
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and preparation of porous polymer particles with polydopamine coating and selective enrichment for biomolecules†
Hao Wangab,
Zihao Qinc,
Yi Liuad,
Xiaoting Liab,
Jianfei Liuad,
Yongfeng Liu*ad,
Dongdong Huangae and
Duolong Di *ad
*ad
aCAS Key Laboratory of Chemistry of Northwestern Plant Resources, Key Laboratory for Natural Medicine of Gansu Province, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, Lanzhou 730000, P. R. China. E-mail: yfliu@licp.cas.cn; didl@licp.cas.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cCenter for Degradable and Flame-Retardant Polymeric Materials, College of Chemistry, State Key Laboratory of Polymer Materials Engineering, National Engineering Laboratory of Eco-Friendly Polymeric Materials (Sichuan), Sichuan University, Chengdu 610064, PR China
dCentre of Resource Chemical and New Material, Qingdao 266100, P. R. China
eCollege of Chemistry and Chemical Engineering, Northwest Normal University, Lanzhou 730070, P. R. China
First published on 22nd September 2017
Abstract
Macroporous adsorption resins are one of the most important polymer particles for diverse potential applications. Typically, gigaporous structures can be generated by the surfactant reverse micelles swelling method, providing polymer particles with ultra-large pore diameter. Herein, we reported the first example of poly(styrene-co-ethylene glycol dimethacrylate) (PSE) as efficient enrichment media for biomolecule enrichment. The PSE possessed a gigaporous structure which was even capable of adsorbing biomacromolecule bovine serum albumin (BSA). A novel and facile strategy of polydopamine (PDOPA) modification was proposed to fabricate porous particles, and hereby PDOPA in ethanol aqueous solution was coated on PSE with well-controlled thickness. Laser scanning confocal microscopy was employed to characterize the internal pore structure and understand the distribution of adsorbate in the porous particles. The PDOPA modified porous particles exhibited a selective adsorption for peptides with different molecular weights based on the sieving effect and other interactions.
Introduction
Macroporous adsorption resins (MAR) have drawn great attention in many fields for their excellent properties, such as large specific surface area, low density and high adsorption capacity.1,2 Due to these unique advantages, MAR have been widely applied in the separation and purification of bioactive compounds from natural resources: flavonoids from the extract of Chinese wolfberry,3 narirutin from extracts of Citrus unshiu peels,4 anthocyanins from grape pomace extracts,5 and tea polyphenols and caffeine from discarded green tea,6 etc. However, the pore size of the MAR mentioned above are approximately 5–10 nm in general. Comparing with protein and peptides molecules with large molecule-size and complex structures, those bioactive compounds mentioned above are much smaller molecules.Peptides are a special bioactive substance between amino acids and proteins, which are divided into endogenous peptides from human and exogenous peptides from animals, plants and microorganisms. Due to their unique biological activities, peptides have been used in variety of fields. Peptides can form amyloid fibers and fibrils have been used as amyloid model structures.7 Peptides as drug-targeting agents can widen the therapeutic window by both increasing drug efficacy and decreasing certain side effects.8 Because of the high specificity of target peptides, they can be adapted for diagnostic applications in the same manner in which antibodies are currently used.9 In addition, peptides have become an important health care product and vaccines.10 Generally, the way to obtain peptides consists of two species: separation and synthesis. Since peptides synthesis is more time-consuming, demanding and expensive, it is a good idea to get them directly from organism. Therefore, separation and purification of biological molecules like proteins and peptides is a greatly challenge.
The pore size plays an essential role in the adsorption and desorption process for biomolecule.2 Preparation of a novel MAR with larger pore size becomes unavoidable for current challenge. Until now, a small minority of methods for preparation of porous particles with relatively large pore size have been reported. Wang and co-workers11 prepared polymers with polyethylene glycol, which showed outstanding adsorption and desorption capabilities with bovine serum albumin and trypsin. Li and co-worker12 studied the lipase distribution, thermal stability, stability, and reusability by adopting giga-/macro-/meso-porous polystyrene microspheres with pore diameter of 314, 104 and 14.7, respectively, and the gigaporous polystyrene microsphere exhibited remarkable advantages as a potential enzyme support compared with other microspheres. Basso and co-worker13–15 prepared porous polymer particles with pore diameter of 200 nm, and porosity could be tuned to make reactants intersperse adequately to ensure the synthesis of peptides. These particles and microspheres with larger pore size have been widely used in the separation and synthesis of biomolecules.
As is known to all, the preparation methods of porous particles are intricate and demanding. Therefore, some literatures have covered all sorts of techniques to prepare porous particles and microspheres, such as suspension,16,17 precipitation,18,19 electrospraying,20 dispersion,21,22 microfluidic23,24 and emulsification25,26 polymerizations. Among these, suspension, precipitation and emulsification polymerization methods are the main techniques. Table 1 listed a brief comparison about these three methods.2,16,18,19,25–27
| Emulsification | Suspension | Precipitation | |
|---|---|---|---|
| Particle size | 500 nm to 1 mm | 100 nm to 5 mm | 100 nm to 5 mm |
| Pore size | 20 nm to 500 nm | 20 nm to 500 nm | 50 nm to 2 μm |
| Advantages | Reaction rapidly; narrow particle size distribution | Adjustable temperature | No suspension agents; no surfactants; adjustable temperature |
| Disadvantages | Removing surfactants hardly and complicatedly | Removing suspension agents hardly and complicatedly | Microspheres less regular; harsh reaction condition; time consuming |
Currently, a new method known as the surfactant reverse micelles swelling method has been proposed,25 and the theory is easy and accessible. The oil phase is fully dispersed by ultrasound assisted dispersion to form reverse micelles. Then, we transferred oil phase to the water phase. Under the stirring, the reverse micelles disperse in water phase, and water molecules will enter into oil droplets on the basis of several interactions to form the water channels. Once polymerization is finished, the water phase is removed by drying under vacuum conditions, and the previous water channels become large pores.26,28 This method exhibits favorable advantages of both suspension and emulsification, which is time-saving, exercisable, temperature controllable and specifically pore sizes tunable. Large pore size is one of vital factors in determining the adsorption and desorption properties for biomolecules, and commonly the ideal size of the pores is three to five times of the biomolecule's size.29 Two porous particles were prepared by this method, however, strong hydrophobicity of poly(styrene-co-divinyl benzene) (PS-DVB) caused undesirable absorption ability, which is partly attributed to poor dispersion of the materials in biomolecule solutions.30 Many researchers polished up these particles by coating and grafting hydrophilic groups on surface of PS-DVB.30,31 Obviously, these methods are complex and time-consuming. To surmount these problems, a novel dopamine-modified poly(styrene-co-ethylene glycol dimethacrylate) (PS-EGDMA) has to be formulated to improve corresponding hydrophilic property. In order to simplify the names of materials later, we abbreviate PS-EGDMA to PSE.
Recently, the adhesive proteins secreted by mussels have gained great attention owing to the unexpected ability of mussels to coat various surfaces, such as noble metals, oxides, polymers, semiconductors, and ceramics.32 The adhesive proteins are found at the adhesive layers between the mussels and matrix, and the proteins primarily consist of a particular amino acid known as 3,4-dihydroxyphenethylamine (dopamine, DOPA) with abundant hydroxyl groups.33 Lee and co-worker have proven that DOPA can self-polymerize under alkaline environment and spontaneously deposit onto surface of substances.32,34 Polydopamine (PDOPA) and PDOPA modified porous particles were biocompatible,32 so PDOPA modified PSE (PSEP) would not affect the bioactivity of peptides if the peptides were enriched or separated by the material. In addition, PDOPA coating with hydroxyl and amino groups may provide potential to endure secondary reactions and functional modification by Michael addition or Schiff base reactions or others, such as immobilization of various inorganic matters at the polymer surface.35,36
Herein, we proposed a novel micro-hydrophilic and PDOPA-coated porous particle with desirable hydrophilicity, high mechanical strength and excellent adsorption ability. A new and facile method was applied to modify relatively hydrophobic porous particles with PDOPA coating with improved coating efficiency. The PSE possessed a gigaporous structure and could enrich biomacromolecule bovine serum albumin (BSA). Due to catechol-rich PDOPA, these coating particles would exhibit superior adsorption property for certain peptides with certain functional groups.
Experimental section
Materials
Styrene (ST, AR) and divinylbenzene (DVB, AR) was acquired from Tianjin Damao Chemical Reagent Factory (Tianjin, China). ST and DVB were distilled under reduced pressure before use. Ethylene glycol dimethacrylate (EGDMA, AR, 97%) was bought from Tokyo Chemical Industry Co (Tokyo, Japanese). The initiator benzoyl peroxide (BPO) was purchased from Beijing Chemical Reagents Co. Hexadecane (HD, LR) was purchased from Wako Pure Chemical Industries, Ltd. and used as a hydrophobic additive to retard the monomer diffusing into the aqueous phase.25 Sorbitan monooleate (Span 80, CP) was purchased from Laiwu Shuangshuang chemical plant (Shandong, China). The stabilizer poly (vinyl alcohol) (PVA, degree of polymerization 1750) was purchased from Sinopharm Chemical Reagent Co., Ltd. (Beijing, China). The inhibitor hydroquinone (HQ, AR) was purchased from Shanghai Zhongqin Chemical Reagent Co., Ltd. (Shanghai, China) and used to prevent the secondary nucleation in the aqueous phase.21 Sodium dodecyl sulfate (SDS, AR) was purchased from Merck (USA) and can associate with PVA in solution and formed a complex at the interface, to decrease interfacial tension and stabilize the droplets.37 Na2CO3 (CP) was purchased from Tianjin Guangfu Science and Technology Development Co., Ltd. (Tianjin, China). Tris (hydroxymethyl) aminomethane (TRIS, CP) and DOPA (98%) was purchased from Shanghai Zhongqin Chemical Reagent Co., Ltd. (Shanghai, China). Ethanol (CP) and acetone (LR) were used to wash the particles. Glutathione (GSH), bradykinin (BK), insulin (INS) and bovine serum albumin (BSA) were obtained from Amresco.Preparation of porous particles
A recipe is shown in Table 2. The aqueous phase consists of the surfactant SDS, electrolyte Na2CO3, inhibitor HQ, stabilizer PVA and deionized water. Before dissolving other reagents, PVA was stirred in water with constant speed of 90 rpm, temperature of 115 °C, and time of 3 h. The admixture of monomer ST, crosslinking agent EDGMA, hydrophobic additive HD, surfactant Span 80, and initiator BPO were used as the oil phase. Next, an emulsion formed by transferring the oil phase into water phase in a four-neck glass flask with an anchor electric whisk, a condenser pipe, nitrogen inlet and exit. Before heating, the emulsion was stirred with 120 rpm for 1 h. Then, with increasing temperature to 75 °C, the polymerization process was lasted for 12 h under nitrogen atmosphere. The porous microspheres were washed with distilled water and ethanol in turn for 4 times respectively. Other residual impurities were removed by extracting with acetone in a Soxhlet apparatus for 6 h. The particles were finally dried in vacuum at 40 °C for 6 h.| Water phase | Weight (g) | Oil phase | Weights (g) |
|---|---|---|---|
| Water | 100 | HD | 0.2 |
| PVA | 1.0 | ST | 3.0 |
| HQ | 0.01 | EDGMA | 1.5 |
| Na2CO3 | 0.02 | BPO | 0.16 |
| SDS | 0.015 | Span 80 | 1.6 |
Preparation of PSEPs
The hydrophilization of PSE porous particle was proceeded by self-polymerization of DOPA in Tris–HCl ethanol-distilled water solution. 3 g Tris, 75 mL 0.1 mol L−1 HCl solution and certain distilled water were added in a volumetric flask of 500 mL and pH value of Tris–HCl solution was adjusted to 8.5. Eight PESPs via varying the additional volumes of ethanol and distilled water in the solution were synthesized. 0.2 g of particles and 0.1 g of DOPA were added in the Tris buffer (40 mL) in 250 mL conical flasks. Then, different ethanol solutions were added (30 mL) to obtain eight PESPs in a constant temperature oscillator. And then PSEPs were obtained with different ethanol concentrations (0, 0.01, 0.02, 0.04, 0.06, 0.11, 0.23 and 0.34 g mL−1, respectively), and denoted as E1–E8, respectively.The temperature and stirring speed were set at 25 °C and 100 rpm for 12 h. According to different concentrations of dopamine hydrochloride, under the same other conditions, ten PSEPs were prepared. The prepared materials were obtained with different dopamine concentrations (0.03, 0.06, 0.1, 0.2, 0.3, 0.6, 0.9, 1.0, 1.2, 1.4 g L−1, respectively), and denoted as D1–D10, accordingly.
Characterization
Scanning electron microscopy (SEM) images of the porous particles were acquired by JSM-6701 SEM instrument. IR spectra were obtained on a Nexus-670 Fourier transform infrared (FTIR) spectrometer in the range of 4000–500 cm−1 with potassium bromide pellets. The specific surface area was determined by the Brunauer–Emmett–Teller (BET) method and the pore volume and size were estimated by the Barrett–Joyner–Halenda (BJH) method using nitrogen as an adsorbate at 77 K. Before the determination, the porous particles were outgassed at 375 K for 24 h on the degas port of the analyzer. Static water contact angle was measured at three different locations on each sample using a contact angle tester, and the average values were recorded. The color analysis was accomplished by using a Minolta CM-3700 spectrometer. The tested samples were pressed in the form of a disc. Color parameters were obtained using the Spectra Match 3.2 software. The XPS spectra were recorded on a Thermon Scientific K-Alpha-surface Analysis X-ray photoelectron spectroscope equipped with X-ray Monochromatisation. The values of zeta potential of suspensions were measured on a Malvern Zeta-sizer Nanosystem with irradiation from a 633 nm He–Ne laser. The solid/liquid ratio of the suspension for the test is 1/200 (w/w), and the samples were scattered with deionized water by high speed stirring. Rheological measurements were carried out using a rotational rheometer with a coaxial double gap flow geometry (DG26.7) under small amplitude oscillatory shear (SAOS) mode at 25 °C, within the linear viscoelastic range. Particles were sliced by using pathological microtome and the 5 μm slices were observed with a TCS SP2 LCSM (Leica).Adsorption experiment for biomolecule on PSE and PSEPs
The porous particles were soaked in ethanol for 3 h before use, then filtrated and washed with distilled water to remove leftover ethanol. 0.1 g different series of PSEPs were used in the adsorption experiment. Biomolecule was used as adsorbate to perform the adsorption experiments. 0.1 g dried particles were contacted with 50 mL sample solution with initial concentrations of 0.5 mg mL−1. After adsorption process reached equilibrium, the concentration of biomolecule in the residue solution was analyzed by HPLC. The adsorption time, temperature and stirring speed were set at 8 h, 25 °C and 100 rpm, respectively.The adsorption capacity was calculated by the following equation:
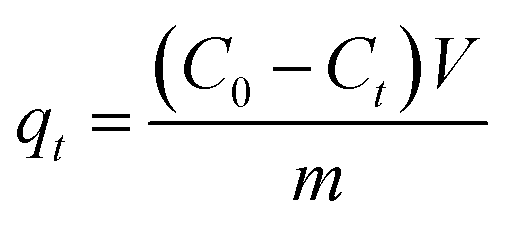
Results and discussion
Effect of particles with different stirring speed
To synthesize PSE, we adopted surfactant reverse micelles with six different stirring speeds (140–240 rpm). The effect of stirring speed on the structure and pore diameters of the particles was investigated and corresponding results can be observed from Table 3.| Sample | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Average pore diameter (nm) | Stirring speeds (rpm) |
|---|---|---|---|---|
| PSE1 | 37.97 ± 0.03 | 0.20 ± 0.02 | 169.48 ± 0.04 | 140 |
| PSE2 | 25.40 ± 0.03 | 0.11 ± 0.01 | 169.41 ± 0.05 | 160 |
| PSE3 | 23.00 ± 0.02 | 0.08 ± 0.03 | 161.60 ± 0.02 | 180 |
| PSE4 | 62.33 ± 0.08 | 0.21 ± 0.02 | 175.94 ± 0.04 | 200 |
| PSE5 | 14.37 ± 0.06 | 0.07 ± 0.01 | 135.23 ± 0.02 | 220 |
| PSE6 | 20.40 ± 0.03 | 0.08 ± 0.01 | 66.73 ± 0.03 | 240 |
| D4 | 39.51 ± 0.07 | 0.15 ± 0.02 | 44.62 ± 0.01 | 200 |
| D6 | 26.50 ± 0.01 | 0.09 ± 0.01 | 30.43 ± 0.03 | 200 |
| D7 | 23.43 ± 0.04 | 0.08 ± 0.01 | 21.66 ± 0.01 | 200 |
| D8 | 15.39 ± 0.08 | 0.07 ± 0.01 | 14.56 ± 0.02 | 200 |
| D9 | 13.29 ± 0.01 | 0.05 ± 0.02 | 12.36 ± 0.04 | 200 |
As mentioned in some literature,26 it is necessary to choose an optimal speed for obtaining expectant particles to separate peptides. As shown in Fig. 1a and 2a, due to low stirring speed, the polymer particles and polymer precursors were inclined to form aggregates and the aggregates were difficult to disperse again in the process of polymerization. In other words, secondary polymerization would happen for part of resin particles precursor.26 Accordingly, PSE1 particles possess a little undesirable gigaporous pores. PSE2 and PSE3 have extremely low pore volume (shown in Table 3), which might be due to that water phase and oil phase are not fully mixed at low stirring speed, and that phenomenon is supported by sound reasons in Fig. 1b, c and 2a. In contrast, stirring speeds of PSE5 and PSE6 are so high that their particles were deformed and destroyed in polymerization process, as shown in Fig. 1e, f and 2a.
 | ||
Fig. 1 The SEM images of the particles prepared at different stirring speeds at a magnification of ×10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (a) PSE1; (b) PSE2; (c) PSE3; (d) PSE4; (e) PSE5; (f) PSE6. 000 (a) PSE1; (b) PSE2; (c) PSE3; (d) PSE4; (e) PSE5; (f) PSE6. | ||
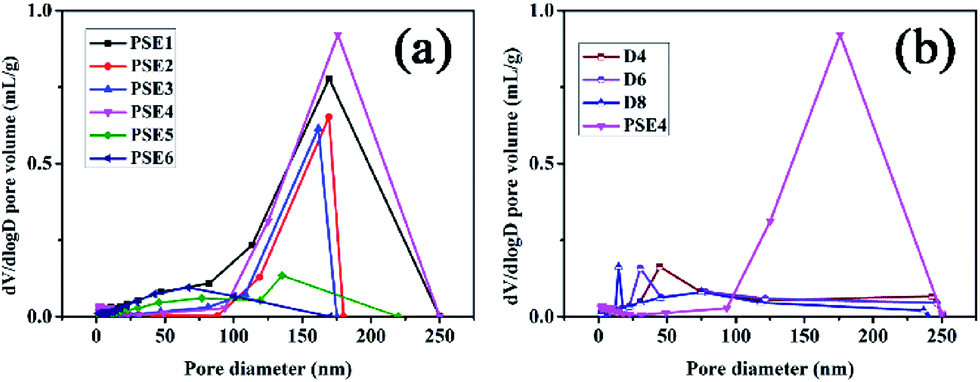 | ||
| Fig. 2 Pore distribution of PSE and PSEPs (a) different stirring speed; (b) different DOPA concentrations. | ||
As can be seen from Table 3, PSE4 surface area is the biggest in the six particles, which is an important factor for an adsorption process. The pore volume of PSE4 is higher than other particles in Table 3. Consequently, PSE4 was selected as optimum particles, and the stirring speed is 200 rpm.
PSE modified by PDOPA
Despite amount of crosslinking agent affecting hydrophilicity of the porous particles, we attempted to improve hydrophilicity of particles by another method, as shown in Fig. 3. The difficulties in coating process may be that PSE scatters poorly in buffer solution due to its hydrophobic property.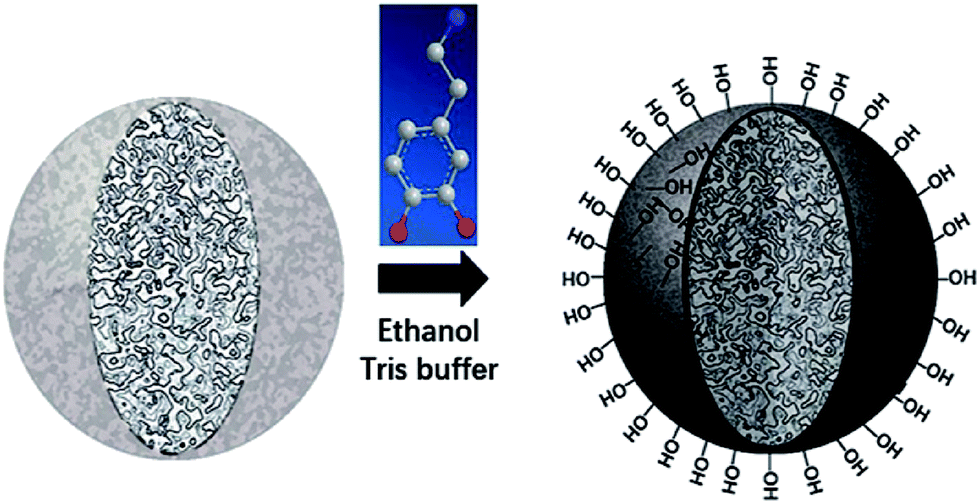 | ||
| Fig. 3 Schematic preparation of porous particles with PDOPA coating. | ||
Anhydrous ethanol was added into Tris buffer to promote the dispersion of the PSE in the solution. The dopamine was oxidized to dopaminequinone and eventually formed PDOPA after a battery of complex reactions.32 The color of E1 without addition of ethanol was gray while that of E7 with addition of ethanol was black (see Fig. S1 in the ESI†). To prove whether the successful coating of PDOPA on the surface of particles, FTIR spectra of PSE4, PDOPA and E7 were obtained, and shown in Fig. 4. In 3450 cm−1, PDOPA and E7 obtained uniform bands.33 On the contrary, bands of PSE4 were adverse and even totally opposite in 3450 cm−1. After coating with PDOPA by the oxidative self-polymerization, the PSEPs show clear adsorption peaks at 3450 cm−1, which was ascribed to the stretching vibration of N–H. At 1740 cm−1, there are distinct peaks for PSE4 and E7, which was ascribed to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. According to this phenomenon, we confirmed that DOPA had been coated on the surface of particles by the self-polymerization. In addition, this result could be verified by XPS spectra (see Fig. S2a in the ESI†). For the original PSE4, the nitrogen atoms entirely absent, but after coating with PDOPA, it is clear that nitrogen is present in XPS spectra.
O. According to this phenomenon, we confirmed that DOPA had been coated on the surface of particles by the self-polymerization. In addition, this result could be verified by XPS spectra (see Fig. S2a in the ESI†). For the original PSE4, the nitrogen atoms entirely absent, but after coating with PDOPA, it is clear that nitrogen is present in XPS spectra.
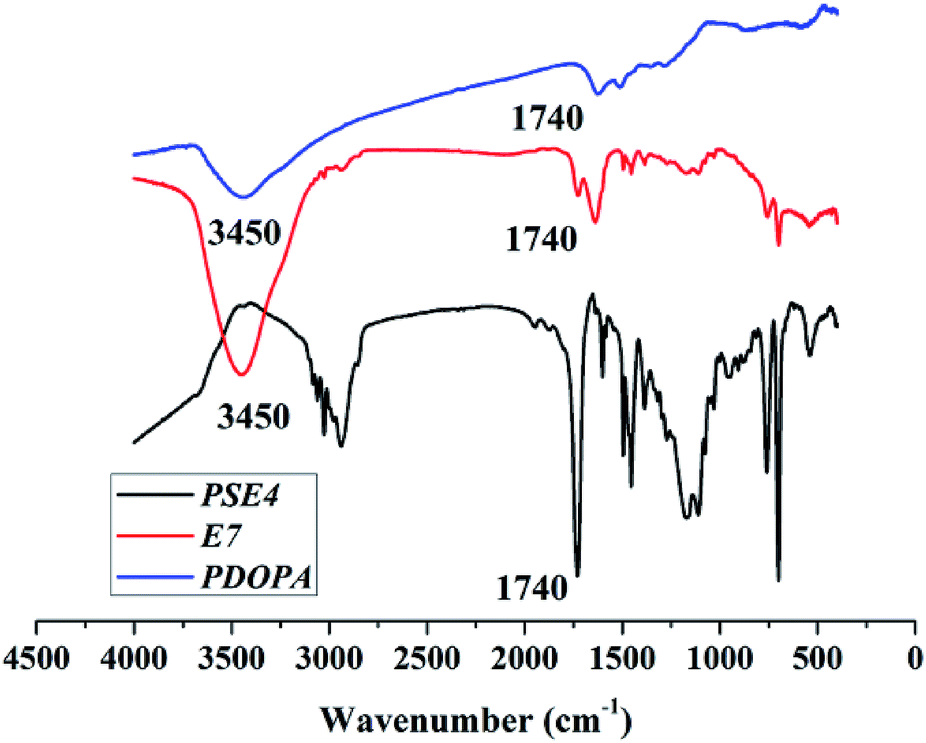 | ||
| Fig. 4 FTIR spectra of PSE4, PDOPA and E7. | ||
Zeta (ζ) potential is a crucial parameter for experimental design in polymeric microfluidic substrate materials.38 The larger the absolute value of polymeric zeta potential is, the better the diffusion degree. The zeta potential values of all PSEPs were negative in Fig. 5a, which also showed nature of negative charge on the surface of particles in the buffer solution. Particles obtain optimal dispersion degree in 0.23 g mL−1 ethanol Tris buffer solution. However, in Fig. 5b, with ethanol concentration increasing, the absolute value of the zeta potential of PDOPA decreased, which shows exorbitant ethanol concentration is not conducive to the scatter of PDOPA. Even so, in E7 buffer solution, adding ethanol indeed facilitated coating of PDOPA, which also could be verified by contact angle testing (see Fig. S3 in the ESI†). For the PSE4 with average contact angle of 106.8°, it indicates that the surface is strongly hydrophobic. If ordinary Tris buffer without ethanol is used, the coating result is not what is expected. Under this circumstance, the contact angle (101.1°) changed only a little compared to PSE4. Instead, there has been a great decrease in contact angle for E7. It's known that the surface of PDOPA possesses abundant hydroxyl groups, a highly desirable hydrophilic group. The contact angle of E7 (18.3°) decreased significantly, indicating more PDOPA coated on the surface.
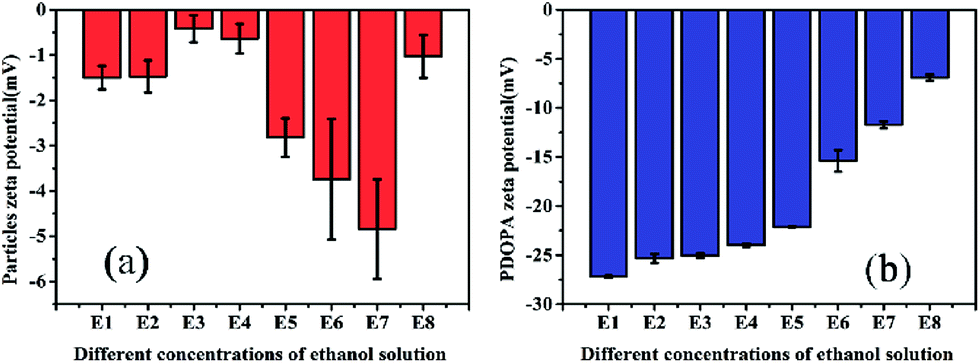 | ||
| Fig. 5 Zeta potential of PSE4 and PDOPA in different concentrations of ethanol solution. | ||
The analysis of the PDOPA coating amount
In the previous experiment, ten PSEPs were prepared via varying dopamine hydrochloride concentration, and the color was measured by distinguishing the color space L*a*b* (CLELAB System), which is one of the most used systems for color measurement.39 In the system, L* reflects the brightness and a* as well as b* are the chromaticity coordinates. According to the color characteristic of PDOPA black (shown in Fig. S4 in the ESI†), the more the amount of PDOPA is, the blacker the surface of the particles is. A value ΔL* defined by the following expression was used to describe degree of blackness, and set ΔL* of PDOPA to zero in the Table 4.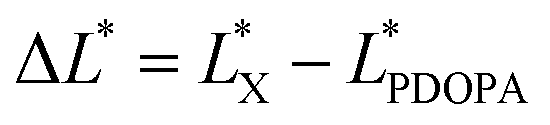
| Sample | L* | ΔL* |
|---|---|---|
| PSE4 | 86.3 | 81.9 |
| D1 | 61.2 | 56.8 |
| D2 | 51.6 | 47.2 |
| D3 | 44.6 | 40.2 |
| D4 | 39.6 | 35.2 |
| D5 | 37.8 | 33.4 |
| D6 | 25.5 | 21.1 |
| D7 | 28.4 | 24.0 |
| D8 | 13.4 | 9.0 |
| D9 | 13.4 | 9.0 |
| D10 | 12.4 | 8.0 |
| PDOPA | 4.4 | 0 |
In which 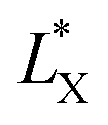 and
and 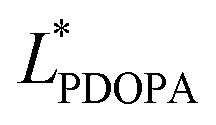 are the brightness of X material and PDOPA, respectively. By comparing the values of ΔL*, the amount of dopamine coated on particles could be indirectly illustrated. The optical properties of PSE4, PDOPA and some PSEPs also were analyzed. With dopamine hydrochloride concentration increasing, the color of PSEP is getting much darker and the reflectance of samples is lower in the UV-Vis reflectance spectra (from 350 to 750 nm) in Fig. S4.†
are the brightness of X material and PDOPA, respectively. By comparing the values of ΔL*, the amount of dopamine coated on particles could be indirectly illustrated. The optical properties of PSE4, PDOPA and some PSEPs also were analyzed. With dopamine hydrochloride concentration increasing, the color of PSEP is getting much darker and the reflectance of samples is lower in the UV-Vis reflectance spectra (from 350 to 750 nm) in Fig. S4.†
To verify further the amount of dopamine coated on the particles, XPS was opted to quantitatively analyze the amount of nitrogen element and quantify indirectly the amount of PDOPA onto the surface of PSE. As can be seen in Fig. S2,† the three particles have different amounts of nitrogen. Nitrogen peak area ratio of D6/D4 and D8/D4 are 1.24 and 1.95 respectively, indicating that with the increase of initial dopamine concentration, the thickness of coating increased, which is consistent with the results of the previous L* value test.
Due to the addition of PDOPA, pore diameters and volumes of particles would decrease as described in Table 3 and Fig. 2b. The particles directly adsorbed FITC, and then were sliced to distinguish the pore structure of PSE and PSEPs by laser scanning confocal microscope (see Fig. S6 in the ESI†). In the initial particles, there are complete and wide channels. In the D4 and D6 particles, although there are some channels, which are less and narrow. In the D8 particles, there are fewer channels. With the increase of the amount of PDOPA, fluorescence inside the particles is getting less and less, which reflects the smaller pore size and volume.
Mechanical properties of particles
The reinforcing effect of PDOPA coated clay33 and epoxy resin34 has been reported. The original particles PSE4 are very fragile which may be not favorable to preserve pore structure. To solve this problem, the particle surface was modified with PDOPA coating. Rheological properties can give information on storage modulus (E) of porous particles, which is an important parameter to investigate the mechanical properties of polymers.40,41 The results of storage modulus are given in Fig. 6, which show that the introduction of PDOPA in the particles surface caused an increase of storage modulus. The reinforcement effect is more obvious for D4 than that for D2, which indicates that the storage modulus is closely related to the amount of PDOPA. It can be deduced that the thickness of PDOPA coating on the surface of D4 is much thicker than D2, as a result mechanical strength of D4 is higher.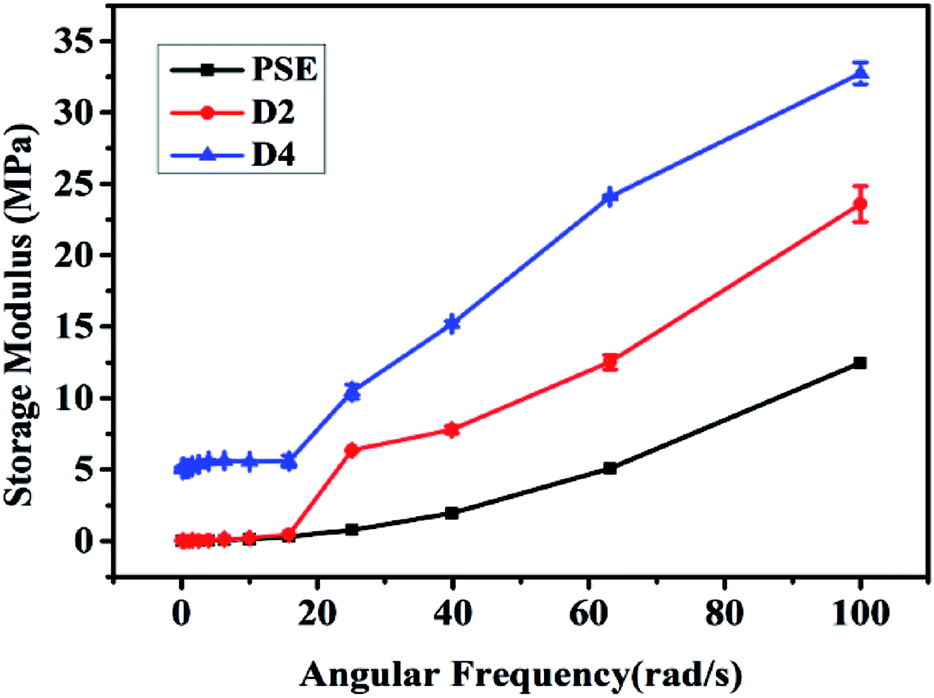 | ||
| Fig. 6 Storage modulus of three particles. | ||
Gas adsorption
Gas adsorption is an important step to test the performance of materials.15,42 By applying nitrogen adsorption onto porous materials at 77 K, the surface areas and porosities of the particles can be calculated by nitrogen adsorption–desorption isotherm.43 Some literatures have reported that porous materials have very good adsorption properties for other several gases, such as CO2,44 CH4 (ref. 45) and volatile organic compounds (VOCs).46 The research about the adsorption characteristic of PSEPs for gases is carried out to explore corresponding potential application. To investigate nitrogen adsorption and desorption of PSE and PSEPs, much information of particles was obtained, and pores size distribution and volume were estimated by the BJH in the Fig. 2, as well as the specific surface area was calculated by the BET in the Table 3.According to the nitrogen adsorption–desorption isotherms of PSE4 and D4 (see Fig. S5a in the ESI†), the adsorption capacity of PSE4 is obviously higher than that of D4 PSEP. In conjunction with the data in the Table 3, part PDOPA may penetrate into the internal pores of polymer particles. In this case, as the concentration of DOPA increasing, the pore diameters and pore volume of the polymer particles would further reduce, which can be verified by the nitrogen adsorption–desorption isotherms of D6 and D8 PSEPs (see Fig. S5a in the ESI†). The nitrogen uptake decreased as further increase in the concentration of DOPA concentration, which was ascribed to the inner space of the polymer particle is occupied by part PDOPA coating.
Biomolecule adsorption
The study on the adsorption capacity and sieving effect of PSE and PSEPs for biomolecule is crucial to explore their potential applications. Some literatures have reported the adsorption efficiencies of columns for various biological molecules such as uracil,47 proteins48 and phosphopeptides.49 In this study, four peptides biomolecules, glutathione (GSH), bradykinin (BK), insulin (INS) and bovine serum albumin (BSA) with molecular weights of 307.32 Da, 1060.23 Da, 5807.69 Da and 66446.01 Da, respectively, were selected to explore the adsorption characteristics of the porous particles. The adsorption capacities on PSE and PSEPs were shown in Fig. 7, and their selective adsorption properties can be tailored by varying dopamine concentration and then pore diameters. The PSE particles displayed significantly higher enrichment efficiency of BSA compared to the PSEPs. It could be explained that the adsorption capacity was controlled primarily by gigaporous adsorption, sieving effect and van der Waals forces between PSE and BSA. In addition, according to the amino acids of BSA and corresponding functional groups, hydrogen bond, π–π stacking interaction and other forces may be also involved in the adsorption process. However, PSE does not have good performance for other peptides with low molecular weight. After dopamine modification, due to a sharp drop in pore diameters, adsorption capacity for BSA decreased sharply. As shown in Fig. S7,† change in adsorption capacity for GSH was not obvious as a result from low molecular weight and molecular size of GSH, and D7 obtains the best adsorption efficiency. The adsorption capacity for BK decreased first and then increased. The phenomenon might be attributed to the π–π stacking interaction between two benzene rings as shown in Fig. S8† and benzene rings on the surface of PSE and hydrogen bonding interaction between BK and rich hydroxyl and amino groups on the particles surface. Due to the addition of PDOPA, the benzene ring exposed on the surface of PSE was less and less. The main functional groups on the surface of PDOPA coating are hydroxyl and amino groups, not the benzene ring, which leads to the decrease in adsorption capacity for BK. With further increase of PDOPA, the benzene rings on the surface of PSEPs were more and more, and thus π–π stacking interaction couldn't be ignored. The maximum adsorption capacity of D9 for BK should be attributed to hydrogen bonding and π–π stacking interaction. In a ward, the optimum particles for GSH, BK, INS and BSA adsorption are D7, D9, D4, and PSE, respectively.
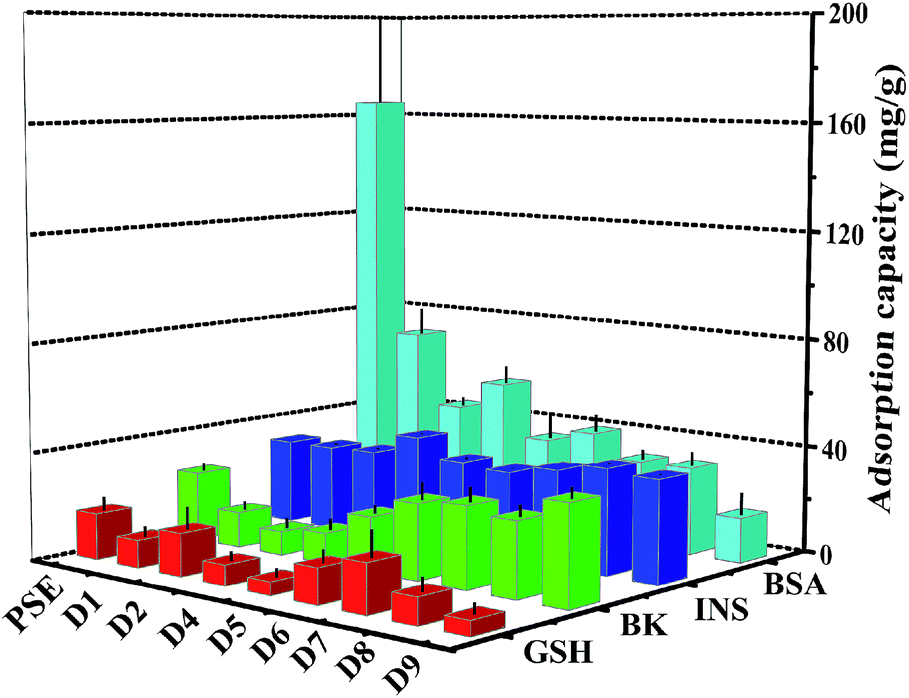 | ||
| Fig. 7 Adsorption amount of biomolecule on PSE and PSEPs. | ||
In order to study the adsorption of biomolecules in the particles inside, FITC-INS adsorption experiment is an excellent method. The slices of polymer particles were sliced (5 μm) by the pathological microtome creatively, and the pore size distribution of the particles could be visualized by laser scanning confocal microscopy (LCSM) in Scheme 1. On the basis of discussion mentioned above, adding appropriate amounts of dopamine will improve the hydrophilicity of the particles. The PSE displayed remarkable adsorption capacity compared to the conventional PS-DVB as shown in Fig. S7c.† The adsorption force between INS and PS-DVB and PSE is van der Waals forces and conjugated effect. Although their pore diameters are large, the adsorption amount of INS is less. After modification by PDOPA, although pore diameters slightly decrease, the formation of hydrogen bonds between adsorbate and adsorbent will improve adsorption capacity. The FITC-INS adsorption experiment gave a straight reflection about adsorption distribution and quantity in particles in Fig. 8. PSE have better hydrophilicity than PS-DVB, there would be more FITC-INS inside the PSE, hence LCSM has stronger fluorescence. With the increase of the amount of PDOPA, adsorption capacity and fluorescence intensity also increased.
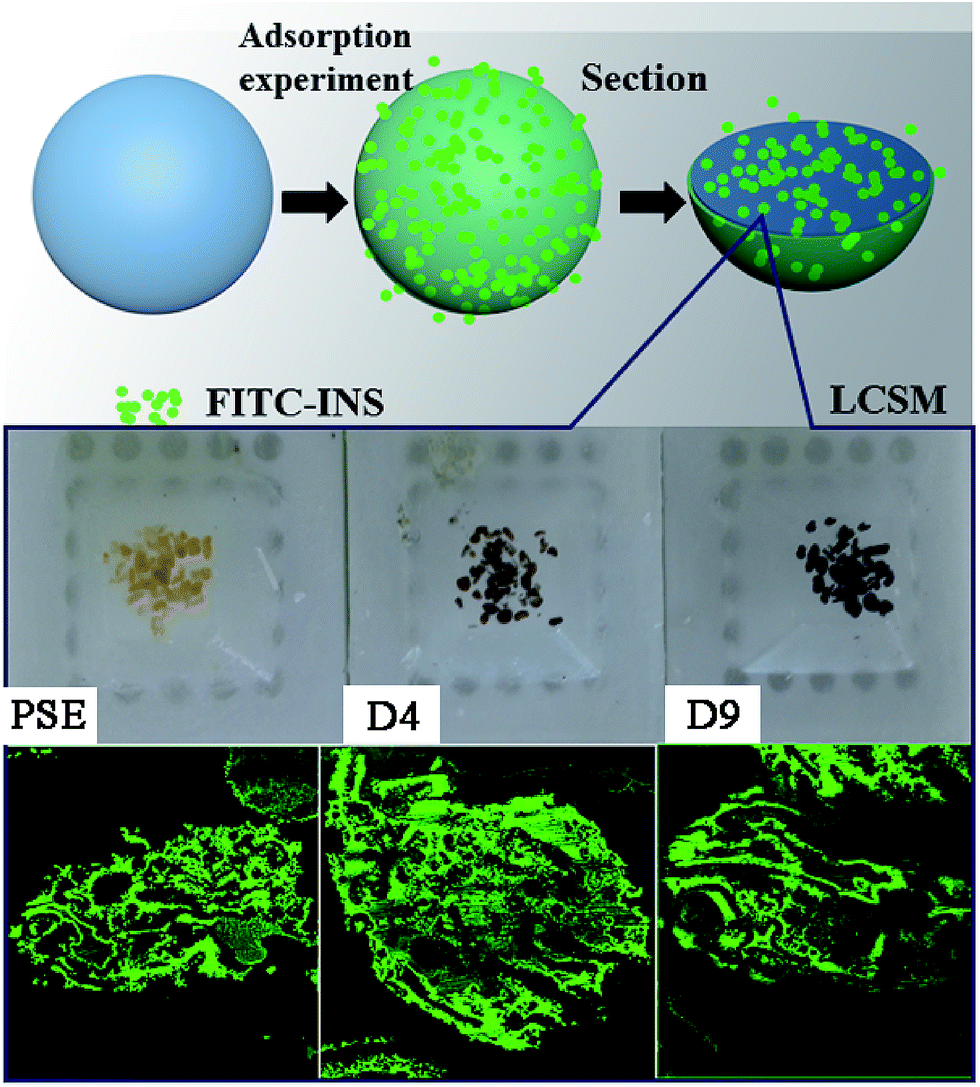 | ||
| Scheme 1 Direct interpretation of the internal pore structure and peptides adsorption distribution of particles by LCSM. | ||
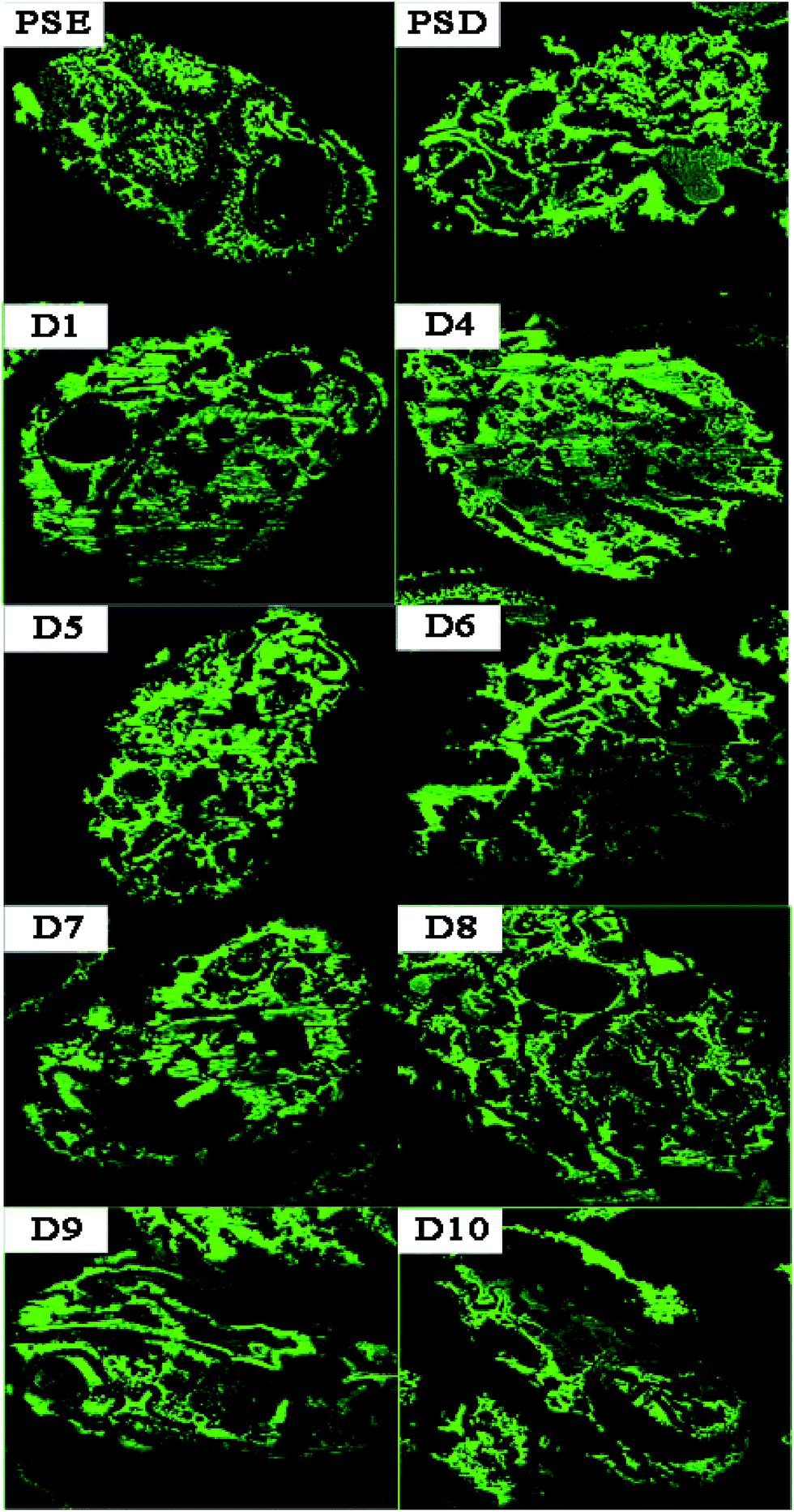 | ||
| Fig. 8 LCSM images of PSD, PSE and PSEPs after FITC-INS adsorption experiment. | ||
Especially, the superiority was palpable for the D4. With further modification, PDOPA occupy a part of the channels, which caused a decrease in pore diameters, and FITC-INS can't reach the adsorption site of pores, leading to decrease in adsorption performance, such as D5 and D6 in Fig. 7, which was confirmed that black area increased inside the particles in Fig. 8. With further modification, from D6 to D8 particles, pore diameters further reduced. Due to abundant hydroxyl groups present on the surface of particles, FITC-INS was enriched by the particle surface, so adsorption capacity rose. It can be seen that the black area and surface fluorescence increases. For D9 and D10, the FTIC-INS uptake may be mainly ascribed to adsorption on the surface of the material while almost no adsorption in the pore channels can be observed in Fig. 8, resulting in the decrease in adsorption capability.
Conclusions
We have reported the first example of the application of PSE for BSA and PSEPs for peptides as efficient enrichment media. A novel strategy with PDOPA modification was proposed for the fabrication of PSEPs with well-controlled thickness. Excellent adsorption capacity of PSE for BSA was observed due to its distinctive gigapores. The resultant PSEPs provided the significant enrichment superiority for BK and INS over commonly used PSE and PS-DVB. We propose that the driving force of the recognition process is mainly sieving effect for four biomolecules. Besides that, van der Waals forces, π–π stacking interaction and hydrogen bonding also play important roles during the adsorption process. The PSE can be applied for the biomacromolecule (like BSA) enrichment. The PSEPs with unique hydroxyl and amino layer processed superior enrichment performance, and according to different molecular size and functional groups of peptides, the selective adsorption properties can be tailored by varying dopamine concentrations. The results of our study indicate that the prospect of PSE for its wide application in porous materials, and PSEPs particles would provide a useful way to selectively separate peptides.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge the financial support of this work by the National Natural Sciences Foundation of China (21605150 and 21544013), the West Light Foundation of the Chinese Academy of Sciences, Natural Science Foundation of Gansu Province of China (1501RJZA011).Notes and references
- A. G. Slater and A. I. Cooper, Science, 2015, 348, 8075–8085 CrossRef PubMed.
- Y. Cai, Y. Chen and X. Hong, Int. J. Nanomed., 2013, 8, 1111–1120 Search PubMed.
- S. Wu, Y. Wang and G. Gong, Food Bioprod. Process., 2015, 93, 148–155 CrossRef CAS.
- M. R. Kim, W. C. Kim, Y. D. Lee and W. C. Kim, J. Food Eng., 2007, 78, 27–32 CrossRef CAS.
- D. Kammerer and J. G. Kljusuric, Eur. Food Res. Technol., 2005, 220, 431–437 CrossRef CAS.
- Y. Liu, Q. Bai and Y. Liu, Eur. Food Res. Technol., 2014, 238, 59–69 CrossRef CAS.
- C. A. Hauser, S. Maurerstroh and I. C. Martins, Chem. Soc. Rev., 2014, 43, 5326–5455 RSC.
- B. P. Gray and K. C. Brown, Chem. Rev., 2014, 114, 1020–1081 CrossRef CAS PubMed.
- S. L. Deutscher, Chem. Rev., 2010, 110, 3196–3323 CrossRef CAS PubMed.
- K. Takahiro, E. Shin and U. Hirotsugu, Nat. Rev. Urol., 2017, 77, 1–10 Search PubMed.
- Y. Zhang, R. W. Wang and H. M. Guang, Chin. J. Process Eng., 2006, 51, 93–99 Search PubMed.
- Y. Li, F. Gao and W. Wei, J. Mol. Catal. B: Enzym., 2010, 66, 182–189 CrossRef CAS.
- A. Basso, P. Braiuca, L. D. Martin, C. Ebert, L. Gardossi, P. Linda, S. Verdelli and A. Tam, Chem.–Eur. J., 2004, 10, 1007–1013 CrossRef CAS PubMed.
- A. Basso, P. Braiuca, C. Ebert, L. Gardossi and P. Linda, J. Chem. Technol. Biotechnol., 2006, 81, 1626–1640 CrossRef CAS.
- M. T. Gokmen and E. F. D. Prez, Prog. Polym. Sci., 2012, 37, 365–405 CrossRef CAS.
- K. Kanamori, J. Hasegawa and K. Nakanishi, Macromolecules, 2008, 41, 7186–7193 CrossRef CAS.
- F. S. Macintyre and D. C. Sherrington, Macromolecules, 2004, 37, 7628–7636 CrossRef CAS.
- F. Limé and K. Irgum, Macromolecules, 2009, 42, 4436–4442 CrossRef.
- A. S. Goldmann, A. Walther, L. Nebhani, R. Joso, D. Ernst, K. Loos, C. Barner-Kowollik, L. Barner and A. Müller, Macromolecules, 2009, 42, 3707–3714 CrossRef CAS.
- Y. Gao, Y. Bai, D. Zhao, M. W. Chang, M. Z. Ahmad and J. S. Li, Polymer, 2015, 7, 2701–2710 CAS.
- H. Minami, A. Kimura and K. Kinoshita, Langmuir, 2010, 26, 6303–6307 CrossRef CAS PubMed.
- J. S. Song, F. Tronc and M. Winnik, J. Am. Chem. Soc., 2004, 126, 6562–6563 CrossRef CAS PubMed.
- C. X. Zhao and A. P. J. Middelberg, Angew. Chem., Int. Ed., 2009, 48, 7208–7211 CrossRef CAS PubMed.
- A. R. Abate and D. A. Weitz, Small, 2009, 5, 2030–2032 CrossRef CAS PubMed.
- W. Q. Zhou, T. Y. Gu and Z. G. Su, Polymer, 2007, 48, 1981–1988 CrossRef CAS.
- D. Zhang, W. Zhou and J. Li, Polymer, 2016, 8, 142–146 CrossRef.
- C. Sayil and O. Okay, Polymer, 2001, 42, 7639–7652 CrossRef CAS.
- W. Zhou, J. Li and W. Wei, Colloids Surf., A, 2011, 384, 549–554 CrossRef CAS.
- S. L. Gao, Y. J. Wang and X. A. Diao, Bioresour. Technol., 2010, 101, 3830–3837 CrossRef CAS PubMed.
- J. B. Qu, W. Q. Zhou and W. Wei, Langmuir, 2008, 24, 13646–13652 CrossRef CAS PubMed.
- Y. Liu, Y. Yin and L. Wang, J. Mater. Chem., 2013, 1, 3888–3896 RSC.
- H. Lee, S. M. Dellatore and W. M. Miller, Science, 2007, 318, 426–429 CrossRef CAS PubMed.
- L. Yang, L. P. Si and J. K. H. Teo, ACS Appl. Mater. Interfaces, 2011, 3, 3026–3032 CAS.
- Y. Ling, W. Li and B. Wang, RSC Adv., 2016, 6, 31037–31045 RSC.
- J. Ou, J. Wang and S. Liu, Appl. Surf. Sci., 2009, 256, 894–899 CrossRef CAS.
- J. Ou, J. Wang and J. Zhou, Prog. Org. Coat., 2010, 68, 244–247 CrossRef CAS.
- H. Yuyama, T. Watanabe and G. H. Ma, Colloids Surf., A, 2000, 168, 159–174 CrossRef CAS.
- B. J. Kirby and E. F. Hasselbrink, Electrophoresis, 2004, 25, 203–213 CrossRef CAS PubMed.
- E. G. V. D. Linden and L. F. B. Malta, Dyes Pigm., 2011, 90, 36–40 CrossRef.
- Y. Jung, Y. H. Son, L. K. Lee, T. X. Phuoc, Y. Soong and M. K. Chyu, ACS Appl. Mater. Interfaces, 2011, 3, 3515–3522 CAS.
- V. Khoshkava and M. R. Kamal, ACS Appl. Mater. Interfaces, 2014, 6, 8146–8157 CAS.
- L. Tan and B. Tan, Chem. Soc. Rev., 2017, 46, 3322–3356 RSC.
- S. Bandyopadhyay, A. G. Anil and A. James, ACS Appl. Mater. Interfaces, 2016, 8, 27669–27678 CAS.
- C. F. Martín, E. Stöckel and R. Clowes, J. Mater. Chem., 2011, 21, 5475–5483 RSC.
- Y. Yang, B. Tan and C. D. Wood, J. Mater. Chem. A, 2016, 4, 15072–15080 CAS.
- C. Lu, S. Liu and J. Xu, Anal. Chim. Acta, 2016, 902, 205–211 CrossRef CAS PubMed.
- J. Urban, F. Svec and J. M. J. Fréchet, Anal. Chem., 2010, 82, 1621–1623 CrossRef CAS PubMed.
- Y. Lv, Z. Lin and T. Tan, J. Chromatogr. A, 2013, 1316, 154–159 CrossRef CAS PubMed.
- A. Saeed, F. Maya and D. J. Xiao, Adv. Funct. Mater., 2014, 24, 5790–5797 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08175h |
| This journal is © The Royal Society of Chemistry 2017 |