DOI:
10.1039/C7RA08188J
(Paper)
RSC Adv., 2017,
7, 46125-46131
Solvent-induced diversity of luminescent metal–organic frameworks based on different secondary building units†
Received
25th July 2017
, Accepted 6th September 2017
First published on 28th September 2017
Abstract
By using a symmetrical V-shaped rigid 5′-carboxyl-(1,1′-3′,1′′-terphenyl)-4,4′′-dicarboxylic acid (H3L), three Cd(II)-based metal–organic frameworks (MOFs), [Cd3(L)2(H2O)4]·DMF (1), [Cd2(L)(SO4)2]·3(Me)2NH2 (2) and [Cd(HL)(H2O)]·0.5H2O (3), have been synthesized under solvothermal conditions. Due to the reactions in different solvent systems, L3−/HL2− in 1–3 show different coordination modes with Cd(II) ions to form various secondary building units (SBUs) in the final structures. The desolvated structure of 1 (1a) contains two shapes of 1D channels with suitable pore sizes. 2 is a 3D dense packing pattern with a three nodal (5,6,7)-connected new topological net, and 3 is a 2D layered (4,4)-connected sql network connected with partly deprotonated HL2− ligands. As a result, 1a possesses not only high CO2 loading but also excellent CO2/CH4 selectivity. In addition, all complexes display solid-state luminescence stemming from ligand-to-metal charge transfer.
1 Introduction
Metal–organic frameworks (MOFs)1 are porous materials showing a wide variety of thrilling chemical and physical properties, and, consequently, can be explored as functional materials in very diverse fields.2 Indeed, both the porosity of MOFs and their fascinating host–guest chemistry lie at the origin of most of these properties.3 Similar to other porous materials,4 MOFs have proven their efficiency as vessels to capture and host small molecules, and, eventually, to separate mixtures of molecules according to their steric and stereochemical features as well as reactivity properties (functional substituent groups).5 Anthropogenic carbon dioxide emissions to the atmosphere are one of the most urgent climate issues of our age, which makes the development of materials for capturing the carbon dioxide produced by fossil fuels and sequestering it away from the atmosphere one of the grand challenges of the 21st century.6 Owing to MOFs diversity and performance, hierarchically porous structures have attracted considerable attention as an important family of functional materials in recent years.7
As the basic building blocks, the organic ligands are vital in the assembly of structures and functions of MOFs. The rigid aromatic multicarboxylate ligands have been proved to be the ideal candidates for building versatile MOFs with rigidity, porosity, thermal and chemical stabilities,8 such as 1,3,5-benzenetricarboxylate, 1,2,4,5-benzenetetracarboxylate, and 4,4′-biphenyldicarboxylate.9 With a more in-depth study, the V-shaped rigid multicarboxylate ligands show plentiful advantages due to their manifold coordination modes, and they are liable to form cluster-based secondary building units (SBUs) with various metal ions.10 In many instances, a V-shaped steric configuration is favourable to form versatile high-dimensional and porous frameworks.11 Up to now, there are many porous MOFs assembled by utilizing V-shaped rigid multicarboxylates,12 such as 3,5-di(3,5-dicarboxylpheny)benzoic acid, 3,3′,5,5′-benzene-1,3-biyl-tetra-benzoic acid, and so on, which possess fascinating structures as well as intriguing properties.
In this work, a symmetrical V-shaped rigid ligand, 5′-carboxyl-(1,1′-3′,1′′-terphenyl)-4,4′′-dicarboxylic acid (H3L) (Scheme 1), is chosen to fabricate porous MOFs based on the following advantages: (i) H3L with three carboxylic groups possesses multiple coordination sites and various coordination modes, which is beneficial to produce porous frameworks; (ii) the multicarboxylic groups in H3L has an inclination to form various metal clusters, which can not only stabilize the structure, but also accelerate the interactions between the framework and CO2.13 Herein, the reaction of H3L with Cd(II) ions gave three new luminescent MOFs, [Cd3(L)2(H2O)4]·DMF (1), [Cd2(L)(SO4)2]·3(Me)2NH2 (2), [Cd(HL)(H2O)]·0.5H2O (3). L3−/HL2− in 1–3 show different coordination modes and can easily form various SBUs in the final structures. Complexes 1–3 reveal the luminescent properties, as well as selective adsorption for CO2 over CH4 for 1.
 |
| | Scheme 1 5′-Carboxyl-(1,1′-3′,1′′-terphenyl)-4,4′′-dicarboxylic acid. | |
2 Experimental section
2.1 Materials and general methods
All the reagents and solvents were purchased from commercial sources and used directly without further purification. Elemental analyses (C, H, and N) were performed on Perkin-Elmer 2400C Elemental Analyzer. Infrared (IR) spectra were acquired with KBr discs in the range of 4000 to 400 cm−1 on Bruker EQUINOX-55 IR spectrometer. Thermogravimetric analyses (TGA) were collected on NETZSCH STA 449C microanalyzer under N2 stream at a heating rate of 5 °C min−1. Powder X-ray diffraction patterns were collected on a Bruker D8 ADVANCE X-ray powder diffractometer (Cu Kα, 1.5418 Å) at room temperature. Gas sorption isotherms were measured by ASAP 2020 M adsorption equipment. The solid state luminescent spectra were collected on a Hitachi F-4500 fluorescence spectrophotometer at room temperature.
2.2 Synthesis of [Cd3(L)2(H2O)4]·DMF (1)
A mixture of Cd(NO3)2·4H2O (0.0308 g, 0.1 mmol), H3L (0.018 g, 0.05 mmol) and DMF/H2O (15.0 mL, v/v = 1/1) (DMF = N,N-dimethylformamide) was mixed in a Teflon-lined stainless steel vessel (25 mL), and then heated at 150 °C for 72 h. After a gradual cooling procedure to room temperature, colourless block crystals were obtained in ∼51.3% yield based on Cd. Anal. calcd for C45H37Cd3NO17: C, 45.0; H, 3.08; N, 1.17. Found: C, 45.52; H, 3.17; N, 1.72%. IR (cm−1): 3423 (w), 3235 (w), 2923 (m), 1652 (s), 1521 (s), 1313 (m), 1182 (w), 1103 (w), 862 (w), 767 (m), 609 (w).
2.3 Synthesis of [Cd2(L)(SO4)2]·3(Me)2NH2 (2)
A mixture of Cd(NO3)2·4H2O (0.0308 g, 0.1 mmol), H3L (0.018 g, 0.05 mmol), DMF/ETOH (15.0 mL, v/v = 4/1) and H2SO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:3) was mixed in a Teflon-lined stainless steel vessel (25 mL), and then heated at 120 °C for 72 h. After a gradual cooling procedure to room temperature, colourless block crystals were obtained in ∼49.0% yield based on Cd. Anal. calcd for C27H35Cd2N3O14S2: C, 35.45; H, 3.83; N, 4.6. Found: C, 34.53; H, 3.17; N, 4.3%. IR (cm−1): 3438 (m), 3027 (s), 2794 (s), 2478 (s), 1594 (s), 1548 (s), 1394 (s), 1106 (s), 1031 (s), 960 (m), 856 (m), 767 (m), 609 (m).
:H2O (1:3) was mixed in a Teflon-lined stainless steel vessel (25 mL), and then heated at 120 °C for 72 h. After a gradual cooling procedure to room temperature, colourless block crystals were obtained in ∼49.0% yield based on Cd. Anal. calcd for C27H35Cd2N3O14S2: C, 35.45; H, 3.83; N, 4.6. Found: C, 34.53; H, 3.17; N, 4.3%. IR (cm−1): 3438 (m), 3027 (s), 2794 (s), 2478 (s), 1594 (s), 1548 (s), 1394 (s), 1106 (s), 1031 (s), 960 (m), 856 (m), 767 (m), 609 (m).
2.4 Synthesis of [Cd(HL)(H2O)]·0.5H2O (3)
A mixture of Cd(NO3)2·4H2O (0.0308 g, 0.1 mmol), H3L (0.018 g, 0.05 mmol) and H2O/CH3CN (14.0 mL, v/v = 2/5) was mixed in a Teflon-lined stainless steel vessel (25 mL), and then heated at 105 °C for 72 h. After a gradual cooling procedure to room temperature, colourless block crystals were obtained in ∼49.7% yield based on Cd. Anal. calcd for C23H18CdO7.5: C, 64.97; H, 3.20. Found: C, 64.52; H, 3.6. IR (cm−1): 3436 (m), 2931 (s), 2794 (s), 1664 (m), 1608 (m), 1405 (s), 1265 (m), 1031 (s), 1110 (m), 935 (w), 858 (w), 769 (m).
2.5 Crystallographic data collection and refinement
Suitable single crystals were tested using a Bruker SMART APEXII CCD diffractometer equipped with Mo-Kα radiation (λ = 0.71073 Å) at room temperature. The structures were solved using direct methods and refined using a full-matrix least-squares refinement on F2 with SHELXL-2014 and olex2.14 Non-hydrogen atoms were refined with anisotropic displacement parameters. The solvent molecules in 1 and 3 were highly disordered and were removed from the diffraction data by using the SQUEEZE routine of PLATON.15 The final formulae of 1 and 3 were determined by single-crystal structures, elemental analysis results and TGA. The relevant crystallographic data are shown in Table 1. Selected bond lengths and angles are listed in Table S1 (ESI†). CCDC numbers are 1561956–1561958 for 1–3, respectively.
Table 1 Crystallographic data of 1–3
| R1 = ∑||Fo| − |Fc||/∑|Fo|. wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2. |
| Complex |
1 |
2 |
3 |
| Formula |
C44H31Cd2NO14 |
C27H35Cd2N3O14S2 |
C23H18CdO7.5 |
| Mr |
1200 |
914.52 |
499 |
| Crystal system |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
C2/c |
C2/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a (Å) |
36.916 |
17.442 |
7.079 |
| b (Å) |
21.399 |
21.243 |
11.288 |
| c (Å) |
7.2173 |
11.468 |
13.130 |
| α (°) |
90 |
90 |
65.420 |
| β (°) |
98.129 |
129.716 |
83.016 |
| γ (°) |
90 |
90 |
85.505 |
| V (Å3) |
5644.1 |
3269 |
946.6 |
| Z |
4 |
4 |
2 |
| Dcalc (g cm−3) |
1.327 |
1.858 |
1.722 |
| F(000) |
2216 |
1832 |
488 |
| Rint |
0.0453 |
0.0715 |
0.0243 |
| GOF on F2 |
1.008 |
1.244 |
1.339 |
| R1a [I > 2σ(I)] |
0.0580 |
0.0796 |
0.0744 |
| wR2b (all data) |
0.2294 |
0.3130 |
0.2995 |
3 Results and discussion
3.1 Crystal structure of [Cd3(L)2(H2O)4]·DMF (1)
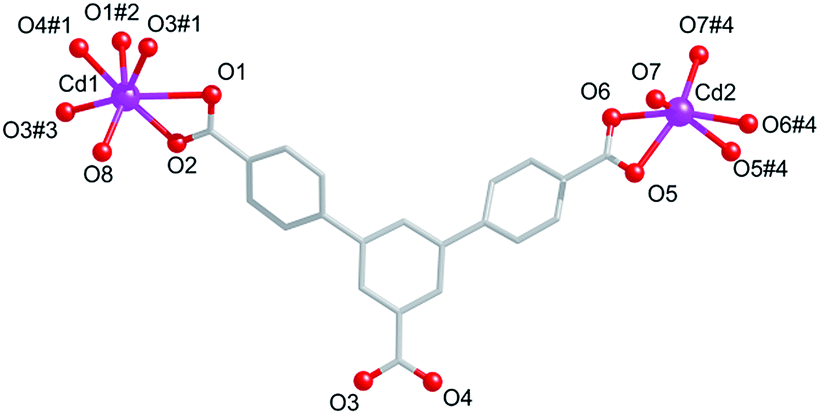
Compound 1 crystallizes in the monoclinic space group C2/c. As shown in the Fig. 1, the asymmetric unit consists of one and a half Cd(II) ions, one fully deprotonated L3− ligand, five coordinated H2O molecules, and one free DMF molecules. Cd1 is seven connected by O atoms from four L3− ligand and one H2O molecules. Different from that of Cd1, Cd2 is six connected by O atoms from two axial L3− ligand and two equatorial H2O molecules. The Cd–O distances vary from 2.241(12) to 2.62(3) Å, and the O–Cd–O angles are in the range from 53.37° to 167.28°.
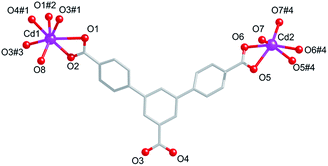 |
| | Fig. 1 Coordination environment around the Cd(II) ions in 1. Symmetry codes: #1: 1.5 − x, 0.5 + y, 2.5 − z, #2: x, 2 − y, 0.5 + z, #3: 1.5 − x, 1.5 − y, 3 − z. #4: 1 − x, y, −0.5 − z. | |
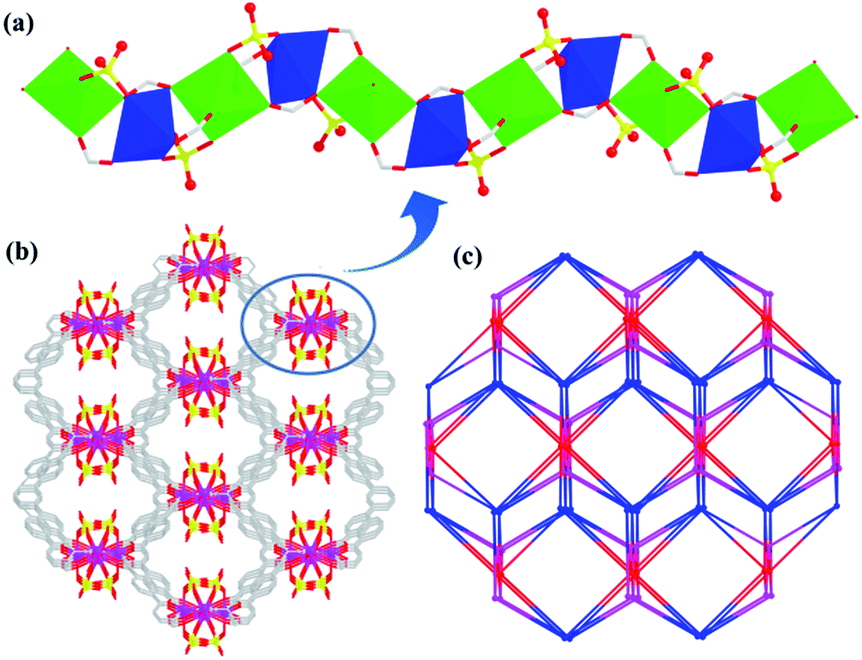
In 1, Cd1 ions take coordination with carboxylates of L3− ligands to form 1D rod-shaped SBUs (Fig. 2a). Then these 1D SBUs are further extended by the Cd2 ions and L3− linkers together to give rise to a 3D porous framework (Fig. 2b). There exist two types of 1D open channels with different shapes along c axis with the effective aperture sizes of 18.1 × 16.2 Å2 and 10.2 × 8.4 Å2, respectively, where the free solvent DMF molecules are located. After the hypothetical removal of the noncoordinated and disordered solvent molecules, the potential solvent area volume of 1a-[Cd3(L)2(H2O)4] is determined to be ∼36.5% out of the total volume by the PLATON program. Topologically, L3− and the Cd1 ions can be considered as five, four-connected nodes, respectively. Thus, the framework of 1 can be view as a (4,5)-connected tcs net with the point symbol of (44·62)(44·66) (Fig. 2c).
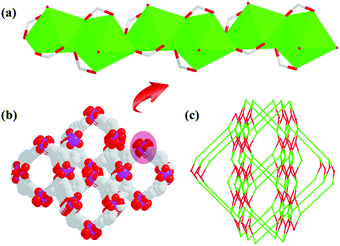 |
| | Fig. 2 (a) The 1D rod-shaped SBU in 1. (b) The 3D microporous framework of 1. (c) The (4,5)-connected topology network. | |
3.2 Crystal structure of [Cd2(L)(SO4)2]·3(Me)2NH2 (2)
Compound 2 is a 3D framework crystallizing in the monoclinic space group C2/c. The asymmetric unit contains two independent Cd(II) ions, one L3− ligand, two coordinated sulphate anions, and three dimethylammonium cations. The Cd1 and Cd2 display the distorted hexahedral pyramidal coordination geometries (Fig. 3). Cd1 is six-coordinate and anchored by four carboxylate groups from four different L3− and two coordinated sulfate anions. Cd2 is also six-coordinated, and anchored by four carboxylate groups from four different ligands and two coordinated sulfate anions. The Cd–O distances vary from 2.446(6) to 2.194(8) Å, and the O–Cd–O angles are in the range from 53.5(3)° to 180.0°.
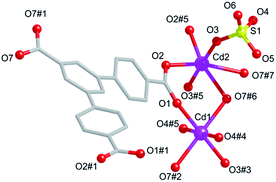 |
| | Fig. 3 Coordination environment around the Cd(II) ions in 2. Symmetry codes: #1: 2 − x, y, 1.5 − z, #2: 2 − x, 1 − y, 2 − z, #3: 1.5 − x, 0.5 − y, 2 − z, #4: 0.5 + x, 0.5 − y, 0.5 + z, #5: 1 − x, y, 1.5 − z, #6: −0.5 + x, −0.5 + y, z, #7: 1.5 − x, −0.5 + y, 1.5 − z. | |
Similar to that of 1, an infinite 1D rod-shaped SBU chain is formed by the carboxylate of L3− and sulfate with Cd(II) ions in 2 (Fig. 4a). These 1D SBUs based on Cd1 and Cd2 are further extended by the organic linker to a 3D porous network, where the dimethylammonium cations and sulfate are included (Fig. 4b). Topologically, the linkers can be viewed as seven-connected nodes, and the centre metal ions (Cd1 and Cd2) can act as 5,6-connected nodes, respectively. Thus, the structure of 2 can be simplified as a 3-nodal (5,6,7)-connected new topological net with a point symbol of (34·43·52·6)(34·43·54·610)(34·44·54·63) (Fig. 4c).
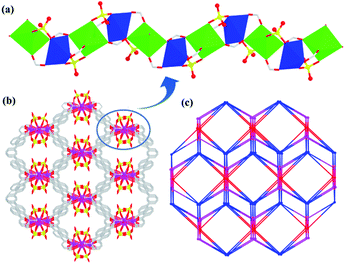 |
| | Fig. 4 (a) The 1D chain SBU in 2. (b) The 3D framework of 2. (c) (5,6,7)-connected topological network. | |
3.3 Crystal structure of [Cd(HL)(H2O)]·0.5H2O (3)
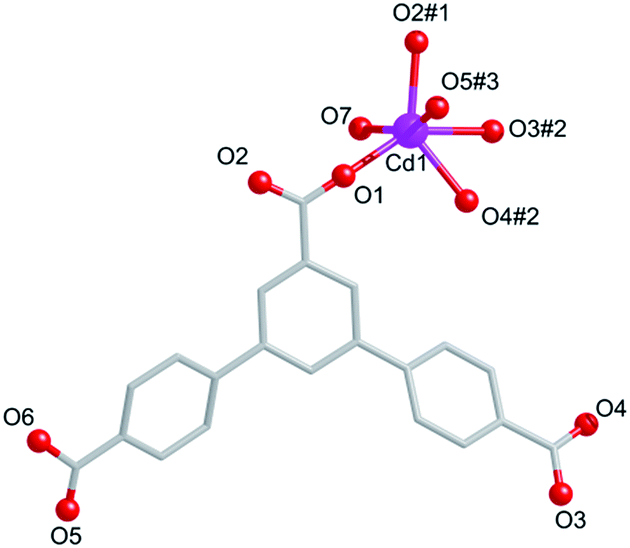
Compound 3 crystallizes in the triclinic space group P. The asymmetric building unit of 3 contains one Cd(II) center, one partly deprotonated HL2− ligand, one coordination water molecule, and a half free water molecule. Each centre Cd(II) is surrounded by six oxygen atoms from four different carboxylate groups of four HL2− ligands and one water molecule (Fig. 5). The lengths of Cd–O bonds are in the range of 2.375(7)–2.194(8) Å.
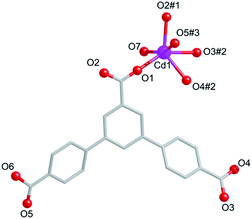 |
| | Fig. 5 Coordination environment of Cd(II) ions in 4. Symmetry codes: #1: −x, −y, −z, #2: −1 − x, −y, 1 − z, #3: −1 + x, −1 + y, z. | |
Due to the existence of deprotonated HL2− ligand in 3, a dimeric SBU is only formed by the carboxylate of HL2− ligands and Cd(II) ions (Fig. S1a, ESI†). The dimeric units are bridged by the linkers to yield a 2D layer (Fig. 6a). Both the linkers and the centre Cd(II) can be viewed as four-coordinated nodes, and the framework forms an uninodal (4,4)-connected sql topology with the point symbol of (44·62) (Fig. 6b). Further, these 2D layers further are stacked together via hydrogen bonding O7–H7B⋯O2 and O7–H7A⋯O3 to construct a 3D supramolecular framework (Fig. S1b, ESI†).
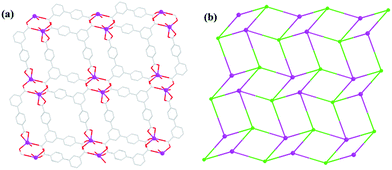 |
| | Fig. 6 (a) The 2D layer of 3. (b) The (4,4)-connected network. | |
3.4 Coordination modes of H3L ligand in complexes 1–3
The comparative structural studies of 1–3 reveal that the coordination mode of H3L is significantly influenced by the central metals and the solvent environment in the formation of frameworks. The H3L in 1 and 2 is fully deprotonated, and connect six metal ions. In contrast, the H3L in 3 is partly deprotonated, and connect four metal ions. As shown in Scheme 2, the three carboxylates of L3− coordination fashions to connect with the centre Cd(II) ions. In 1, the three carboxylates of L3− exhibit two different coordination modes: chelating bidentate (η2μ1χ2) and bridging tridentate (η2μ2χ3). In 2, the three carboxylates of L3− present two different coordination modes: bridging bidentate (η2μ2χ2) and bridging tetradentate (η2μ3χ4) modes. In 3, the three carboxylates of HL2− exhibit three different coordination modes: monodentate (η1μ1χ1) chelating monodentate (η2μ1χ2) and bridging bidentate (η2μ2χ2). The structures of 1–3 suggest that not only H3L coordinate with the different number of metal ions, the different coordination modes of carboxylates play an important role in determining the distinction of final structural topologies. In addition, the coordinated solvents also significantly influence the structures of the frameworks, that is, there is one coordinated SO42− anion in 2 and four H2O molecules in 1. The difference of the coordination solvents affects the coordination environment of the central metals and coordination modes of ligands, leading to different structures.
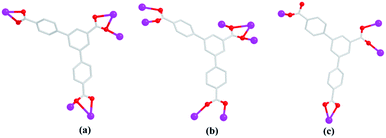 |
| | Scheme 2 The different coordination modes of 1 (a), 2 (b) and 3 (c). | |
3.5 PXRD and thermal analyses
The PXRD patterns show that the peak positions of the obtained crystalline samples of 1–3 match well with the simulated from the single-crystal data, manifesting the phase purity of the synthesized samples (Fig. S2, ESI†). TGA on 1–3 were performed in a N2 atmosphere from 30 to 700 °C (Fig. S3, ESI†). Complex 1 indicates a weight loss of 12.6% in the range of 30–130 °C, which consists of the release of one DMF and four coordinated H2O molecule per formula unit (calcd 12.1%). 2 released two coordinated SO42− anion and three H2N(Me)2 cations between 30 and 375 °C with a weight loss of 36.6% (calcd 36.2%). For 3, it lost one coordinated water and half lattice water molecule from 30 to 110 °C with a weight loss of 5.1% (calcd 5.5%), and the major framework is stable up to 365 °C and then begins to collapse.
3.6 Gas adsorption and discussion
To obtain the guest-free phase of 1a, 1 was soaked in CH2Cl2 for 7 days and subsequent heating at 150 °C under vacuum for 6 h, which is evidenced by the TGA curve of 1a (Fig. S3a, ESI†). The IR of 1a (Fig. S4, ESI†) shows that the lack of characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration of DMF reveals that the disordered solvent has been removed from the nanotubes. The structure integrity of 1a has also been verified by the PXRD experiment (Fig. S2a, ESI†).
O vibration of DMF reveals that the disordered solvent has been removed from the nanotubes. The structure integrity of 1a has also been verified by the PXRD experiment (Fig. S2a, ESI†).
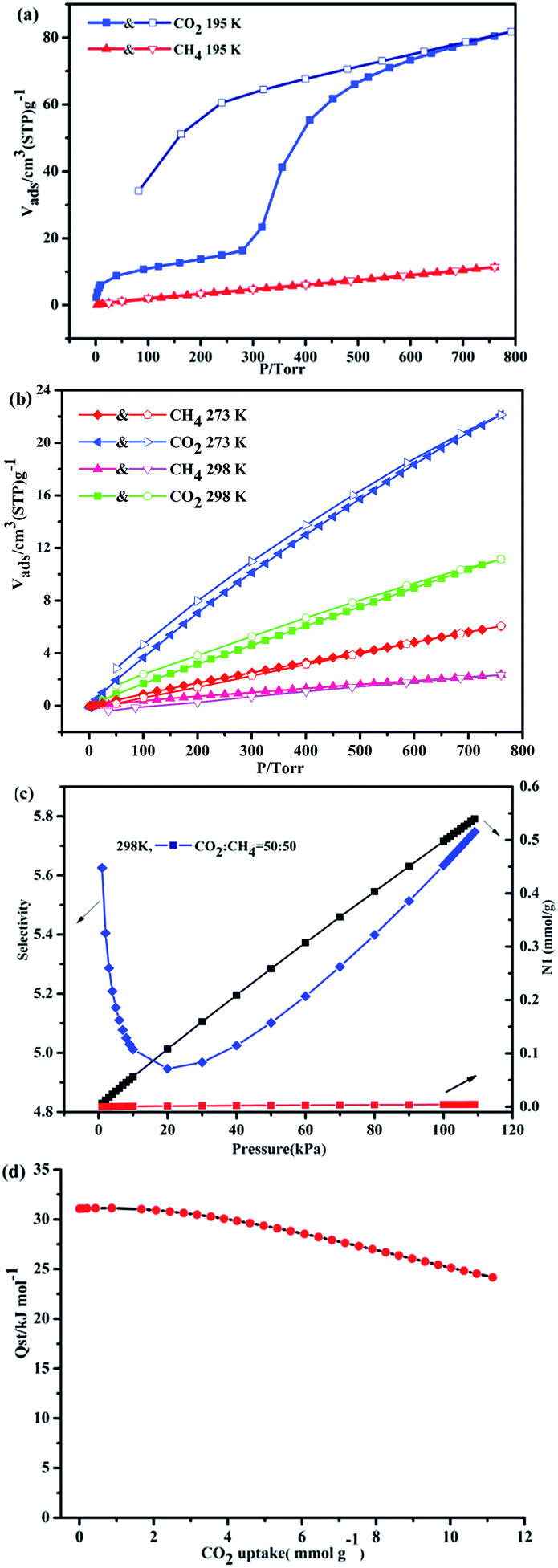
To verify the porosity of 1a, the sorption isotherms of N2, CO2, and CH4 were measured at different temperatures. As shown in Fig. S6 (ESI†), the lower N2 uptake (28.69 cm3 g−1) of 1a at 77 K and 1 bar may be due to the strong interactions of incoming guest molecules with the pore windows at 77 K, which prevents the next incoming guest molecules from entering into the pore.16 On the basis of the N2 adsorption isotherm, the Brunauer–Emmett–Teller (BET) and Langmuir surface areas are 17.06 m2 g−1 and 26.22 m2 g−1, respectively. At 195 K, 1a exhibits the stepwise gas adsorption toward CO2 gas, and the second uptake occurs around P/Torr = 300 mmHg, showing an uptake of 81.8 cm3 g−1 (16.67 wt%) which is more than that of the CH4 uptake (11.39 cm3 g−1, 0.8 wt%). Such dynamic behaviors of layers are responsible for adsorption-induced gate-opening effects in MOF materials, which is associated with the flexibility of the frameworks. Thus, the activated sample of 1a appears to be the semi-closed form, and then undergoes the structural transformation to the open or as-synthesized form once the material obtains sufficient energy from gas adsorbate molecules to overcome energy barrier.17,18 Meanwhile, there is a marked broad hysteresis, confirming that the adsorbed CO2 is not immediately released on reducing the external pressure and is thus trapped within the framework at very low pressures. The possible reason for the stepwise and hysteretic sorption isotherms of the material is related to the framework of 1a with the shrunken pores, and the host structure may expand above the gate opening pressure, which should be another typical example of the breathing MOFs.19 Such adsorption selectivity is also more evident at high temperatures. The CO2 uptake are 22.12 cm3 g−1 (4.31 wt%) at 273 K and 11.14 cm3 g−1 (2.21 wt%) at 298 K and 1 bar. Meanwhile, the CH4 uptakes are 6.0 cm3 g−1 (0.41 wt%) at 273 K and 2.37 (0.17 wt%) at 298 K and 1 bar (Fig. 7b).
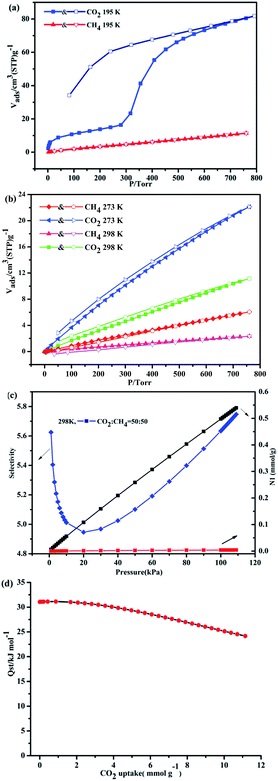 |
| | Fig. 7 (a) The adsorption isotherms of 1a (CO2 and CH4) at 195 K. (b) The adsorption isotherms of CO2 and CH4 at 273 and 298 K. (c) IAST adsorption selectivity of 1a for equimolar mixtures of CO2 and CH4 at 298 K. (d) Isosteric heat of CO2 adsorption for 1a by the virial equation from the adsorption isotherms at 273 and 298 K. | |
Most interestingly, the examination of CO2 and CH4 sorption reveals that there is indeed existence of selectivity for respective sorbate molecules at different temperature. 1a shows high adsorption selectivity for CO2/CH4 at 298 K, as shown in Fig. 7c. The adsorption selectivities are calculated via the ideal adsorption solution theory (IAST)20–22 (Fig. S7, ESI†). For an equimolar mixture, the CO2/CH4 selectivity is estimated as 5.6 at 298 K. The isosteric heat (Qst) of CO2 adsorption was calculated by the virial equation from the sorption isotherms at 298 K. At initial coverage, Qst start at 30.0 kJ mol−1, however, as the CO2 uptakes rise up to 11.16 mmol g−1, the Qst reaches to 24.1 kJ mol−1 (Fig. 7d). This value is superior to those MOFs decorated by typical active site such as Lewis basic sites (LBSs) and OMSs, for example Cu(bcppm) (29 kJ mol−1),23 PCN-88 (27 kJ mol−1),24 PCN-16 (22.5 kJ mol−1) and Zn-MOF-74 (30 kJ mol−1),25 but much lower than those of some high polarity functionalized MOFs, such as CuBTTri-en (80 kJ mol−1),26 MIL-100 (60 kJ mol−1), and MIL-101 (45 kJ mol−1).27 The high enthalpies of CO2 enthalpies is mainly due to that the frameworks owns highly polar and specific interactions with CO2 because of its large quadrupole moment. Overall, this study provided theoretical prediction in CO2 capture and the fact that 1a has high selectivity of CO2, indicating that this material may be a promising adsorbent in the biogas treatment and natural gas clean up.
3.7 Luminescence properties
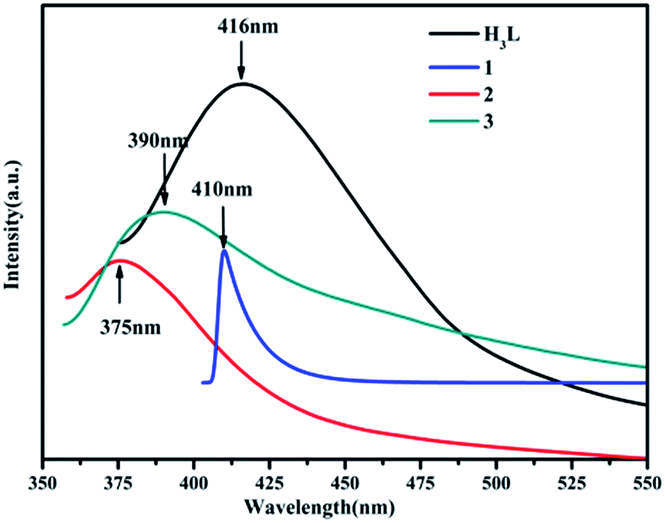
MOFs including d10 ions have been become the potential fluorescent materials in the optical field.28 Thus, the solid-state luminescence properties of the three MOFs and H3L ligand were studied at room temperature (Fig. 8). The maximum emission peaks of the free H3L ligand and 1–3 are observed at ∼416 nm for H3L ligand (λex = 356 nm), ∼410 nm for 1 (λex = 350 nm), ∼375 nm for 2 (λex = 338 nm) and ∼390 nm for 3 (λex = 337 nm), respectively. The maximum emissions of 2 and 3 are obviously blue shifted compared to those of H3L ligand, which may arise from ligand-to-metal charge transfer (LMCT). Furthermore, the luminescence of 1 is similar to that of the free ligand, which is probably attributed to the intraligand transitions modified by metal coordination.29
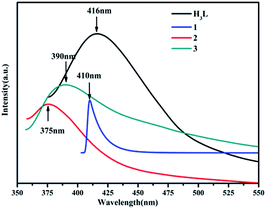 |
| | Fig. 8 Luminescent emission spectra of the free ligand H3L (black), 1 (blue), 2 (red), and 3 (green) in the solid state at room temperature. | |
4 Conclusion
In summary, three new luminescent MOFs including Cd(II) ions have been successfully assembled via a symmetrical tricarboxylate H3L ligand. Because of the reactions in different solvent systems, L3−/HL2− in 1–3 show the different coordination modes to form various SBUs in the final structures. Interestingly, complex 1 possesses two 1D open channels with different shapes. More importantly, the desolvated framework 1a displays permanent porosity with suitable pore size, which affords CO2 loading and selective capture for CO2 over CH4 at 273 K. Herein, this work indicates that the unique structures and gas selectivity for CO2 may draw attention to the fabrication of functional MOF materials.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful for financial support from the NSFC (21531007, 21371142, and 21201139), NSF of Shaanxi Province (2017KJXX-59), the China Postdoctoral Science Foundation (2016M600807, and 2017T100765), and the Postdoctoral Science Foundation of Northwest University (334100049).
Notes and references
-
(a) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed;
(b) Y. Cui, B. Li, H. He, W. Zhou, B. Chen and G. Qian, Acc. Chem. Res., 2016, 49, 483–493 CrossRef CAS PubMed;
(c) G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC;
(d) Y. Wu, G.-P. Yang, Y. Zhao, W.-P. Wu, B. Liu and Y.-Y. Wang, Dalton Trans., 2015, 44, 3271–3277 RSC;
(e) M. Mon, J. Ferrando-Soria, T. Grancha, F. R. Fortea-Pérez, J. Gascon, A. Leyva-Pérez, D. Armentano and E. Pardo, J. Am. Chem. Soc., 2016, 138, 7864–7867 CrossRef CAS PubMed.
-
(a) A. C. McKinlay, R. E. Morris, P. Horcajada, G. Férey, R. Gref, P. Couvreur and C. Serre, Angew. Chem., Int. Ed., 2010, 49, 6260–6266 CrossRef CAS PubMed;
(b) S. Horike, D. Umeyama and S. Kitagawa, Acc. Chem. Res., 2013, 46, 2376–2384 CrossRef CAS PubMed;
(c) J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed;
(d) T. Grancha, J. Ferrando-Soria, M. Castellano, M. Julve, J. Pasán, D. Armentanoc and E. Pardo, Chem. Commun., 2014, 50, 7569–7585 RSC.
- Y. Inokuma, T. Arai and M. Fujita, Nat. Chem., 2010, 2, 780–783 CrossRef CAS PubMed.
-
(a) A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed;
(b) A. G. Slater and A. I. Cooper, Science, 2015, 348, 8075 CrossRef PubMed.
-
(a) D. Alezi, A. M. P. Peedikakkal, Ł. J. Weseliński, V. Guillerm, Y. Belmabkhout, A. J. Cairns, Z. Chen, Ł. Wojtas and M. Eddaoudi, J. Am. Chem. Soc., 2015, 137, 5421–5430 CrossRef CAS PubMed;
(b) S. Yang, A. J. Ramirez-Cuesta, R. Newby, V. Garcia-Sakai, P. Manuel, S. K. Callear, S. I. Campbell, C. C. Tang and M. Schröder, Nat. Chem., 2015, 7, 121–129 CrossRef CAS PubMed;
(c) R. Haldar, M. Inukai, S. Horike, K. Uemura, S. Kitagawa and T. K. Maji, Inorg. Chem., 2016, 55, 4166–4172 CrossRef CAS PubMed.
- J. Yu, L.-H. Xie, J.-R. Li, Y. Ma, J. M. Seminario and P. B. Balbuena, Chem. Rev., 2017, 117(14), 9674–9754 CrossRef CAS PubMed.
-
(a) M.-H. Sun, S.-Z. Huang, L.-H. Chen, Y. Li, X.-Y. Yang, Z.-Y. Yuan and B.-L. Su, Chem. Soc. Rev., 2016, 45, 3479–3563 RSC;
(b) D. Bradshaw, S. El-Hankari and L. Lupica-Spagnolo, Chem. Soc. Rev., 2014, 43, 5431–5443 RSC;
(c) H.-C. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC;
(d) T. T. Xu, C. F Xue, Z. L. Zhang and X. G. Hao, Prog.
Chem., 2014, 26, 1924–1929 CAS.
-
(a) P.-Q. Liao, N.-Y. Huang, W.-X. Zhang, J.-P. Zhang and X.-M. Chen, Science, 2017, 356, 1193–1196 CrossRef CAS PubMed;
(b) M. J. Cliffe, E. Castillo-Martínez, Y. Wu, J. Lee, A. C. Forse, F. C. N. Firth, P. Z. Moghadam, D. Fairen-Jimenez, M. W. Gaultois, J. A. Hill, O. V. Magdysyuk, B. Slater, A. L. Goodwin and C. P. Grey, J. Am. Chem. Soc., 2017, 139, 5397–5404 CrossRef CAS PubMed;
(c) Y.-L. Huang, Y.-N. Gong, L. Jiang and T.-B. Lu, Chem. Commun., 2013, 49, 1753–1755 RSC;
(d) Y.-N. Gong and T.-B. Lu, Chem. Commun., 2013, 49, 7711–7713 RSC.
-
(a) G. Ji, J. Liu, X. Gao, W. Sun, J. Wang, S. Zhao and Z. Liu, J. Mater. Chem. A, 2017, 5, 10200–10205 RSC;
(b) H.-S. Lu, L.-L. Bai, W.-W. Xiong, P.-Z. Li, J.-F. Ding, G.-D. Zhang, T. Wu, Y.-L. Zhao, J. M. Lee, Y.-H. Yang, B.-Y. Geng and Q.-C. Zhang, Inorg. Chem., 2014, 53, 8529–8537 CrossRef CAS PubMed;
(c) Z.-J. Zhang, W. Lukasz and J. Z. Michael, Cryst. Growth Des., 2014, 14, 1526–1530 CrossRef CAS.
-
(a) T. Grancha, X. Qu, M. Julve, J. Ferrando-Soria, D. Armentano and E. Pardo, Inorg. Chem., 2017, 56, 6551–6557 CrossRef CAS PubMed;
(b) S.-H. Huang, C.-H. Lin and W.-C. Wu, Angew. Chem., Int. Ed., 2009, 48, 6124–6127 CrossRef CAS PubMed;
(c) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- L.-L. Liu, C.-X. Yu, F.-J. Ma, Y.-R. Li, J.-J. Han, L. Lin and L.-F. Ma, Dalton Trans., 2015, 44, 1636–1645 RSC.
-
(a) D. Zhao, D. Yuan, D. Sun and H.-C. Zhou, J. Am. Chem. Soc., 2009, 131, 9186–9188 CrossRef CAS PubMed;
(b) Y.-W. Li, J. Xu, D.-C. Li, J.-M. Dou, H. Yan, T.-L. Hu and X.-H. Bu, Chem. Commun., 2015, 51, 14211–14214 RSC;
(c) L. Qin, J.-S. Hu, Y.-Z. Li and H.-G. Zheng, Cryst. Growth Des., 2011, 11, 3115–3121 CrossRef CAS;
(d) M. Zhang, Q. Wang, Z. Lu, H. Liu, W. Liu and J. Bai, CrystEngComm, 2014, 16, 6287–6290 RSC.
-
(a) T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef CAS PubMed;
(b) Y. He, S. Xiang, Z. Zhang, S. Xiong, F. R. Fronczek, R. Krishna, M. O'Keeffe and B. Chen, Chem. Commun., 2012, 48, 10856–10858 RSC;
(c) W.-M. Chen, X.-L. Meng, G.-L. Zhuang, Z. Wang, M. Kurmoo, Q.-Q. Zhao, X.-P. Wang, B. Shan, C.-H. Tung and D. Sun, J. Mater. Chem. A, 2017, 5, 13079–13085 RSC.
-
(a) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS;
(b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- B. Liu, R. Zhao, G. Yang, L. Hou, Y.-Y. Wang and Q.-Z. Shi, CrystEngComm, 2013, 15, 2057–2060 RSC.
-
(a) C.-S. Liu, Z.-H. Zhang, M. Chen, H. Zhao, F.-H. Duan, D.-M. Chen, M.-H. Wang, S. Zhang and M. Du, Chem. Commun., 2017, 53, 3941–3944 RSC;
(b) Q. Chen, Y. Ma, W. Song, Z. Chang, J.-R. Li, J. Zhang, H.-W. Sun, P. Balbuena and X.-H. Bu, Inorg. Chem., 2017, 56, 2614–2620 CrossRef CAS PubMed;
(c) Z. Shi, L. Qin and H. G. Zheng, Dalton Trans., 2017, 46, 4589–4594 RSC;
(d) S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed.
- J. Duan, M. Higuchi, M. L. Foo, S. Horike, K. P. Rao and S. Kitagawa, Inorg. Chem., 2013, 52, 8244–8249 CrossRef CAS PubMed.
- G.-P. Li, G. Liu, Y.-Z. Li, L. Hou, Y.-Y. Wang and Z. Zhu, Inorg. Chem., 2016, 55, 3952–3959 CrossRef CAS PubMed.
-
(a) D. Wang, B. Liu, S. Yao, T. Wang, G. Li, Q. Huo and Y. Liu, Chem. Commun., 2015, 51, 15287–15289 RSC;
(b) A. G. Wong-Foy, O. Lebel and A. J. Matzger, J. Am. Chem. Soc., 2007, 129, 15740–15741 CrossRef CAS PubMed.
-
(a) T. Pham, K. A. Forrest, B. Tudor, S. K. Elsaidi, M. H. Mohamed, K. McLaughlin, C. R. Cioce, M. J. Zaworotko and B. Space, Langmuir, 2014, 30, 6454–6462 CrossRef CAS PubMed;
(b) Y.-L. Wu, J. Qian, G.-P. Yang, F. Yang, Y.-T. Liang, W.-Y. Zhang and Y.-Y. Wang, Inorg. Chem., 2017, 56, 908–913 CrossRef CAS PubMed.
- Y. Xie, H. Yang, Z. U. Wang, Y. Liu, H.-C. Zhou and J.-R. Li, Chem. Commun., 2014, 50, 563–565 RSC.
- J. Zhao, Y. Wang, W. Dong, Y. Wu, D. S. Li, B. Liu and Q. Zhang, Chem. Commun., 2015, 51, 9479–9482 RSC.
- J.-R. Li, J. Yu, W. Lu, L.-B. Sun, J. Sculley, P. B. Balbuena and H.-C. Zhou, Nat. Commun., 2013, 4, 1–8 CAS.
- J. M. Simmons, H. Wu, W. Zhou and T. Yildirim, Energy Environ. Sci., 2011, 4, 2177–2185 CAS.
- H.-R. Fu, F. Wang and J. Zhang, Dalton Trans., 2015, 44, 2893–2896 RSC.
- A. Santra and P. K. Bharadwaj, Cryst. Growth Des., 2014, 14, 1476–1485 CAS.
-
(a) L. Luo, K. Chen, Q. Liu, Y. Lu, T.-A. Okamura, G.-C. Lv, Y. Zhao and W.-Y. Sun, Cryst. Growth Des., 2013, 13, 2312–2321 CrossRef CAS;
(b) P. Lama and P. K. Bharadwaj, Cryst. Growth Des., 2011, 11, 5434–5440 CrossRef CAS;
(c) R. Singh and P. K. Bharadwaj, Cryst. Growth Des., 2013, 13, 3722–3733 CrossRef CAS;
(d) J. Qian, P. Yu and S. Huang, CrystEngComm, 2016, 18, 7955–7958 RSC;
(e) Y. Zhang, J. Han and C. Liao, CrystEngComm, 2016, 18, 9026–9032 RSC.
- B.-B. Kang, N. Wei and Z.-B. Han, RSC Adv., 2015, 5, 1605–1611 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Some figures of single-crystals, PXRD, IR, TGA, selected bond length/angle tables. CCDC 1561956–1561958. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra08188j |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *,
Yang-Tian Yan,
Jing Jin,
Yan Ning and
Yao-Yu Wang
*,
Yang-Tian Yan,
Jing Jin,
Yan Ning and
Yao-Yu Wang