
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Is nano ZrO2 a better photocatalyst than nano TiO2 for degradation of plastics?
W. R. L. Nisansala Bandaraab,
Rohini M. de Silva *a,
K. M. Nalin de Silvaab,
Damayanthi Dahanayakeb,
Sunanda Gunasekarab and
Kulatheepan Thanabalasingamb
*a,
K. M. Nalin de Silvaab,
Damayanthi Dahanayakeb,
Sunanda Gunasekarab and
Kulatheepan Thanabalasingamb
aDepartment of Chemistry, Faculty of Science, University of Colombo, Colombo 03, Sri Lanka. E-mail: rohini@chem.cmb.ac.lk
bSri Lanka Institute of Nanotechnology (SLINTEC), Nanotechnology and Science Park, Mahenwatta, Pitipana, Homagama, Sri Lanka
First published on 28th September 2017
Abstract
The environmental accumulation of plastic is a huge problem due to its low degradability. There are solutions to this problem such as reusing, recycling and the use of biodegradable plastics. However, a complete solution to this problem has not yet been achieved. In this study, photocatalytic degradation of polyethylene and polyropylene was investigated. The effect of ZrO2 nanoparticles in comparison with TiO2 nanoparticles for photocatalytic degradation was studied. TiO2 nanoparticles were synthesized by a sol gel method and ZrO2 nanoparticles were synthesized by a sonochemical method. The TiO2 and ZrO2 particles were characterized using FTIR, XRD, UV visible spectroscopy, EDX, SEM, and TEM. Both types of particles were approximately 50 nm in size. TiO2 nanoparticles were tetragonal and in the anatase phase. ZrO2 particles were tetragonal and nano porous. The application of these particles to polyethylene and polypropylene was performed using nanoparticle suspensions in a THF medium. The degradation of the plastics was studied by investigating chemical changes using FTIR and morphological changes using SEM. Optimization of the concentration and exposure time was performed under laboratory conditions using a sun simulator. Polyethylene and polypropylene were treated under the sun simulator as well as under the real sun light conditions. In both treatment conditions, it was found that there is a significant difference in the degradation of plastics and ZrO2 nanoparticle suspension treated polyethylene and polypropylene showed higher degradation than the TiO2 nanoparticle suspension treated samples at 95% confidence levels.
1. Introduction
Plastics, including polyolefins, are manmade long chain polymer molecules, which are widely used in manufacturing kitchen appliances, furniture, toys, etc. With the increasing demand for plastic products, these materials are being synthesized on a large scale. In addition, properties such as light weight, hydrophobicity, transparency, chemical resistance, durability, flexibility, and ease of cleaning have made these materials attractive for various applications. Currently plastics have become the most widely manufactured materials in the world due to these properties.Basic raw materials for the synthesis of plastics come from extractions from oil, coal and natural gases. Plastics have been categorized into different types such as polyethylene (PE), polypropylene (PP), polystyrene (PS), polyvinylchloride (PVC), polyurethane (PU), polyethylene terephthalate (PET), polybutylene terephthalate (PBT) and nylons, depending on the monomer unit present in the polymer.1
Plastics have poor biodegradability and their waste persists in the environment for many years. This leads to a major environmental threat in many parts of the world.2 Accumulated plastic waste may have adverse health effects, and minimizes the efficient use of land. Plastic pollution in the terrestrial and the marine environment presents a direct threat to wildlife and marine life.
Land filling has been a traditional solution to plastic waste management.3 However, this has been identified as a temporarily solution, because of the increasing rate of plastic waste accumulation. Therefore attention has been given to recycling, reusing and reducing the use of plastics. Although these kinds of steps have been taken as solutions to the plastic waste problem, they do not adequately address the ever growing amount of plastic waste originating from households. As a result, bio-degradable plastics were developed.4 Nevertheless plastic pollution is still a great threat to the environment. Photocatalytic degradation of plastic can be a viable option. This is a low temperature process with economic advantages.5
TiO2 is an inorganic oxide which exists in three different phases, namely, anatase, rutile, and brookite. Anatase and rutile are the active crystalline phases of TiO2.6 TiO2 has a band gap varying from 3.0 eV to 3.2 eV (ref. 7) and it is a nontoxic semiconductor. Because of the high photo catalytic activity of TiO2, it has been used for various applications such as for water and air purification,8 degradation of organic compounds,9 dye sensitized solar cells,10 sterilization,11 disinfection,12 self-cleaning13 fabrics, and photo induced water splitting.14 The photo catalytic activity of TiO2 depends on the crystalline structure, crystalline size and the morphology. Due to characteristics such as inexpensiveness, good photo stability, non-toxicity and high reactivity, TiO2 has been generally regarded as a good photocatalyst.15
ZrO2 is a transition metal oxide which is nontoxic, chemically inert and thermally stable. Pure ZrO2 exists in three crystallographic phases namely monoclinic, tetragonal and cubic.16 ZrO2, has a wide band gap. The band gap energy of nano ZrO2 particles varies from 3.3 eV to 5.1 eV.17 Zirconia has useful chemical and physical properties such as high thermal and chemical stability, low thermal conductivity, high corrosion resistance, high strength and fracture toughness. Hence zirconia is used in oxygen sensors,18 fuel cells,19 catalyst and catalytic supports,20–23 high dielectric material24 for large scale integrated circuits and as gate dielectric in metal oxide semiconductors.
Currently nanomaterials have been identified as potential catalysts to degrade plastics with enhanced properties. Metal oxide nanoparticles have been blended with the plastics in order to enhance the degradation by the photo catalytic effect. Use of TiO2 in degradation of plastics is well known. However, use of pure ZrO2 has not been recorded before. This study is focused on the investigation of the catalytic ability of ZrO2 as compared to TiO2, towards the degradation of polyethylene and polypropylene.
2. Experimental section
2.1 Materials
Polyethylene (gauge size 80) and polypropylene (gauge size 200) samples were purchased from the local market, Sri Lanka. Titanium tetraisopropoxide (TTIP, 97%), zirconium nitrate (99%), potassium iodide (99%), phthalic acid (99.5%), tetrahydrofuran (THF, 99.9%), methanol (99.9%), ethanol (99.8%), and nitric acid (70%) were all analytical grade and purchased from Sigma Aldrich.2.2 Synthesis of TiO2 nanoparticles
A volume of 5 ml TTIP (molecular weight 283.8 g mol−1) was dissolved in 15 ml of isopropanol and stirred for 1 hour at 1000 rpm at room temperature. A solution of nitric acid at pH 2 (250 ml) was prepared using deionized water and HNO3. The acid solution was added drop wise to the mixture while stirring. The white slurry was aged for 48 hours. This suspension was centrifuged at 9000 rpm for 30 minutes. The resulting precipitate was washed twice with ethanol. The resulting precipitate was heated for 2 hours at 100 °C. Then the solid was heated at 450 °C for 1 hour.62.3 Synthesis of ZrO2 nanoparticles
Zr(NO3)4·3H2O (10.17 g, 30 mmol) and KI (5.1 g, 30 mmol) were mixed with 30 ml of methanol. Isophthalic acid (5.1 g, 10 mmol) was also dissolved in 30 ml of methanol. Both solutions were sonicated separately for 30 minutes. Then the two solutions were mixed together and sonicated for further 30 minutes. The mixture was centrifuged for 15 minutes at 400 rpm. The resulting yellow precipitate was washed twice with deionized water. Then it was washed with acetone. The precipitate was decomposed at 220 °C for 1 hour. Then it was heated at 700 °C for 4 hours in the muffle furnace.252.4 Preparation of nanoparticle dispersion
The TiO2 and ZrO2 nanoparticle solutions were prepared to following concentrations: 6000 ppm, 7000 ppm, 8000 ppm, 9000 ppm, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm, 11000 ppm and 20000 ppm. This was done by dispersing 6, 7, 8, 9, 10, 11, and 20 mg of nanoparticles, respectively, in 1.0 ml of THF, and sonicating for 30 min.
000 ppm, 11000 ppm and 20000 ppm. This was done by dispersing 6, 7, 8, 9, 10, 11, and 20 mg of nanoparticles, respectively, in 1.0 ml of THF, and sonicating for 30 min.
2.5 Application of the dispersion of nanoparticles to polyethylene and polypropylene samples
Polyethylene and polypropylene samples were cut into 1.5 cm × 1.5 cm squares. A volume of 200 μL of each dispersion of nanoparticles was spread on a plastic square. The nanoparticle suspensions with concentrations of 6000 ppm, 7000 ppm, 8000 ppm, 9000 ppm, 10000 ppm, 11000 ppm and 20000 ppm were used to find the optimize concentration. After the evaporation of the tetrahydrofuran, the treated samples were kept under the sun simulator and under the real sunlight. The samples were kept for 20 hours, 40 hours, 60 hours, 80 hours and 100 hours under the sun simulator for time optimization. One hour under the sun simulator is equivalent to 10 hours under the real sunlight. The samples were treated under the real sunlight, 4 hours per day (9 am to 1 pm) for consecutive days until it completes 20 hours and 100 hours. The applied nanoparticle coating was washed by the tap water before taking the FTIR measurements.
2.6 Determination of plastic degradation
The qualitative and quantitative detection of the carbonyl group which appears as the degradation of polymer was done by the absorption mode of FTIR studies on each treated plastic sample using FTIR in ATR-IR mode. For each sample, a number of 64 scans with resolution 4 cm−1 were recorded in absorbance units from 4000 cm−1 to 600 cm−1. The Carbonyl Index (CI) of each plastic sample was calculated. The morphological studies of the polymer surface was done by obtaining SEM images of the samples.2.7 Characterization
The sizes and morphologies of the synthesized TiO2 and ZrO2 nanoparticles were analyzed using a Hitachi SU6600 Scanning Electron Microscope with EDX system (Thermo Scientific) and JEOL JEM-2100 Transmission Electron Microscope at 200 kV.The phase purity of the products was verified by a Bruker D8 PXRD using Cu Kα−1 radiation.
Bruker FTIR spectrometer of the vertex series was used to characterize the nanoparticles including the determination of degradation of plastic using absorption spectrum. Sixty-four scans of symmetrical interferograms were averaged and the spectrum was calculated from 4000 to 600 cm−1 at a spectral resolution of 4 cm−1. For the integration measurements of the carbonyl peak and the CH2 peak OPUS version 6.0 software was used.
US 900 Sun simulator was used to simulate the sun light.
A Shimadzu UV visible spectrophotometer was used for the determination of the band gap energy of the prepared nanoparticles.
3. Results and discussion
3.1 Characterization of TiO2 and ZrO2 nanoparticles
In the FTIR spectrum of TiO2 the band centred around 3400 cm−1 and 1650 cm−1 in Fig. 1(a) are due to the stretching and bending vibrations of hydroxyl groups. The peak around 3400 cm−1 is ascribed to the hydroxyl group of Ti–OH. The weak peak around 1650 cm−1 is associated with the deformation vibration of H–O–H bonds which results due to the fact the spectra were recorded in situ and re-desorption of water from the atmosphere is possible.26–28 The peak centred around 550 cm−1 is due to the stretching vibration of Ti–O bond. The peaks around 1450 cm−1 are a result of Ti–O–Ti stretching vibrations.6 This is in good agreement with the published data.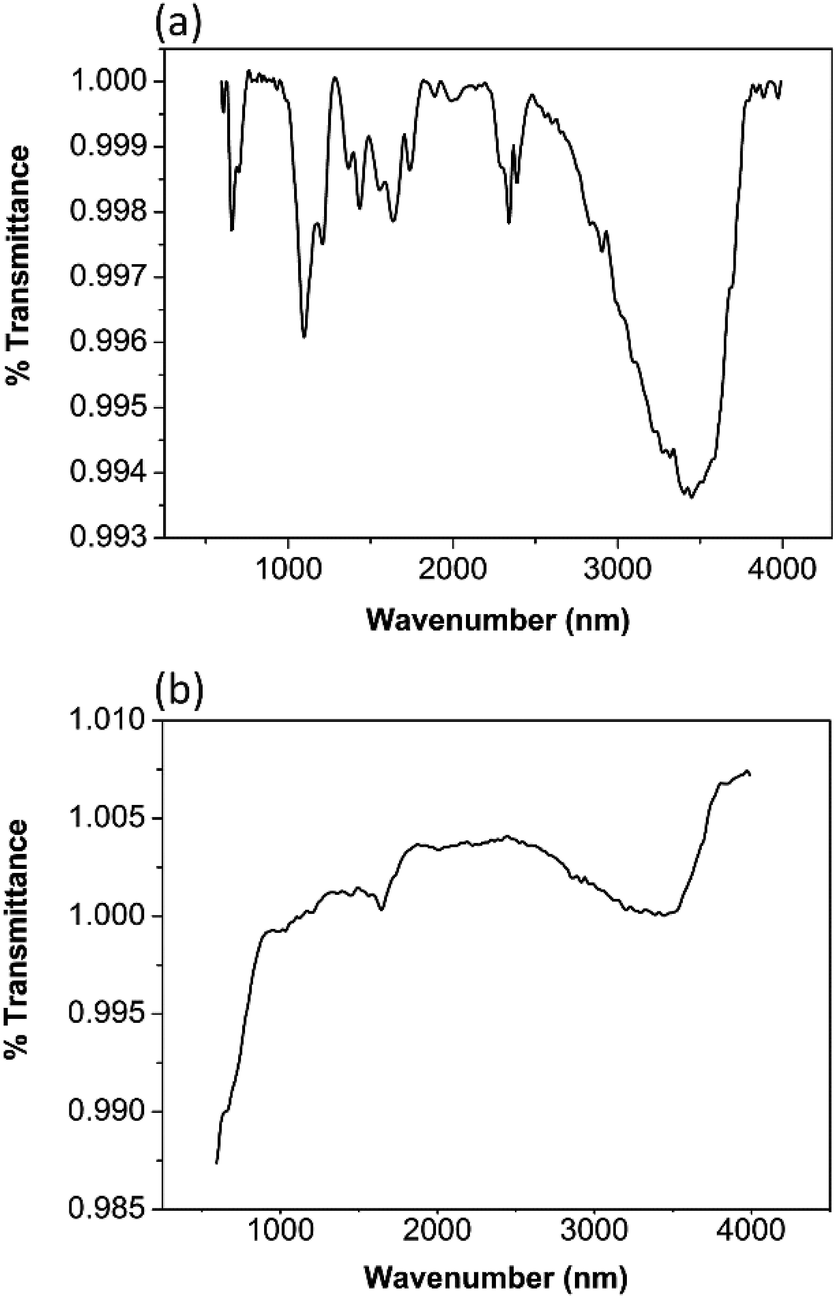 | ||
| Fig. 1 FTIR of (a) TiO2 (b) ZrO2. | ||
In the FTIR spectra of ZrO2 the broad peak around 3400 cm−1 and the sharp peaks around 1650 cm−1 in Fig. 1(b) are due to the stretching and bending vibrations of the hydroxyl groups resulting from the absorption of the water molecules. The band around 1400 cm−1 is due to the adsorption of non-bridging OH groups.29,30 The peaks around 540 cm−1 and 700 cm−1 arise from the Zr–O vibrations of the tetragonal ZrO2.29,31 A sharp band around 750 cm−1 is characteristic for monoclinic ZrO2.30 Interestingly, a peak near 750 cm−1 cannot be seen in this spectrum. Therefore, it can be presumed that monoclinic ZrO2 is not present in the sample.
The X-ray diffractogram of a nano TiO2 sample in Fig. 2(a) exhibits the pattern that is similar to anatase TiO2. All the peaks are in good agreement with JCPDS #89-4921. The XRD pattern of the prepared sample showed peaks at 25.1°, 37.7°, 47.7°, and 53.8° corresponding to the tetragonal anatase crystal planes of (101), (004), (200) and (105) respectively.6,14 It is noted that the XRD peaks of nano TiO2 are broader than those of bulk TiO2 as expected, due to the smaller crystalline size compared to the bulk.32 The average crystallite size of the tetragonal phase of anatase TiO2 calculated from the (101) diffraction peak was found to be 8.20 nm. The d spacing value for the prominent peak of the 101 plane is 0.35 nm.
 | ||
| Fig. 2 XRD of (a) TiO2 (b) ZrO2. | ||
The X-ray diffractogram of a prepared ZrO2 sample in Fig. 2(b) is phase pure, agreeing well with JCPDS #80-0965. High diffraction peaks at 30.1°, 35.1°, 50.2° and 60.4° correspond to the (101), (110), (112) and (211) planes, and low intensity peaks at 33.6°, 50.8°, and 59.3° correspond to the (002), (200), and (103) tetragonal phase of ZrO2 respectively. However, the assignment of cubic and tetragonal structures based solely on the X-ray diffraction analysis can be misleading as the diffraction peaks of cubic phase coincide with the major peaks in the tetragonal phase.33 It can be verified that the prepared sample contains tetragonal phase with the presence of characteristic splitting of the diffraction peaks, whereas the cubic phase exhibits only single peaks. Thus our ZrO2 sample has a tetragonal phase or it might be a mixture of tetragonal phase and cubic phase.33 The average crystallite size of the prepared ZrO2 calculated from (101) diffraction peak was estimated as 10.97 nm. The d spacing value of the prominent (101) plane is 0.29 nm.
According to the UV visible spectrum of TiO2 nanoparticles in Fig. 3(a), it has an absorbance due to band gap transition at 413 nm. The band gap energy was calculated as 3.0 eV.34 It is reported in the literature that the anatase phase has a band gap of 3.2 eV.7,35
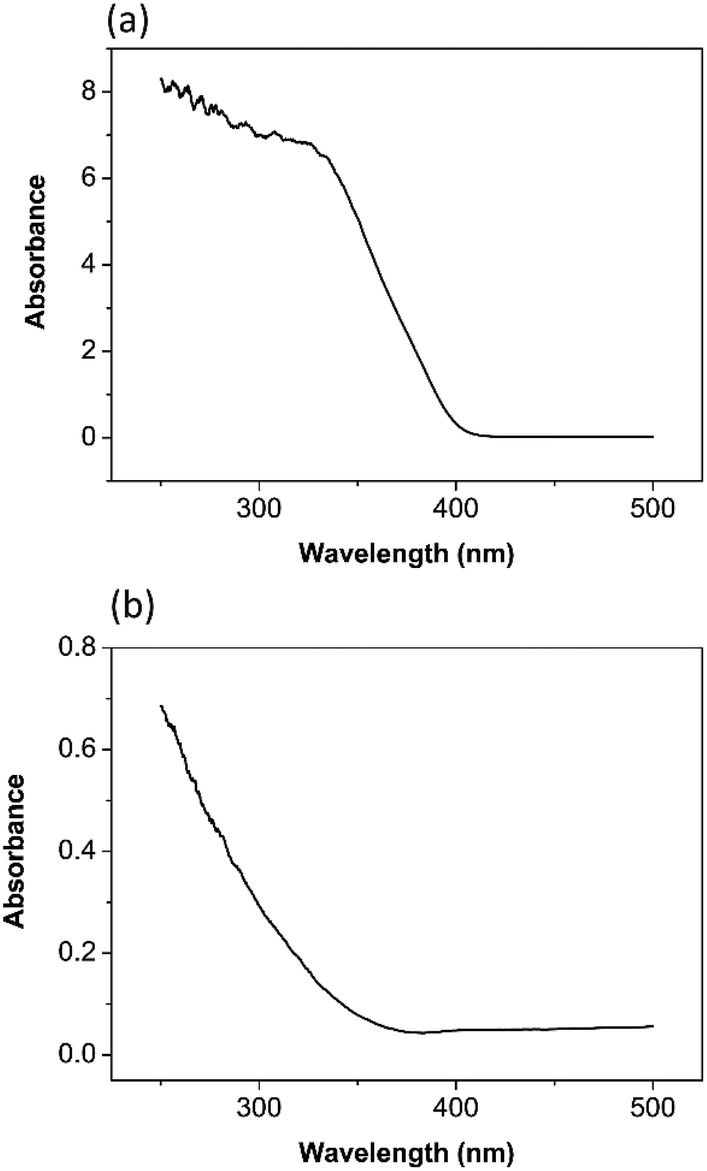 | ||
| Fig. 3 UV/vis spectroscopy of (a) TiO2 (b) ZrO2. | ||
The UV visible spectrum of ZrO2 nanoparticles in Fig. 3(b) has an absorbance due to band gap transition at 365 nm. The band gap energy was calculated as 3.4 eV.36 Basahel33 estimated the band gap energy for monoclinic ZrO2 as 3.25 eV, tetragonal ZrO2 as 3.58 eV, and cubic ZrO2 as 4.33 eV. Comparison with this literature data provides evidence that the tetragonal phase of ZrO2, rather than the cubic, has been synthesized by the procedure described above.
According to the SEM micrograph of the prepared TiO2 nanoparticles in Fig. 4(a) and ZrO2 nanoparticles in Fig. 4(c) it can be confirmed that the particle size is less than 100 nm. Heavy agglomeration was observed in both particles, which may be due to the calcination process, as previously observed.37
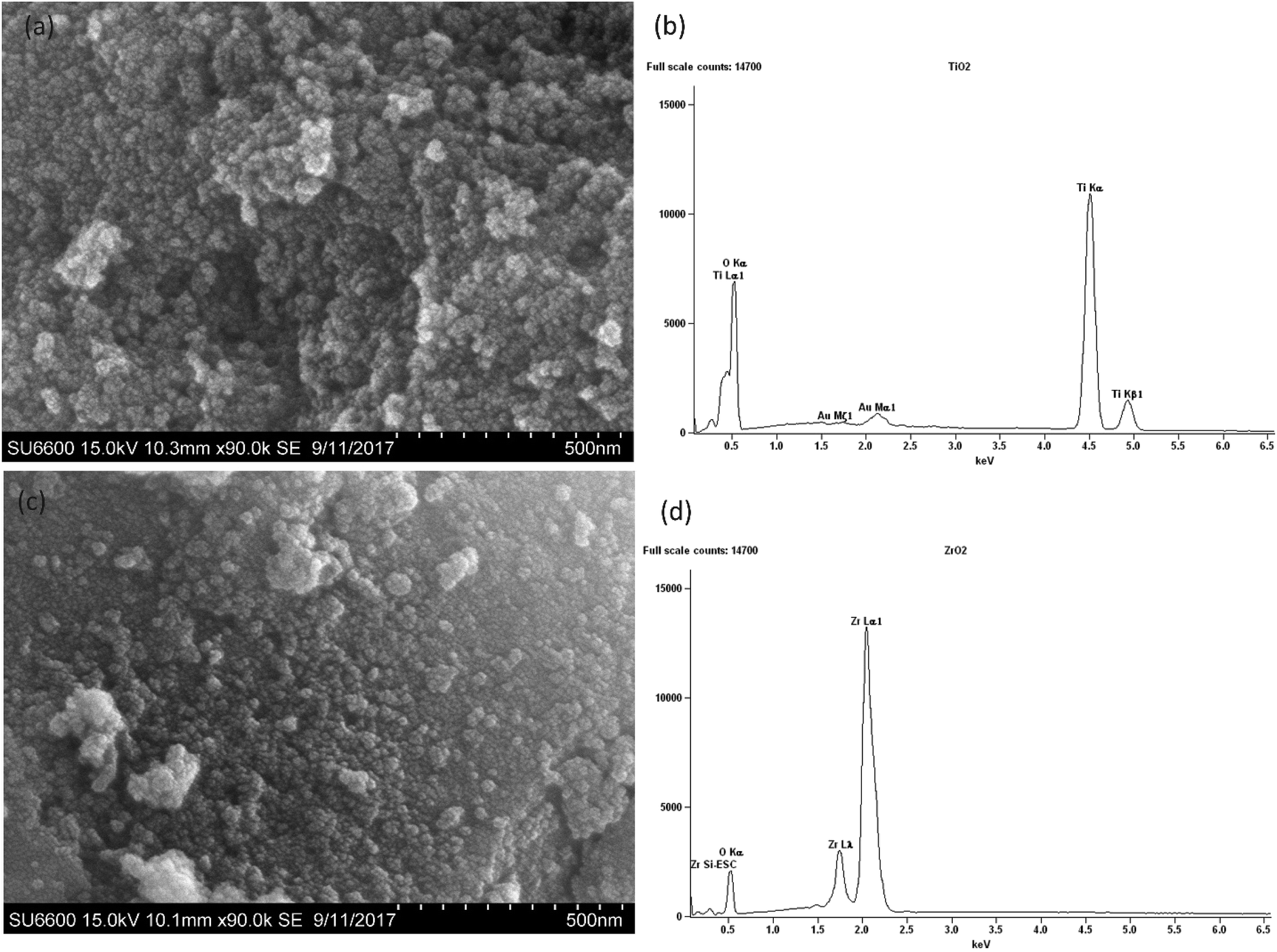 | ||
| Fig. 4 SEM images of nano (a) TiO2 (c) ZrO2, EDX analysis of (b) TiO2 (d) ZrO2. | ||
However it can be observed that the majority of TiO2 particles and ZrO2 particles are around 50 nm in size.
The EDX spectra of the prepared nanoparticles show the elemental composition of the sample. The spectrum of TiO2 in Fig. 4(b) confirms the presence of Ti and O,38 while the spectrum of ZrO2 in Fig. 4(d) confirms the presence of Zr and O.38
According to the TEM images shown in Fig. 5(a) and (b) both TiO2 and ZrO2 particle shapes are irregular and agglomerations are clearly visible. The particle sizes of TiO2 and ZrO2 vary from 15 nm to 55 nm and 5 nm to 60 nm respectively. High resolution TEM image of TiO2 and ZrO2 nanoparticles are given in the Fig. 5(c) and (d) which clearly show the well resolved equidistant lattice fringes. The atomic interlayer distance of TiO2 was calculated to be 0.36 nm (Fig. 5(e)) which can be attributed to the interplanar spacing corresponding to the 101 plane of the anatase phase of TiO2. The atomic interlayer distance of ZrO2 was calculated to be 0.29 nm (Fig. 5(f)) which can be attributed to the interplanar spacing corresponding to the 101 plane of the tetragonal phase of ZrO2. It can be clearly seen in Fig. 5(b) that ZrO2 has a nanoporous structure and the average pore size varies from 3 nm to 10 nm.
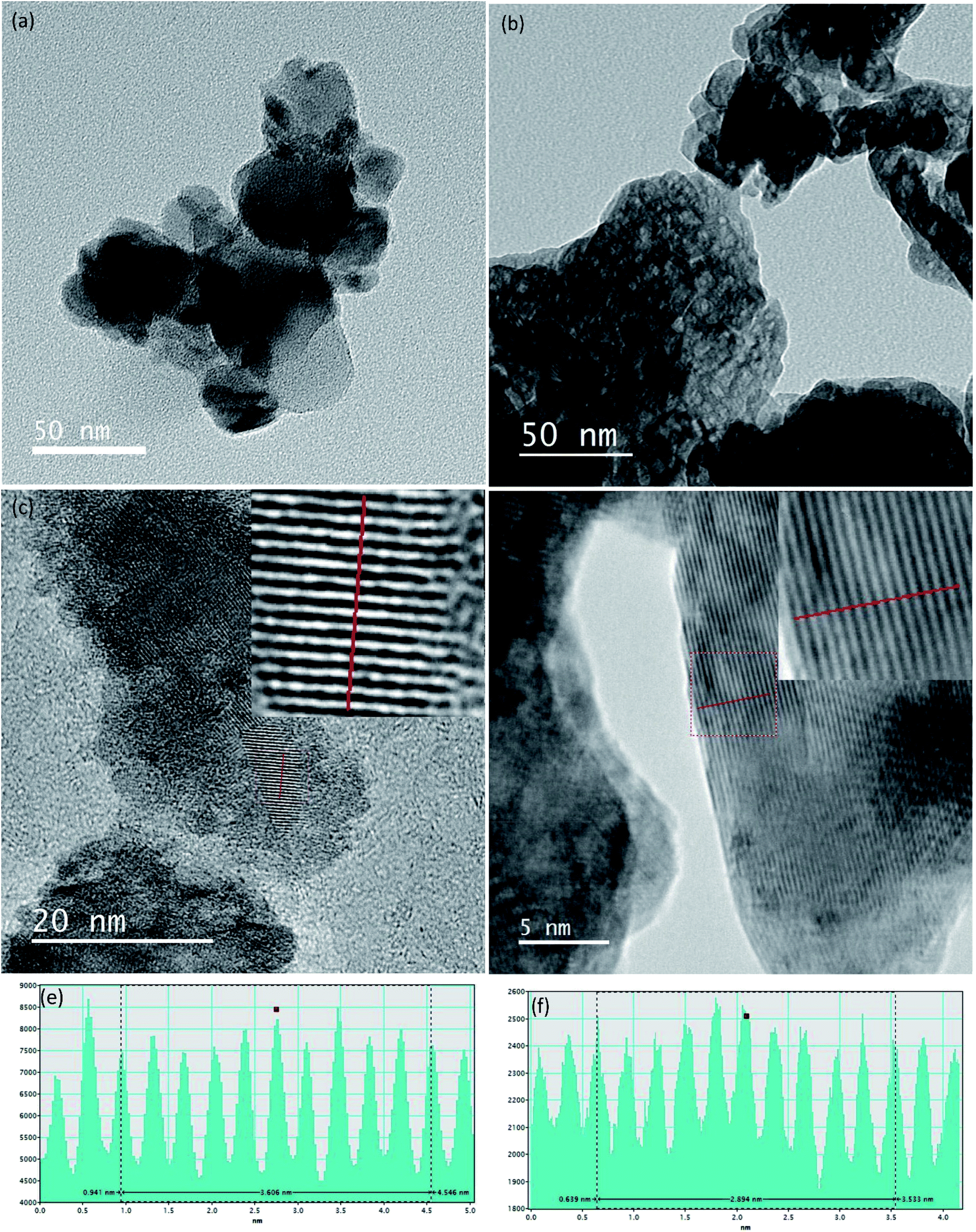 | ||
| Fig. 5 TEM images of nano (a) TiO2 (b) ZrO2, HRTEM image of nano (c) TiO2 (d) ZrO2, line profile of the HRTEM image (e) TiO2 (f) ZrO2. | ||
3.2 Degradation of polyethylene (PE) and polypropylene (PP)
The degradation of PE and PP was determined by morphological studies and statistical studies. The morphology of the PE and PP was studied using SEM images. The carbonyl index was calculated using FTIR data.000 ppm TiO2 suspension and kept under the sun simulator for 100 hours (Fig. 6(h)) showed more degradation than the surface of the PE sample in Fig. 6(g) which kept under the same light source for 20 hours. Thus the damage to the PE surface increases with increased exposure to the light source.
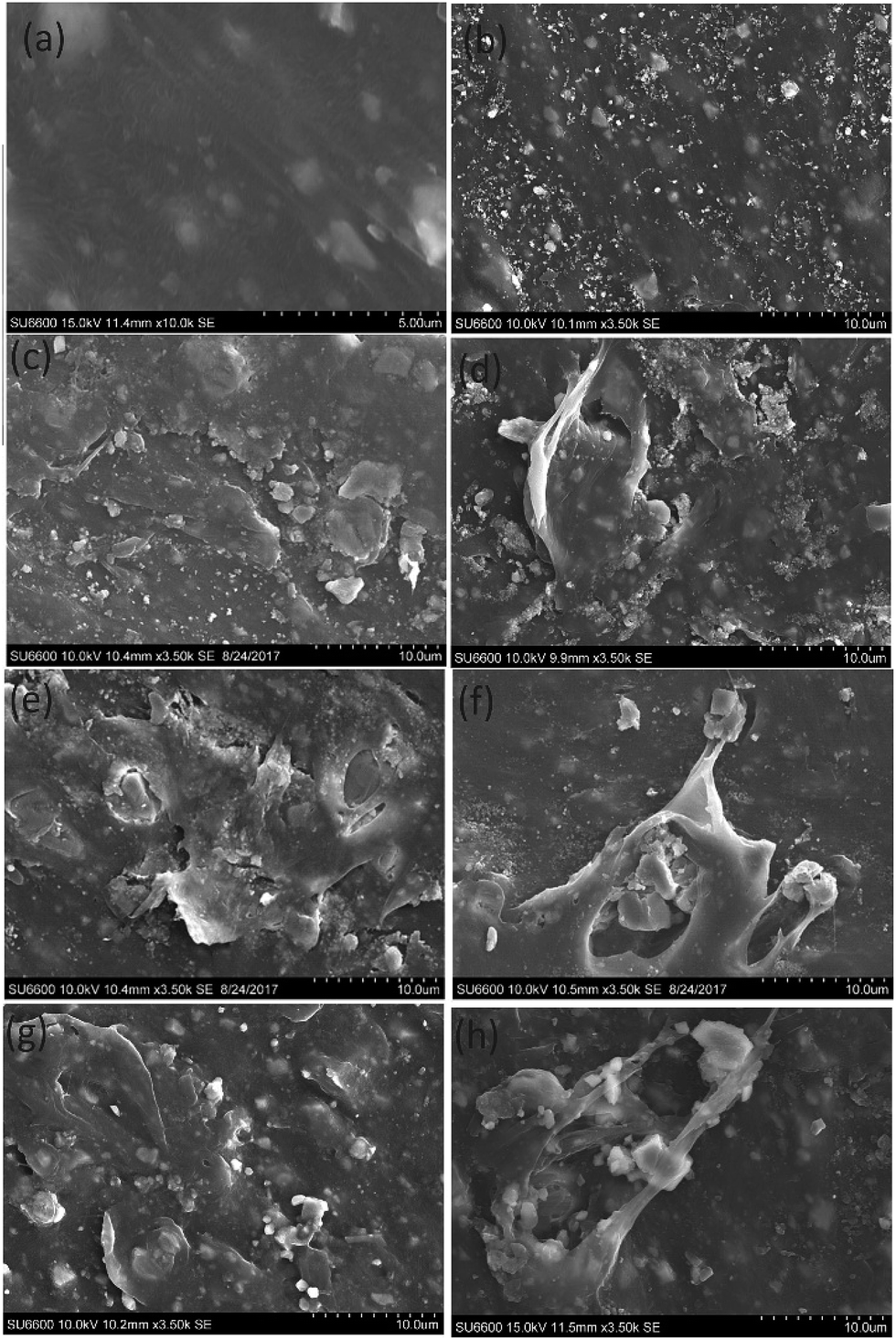 | ||
| Fig. 6 (a) SEM images of pure PE film, (b–f) SEM images of PE film treated with (b) 5000 ppm, (c) 8000 ppm, (d) 10000 ppm (e) 11000 ppm (f) 20000 ppm of ZrO2 for 20 hours under sun simulator (g and h) SEM images of PE films treated with 10000 ppm of TiO2 under sun simulator (g) for 20 hours (h) for 100 hours. | ||
The SEM image of PP film in Fig. 7(c), which was treated with 10000 ppm ZrO2 suspension and kept under real sun light for 20 hours, shows more damage than the PP film in Fig. 7(b) which was treated with 10000 ppm TiO2 suspension and kept under the same conditions. Likewise, the SEM image of PE film in Fig. 7(e) which was treated with 10000 ppm ZrO2 suspension and kept under real sun light for 20 hours shows more damage than the PE film in Fig. 7(d) which was treated with 10000 ppm TiO2 suspension and kept under the same conditions.
 | ||
| Fig. 7 (a) SEM image of pure PP, (b and c) SEM images of PP films treated with 10000 ppm (b) TiO2 (c) ZrO2 for 20 hours under real sun light, (d and e) SEM images of PE films treated with 10000 ppm (d) TiO2 (e) ZrO2 for 20 hours under real sun light. | ||
In order to determine the best nanoparticle concentration for the given mass of PE, the degradation pattern for the PE was observed by measuring the Carbonyl Index (CI).41
| Carbonyl Index = A1/A2 |
For polyethylene
A1 = peak area of the carbonyl group in absorption spectrum (1770–1820 cm−1).
A2 = peak area of the CH stretching group in absorption spectrum (2820–2960 cm−1).
For polypropylene
A1 = peak area of the carbonyl group in absorption spectrum (1719–1769 cm−1).
A2 = peak area of the CH stretching group in absorption spectrum (2744–3004 cm−1).
The nanoparticle suspensions with concentrations of 6000, 7000, 8000, 9000, 10000, 11000, and 20000 ppm, respectively, were used to find a concentration which shows sufficient degradation of PE and PP, in order to compare the degradation caused by TiO2 and ZrO2.
All the CI values were calculated using the above formulae and the peak areas were normalized by using the peak area of CH stretching peak
It was observed from Fig. 8, that the CI is increasing when increasing the nanoparticle concentration from 6000 to 11000 ppm. When the concentration of TiO2 and ZrO2 increases from 11000 ppm to 20000 ppm the carbonyl index decreases. That may be due to the saturation of the PE surface by the nanoparticles. Therefore 10000 ppm was selected as the nanoparticle concentration for the further studies.
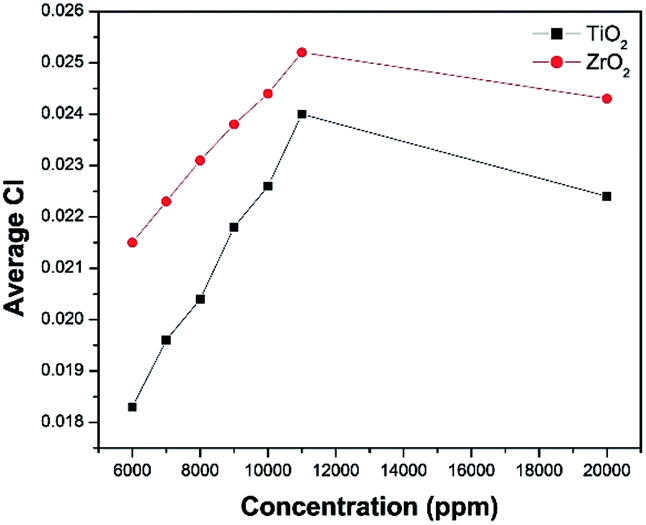 | ||
| Fig. 8 Average CI vs. TiO2 and ZrO2 concentration. | ||
The optimum time required to degrade PE with TiO2 nanoparticles and ZrO2 nanoparticles was determined by replicate measurements at 20, 40, 60, 80, and 100 h, at 10000 ppm concentration using the sun simulator (Table 1).
| Concentration/ppm | Average CI, TiO2 | Average CI, ZrO2 |
|---|---|---|
| 20000 |
0.0224 | 0.0243 |
| 11000 |
0.0240 | 0.0252 |
| 10000 |
0.0226 | 0.0244 |
| 9000 | 0.0218 | 0.0238 |
| 8000 | 0.0204 | 0.0231 |
| 7000 | 0.0196 | 0.0223 |
| 6000 | 0.0183 | 0.0215 |
| 0 | 0.009 | 0.0090 |
The average CI was plotted against the exposure time from 0 to 100 hours (Table 2). When the time is increasing from 0 hours to 20 hours there is a gradual increase in the CI. Within the time range from 20 to 100 hours, there was no significant change in the CI at the 95% confidence level. As there was a considerable degradation of PE and PP when exposed for 20 hours to the sun simulator 20 hours was selected as the exposure time for the further studies to compare the degradation of TiO2 and ZrO2.
| Time (hours) | Average CI |
|---|---|
| 0 | 0.0091 |
| 20 | 0.0225 |
| 40 | 0.0227 |
| 60 | 0.0230 |
| 80 | 0.0232 |
| 100 | 0.0234 |
In both treatment conditions (under sun simulator and real sun light) it was found that, at 95% confidence levels, there is a significant difference between the degradation of selected plastics by ZrO2 and TiO2. ZrO2 nanoparticle suspension treated PE and PP samples showed higher degradation than that of the TiO2 treatment (Table 3) (Fig. 9). TiO2 nano particles do not possess a mesoporous characteristic which can result in more efficient light scattering sites inside the mesopore structure.42 But ZrO2 has a mesoporous structure. That may be one reason for the increased photocatalytic activity of ZrO2 than TiO2. Sreethawong42 has reported that the mesoporous assembled ZrO2 nano particles showed a comparatively higher degradation of Methyl Orange than commercial P-25 TiO2 nano particles. The presence of oxygen vacancies and their relative abundance in the surface region is considered to be the reason for the photocatalytic activity of both TiO2 and ZrO2.43 ZrO2 has more capability for stabilizing oxygen vacancies than TiO2.43 Therefore ZrO2 shows more photocatalytic activity than TiO2. It is generally accepted that a higher band gap corresponds to a higher redox ability. According to the UV visible data ZrO2 has a higher band gap energy than TiO2.44 Thus, due to mesoporous structure, greater capability for stabilizing oxygen vacancies, and the higher band gap, ZrO2 has higher photocatalytic activity than TiO2, and it shows higher degradation of PE and PP than TiO2 under the given experimental conditions.
| Samples | Polyethylene | Polypropylene | ||
|---|---|---|---|---|
| Treated | Sun simulator | Real conditions | Sun simulator | Real conditions |
| Pure | 0.0090 | 0.0090 | 0.0072 | 0.0072 |
| Untreated | 0.0097 | 0.0175 | 0.0074 | 0.0079 |
| THF | 0.0106 | 0.0183 | 0.0081 | 0.0087 |
| TiO2 | 0.0226 | 0.0260 | 0.0112 | 0.0124 |
| ZrO2 | 0.0244 | 0.0382 | 0.0149 | 0.0190 |
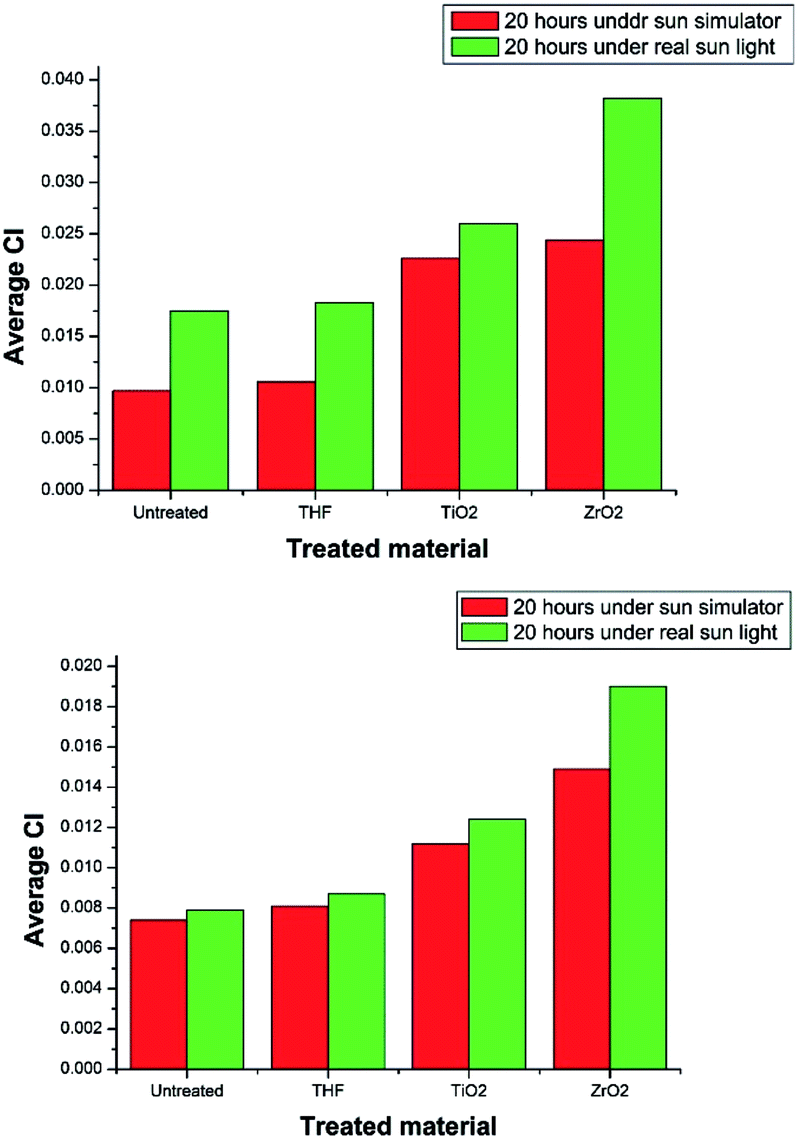 | ||
| Fig. 9 Average CI variation of (a) PE (b) PP under laboratory conditions and the real conditions. | ||
The degradation of treated PE and PP films in real environmental conditions is superior to that of laboratory conditions. The variation in degradation may result because of the samples were not continuously treated under the direct sun light. They were treated four hours per day and kept in the dark for the rest of the time in order to supply almost a constant intensity of light all the time. When the samples were exposed to the sun light for the first time, the photo catalytic degradation of PE and PP is initiated. As a result, free radicals are formed. That could be the reason for the higher degradation of the PE and PP samples kept under the direct sun light more than the samples kept under the sun simulator. The purpose of treating under the real conditions was to check the eligibility of degradation of PE and PP under direct sun light.
4. Conclusion
Successful synthesis of TiO2 and ZrO2 nanoparticles were carried out in this study. TiO2 nanoparticles were tetragonal and they were in the anatase phase. ZrO2 nanoparticles were in the tetragonal phase or a mixture of tetragonal and cubic phases. Concentration optimization and time optimization were done in laboratory conditions under the sun simulator. For both TiO2 and ZrO2 suspensions the optimum concentration was found to be 10000 ppm, while 20 hours was taken as the optimum time. PE and PP were treated under the sun simulator as well under the real sun light. In both treatment conditions it was found that, with 95% confidence, there is a significant difference between the degradation of selected plastics by ZrO2 and TiO2. ZrO2 nanoparticle suspension treated PE and PP samples showed higher degradation than similar samples treated with the TiO2 nanoparticle suspension. Under these experimental conditions it can be concluded that nano ZrO2 can cause higher photocatalytic degradation than nano TiO2.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors would like to thank Dr Ranil D. Gunarathne for comments that greatly improved the manuscript.References
- A. A. Shah, F. Hasan, A. Hameed and S. Ahmed, Biotechnol. Adv., 2008, 26, 246–265 CrossRef CAS PubMed.
- H. Webb, J. Arnott, R. Crawford and E. Ivanova, Polymers, 2013, 5, 1 CrossRef.
- P. Bansal, P. Sharma and V. Goyal, Biol. Plant., 2002, 45, 125–127 CrossRef CAS.
- R. Mohee, G. D. Unmar, A. Mudhoo and P. Khadoo, Waste Manag., 2008, 28, 1624–1629 CrossRef CAS PubMed.
- S. Horikoshi, N. Serpone, Y. Hisamatsu and H. Hidaka, Environ. Sci. Technol., 1998, 32, 4010–4016 CrossRef CAS.
- S. K. Kassahun, Z. Kiflie, D. W. Shin, S. S. Park, W. Y. Jung and Y. R. Chung, J. Sol-Gel Sci. Technol., 2017, 82, 322–334 CrossRef CAS.
- J. Zhang, P. Zhou, J. Liu and J. Yu, Phys. Chem. Chem. Phys., 2014, 16, 20382–20386 RSC.
- M. N. Chong, B. Jin, C. W. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed.
- B. Neppolian, S. Mine, Y. Horiuchi, C. Bianchi, M. Matsuoka, D. Dionysiou and M. Anpo, Chemosphere, 2016, 153, 237–243 CrossRef CAS PubMed.
- M. Dissanayake, H. Divarathna, C. Dissanayake, G. Senadeera, P. Ekanayake and C. Thotawattage, J. Photochem. Photobiol., A, 2016, 322, 110–118 CrossRef.
- I. Junkar, M. Kulkarni, B. Drašler, N. Rugelj, A. Mazare, A. Flašker, D. Drobne, P. Humpolíček, M. Resnik and P. Schmuki, Bioelectrochemistry, 2016, 109, 79–86 CrossRef CAS PubMed.
- K. Gulati, L. Johnson, R. Karunagaran, D. Findlay and D. Losic, Biomacromolecules, 2016, 17, 1261–1271 CrossRef CAS PubMed.
- R. N. Wijesena, N. D. Tissera, R. Perera, K. N. de Silva and G. A. Amaratunga, J. Mol. Catal. A: Chem., 2015, 398, 107–114 CrossRef CAS.
- A. Kadam, R. Dhabbe, M. Kokate, Y. Gaikwad and K. Garadkar, Spectrochim. Acta, Part A, 2014, 133, 669–676 CrossRef CAS PubMed.
- X.-H. Ning, Q.-L. Meng, Y.-L. Han, D.-Y. Zhou, L. Li, L. Cao, Z.-K. Weng, R. Ding and Z.-B. Wang, RSC Adv., 2017, 7, 34907–34911 RSC.
- A. Singhania and S. M. Gupta, Beilstein J. Nanotechnol., 2017, 8, 264 CrossRef CAS PubMed.
- J. A. Navío, M. C. Hidalgo, G. Colón, S. G. Botta and M. I. Litter, Langmuir, 2001, 17, 202–210 CrossRef.
- G.-L. Tan and X.-J. Wu, Thin Solid Films, 1998, 330, 59–61 CrossRef CAS.
- Y. Chu, Y. Chen, N. Chen, F. Wang and H. Zhu, RSC Adv., 2016, 6, 96768–96777 RSC.
- Z. Guerra-Que, G. Torres-Torres, H. Perez-Vidal, I. Cuauhtemoc-Lopez, A. Espinosa de los Monteros, J. N. Beltramini and D. M. Frias-Marquez, RSC Adv., 2017, 7, 3599–3610 RSC.
- S.-y. Li, Y. Wang, J.-G. Wu, L.-f. Guo, M. Ye, Y.-H. Shao, R. Wang, C.-e. Zhao and A. Wei, RSC Adv., 2016, 6, 72037–72043 RSC.
- T. A. Maia and E. M. Assaf, RSC Adv., 2014, 4, 31142–31154 RSC.
- M. Tamura, T. Kitanaka, Y. Nakagawa and K. Tomishige, ACS Catal., 2016, 6, 376–380 CrossRef CAS.
- T. A. Cheema and G. Garnweitner, CrystEngComm, 2014, 16, 3366–3375 RSC.
- M. Ranjbar, M. Yousefi, M. Lahooti and A. Malekzadeh, Int. J. Biomed. Nanosci. Nanotechnol., 2012, 8, 191–196 Search PubMed.
- L. Zhou, A. Zhao, Z. Wang, Z. Chen, J. Ren and X. Qu, ACS Appl. Mater. Interfaces, 2015, 7, 2905–2911 CAS.
- C. Khatua, I. Chinya, D. Saha, S. Das, R. Sen and A. Dhara, Int. J. Smart Sens. Intell. Syst., 2015, 8, 1424–1442 Search PubMed.
- S. Mugundan, B. Rajamannan, G. Viruthagiri, N. Shanmugam, R. Gobi and P. Praveen, Appl. Nanosci., 2015, 5, 449–456 CrossRef CAS.
- K. Anandan and V. Rajendran, J. Phys. Sci., 2013, 17, 179–184 Search PubMed.
- S. Jayakumar, P. Ananthapadmanabhan, T. Thiyagarajan, K. Perumal, S. Mishra, G. Suresh, L. Su and A. Tok, Mater. Chem. Phys., 2013, 140, 176–182 CrossRef CAS.
- Y. Yu, X. Wang, Y. Cao and X. Hu, Appl. Surf. Sci., 2001, 172, 260–264 CrossRef CAS.
- J. M. Pettibone, D. M. Cwiertny, M. Scherer and V. H. Grassian, Langmuir, 2008, 24, 6659–6667 CrossRef CAS PubMed.
- S. N. Basahel, T. T. Ali, M. Mokhtar and K. Narasimharao, Nanoscale Res. Lett., 2015, 10, 73 CrossRef PubMed.
- X. Meng, D.-W. Shin, S. M. Yu, J. H. Jung, H. I. Kim, H. M. Lee, Y.-H. Han, V. Bhoraskar and J.-B. Yoo, CrystEngComm, 2011, 13, 3021–3029 RSC.
- C. Dette, M. A. Pérez-Osorio, C. S. Kley, P. Punke, C. E. Patrick, P. Jacobson, F. Giustino, S. J. Jung and K. Kern, Nano Lett., 2014, 14, 6533–6538 CrossRef CAS PubMed.
- J.-P. Xu, R.-J. Zhang, Y. Zhang, Z.-Y. Wang, L. Chen, Q.-H. Huang, H.-L. Lu, S.-Y. Wang, Y.-X. Zheng and L.-Y. Chen, Phys. Chem. Chem. Phys., 2016, 18, 3316–3321 RSC.
- S. Liu, W. Wang, J. Chen, J.-G. Li, X. Li, X. Sun and Y. Dong, J. Mater. Chem. A, 2015, 3, 17837–17848 CAS.
- H. M. Moghaddam and S. Nasirian, S. Afr. J. Sci., 2011, 107, 01–05 Search PubMed.
- A. L. Forster, A. M. Forster, J. W. Chin, J.-S. Peng, C.-C. Lin, S. Petit, K.-L. Kang, N. Paulter, M. A. Riley and K. D. Rice, Polym. Degrad. Stab., 2015, 114, 45–51 CrossRef CAS.
- D. J. Nagle, G. A. George, L. Rintoul and P. M. Fredericks, Vib. Spectrosc., 2010, 53, 24–27 CrossRef CAS.
- A. Eshraghi, H. Khademieslam, I. Ghasemi and M. Talaiepoor, BioResources, 2012, 8, 201–210 CrossRef.
- T. Sreethawong, S. Ngamsinlapasathian and S. Yoshikawa, Chem. Eng. J., 2013, 228, 256–262 CrossRef CAS.
- S. Tosoni, H. Y. T. Chen and G. Pacchioni, ChemPhysChem, 2015, 16, 3642–3651 CrossRef CAS PubMed.
- B. M. Pirzada, N. A. Mir, N. Qutub, O. Mehraj, S. Sabir and M. Muneer, Mater. Sci. Eng., B, 2015, 193, 137–145 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |