.
- O. Nakabeppu and T. Suzuki, J. Therm. Anal. Calorim., 2002, 69, 727–737 CrossRef CAS.
- K. Kim, J. Chung, G. Hwang, O. Kwon and J. S. Lee, ACS Nano, 2011, 5, 8700–8709 CrossRef CAS PubMed.
- A. Bontempi, T. P. Nguyen, R. Salut, L. Thiery, D. Teyssieux and P. Vairac, Rev. Sci. Instrum., 2016, 87, 063702 CrossRef PubMed.
- A. Majumdar and J. Varesi, J. Heat Transfer, 1998, 120, 297–305 CrossRef CAS.
- A. Dazzi, R. Prazeres, F. Glotin and J. M. Ortega, Opt. Lett., 2005, 30, 2388–2390 CrossRef CAS PubMed.
- S. Lefèvre, J. B. Saulnier, C. Fuentes and S. Volz, Superlattices Microstruct., 2004, 35, 283–288 CrossRef.
- Y. Ezzahri, L. D. Patiño Lopez, O. Chapuis, S. Dilhaire, S. Grauby, W. Claeys and S. Volz, Superlattices Microstruct., 2005, 38, 69–75 CrossRef CAS.
- P. S. Dobson, J. M. R. Weaver and G. Mills, IEEE Sensors, 2007, pp. 708–711, DOI:10.1109/ICSENS.2007.4388498.
- P. Tovee, M. Pumarol, D. Zeze, K. Kjoller and O. Kolosov, J. Appl. Phys., 2012, 112, 114317 CrossRef.
- E. Puyoo, S. Grauby, J.-M. Rampnoux, E. Rouvière and S. Dilhaire, J. Appl. Phys., 2011, 109, 024302 CrossRef.
- M. Timofeeva, A. Bolshakov, P. D. Tovee, D. A. Zeze, V. G. Dubrovskii and O. V. Kolosov, Ultramicroscopy, 2016, 162, 42–51 CrossRef CAS PubMed.
- F. Menges, P. Mensch, H. Schmid, H. Riel, A. Stemmer and B. Gotsmann, Nat. Commun., 2016, 7, 10874 CrossRef CAS PubMed.
- L. D. P. Lopez, S. Grauby, S. Dilhaire, M. Amine Salhi, W. Claeys, S. Lefèvre and S. Volz, Microelectron. J., 2004, 35, 797–803 CrossRef CAS.
- G. Hwang and O. Kwon, Int. J. Therm. Sci., 2016, 108, 81–88 CrossRef CAS.
- S. Gomes, P. O. Chapuis, F. Nepveu, N. Trannoy, S. Volz, B. Charlot, G. Tessier, S. Dilhaire, B. Cretin and P. Vairac, IEEE Trans. Compon. Packag. Technol., 2007, 30, 424–431 CrossRef CAS.
- G. B. M. Fiege, F. J. Niedernostheide, H. J. Schulze, R. Barthelmeß and L. J. Balk, Microelectron. Reliab., 1999, 39, 1149–1152 CrossRef.
- M. Tortello, S. Colonna, M. Bernal, J. Gomez, M. Pavese, C. Novara, F. Giorgis, M. Maggio, G. Guerra, G. Saracco, R. S. Gonnelli and A. Fina, Carbon, 2016, 109, 390–401 CrossRef CAS.
- L. Shi, J. Zhou, P. Kim, A. Bachtold, A. Majumdar and P. L. McEuen, J. Appl. Phys., 2009, 105, 104306 CrossRef.
- Y.-J. Yu, M. Y. Han, S. Berciaud, A. B. Georgescu, T. F. Heinz, L. E. Brus, K. S. Kim and P. Kim, Appl. Phys. Lett., 2011, 99, 183105 CrossRef.
- F. Menges, H. Riel, A. Stemmer and B. Gotsmann, Nano Lett., 2012, 12, 596–601 CrossRef CAS PubMed.
- S. S. Kharintsev, E. A. Chernykh, A. I. Fishman, S. K. Saikin, A. M. Alekseev and M. K. Salakhov, J. Phys. Chem. C, 2017, 121, 3007–3012 CAS.
- S. Volz, X. Feng, C. Fuentes, P. Guérin and M. Jaouen, Int. J. Thermophys., 2002, 23, 1645–1657 CrossRef CAS.
- Y. Zhang, E. E. Castillo, R. J. Mehta, G. Ramanath and T. Borca-Tasciuc, Rev. Sci. Instrum., 2011, 82, 024902 CrossRef PubMed.
- T. Borca-Tasciuc, Annu. Rev. Heat Transfer, 2013, 16, 211–258 CrossRef.
- P. J. Newby, B. Canut, J.-M. Bluet, S. Gomès, M. Isaiev, R. Burbelo, K. Termentzidis, P. Chantrenne, L. G. Fréchette and V. Lysenko, J. Appl. Phys., 2013, 114, 014903 CrossRef.
- S. Gomès, L. David, V. Lysenko, A. Descamps, T. Nychyporuk and M. Raynaud, J. Phys. D: Appl. Phys., 2007, 40, 6677 CrossRef.
- M. Luna, J. Colchero and A. M. Baró, J. Phys. Chem. B, 1999, 103, 9576–9581 CrossRef CAS.
- M. Chirtoc, J. Gibkes, R. Wernhardt, J. Pelzl and A. Wieck, Rev. Sci. Instrum., 2008, 79, 8 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2017 |