
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A pair of novel Zn(II) enantiomeric coordination polymers based on a chiral multicarboxylate ligand: synthesis, crystal structures and molecular recognition properties†
Lei Gua,
Han-Zhong Zhanga,
Wen-Hong Jianga,
Guang-Feng Houb,
Ying-Hui Yu a and
Dong-Sheng Ma*a
a and
Dong-Sheng Ma*a
aSchool of Chemistry and Materials Science, Heilongjiang University, Harbin 150080, PR China. E-mail: madongsheng@hlju.edu.cn; Fax: +86 451 86609151; Tel: +86 451 86609001
bAgricultural College, Heilongjiang University, Harbin 150080, PR China
First published on 26th September 2017
Abstract
A pair of Zn(II) enantiomeric coordination polymers, 1-R and 1-S, namely, [Zn4((R)-H3L)2(OH)2(H2O)2]·3H2O {H3L= (R)- or (S)-5-(1-carboxyethoxymethyl)-isophthalic acid}, were synthesized via solvothermal reactions. Their structures were determined by single-crystal X-ray diffraction analysis and further characterized by IR spectra, elemental analyses, thermogravimetric analysis (TGA), solid-state circular dichroism (CD) and powder X-ray diffraction (PXRD). The pair of enantiomers crystallized in the same chiral space group, featuring a 3,6-c net and kgd topological type structure with a point symbol of (43)2(46·66·83). Their CD spectra exhibit obvious positive or negative Cotton effects, indicating their homochirality. 1-R and 1-S also proved to be second-harmonic generation (SHG) active. Moreover, their fluorescent selective recognition towards nitro aromatic compounds and metal cations were also investigated.
Introduction
As we all know, chirality is an intrinsic universal feature of various levels of matter. Molecular chirality plays a vital role in many areas.1 In particular, chiral coordination polymers (CCPs) have aroused considerable interest recently as potential candidates for enantiomorphic recognition and separation, asymmetric catalysis, chiral drug delivery, nonlinear optics (NLO), luminescence, and so on.2Currently, adopting homochiral organic ligands as building blocks by the self-assembly process is a reliable and common approach in CCP construction, as the chirality of the ligand could be effectively introduced and retained in the CCPs.3 Accordingly, many efforts have been devoted to the design and synthesis of enantiopure ligands. The carboxylic ligands, especially multicarboxylic ligands have been widely adopted as the organic linkers to construct coordination polymers, as carboxylate donor ligands with various coordination modes could satisfy various geometric requirements of metal centers for the generations of fascinating structures with high dimensions, good thermal stabilities, and versatile topologies.4
As one of ongoing research in our laboratory, several different chiral carboxylate based ligands have been synthesized and investigated.5 Inspired by the aforementioned considerations, we synthesized a pair of chiral tridentate carboxylic acid ligands: (R)- and (S)-5-(1-carboxyethoxymethyl)-isophthalic acid (H3L) (Scheme 1). Choosing a pair of enantiomorphic H3L ligands as the organic spacers is mainly based on the following consideration: (a) as a multicarboxylic ligand with one flexible carboxylic group and two rigid carboxylic groups, the multiple bridging modes of carboxylic moieties increased the possibility of the generation of fascinating structures with high dimensions. (b) A pair of enantiomerically pure ligands ((R)-H3L and (S)-H3L) could efficiently ensure the production of a pair of enantiomorphic CCPs, the comparative investigation of whose structures and functional properties might be interesting and meaningful.
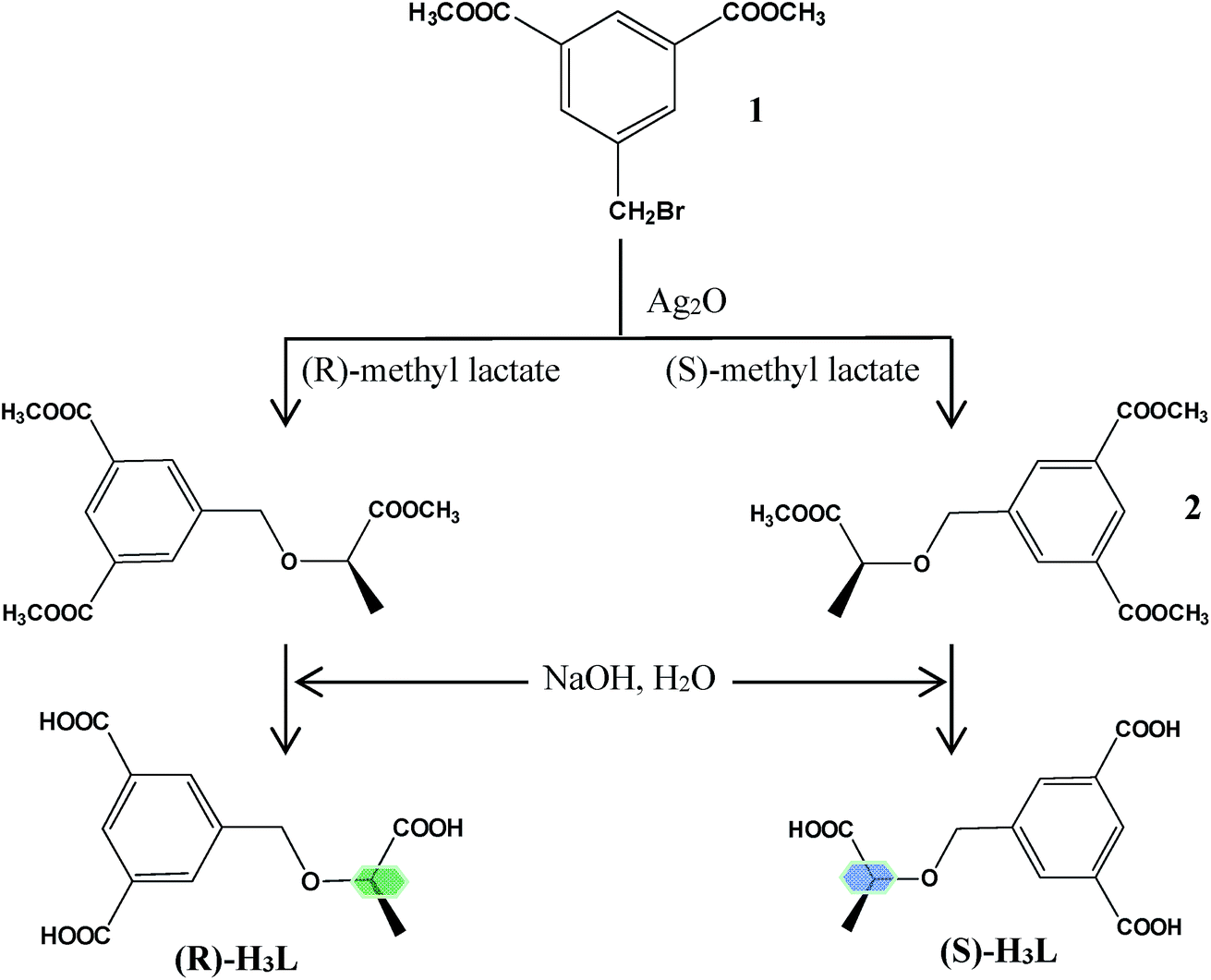 | ||
| Scheme 1 Synthesis route of the enantiopure ligands: (R)-H3L and (S)-H3L. | ||
Based on the above two enantiomers and zinc as the metal center, a pair of CCPs, namely, [Zn4((R)-H3L)2(OH)2(H2O)2]·3H2O (1-R) and [Zn4((S)-H3L)2(OH)2(H2O)2]·2H2O (1-S) were synthesized under hydrothermal conditions. Their crystal structures, thermal stabilities, circular dichroism (CD) spectra, nonlinear optical (NLO) and fluorescent sensing properties were studied.
Experimental section
General methods and materials
All other reagents and solvents were purchased commercially without further purification. The IR spectra were obtained on a Perkin Elmer Spectrum 100 FT-IR spectrometer equipped with a DGTS detector (32 scans) using KBr disks in the range 4000–500 cm−1. Elemental analysis for C, H were performed on a Perkin-Elmer 240 elemental analyzer. Thermal analyses were performed on a SDT 2960 thermal analyzer from room temperature to 800 °C at a heating rate of 20 °C min−1 under nitrogen flow. Powder X-ray diffraction (PXRD) data were collected on a Bruker-D8 Advance X-ray diffractometer with Cu Kα radiation (λ = 1.5406 Å) and 2θ ranging from 5 to 50°. Circular dichroism (CD) spectra of solid samples were recorded at room temperature with a MOS-450 spectrometer (KBr pellets). The second-harmonic generation (SHG) intensity was approximately estimated by measuring microcrystalline samples relative to urea. Solid UV-visible spectra were obtained in the 200–800 nm range on a JASCO UVIDEC-660 spectrophotometer. A pulsed Q-switched Nd:YAG laser at a wavelength of 1064 nm was used to generate a SHG signal from powder samples. The backscattered SHG light was collected by a spherical concave mirror and passed through a filter that transmits only 532 nm radiation.6 The luminescent spectrum was taken an Edinburgh FLS 920 fluorescence spectrophotometer at room temperature.| 1-R | 1-S | |
|---|---|---|
| a R1 = ∑||Fo| − |Fc||/∑|Fo|.b wR2 = {∑[w(Fo2 − Fc2)2/∑w(Fo2)2]}1/2. | ||
| Empirical formula | C24H29Zn4O20.5 | C24H29Zn4O20 |
| Fw | 906.95 | 897.94 |
| Crystal system | Triclinic | Triclinic |
| Space group | P1 | P1 |
| a (Å) | 8.1196(5) | 8.1097(3) |
| b (Å) | 9.4949(7) | 9.5040(4) |
| c (Å) | 10.9553(7) | 10.9547(4) |
| α (deg) | 105.246(6) | 105.0820(10) |
| β (deg) | 108.647(6) | 108.5090(10) |
| γ (deg) | 95.760(6) | 95.9140(10) |
| V (Å3) | 756.22(9) | 757.02(5) |
| Z | 1 | 1 |
| Dcalc (g cm−3) | 1.992 | 1.970 |
| μ (mm−1) | 3.227 | 3.221 |
| F(000) | 457 | 452 |
| Collected/unique | 5327/4333 | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 709/5278 709/5278 |
| R(int) | 0.0198 | 0.0299 |
| GOF on F2 | 1.005 | 1.018 |
| R1a [I > 2σ(I)] | 0.0283 | 0.0267 |
| wR2b [I > σ(I)] | 0.0577 | 0.0666 |
| R1a (all) | 0.0359 | 0.0379 |
| wR2b (all) | 0.0605 | 0.0718 |
| Flack parameters | 0.04(2) | −0.01(2) |
Results and discussion
Structural description
CCPs 1-R and 1-S crystallized in a same chiral space group P1 with a Flack parameter of 0.09(4) and single-crystal XRD analyses revealed that the two compounds are isostructural, thus only the structure of 1-R is described in detail. In an asymmetric unit, there are four crystallographically unique Zn(II) atoms, two deprotonated R-H3L3− ligands, two coordinated water molecules, two μ3-bridging hydroxyl anions and three guest water molecules. The chiral carbon in CCP 1-R retains the same R configuration as that in the starting material of (R)-H3L. The Zn1 ion is 6-coordinated in a distorted octahedron defined by three oxygen atoms from three carboxylate groups from three R-H3L3− ligands, one oxygen atom from ether group of R-H3L3− ligand and two μ3-bridging hydroxyl anions. The Zn2 ion is also in a 6-coordinated distorted octahedron by four oxygen atoms from four carboxylate groups of R-H3L3− ligands, one μ3-bridging hydroxyl anion and one guest water molecule. The coordination forms of Zn3 and Zn4 ion are same as those of Zn1 and Zn2, respectively (Fig. 1(a)). The Zn–O bond lengths range from 1.975(6) to 2.337(7) Å and the O–Zn–O bond angles range from 70.8(3) to 179.4(3), which are all in the normal ranges (Table S1†).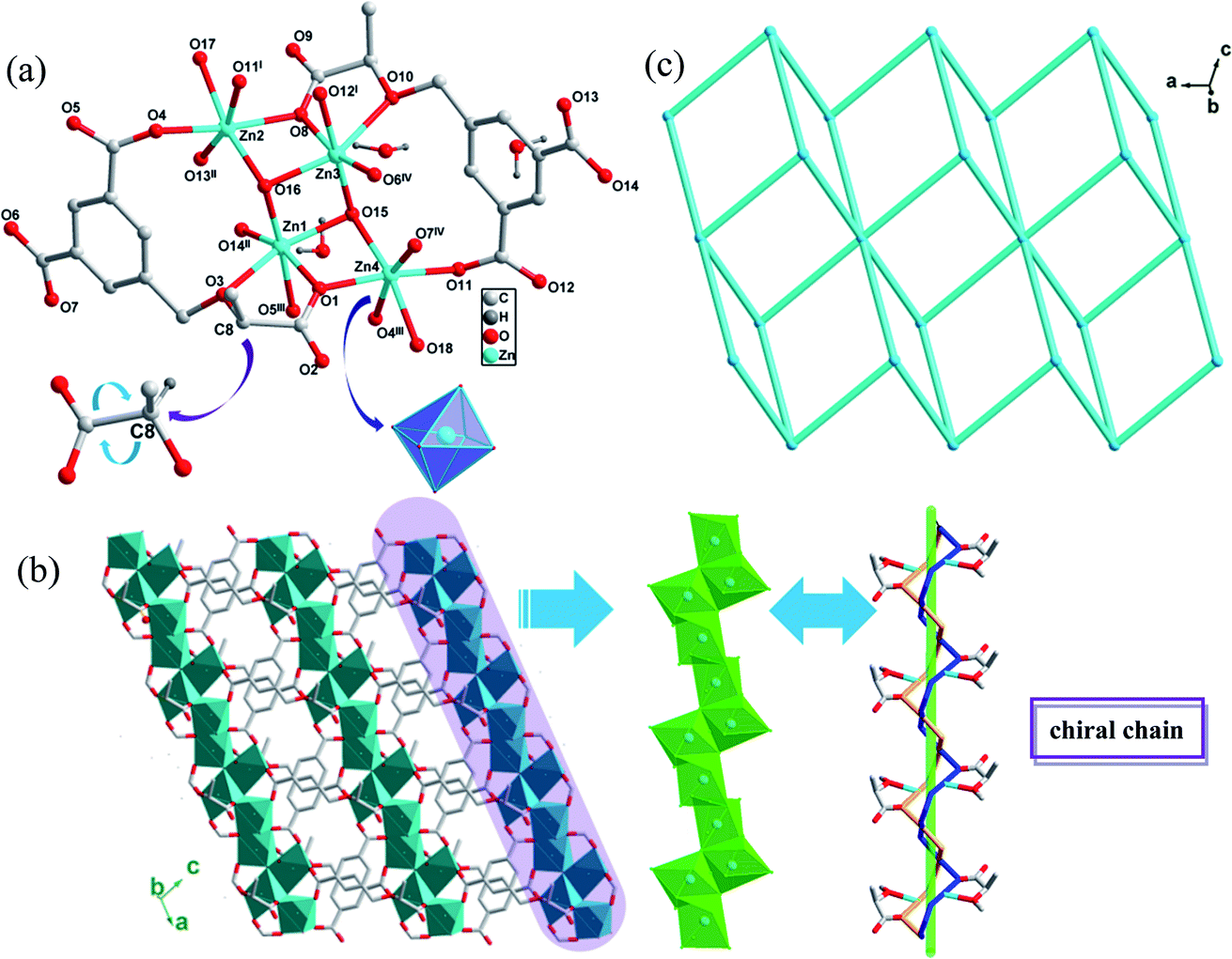 | ||
| Fig. 1 (a) Stick-ball representation of the asymmetric unit of CCP 1-R. (b) 2D framework of 1-R constructed from chiral chains. (c) Schematic view of 2D (3,6)-connected framework with a point symbol of (43)2(46·66·83). Symmetry codes: (I) −1 + x, y, z; (II) −1 + x, y, 1 + z; (III) 1 + x, y, z; (IV) 1 + x, y, 1 + z. | ||
There is an infinite chiral chain in 1-R and 1-S, which is constructed by Zn ions linking carboxylate groups of (R)-H3L or (S)-H3L ligands and μ3-bridging hydroxyl anions. In one [Zn-carboxylate groups] helical tube, there are R direction [Zn2–O9–Zn3–O6–C12–O7–Zn4–O4] fragment, with the Zn–Zn distance between each fragment of 3.5337 Å and the pitch of the helix of 8.12 Å. Another S direction [Zn2–O13–C20–O14–Zn1–O1–Zn4–O11] fragment shares the same pitch of the helix as R direction fragment, with the chiral carbons of (R)-H3L ligands keeping the original direction. Interestingly, the Zn ions are closely connected by μ3-bridging hydroxyl anions, which are indispensable during the construction of chiral chains. The experimental results also confirmed this inference, as it was found that the CCPs could not be generated without the presence of NaOH. Furthermore, the chiral chains connected with each other by the (R)-H3L ligands and expanded indefinitely, resulting in a 2D network structure(Fig. 1(b)). In order to further analyze and understand this 2D structure of 1-R, topological analysis is carried out. If the (R)-H3L3− ligands are considered as 3-connected node and Zn ions are considered as 6-connected notes, the 2D structure of CCP 1-R can be regarded as a 3,6-c net and kgd topological type with a point symbol of (43)2(46·66·83) (Fig. 1(c)).
PXRD patterns and thermal stability analysis
For further confirming the phase purity of the crystalline, the experimental and simulated PXRD patterns of 1-R and 1-S are compared (Fig. S1†). The experimental PXRD patterns corroborate the simulated patterns generated from single-crystal diffraction data, indicating the purity of the crustal samples. The differences in intensity may be due to the preferred orientation of the powder samples. When studying the stability of the CCPs, the existence of bulk water in 1-R and 1-S is also observed by TGA under air atmosphere (Fig. S2†). The weight loss process for 1-R can be divided into two steps. The first weight loss of 9.30% occurring at 50–270 °C corresponds to the total loss of the five water molecules (calcd 9.93%). Then the weight loss of 57.96% occurring at 270–690 °C could be ascribed to the total loss of the two ligand molecules (calcd 59.16). The remaining weight of 32.74% corresponds to the formation of ZnO. The weight loss process for 1-S is similar to that of 1-R (Fig. S3†).The solid-state circular dichroism (CD) spectra analysis
In order to further demonstrate the homochirality of CCPs 1-R and 1-S, solid-state circular dichroism (CD) analysis has been conducted. As shown in Fig. 2, the CD spectrum for the bulk sample of 1-R exhibits a positive Cotton effect with peak at 332 nm, while the bulk sample of 1-S exhibits negative Cotton effect at the same wavelength. The result confirms that the CCPs 1-R and 1-S are homochiral enatiomers.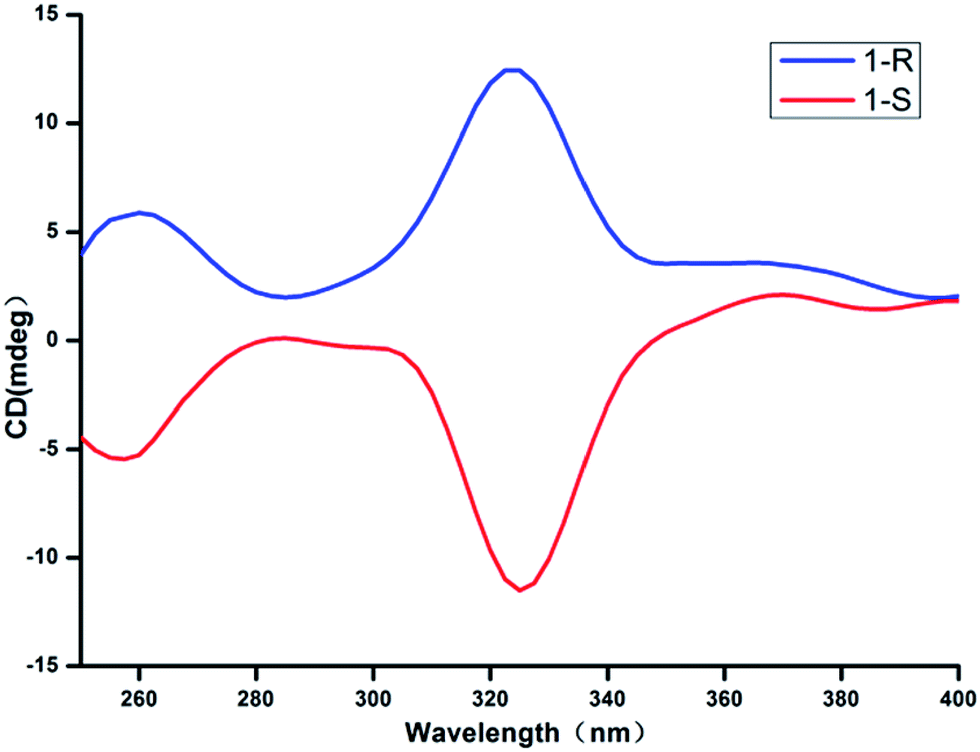 | ||
| Fig. 2 Solid-state CD spectra of CCPs 1-R and 1-S. | ||
Second-harmonic generation (SHG) properties
Nonlinear optical (NLO) active materials have been widely studied and applied in laser frequency conversion and electro-optical devices at present. As one of the most common NLO behaviors, second-harmonic generation (SHG) have been of increasing interest to synthetic chemists.9 SHG active materials has found the most common use in the laser industry. Coordination polymers have been regarded as an attractive category of SHG active materials, as the structures of which could be fine tuned on molecular level. The rational design and synthesis of CCPs in a systematic way might give rise to potential SHG active materials. Supramolecular aggregates and chirality could significantly enhance the nonlinear optical properties.10 And d10 metal, such as Zn and Cd are most adopted for the construction of coordination polymers with SHG activities.11 Due to the fact that the pair of CCPs 1-R and 1-S are crystallized in the non-centrosymmetric chiral space groups and constructed from metal Zn, their SHG activities are investigated. Their SHG properties have been carried out by the Kurtz–Perry method using a powder technique.12 Preliminary experimental results show that 1-R and 1-S are all 0.3 times as much as that of urea. CCPs 1-R and 1-S exhibit lower SHG response than that of urea, which might be resulted from the lacking of a donor–acceptor system in final structures.Fluorescent selective recognition
It was ever reported that the fluorescence of some coordination polymers could be quenched by nitroaromatic compounds under certain conditions, suggesting their potential applications for recognizing nitroaromatic explosives.13 In our investigation, trinitrotoluene (TNT) are used to investigate the luminescent quenching effect of CCPs 1-R and 1-S. The pair of enatiomers 1-R and 1-S show very similar results.The sample of CCP 1-R was dispersed in DMF solution so that the TNT molecules could adhere to the particle surface of CCP, facilitating possible host–guest interactions. As shown in Fig. 3, with the concentration of TNT gradually increased, the luminescence intensity of CCP 1-R decreased significantly. According to the previous reports, such luminescence quenching might be related to the donor–acceptor electron-transfer mechanism. The electrons transfers from an excited CCP 1-R to the electron deficient TNT, resulting in the occurrence of fluorescence quenching of CCP 1-R.14
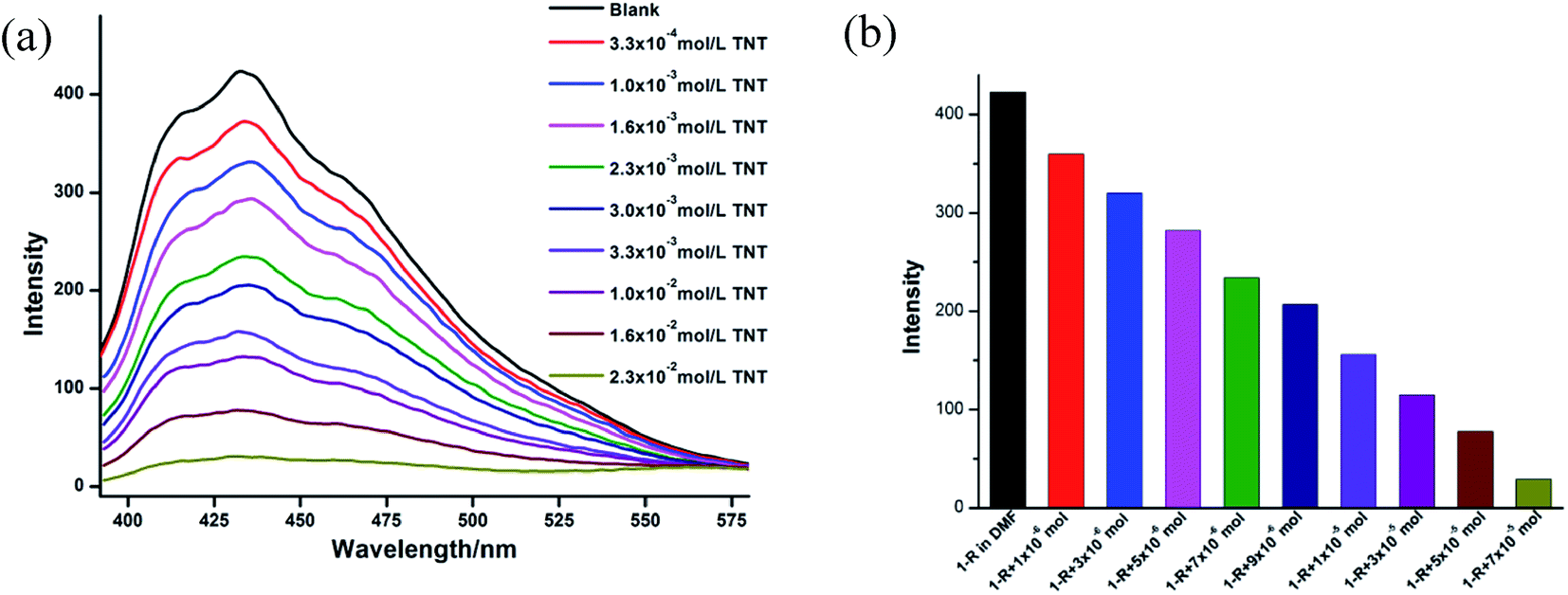 | ||
| Fig. 3 (a) The fluorescent spectra of 1-R in DMF with different amount of TNT. (b) Luminescent intensity diversity at 441 nm of 1-R in DMF with different amount of TNT. | ||
According to some reports, not only nitro aromatic compounds but also metal cations could have an quenching effect toward some coordination polymers.15 In order to further study the fluorescent selective recognition of CCPs, different metal cations are screened to investigate their effect on the fluorescence of CCPs 1-R and 1-S. As a result, CCPs 1-R and 1-S also show very similar selectivity toward the metal cations.
CCP 1-S was added in DMF and stirred for 24 h, so that 1-S was sufficiently dispersed in DMF solution. For the purpose of verifying the selectivity of CCP-1 toward the metal cations, nine metal nitrates, M(NO3)x (M = Ba2+, Cd2+, Pb2+, Co2+, K+, Ni2+, Ag+, Cu2+, Fe3+) are used for screening. The aqueous solution containing 1 × 10−3 mol L−1 M(NO3)x was added to the suspension of CCP 1-S, respectively. The results are shown in Fig. 4. It was found that Fe3+ have the most obviously inhibiting effect on the luminescence of CCP 1-S when adding the same molar amount of various metal cations. The results indicate that CCP 1-S has an obvious selection effect toward Fe3+.
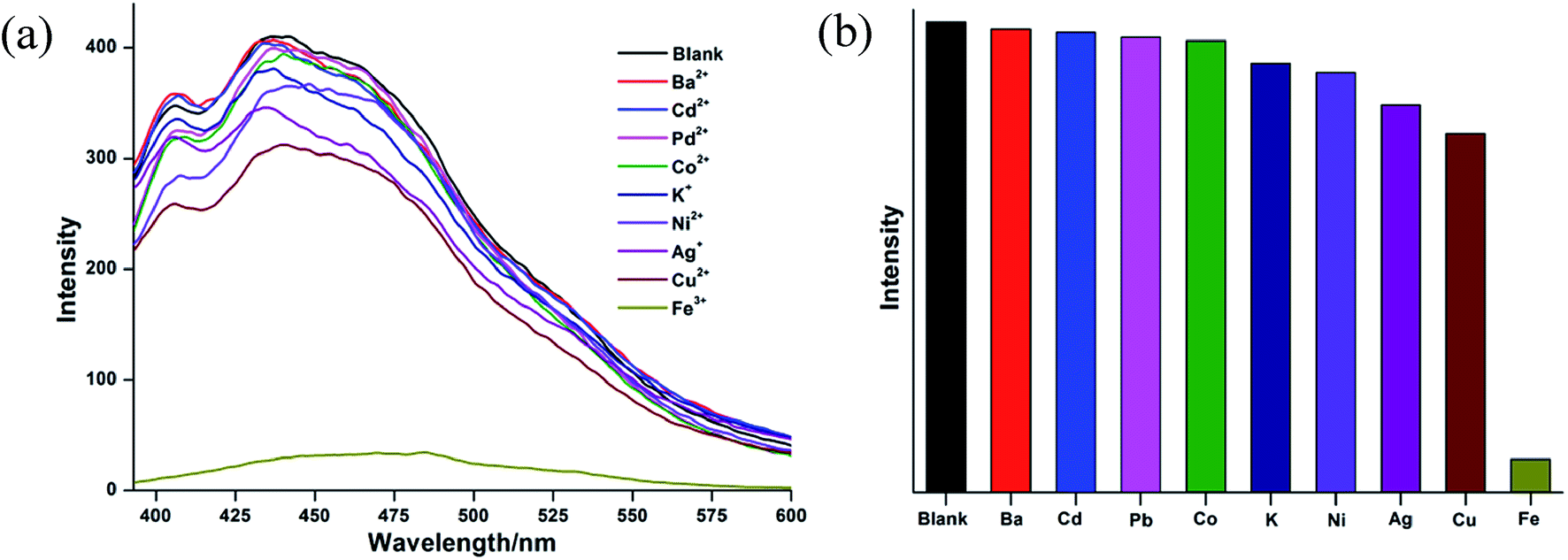 | ||
| Fig. 4 (a) Luminescent spectra of 1-S in the presence of metal cations of 1 × 10−3 mol L−1. (b) Luminescent intensity diversity at 441 nm of 1-S when treated with various cations of 1 × 10−3 mol L−1. | ||
As shown in Fig. 5, as we increase the amount of Fe3+, the luminescence intensity of CCP 1-S decreased obviously. In previous reports, the mechanisms for luminescence quenching effect of Fe3+ ion were proposed as the collapse of the framework, cationic exchange, and the physical interactions between Fe3+ ion and carboxylic acid.16 To further probe the possible luminescence quenching mechanism of Fe3+ ion, we removed the Fe3+ ion from the suspension of CCP 1-S by filtrating and washing it with water and ethanol for several times. The resulted sample was used for powder XRD and luminescence analysis. The results show that the framework of CCP 1-S was retained the same as the original and the luminescence also returned to normal. We inferred that the luminescence quenching mechanism of Fe3+ ion toward CCP 1-S might be ascribed to the physical interactions between Fe3+ ion and carboxylic group of CCP 1-S. This also indicates that CCP 1-S could be recycled in metal recognition.
 | ||
| Fig. 5 (a) The fluorescent spectra of 1-S in DMF with different amount of Fe3+. (b) Luminescent intensity diversity at 441 nm of 1-S in DMF with different amount of Fe3+. | ||
Conclusion
In summary, we synthesized a pair of novel Zn(II) enantiomeric CCPs based on a pair of chiral multicarboxylate ligand, (R)- or (S)-5-(1-carboxyethoxymethyl)-isophthalic acid. The two enantiomeric CCPs 1-R and 1-S are isostructural and also exhibit very similar functional properties. The SHG optical results show that CCPs 1-R and 1-S have medium SHG activities which could be used as potential optical materials. Furthermore, the luminescence of CCP 1 could be effectively quenched by trinitrotoluene and Fe3+, indicating their potential application as sensory materials. At the same time, it was also found that the luminescence of CCP 1 could return to normal after the Fe3+ ion being completely removed from CCP 1, which indicating that CCP 1 could be recycled and used in metal recognition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (No. 21371052 & 21272061).References
- (a) R. Hoffmann, The Same and Not the Same, Columbia University Press, New York, 1995 Search PubMed; (b) G. Keliang and W. Dongzhi, Appl. Microbiol. Biotechnol., 2006, 70, 135–139 CrossRef PubMed; (c) S. Sharma, M. Chauhan, A. Jamsheera, S. Tabassum and F. Arjmand, Inorg. Chim. Acta, 2017, 458, 8–27 CrossRef CAS; (d) M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed; (e) J.-Z. Gu, Y.-H. Cui, J. Wu and A. M. Kirillov, RSC Adv., 2015, 5, 78889–78901 RSC; (f) N. Chen, M.-X. Li, P. Yang, X. He, M. Shao and S.-R. Zhu, Cryst. Growth Des., 2013, 13, 2650–2660 CrossRef CAS; (g) M. Yadav, A. Bhunia, S. K. Jana and P. W. Roesky, Inorg. Chem., 2016, 55, 2701–2708 CrossRef CAS PubMed.
- (a) D. W. Lim, J. W. Yoon, K. Y. Ryu and M. P. Suh, Angew. Chem., Int. Ed., 2012, 51, 9952 CrossRef; (b) C. Wang, T. Zhang and W.-B. Lin, Chem. Rev., 2012, 112, 1084 CrossRef CAS PubMed; (c) J. Zhang and X.-H. Bu, Angew. Chem., Int. Ed., 2007, 119, 6227 CrossRef; (d) J. Crassous, Chem. Soc. Rev., 2009, 38, 830 RSC; (e) T. Hang, D.-W. Fu, Q. Ye and R.-G. Xiong, Cryst. Growth Des., 2009, 9, 2026 CrossRef CAS; (f) X.-L. Zhao, D. Tian, Q. Gao, H.-W. Sun, J. Xu and X.-H. Bu, Dalton Trans., 2016, 45, 1040 RSC.
- (a) G. Echue, I. Hamley, G. C. Lloyd Jones and C. F. Faul, Langmuir, 2016, 32, 9023–9032 CrossRef CAS PubMed; (b) D.-W. Gao, Q. Gu and S.-L. You, ACS Catal., 2014, 4, 2741–2745 CrossRef CAS; (c) S. Yoshioka, Y. Inokuma, M. Hoshino, T. Sato and M. Fujita, Chem. Sci., 2015, 6, 3765–3768 RSC; (d) L. Zhang, T. Wang, Z. Shen and M. Liu, Adv. Mater., 2016, 28, 1044–1059 CrossRef CAS PubMed; (e) D. N. Dybtsev, M. P. Yutkin, E. V. Peresypkina, A. V. Virovets, C. Serre, G. Férey and V. P. Fedin, Inorg. Chem., 2007, 46, 6843 CrossRef CAS PubMed; (f) J. Zhang and X.-H. Bu, Chem. Commun., 2008, 1756 RSC; (g) J. Zhang and X.-H. Bu, Angew. Chem., Int. Ed., 2007, 46, 6115 CrossRef CAS PubMed; (h) J. J. Bodwin, A. D. Cutland, R. G. Malkani and V. L. Pecoraro, Coord. Chem. Rev., 2001, 216–217, 489–512 CrossRef CAS.
- (a) M. Jung, H. Kim, K. Baek and K. Kim, Angew. Chem., Int. Ed., 2008, 47, 5755 CrossRef CAS PubMed; (b) L.-H. Cao, Y.-L. Wei, Y. Yang, H. Xu, S.-Q. Zang, H.-W. Hou and T. C. W. Mak, Cryst. Growth Des., 2014, 14, 1827–1838 CrossRef CAS; (c) Z.-J. Lin, J. Lu, M. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867–5895 RSC; (d) L.-B. Sun, J.-R. Li, W.-G. Lu, Z.-Y. Gu, Z.-P. Luo and H.-C. Zhou, J. Am. Chem. Soc., 2012, 134, 15923 CrossRef CAS PubMed; (e) W.-J. Zhuang, D.-Q. Yuan, D.-H. Liu, C.-L. Zhong, J.-R. Li and H.-C. Zhou, Chem. Mater., 2012, 24, 18 CrossRef CAS; (f) H. Furukawa, J. Kim, K. E. Plass and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 8398 CrossRef CAS PubMed.
- (a) H.-T. Ye, C.-Y. Ren, G.-F. Hou, Y.-H. Yu, X. Xu, J.-S. Gao, P.-F. Yan and S.-W. Ng, Cryst. Growth Des., 2014, 14, 3309–3318 CrossRef CAS; (b) Y.-H. Yu, H.-T. Ye, G.-F. Hou, C.-Y. Ren, J.-S. Gao and P.-F. Yan, Cryst. Growth Des., 2016, 16, 5669–5677 CrossRef CAS; (c) T. Liu, Y.-H. Yu, H.-Z. Zhang, W.-H. Jiang, J.-S. Gao and G.-F. Hou, Cryst. Growth Des., 2017, 17, 1788–1795 CrossRef CAS; (d) J.-H. Huang, G.-F. Hou, D.-S. Ma, Y.-H. Yu, W.-H. Jiang, Q. Huang and J.-S. Gao, RSC Adv., 2017, 7, 18650–18657 RSC.
- R.-G. Xiong, X. Xue, H. Zhao, X.-Z. You, B. F. Abrahams and Z.-L. Xue, Angew. Chem., Int. Ed., 2002, 41, 3800 CrossRef CAS PubMed.
- (a) J. Xu, R. Wang, Y. Li, Z. Gao, R. Yao, S. Wang and B. Wu, Eur. J. Inorg. Chem., 2012, 3349–3360 CrossRef; (b) T. Routasalo, J. Helaja, J. Kavakka and A. M. P. Koskinen, Eur. J. Org. Chem., 2008, 3190 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- (a) E. Cariati, G. Cavallo, A. Forni, G. Leem, P. Metrangolo, F. Meyer, T. Pilati, G. Resnati, S. Righetto, G. Terraneo and E. Tordin, Cryst. Growth Des., 2011, 11, 5642–5648 CrossRef CAS; (b) S. Rosenne, E. Grinvald, E. Shirman, L. Neeman, S. Dutta, O. Bar-Elli, R. Ben-Zvi, E. Oksenberg, P. Milko, V. Kalchenko, H. Weissman, D. Oron and B. Rybtchinski, Nano Lett., 2015, 15, 7232–7237 CrossRef CAS PubMed.
- (a) T. Verbiest, J. Mater. Chem., 1999, 9, 2005–2012 RSC; (b) T. Verbiest, Science, 1998, 282, 913–915 CrossRef CAS PubMed.
- (a) S.-Q. Zang, Y. Su, Y.-Z. Li and Z.-P. Ni, Inorg. Chem., 2006, 45, 174–180 CrossRef CAS PubMed; (b) S.-L. Zheng, J.-H. Yang, X.-L. Yu, X.-M. Chen and W.-T. Wong, Inorg. Chem., 2004, 43, 830–838 CrossRef CAS PubMed.
- (a) N. Nwaji, J. Mack, J. Britton and T. Nyokong, New J. Chem., 2017, 41, 2020–2028 RSC; (b) S. Kujala, A. Mannila, L. Karvonen, K. Kieu and Z. Sun, Sci. Rep., 2016, 6, 223 Search PubMed.
- (a) S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153–4155 CrossRef CAS PubMed; (b) C. Zhang, Y. Yan, L. Sun, Z. Liang and J. Li, CrystEngComm, 2016, 18, 4102–4108 RSC; (c) M. E. Germain and M. J. Knapp, Chem. Soc. Rev., 2009, 38, 2543–2555 RSC.
- (a) Q. Zheng, F. Yang, M. Deng, Y. Ling, X. Liu, Z. Chen, Y. Wang, L. Weng and Y. Zhou, Inorg. Chem., 2013, 52, 10368 CrossRef CAS PubMed; (b) B. Gole, A. K. Bar and P. S. Mukherjee, Chem. Commun., 2011, 47, 12137–12139 RSC; (c) G. Y. Wang, L. L. Yang, Y. Li, H. Song, W. J. Ruan, Z. Chang and X. H. Bu, Dalton Trans., 2013, 42, 12865–12868 RSC.
- (a) Q. Tang, S. Liu, Y. Liu, J. Miao, S. Li, L. Zhang, Z. Shi and Z. Zheng, Inorg. Chem., 2013, 52, 2799–2801 CrossRef CAS PubMed; (b) Y. J. Cui, Y. F. Yue, G. D. Qian and B. L. Chen, Luminescent functional metal–organic frameworks, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed.
- (a) M. Zheng, H. Tan, Z. Xie, L. Zhang, X. Jing and Z. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 1078–1083 CrossRef CAS PubMed; (b) G. P. Li, G. Liu, Y. Z. Li, L. Hou, Y. Y. Wang and Z. Zhu, Inorg. Chem., 2016, 55, 3952–3959 CrossRef CAS PubMed; (c) M.-M. Chen, X. Zhou, H.-X. Li, X.-X. Yang and J.-P. Lang, Cryst. Growth Des., 2015, 15, 2753–2760 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 and Fig. S1–S6. CCDC 1565357 and 1565358 for 1-R and 1-S. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra08433a |
| This journal is © The Royal Society of Chemistry 2017 |