
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring sesquiterpene alkaloid-like scaffolds via Beckmann-transannular remodelling of beta-caryophyllene†
Shuang-Jiang Maa,
Jie Yu a,
Hua-Fang Fana,
Zi-Han Lic,
An-Ling Zhang*a and
Qiang Zhang*ab
a,
Hua-Fang Fana,
Zi-Han Lic,
An-Ling Zhang*a and
Qiang Zhang*ab
aShaanxi Key Laboratory of Natural Products & Chemical Biology, College of Chemistry & Pharmacy, Northwest A&F University, Yangling 712100, PR China. E-mail: zhangq@nwsuaf.edu.cn; anlingzh@nwsuaf.edu.cn
bState Key Laboratory of Medicinal Chemical Biology, Nankai University, Tianjin 300071, PR China
cCollege of Life Sciences, Northwest A&F University, Yangling 712100, PR China
First published on 18th August 2017
Abstract
High-throughput screening (HTS) is the dominant approach to identify lead compounds in drug development. However, most compound-screening collections provide little structural or stereochemical complexity, which do not offer enough diversity to merit the modulation of many drug targets. Here we describe a facile strategy for the creation of diverse compounds with high structural and stereochemical complexity using readily available natural products, β-caryophyllene, as a synthetic starting point. Our findings demonstrate that a cascaded Beckmann-transannular protocol transforms macrocyclic natural products into poly-heterocyclic unnatural skeletal types. These compounds are significantly more complex and diverse than those in standard screening collections.
Introduction
Natural products (NPs) have played an essential role in developing novel drugs due to their structural diversity.1–4 Nevertheless, pharmaceutical research into NPs has recently declined owing to factors such as the difficulty of collecting novel compounds containing privileged structures.2 Thus, new strategies to augmenting chemical diversity are crucial for retaining the utility of natural products and their derivatives. Recently, creating complex small molecules directly from NPs has appeared as a convenience approach in diversity-oriented synthesis (DOS) area.5–9 NPs are being considered as exceptional starting points due to the high abundance and structural variation. Additionally, NPs provide an excellent framework for site selective and stereoselective transformations.The construction of heterocyclic molecules with potentially interesting biological activities represents an important part of the drug discovery process. Especially there has been a growing interest in the synthesis of medium-sized rings because of their many applications.10 Among them, poly-N-heterocyclic NPs displayed excellent bioactivities, such as homoharringtonine (topoisomerase inhibition, IC50 9.0 nM),11 stenine B (acetylcholinesterase inhibition IC50 2.1 μM),12 sessilistemonamine E (acetylcholinesterase inhibition IC50 9.1 μM),13 and cryptopleurin (NF κB, IC50 1.4 nM)14 (Fig. 1). Compounds of this type have been reported to show a wide range of potential applications in antitumor, anti-inflammatory and Alzheimer's disease therapy.
 | ||
| Fig. 1 Bioactive natural products with N-containing poly-heterocyclic skeleton. | ||
The propensity of macrocyclic precursors to undergo transannular cyclization has been deftly harnessed by nature to build thousands of polycyclic terpenoids and alkaloids with medium-sized rings.9,15–17 Our previous diversity-oriented transformation demonstrated the convenience protocol to build natural-like libraries containing medium-sized rings from macrocyclic caryophyllene.18,19 In this paper, we consider applying Beckmann rearrangement associated with transannular cyclization to explore chemical space covering poly-N-heterocyclic skeletons.
The natural sesquiterpenes, such as β-caryophyllene and its epoxide (1), are readily abundant to be ideal starting points to build diverse natural-like scaffolds along NP-driven DOS strategy. Due to the tensional cyclobutane, macrocyclic moiety and exocyclic olefin, compound 1 plays as a key precursor in nature to form diverse tricyclic sesquiterpenes by transannular rearrangements.20 In this paper, we reported a concise procedure to build natural-like skeletons with N-containing poly-heterocyclic skeletons via Beckmann-transannular rearrangement on the macrocycle of caryophyllene (Scheme 1). To start Beckmann rearrangements on the macrocycle, compound 1 was introduced oxime group at C-8 as starting site (Scheme 1).
 | ||
| Scheme 1 Beckmann-transannular strategy to NP-like library. | ||
Results and discussion
The end olefin at C-8 in 1 was cleaved by RuCl3/NaIO4 oxidation to yield a 13-nor product (2) with carbonyl function group. The key intermediate oxime derivative (3) was prepared via condensation with hydroxylamine. In the next step, Lewis acid LiBF4/MeCN inspired 3 into a DOS library containing five compounds 4–8 (Scheme 2).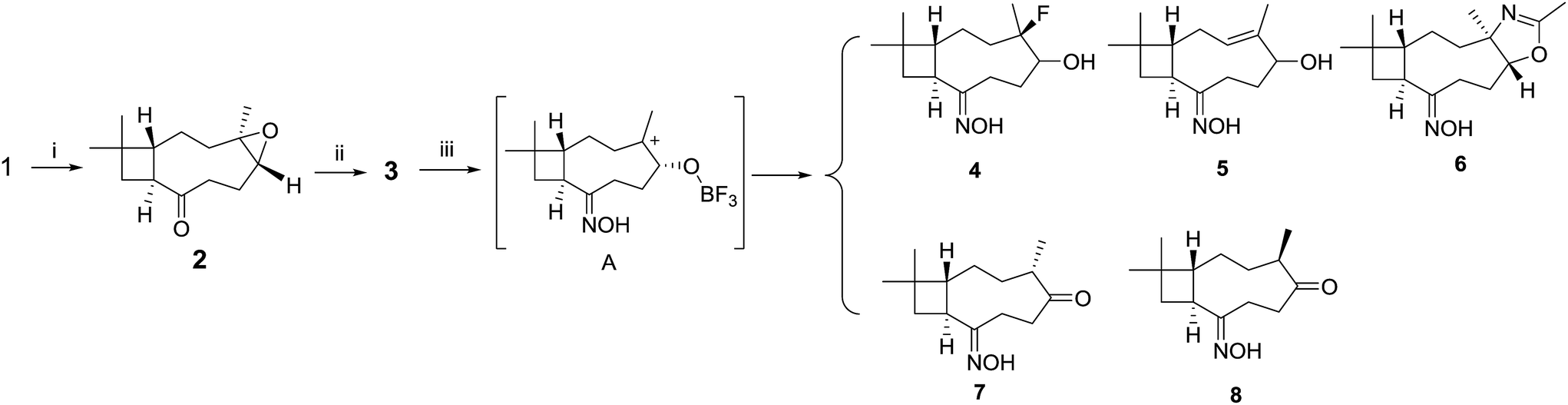 | ||
| Scheme 2 Diversity-oriented transformation to oxime. Reaction condition: (i) NaIO4, RuCl3, MeCN–EtOAc–H2O; (ii) NH2OH·HCl, NaOH; (iii) LiBF4, MeCN, 45 °C. | ||
The structures of compounds 4–8 were unambiguously elucidated using 1D and 2D NMR and MS spectra. As a representative example, the structural analysis of 6 was elucidated in detail herein. Its molecular formula (MF) was assigned as C16H26N2O2 by HRESIMS, indicating 5 degrees of unsaturation. Its 13C and DEPT NMR spectroscopic data revealed the presence of two imine groups (δC 161.1, 162.4) and other 15 aliphatic carbons, including three methyls, five methenes, three methines and two quaternary carbon atoms. Apart from those two degrees of unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds, the remaining implied that 6 possesses three rings in the structure.
N bonds, the remaining implied that 6 possesses three rings in the structure.
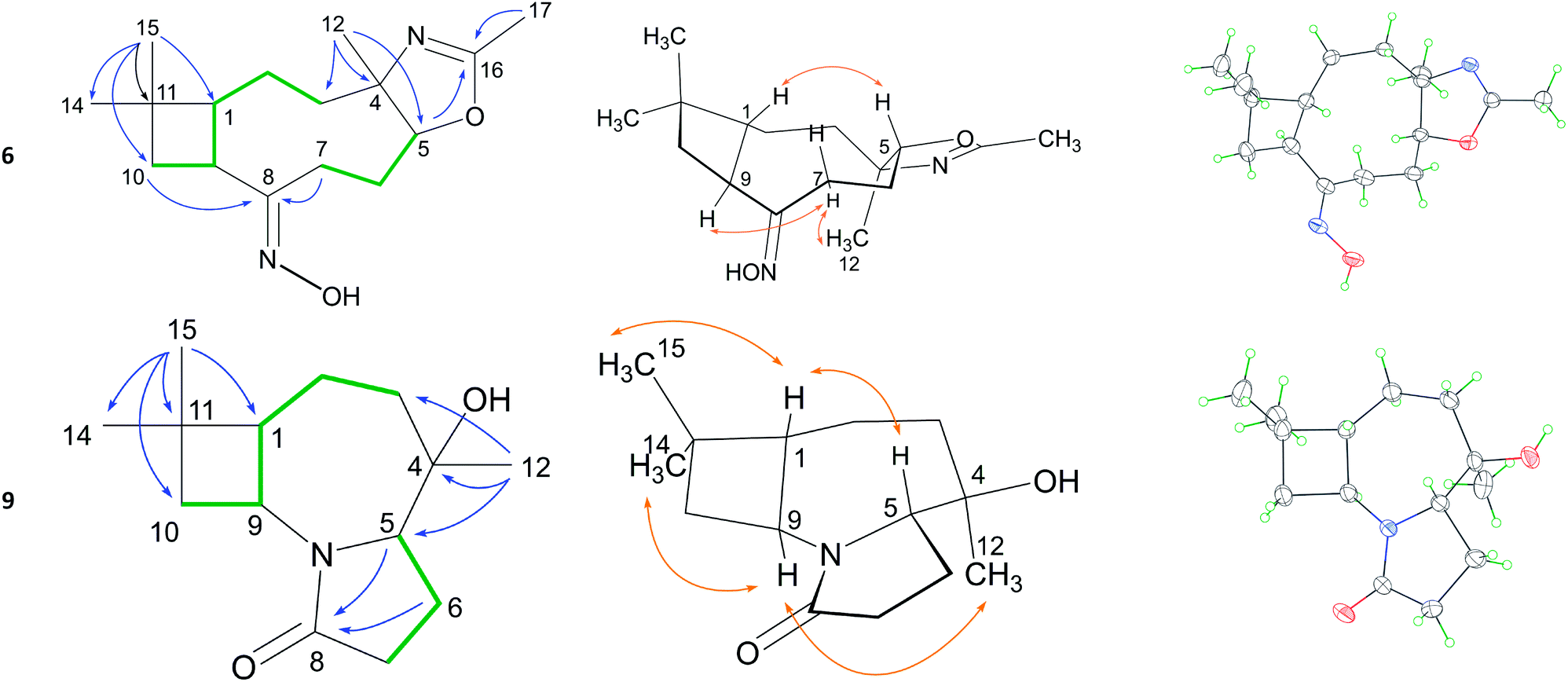
Extensive analysis of the HSQC, 1H–1H COSY, and HMBC correlations (Table 1) established the whole planar structure. 1H–1H COSY correlations disclosed two isolated spin systems. HMBC correlations from H3-15 to C-1, C-10, C-11 and C-14 indicated a moiety of cyclobutane with a gem-dimethyl group. The HMBC correlations from H2-10 and H2-7 to C-8 indicated that the C-7 and C-9 are linked up at C-8. The HMBC correlations from H3-12 to C-3, C-4 and C-5 disclosed a 9 membered ring. The dihydrooxazole ring with a methyl was identified by the HMBC correlations of H-5/C-16 and H3-17/C-16. The relative configuration were analysed on the basis of NOESY correlations (Table 1). The NOE correlation of H-1/H-5 indicated these H atoms are in β-orientation. The NOE correlations of H-9/Hα-7/H3-12 indicated the H-9 and H3C-12 are in α-orientation. The relative configuration were also confirmed by Cu Kα X-ray diffraction analysis (Table 1). However, high values of Flack parameters (0.03) indicated that the X-ray data did not provide unambiguous information about absolute configuration. Alternatively, based on the unchanged (1R) and (9S) configurations, the absolute configuration was deduced as therein on the basis of NOESY correlations related to H-1 and H-9.
|
|---|
| a See Table S1 in ESI for the 2D NMR correlations of compounds 4–15. |
 |

Compound 6 possesses a 2-oxazoline moiety which was generated from epoxyethane moiety in 3 and the solvent acetonitrile. The 2-oxazoline ring was reported to be synthesized in good yields catalyst by BF3–OEt2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 or SiF4.22 In the current reaction, LiBF4 was considered to decompose into BF3 and LiF. Compound 3 was transformed into carbonium ion A catalysed by BF3 (Scheme 2). The intermediate A was attacked by F− or MeCN to yield fluoride 4 and 2-oxazoline derivate 6. Simultaneously, elimination reactions of A afforded products 5, 7 and 8.
21 or SiF4.22 In the current reaction, LiBF4 was considered to decompose into BF3 and LiF. Compound 3 was transformed into carbonium ion A catalysed by BF3 (Scheme 2). The intermediate A was attacked by F− or MeCN to yield fluoride 4 and 2-oxazoline derivate 6. Simultaneously, elimination reactions of A afforded products 5, 7 and 8.
These findings showed that the LiBF4 did not provide access to the targeted remodeling skeletons. Pleasingly, we successfully formed the 2-oxazoline moiety at milder condition than that previously reported.21,22 To fully exploit build-in reactivity and increase rigid polycyclic molecules, Beckmann rearrangements of compound 3 were investigated with catalyst p-TsCl (Scheme 3). Compound 3 gave entirely different cyclization products 9–11, with polycyclic lactam skeletons. To acquire similar rearranged derivatives, the epoxyethane moiety in the starting material 3 was reduced into an endocyclic olefin. A following Beckmann rearrangement afforded a macrolactam 14 and two other polycyclic products 10 and 15.
 | ||
| Scheme 3 Beckmann-transannular remodelling. (i) NH2OH·HCl, NaOH; (ii) p-TsCl, Et3N, DMAP (cat.), DCM, 3 h, reflux; (iii) Zn powder; (iv) NH2OH·HCl, NaOH. | ||
To verify the skeleton remodeling, the structures of products 9–11, 14 and 15 were investigated by HRMS, IR, 1D and 2D NMR. All the detailed correlations in 2D NMR were summarized in the Table S1 in ESI material.† For example, the MF of compound 9, C14H23NO2, was determined by HRESIMS. Two isolated spin systems were recognized by the COSY correlations as shown in Table 1. The HMBC correlations of H-5 and H2-6 to C-8 revealed cascaded Beckman-transannular remodeling on the caryophyllene's macrocycle. Furthermore, the X-ray analysis using Cu Kα diffraction disclosed the N-containing 4/7/5 scaffold 9. The configuration was determined by NOESY correlations. The H-1(S) was selected as the reference group whose absolute configuration kept unchanged in the reaction process. The NOESY correlations among H-1, H3-15 and H-5 indicated that the newly formed chiral centre C-5 is S-configuration. The NOESY correlations from H-9 to H3-14 and H3-12 indicated that the chiral C-4 is R-configuration. The whole configuration was also confirmed by the X-ray diffraction as shown in Table 1.
Based on the observed stereochemistry, a proposed mechanism for the formation of 9–11 is shown in Scheme 4. In the Beckmann process, the departure of the leaving group, TsO–, was accompanied by the [1,2]-shift of the C-7 or C-8 to afford the lactam tautomers B or C. In the next step, transannular rearrangements occurred between the imines in B or C with the epo C-4 or C-5 to form the polycyclic skeletons of 9–11. Their structures indicated a cascaded Beckmann-transannular rearrangement in the process.
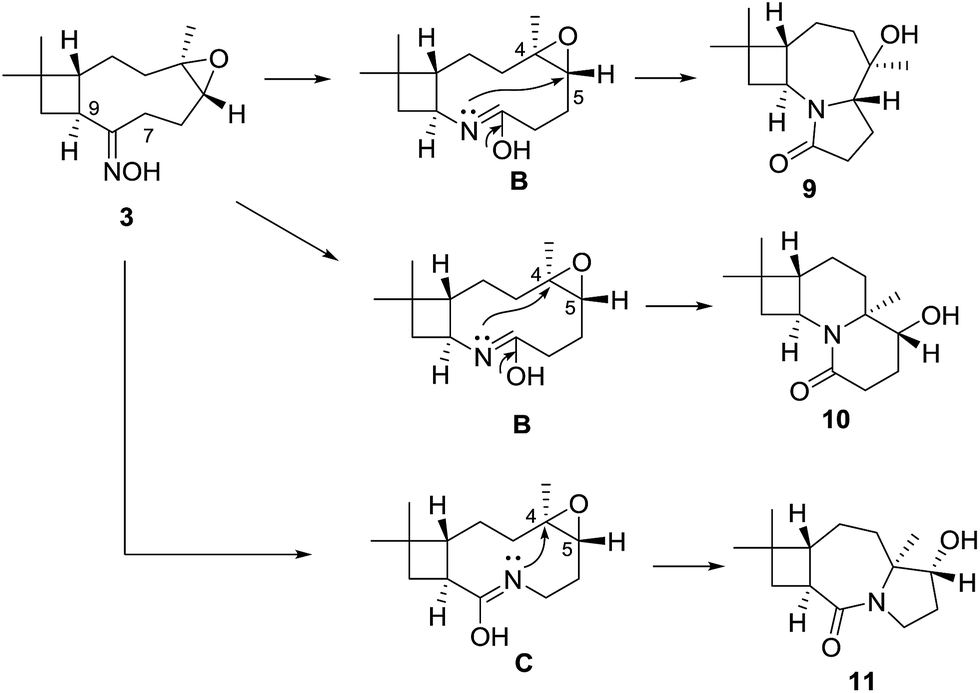 | ||
| Scheme 4 Proposed mechanism for the Beckmann-transannular rearrangements to 9–11. | ||
To evaluate the biological relevancy of product library, butyrylcholinesterase (BChE) was selected as bioassay target. Previous relationship observed between cholinergic dysfunction and Alzheimer's disease (AD) severity provides a rationale for the therapeutic use of cholinesterase inhibitors (ChEIs).23 Administration of ChEIs to increase the amount of cholines available for neural transmission has proven successful in relieving some cognitive as well as behavioral symptoms of AD.24,25 BChE inhibition was found to elevate brain acetylcholine, augment learning and reduce Alzheimer β-amyloid peptide.26 The bioassay indicated a weak inhibition of compound 15 (IC50 134.6 μM), with positive control, galanthamine (IC50 82.6 μM).
Conclusions
In conclusion, highly functionalized epoxy-macrocyclic sesquiterpenes, such as β-caryophyllene epoxide, endowed with a potential to generate rigid polycyclic structures not easily accessed by other methods. Our findings demonstrated that a Beckmann-transannular rearrangement on such a macrocycle is a concise approach to generate NP-like library with a lactam unit. In addition, β-caryophyllene and its epoxide are readily plentiful in natural source. Thus they can be served as ideal staring points to construct complex cyclic scaffolds covering unknown chemical space or bioactive space. On the basis of the present findings, combination of macrocyles with other naturally occurring bioactive blocker, will yield highly diverse products with NP-like scaffolds.Experimental procedures
General procedures
UV spectra were obtained using an Evolution 300 UV-vis Spectrophotometer (Thermo Fisher Scientific Inc., USA). IR were recorded on a Bruker Tensor 27 spectrophotometer with KBr disks (Bruker Corp., German). Optical rotations were measured on an Autopol III automatic polarimeter (Rudolph Research Analytical, USA). ECD spectra were obtained on a Chirascan CD Spectrometer (Applied Photophysics Ltd., United Kingdom). ESI-MS were performed on an LTQ Fleet instrument (Thermo Fisher Scientific Inc., USA). HR ESIMS data were obtained by an AB Sciex Triple TOF 4600 system (AB Sciex Pte. Ltd., USA). 1D and 2D NMR spectra were recorded on an AVANCE III (500 MHz) instrument (Bruker Corp., Germany). Chemical shifts were reported using solvent residual as the internal standard. Crystallographic data collection was carried out on a Bruker Smart Apex CCD area detector diffractometer with graphite-monochromated Cu Kα radiation (λ = 1.54184 Å) at 293(2) K using ω-scan technique. The diffraction data were integrated by using the SAINT program, which was also used for the intensity corrections for the Lorentz and polarization effects. Semiempirical absorption corrections were applied using SADABS program. Analytic and semi-preparative HPLC were performed on a Waters 1525 instrument (Waters Corp., USA). Column chromatography was performed on silica gel (90–150 μm; Qingdao Marine Chemical Inc., China), Sephadex LH-20 (40–70 μm; Amersham Pharmacia Biotech AB, Sweden), and Chromatorex C18 gel (40–75 μm; Fuji Silysia Chemical LTD., JPN). GF254 plates (Qingdao Marine Chemical Inc., China) were used for thin-layer chromatography (TLC).Synthesis of oxime 3
(−)-β-Caryophyllene epoxide (5.0 g, 22.7 mmol), NaIO4 (36.4 g, 170.25 mmol, 7.5 eq) was added to a mixed solvent (25 ml MeCN, 25 ml EtOAc and 37.5 H2O) and RuCl3 (2.2% mol) in a 250 ml round-bottom flask equipped with a stir bar. After being stirred for 6 h, the reaction mixture was filtered. The filtrate was extracted with EtOAc (100 ml, 3 times). The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified on column chromatography (silica gel, petroleum ether:EtOAc = 10:1, v/v) to give the product 2 (3.1 g, 61% yield).
A solution of 2 (1.11 g, 5 mmol in 15 ml EtOH) was added to a stirred solution of NH2OH HCl (2.08 g, 30 mmol) in 15 ml 2 N aqueous NaOH. After being stirred for 30 min at room temperature, 60 ml H2O was added. Then the reaction mixture was extracted with DCM (100 ml, 3 times). The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure to afford the crude residue. A further purification on column chromatography (silica gel, petroleum–EtOAc 20:1, v/v) to yield the product 3 (1.04 g, 87% yield).
DOS procedure catalysed by LiBF4
LiBF4 (164.3 mg, 1.75 mmol) was added slowly to a stirred solution of 3 (1.04 g, 4.38 mmol) in 10 ml MeCN. The reaction mixture was stirred at 45 °C for 20 h. The reaction was quenched with water (30 ml). And the aqueous phase was extracted with DCM for 3 times. The combined organic layers were dried over Na2SO4, and concentrated under reduced pressure to give a crude product (0.717 g).The crude was chromatographed on a silica gel column being eluted with CHCl3–MeOH (from 100:1 to 1:1) to give three fractions (Fr.1–Fr.3). Fr.1 (110 mg) was separated on an HPLC C18 column (Thermo BDS Hypersil 250 × 10 mm) with 80% methanol to yield compounds 7 (15 mg) and 8 (14 mg). Fr.2 (330 mg) was separated by a silica gel column chromatography (CHCl3–MOH 100:1) to afford compounds 4 (200 mg). Fr.3 (260 mg) was isolated on an HPLC C18 column with elution of 60% MeOH to yield 5 (160 mg) and 6 (40 mg).
Beckmann remodeling of compound 3
To a solution of compound 3 (1.00 g, 4.2 mmol) in 10 ml DCM, a solution of paratoluenesulfonyl chloride (p-TsCl, 2.00 g, 10.54 mmol), Et3N (1.5 ml, 10.54 mmol) and DMAP (10.3 mg, 2 mol%) in DCM (20 ml) was added dropwise. After being stirred for 3 h at room temperature, the reaction mixture was diluted with 20 ml DCM and washed with water (2 × 100 ml) and brine (2 × 100 ml). After being concentrated in vacuum and dried over Na2SO4, the crude product (820 mg) was yielded.The crude product (820 mg) was run on a silica gel column (gradient petroleum–EtOAc from 20:1 to 1:2) and a semi-preparative HPLC C18 column (Thermo BDS Hypersil 250 × 10 mm, 60% methanol) to yield compounds 9 (80 mg), 10 (30 mg) and 11 (8 mg).
Synthesis of compound 13
To a solution of compound 2 (2.00 g, 9 mmol) in anhydrous EtOH (90 ml), activated zinc powder (100 g) was added. The reaction mixture was refluxed for 48 h. Then the reaction mixture was cooled to room temperature and filtered. The residue was washed with water, and then extracted with EtOAc (3 × 120 ml). The combined extraction were dried over Na2SO4 and concentrated under reduced pressure to afford crude product. The crude product was purified on a silica gel column (petroleum–EtOAc 60:1) to yield the compound 12 (1.00 g, 54% yield).
To a stirred solution of NH2OH HCl (2.00 g, 28.8 mmol) in 14.5 ml aqueous NaOH (2 M), a solution of compound 12 (1.00 g, 4.8 mmol) in ethanol (15 ml). After being stirred for 30 min at room temperature, 60 ml water was added. The reaction mixture was extracted with DCM (100 ml) for 3 times. The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure to yield the crude product. The crude product was run on a silica gel column chromatography (petroleum–EtOAc 20:1) to afford compound 13 (960 mg, 90% yield).
Beckmann remodeling of 13
To a 10 ml DCM solution of compound 13 (0.95 g, 4.3 mmol), a 10 ml DCM solution of p-TsCl (2.10 g, 10.75 mmol), Et3N (1.5 ml, 10.75 mmol) and DMAP (10 mg) was added dropwise. After being stirred for 3 h at room temperature, the mixture was diluted with 20 ml DCM and washed with water (2 × 100 ml) and brine (2 × 100 ml). The organic layer was dried over Na2SO4 and concentrated in vacuum to afford the crude product (820 mg). Remaining reactants were removed on a silica gel column (petroleum–EtOAc 20:1–1:2). A further HPLC C18 chromatography (Thermo BDS Hypersil 250 × 10 mm, 60% MeOH) afforded compounds 10 (8 mg), 14 (90 mg) and 15 (16 mg).
Bioassay
Butyrylcholine esterase inhibitory activity was measured by spectrophotometric method reported previously.27 The reaction mixture contained 120 μl PBS buffer (pH 7.4), 10 μl butyrylcholinesterase solution, 10 μl of test compound solution, which were mixed and incubated for 10 min (37 °C). The reaction was initiated by the addition of 30 μl S-butyrylthiocholine chloride and 30 μl DTBN. The hydrolysis of butyrylthiocholine iodide was monitored at 412 nm after 30 min. Galanthamine was used as positive control. All the reactions were performed in triplicate. The percentage inhibition was calculated as follows:| Inhibition (%) = (1 − Ai) × 100/A0 |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financially supported by State Key Laboratory of Medicinal Chemical Biology, Nankai University (No. 201502003) and the Natural Science and Basic Research Plan of Shaanxi Province (No. 2016JM2002).Notes and references
- T. Rodrigues, D. Reker, P. Schneider and G. Schneider, Nat. Chem., 2016, 8, 531 CrossRef CAS PubMed.
- J. M. Gao, W. J. Wu, J. W. Zhang and Y. Konishi, Nat. Prod. Rep., 2007, 24, 1153 RSC.
- J. M. Gao, S. X. Yang and J. C. Qin, Chem. Rev., 2013, 113, 4755 CrossRef CAS PubMed.
- J. W.-H. Li and J. C. Vederas, Science, 2009, 325, 161 CrossRef PubMed.
- K. C. Morrison and P. J. Hergenrother, Nat. Prod. Rep., 2014, 31, 6 RSC.
- G. Prabhu, S. Agarwal, V. Sharma, S. M. Madurkar, P. Munshi, S. Singh and S. Sen, Eur. J. Med. Chem., 2015, 95, 41 CrossRef CAS PubMed.
- J. Kim, H. Kim and S. B. Park, J. Am. Chem. Soc., 2014, 136, 14629 CrossRef CAS PubMed.
- W. R. J. D. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1, 80 Search PubMed.
- H. Kikuchi, T. Nishimura, E. Kwon, J. Kawai and Y. Oshima, Chem.–Eur. J., 2016, 22, 15819 CrossRef CAS PubMed.
- L. Huang, L.-X. Dai and S.-L. You, J. Am. Chem. Soc., 2016, 138, 5793 CrossRef CAS PubMed.
- K. Burger, B. Mühl, T. Harasim, M. Rohrmoser, A. Malamoussi, M. Orban, M. Kellner, A. Gruber-Eber, E. Kremmer, M. Hölzel and D. Eick, J. Biol. Chem., 2010, 285, 12416 CrossRef CAS PubMed.
- D.-H. Lai, Z.-D. Yang, W.-W. Xue, J. Sheng, Y. Shi and X.-J. Yao, Fitoterapia, 2013, 89, 257 CrossRef CAS PubMed.
- R. A. Ramli, W. Lie and S. G. Pyne, J. Nat. Prod., 2014, 77, 894 CrossRef CAS PubMed.
- W. Gao, A. P.-C. Chen, C.-H. Leung, E. A. Gullen, A. Fürstner, Q. Shi, L. Wei, K.-H. Lee and Y.-C. Cheng, Bioorg. Med. Chem. Lett., 2008, 18, 704 CrossRef CAS PubMed.
- E. Reyes, U. Uria, L. Carrillo and J. L. Vicario, Tetrahedron, 2014, 70, 9461 CrossRef CAS.
- B. R. Balthaser, M. C. Maloney, A. B. Beeler, J. A. Porco and J. K. Snyder, Nat. Chem., 2011, 3, 969 CrossRef CAS PubMed.
- M. Mewald, J. W. Medley and M. Movassaghi, Angew. Chem., Int. Ed., 2014, 53, 11634 CrossRef CAS PubMed.
- Q. Zhang, H.-Y. Tang, M. Chen, J. Yu, H. Li and J.-M. Gao, Org. Biomol. Chem., 2017, 15, 4456 CAS.
- H.-Y. Tang, J.-M. Gao and Q. Zhang, RSC Adv., 2015, 5, 72433 RSC.
- I. G. Collado, J. R. Hanson and A. J. Macías-Sánchez, Nat. Prod. Rep., 1998, 15, 187 RSC.
- J. L. García Ruano and C. García Paredes, Tetrahedron Lett., 2000, 41, 5357 CrossRef.
- M. Shimizu and H. Yoshioka, Heterocycles, 1988, 27, 2527 CrossRef CAS.
- J. C. Morris, Am. Fam. Physician, 2006, 74, 747 Search PubMed.
- S. Stepankova and K. Komers, Curr. Enzyme Inhib., 2008, 58, 160 CrossRef.
- J. Grutzendler and J. C. Morris, Drugs, 2001, 61, 41 CrossRef CAS PubMed.
- N. H. Greig, T. Utsuki, D. K. Ingram, Y. Wang, G. Pepeu, C. Scali, Q.-S. Yu, J. Mamczarz, H. W. Holloway, T. Giordano, D. Chen, K. Furukawa, K. Sambamurti, A. Brossi and D. K. Lahiri, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17213 CrossRef CAS PubMed.
- M. Danish, M. A. Raza, Sehrish and N. Anjum, Asian J. Chem., 2015, 27, 3925 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, characterization data, X-ray structures of compounds 6 and 9 and NMR spectra. CCDC 1552864 and 1519447. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra08196k |
| This journal is © The Royal Society of Chemistry 2017 |