DOI:
10.1039/C7RA08106E
(Paper)
RSC Adv., 2017,
7, 44773-44779
Utility of 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole for development of a highly sensitive stability indicating spectrofluorimetric method for determination of salmeterol xinafoate; application to human plasma
Received
22nd July 2017
, Accepted 13th September 2017
First published on 19th September 2017
Abstract
A new, simple and rapid spectrofluorimetric method was developed and validated for determination of salmeterol xinafoate in dosage form and spiked plasma. The method is considered as the first attempt for the spectrofluorimetric determination of the investigated drug. The proposed method is based on nucleophilic substitution reaction between salmeterol xinafoate and 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole (NBD-Cl) at pH (9) to form a highly fluorescent product that can be measured at 543 nm after excitation at 473 nm. The fluorescence–concentration plot was rectilinear over the concentration range (100–1500 ng mL−1) with a detection limit of 19 ng mL−1. The suggested method was validated according to ICH guidelines and successfully applied for determination of salmeterol xinafoate in its pharmaceutical dosage form and spiked human plasma. The developed method was further extended for a stability study of salmeterol xinafoate under different stress circumstances including alkali, acid, oxidative and photodegradation conditions and the method was proved to be able to determine the intact drug in the presence of its degradation products.
1. Introduction
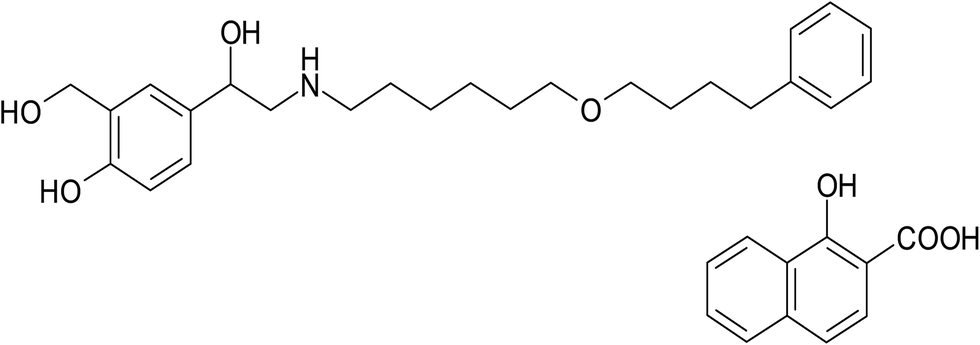
Salmeterol xinafoate (SAL), (RS)-1-[4-hydroxy-3-(hydroxymethyl)phenyl]-2-[[6-(4-phenylbutoxy)hexyl]amino]ethanol-1-hydroxynaphthalene-2-carboxylate (Fig. 1), is a direct-acting sympathomimetic agent with β-adrenoceptor stimulant activity and a selective action on β2 receptors. When given by oral inhalation, it acts as a bronchodilator with a long acting action up to 12 hours. It is used for the treatment of asthma and chronic obstructive pulmonary disease.1 Many analytical techniques in the literature have been reported for the determination of salmeterol xinafoate including spectrophotometric,2–8 HPLC,9,10 and HPTLC.11 These methods were associated with some major drawbacks such as inadequate sensitivity, being time consuming and/or using sophisticated and expensive instruments. On the other hand, spectrofluorimetric methods have the advantages of being simple, highly sensitive and time saving technique.12–16 To our knowledge, there is no reported spectrofluorimetric method for determination of salmeterol xinafoate up till now. In this study, we investigate the reaction of salmeterol xinafoate that contains a basic secondary amine with NBD-Cl which has been widely used for the determination of many pharmaceuticals containing primary and secondary amino groups17–22 in order to develop new, simple, sensitive and rapid spectrofluorimetric method for determination of the studied drug in its dosage form, spiked plasma and in the presence of its degradation products. Thus, the proposed method can be considered as a simple and fast alternative to the already existing stability-indicating HPLC procedures.
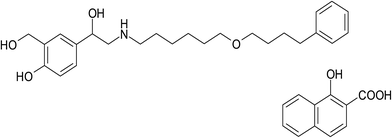 |
| | Fig. 1 Chemical structure of salmeterol xinafoate (SAL). | |
2. Experimental
2.1. Apparatus
Fluorescence spectrometer FS-2 (Scinco, Korea), connected to Dell PC, equipped with 1 cm quartz cell, grating excitation and emission monochromators with slit widths set at 5 nm, PMT voltage 400 volt, Jenway pH meter model 350 (E.U), Laboratory centrifuge (Bremsen ECCO, Germany), A Shimadzu UV-1601 PC spectrophotometer (Tokyo, Japan) with 1 cm quartz cell, MLW type thermostatically controlled water bath (Memmert GmbH, Schwabach, Germany) and Digital analytical balance (AG 29, Mettler Toledo, Glattbrugg, Switzerland).
2.2. Materials and reagents
All materials and reagents used through this study were of analytical reagent grade salmeterol xinafoate (SAL) pure sample (99.8%) was obtained from The Arab Drug Company for pharmaceutical and chemical industries CO., ADCO, (Cairo, Egypt). Metrovent® inhaler (batch no. 490738) (Arab Drug Company for pharmaceutical and chemical industries CO., ADCO, Cairo, Egypt) labeled to contain 25 μg salmeterol xinafoate per actuation.
4-Chloro-7-nitrobenzo-2-oxa-1,3 diazole (NBD-Cl) was purchased from Sigma Aldrich Chemie GmbH (Steinheim, Germany) and NBD-Cl reagent (2.5 × 10−3 M) was prepared in methanol. All other reagents used such as methanol, ethanol, acetone, acetonitrile, N,N-dimethylformamide (DMF), phosphoric acid, citric acid, boric acid, borax, acetic acid, sodium hydroxide and hydrochloric acid were purchased from El-Nasr chemical CO., (Cairo, Egypt). Different types of buffer solutions such as Teorell and Stenhagen, 0.1 M borate and phosphate buffers of different pH ranges were prepared in distilled water.23 Human plasma samples were obtained from healthy volunteers at Assiut University Hospital according to institutional guidelines. In all cases, informed consent was obtained from all participants and plasma samples were kept frozen at −20 °C until assay after gentle thawing.
2.3. Standard drug solution
Stock standard solution (100.0 μg mL−1) of salmeterol xinafoate was prepared in methanol. Further dilutions with methanol were made as appropriately to obtain final concentration range of (100–1500) ng mL−1.
2.4. General analytical procedure
Into a series of 10 mL stoppered test tubes, different volumes of (SAL) working solution were added followed by 2.0 mL of borate buffer and 0.5 mL of NBD-Cl reagent (2.5 × 10−3 M), the tubes were heated at thermostatically controlled water bath fixed at 80 °C for 20 min. After that the tubes were cooled in ice bath and 1.0 mL of 2 M HCl was added, the contents of the tubes were quantitatively transferred into 10 mL volumetric flasks and completed to the volume with methanol. The fluorescence intensity of the reaction product was measured at 543 nm after excitation at 473 nm; blank experiment was performed similarly omitting the drug.
2.5. Application of the proposed method for determination of SAL in its pharmaceutical formulation
Metrovent® inhaler labeled to contain 25.0 μg SAL per actuation was shaken well and 200 actuations (the whole content of the inhaler) were actuated in 100 mL beaker containing 40 mL of methanol. The mouth piece of the inhaler was immersed beneath the methanol to ensure complete delivery of the drug without any waste. The contents of the beaker were gently warmed at 50 °C in thermostatically controlled water bath to expel propellants.9 The contents of the beaker were then transferred into 50 mL volumetric flask and completed to the volume with methanol to obtain final concentration of 100 μg mL−1 and then general analytical procedure was followed.
2.6. Procedure for spiked human plasma
A sample of 5.0 mL of drug-free human blood sample was taken from healthy volunteer into a heparinized tube, centrifuged at 4000 rpm for 30 min. Into a 10 mL stoppered calibrated tube, 1.0 mL of the drug-free plasma (supernatant) was spiked with 1.0 mL of the drug standard working solution. Three milliliters of acetonitrile were added for precipitation of protein13,16,24 and the resultant mixture was diluted to 10 mL with methanol and then centrifuged at 4000 rpm for 30 min, adequate volumes of the supernatant were taken to obtain solutions within the concentration range of the studied drug, and then general procedure was applied, and after that blank value was determined by treating the drug-free blood sample in the same manner without drug addition.
2.7. Procedure for stability indicating study
2.7.1. Acid induced degradation. Into 10 mL test tube, 1.0 mL of stock drug solution was transferred and heated at 80 °C with 1.0 mL of 2 M HCl for 2 hours. After the specified time, the solution was cooled and neutralized to pH 7 using 2 M NaOH solution; the solution was transferred quantitatively into 10 mL volumetric flask and completed to the volume with methanol. Then 1.0 mL from the solution was taken and the general analytical procedure was applied to detect the remaining intact drug content.
2.7.2. Base induced degradation. Into 10 mL test tube, 1.0 mL of stock drug solution was transferred and 1.0 mL of 2 M NaOH was added. The solution was heated at 80 °C for 2 hours and after the specified time the solution was cooled and neutralized for pH 7 using 1.0 mL of 2 M HCl. The solution was transferred quantitatively into 10 mL volumetric flask and completed to the volume with methanol. Then 1.0 mL from the solution was taken and the general analytical procedure was applied to detect the remaining intact drug content.
2.7.3. Oxidative degradation. Oxidative induced degradation with hydrogen peroxide was carried out by transferring 1.0 mL of stock drug solution into 10 mL test tube then 1.0 mL of hydrogen peroxide (30.0% v/v) was added. The solution was heated at 80 °C for 2 hours. The volume was completed to 10 mL with methanol. Then 1.0 mL from the solution was taken and the general analytical procedure was applied to detect the remaining intact drug content.
2.7.4. Photochemical degradation. The photochemical stability of the drug was studied by exposing stock solution to direct artificial day light for 12 hours.
3. Results and discussion
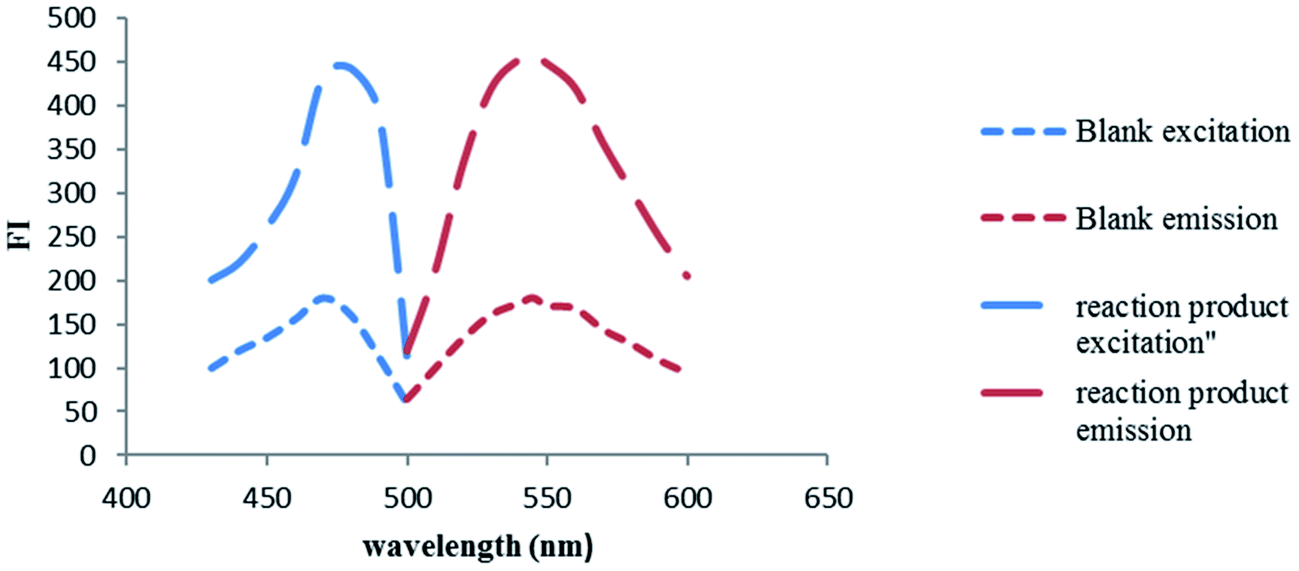
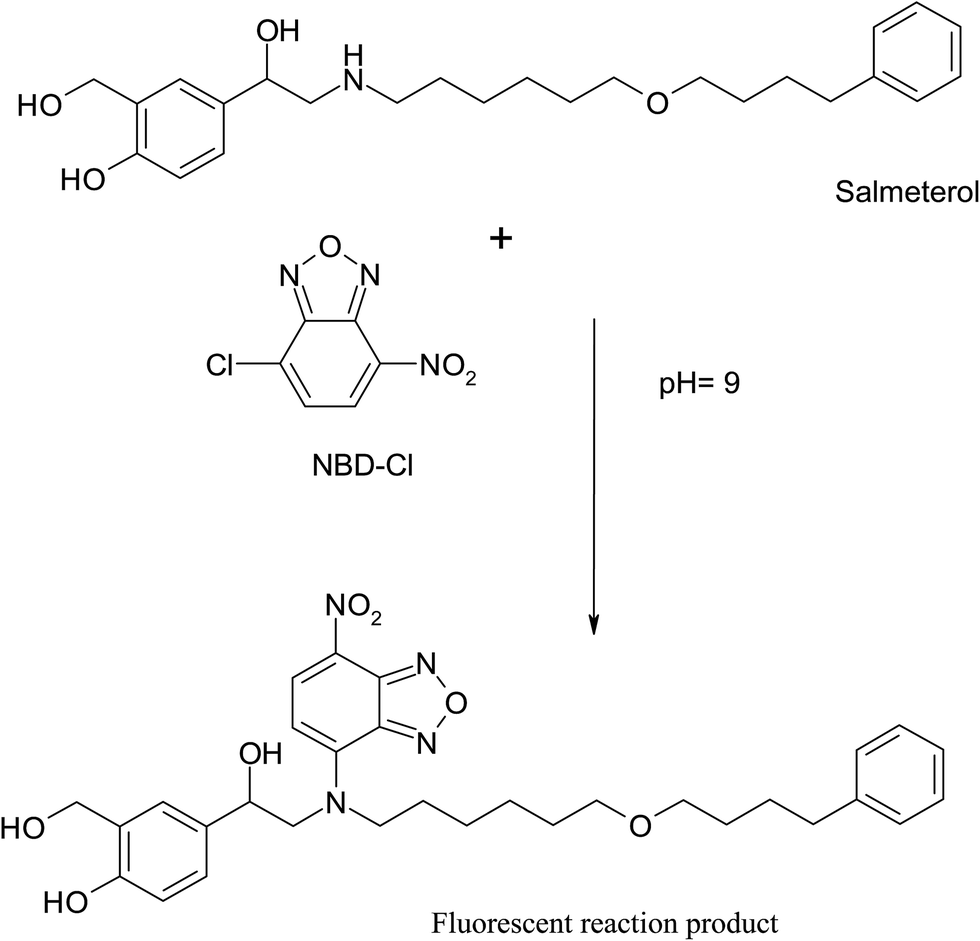
NBD-Cl is known to form highly fluorescent derivatives with primary and secondary amines under relatively mild alkaline conditions.17–22 In this work, we investigated the reaction between SAL & NBD-Cl at 80 °C pH (9) to develop the first spectrofluorimetric derivatizing method for determination of salmeterol xinafoate that gives yellow reaction product with high fluorescence intensity at 543 nm after excitation at 473 nm, Fig. 2. The derivatization reaction mechanism was supposed to proceed through secondary aliphatic amine,17–22 Fig. 3.
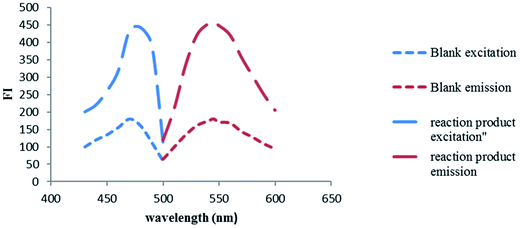 |
| | Fig. 2 Excitation and emission fluorescence spectra of the reaction product of SAL (1.0 μg mL−1) and NBD-Cl. | |
 |
| | Fig. 3 Suggested reaction mechanism between SAL and NBD-Cl. | |
3.1. Optimization of variables
The different experimental parameters affecting the reaction development and stability of the fluorescent product were carefully studied and optimized. These factors were changed individually, while the others were kept unchanged. The studied factors include pH, type of buffers, volume of buffer, NBD-Cl volume, temperature, reaction time and diluting solvent.
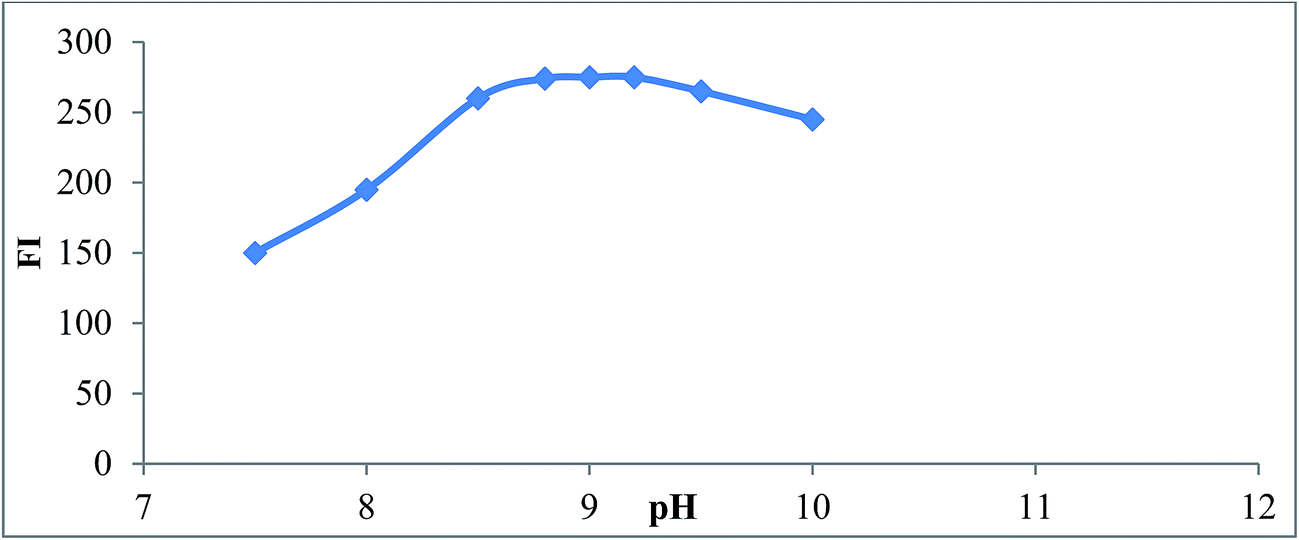
3.1.1. Effect of pH and buffer type. The fluorescence intensity of reaction product of SAL with NBD-Cl was studied using different types of buffer solution as (borate, phosphate, Teorell and Stenhagen buffers) over pH range (7.0–10). Maximum fluorescence intensity obtained at pH (9 ± 0.2) using borate buffer after which the fluorescence intensity started to decline due to increase in the concentration of hydroxide ions which hold back the reaction between SAL and NBD-Cl,25 Fig. 4. Type of buffer was also investigated. Borate buffer was found to be the best buffer that gave maximum fluorescence intensity. Other buffers such as phosphate, Teorell and Stenhagen buffer at same pH did not give high fluorescence intensity as borate buffer. This probably because of the slow rate of hydrolysis of NBD-Cl to NBD-OH. This result is in agreement with Miyano et al.25
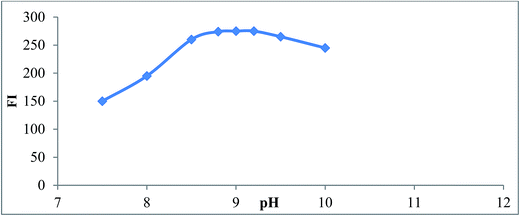 |
| | Fig. 4 Effect of pH on the reaction product of SAL (1.0 μg mL−1) and NBD-Cl, using borate buffer. | |
3.1.2. Effect of buffer volume. The volume of borate buffer pH 9 and its influence on fluorescence intensity was also investigated. It was found that the increase in volume of buffer gradually increased the fluorescence intensity of the reaction product and maximum fluorescence intensity was obtained upon using 2 ± 0.5 mL borate buffer pH 9.
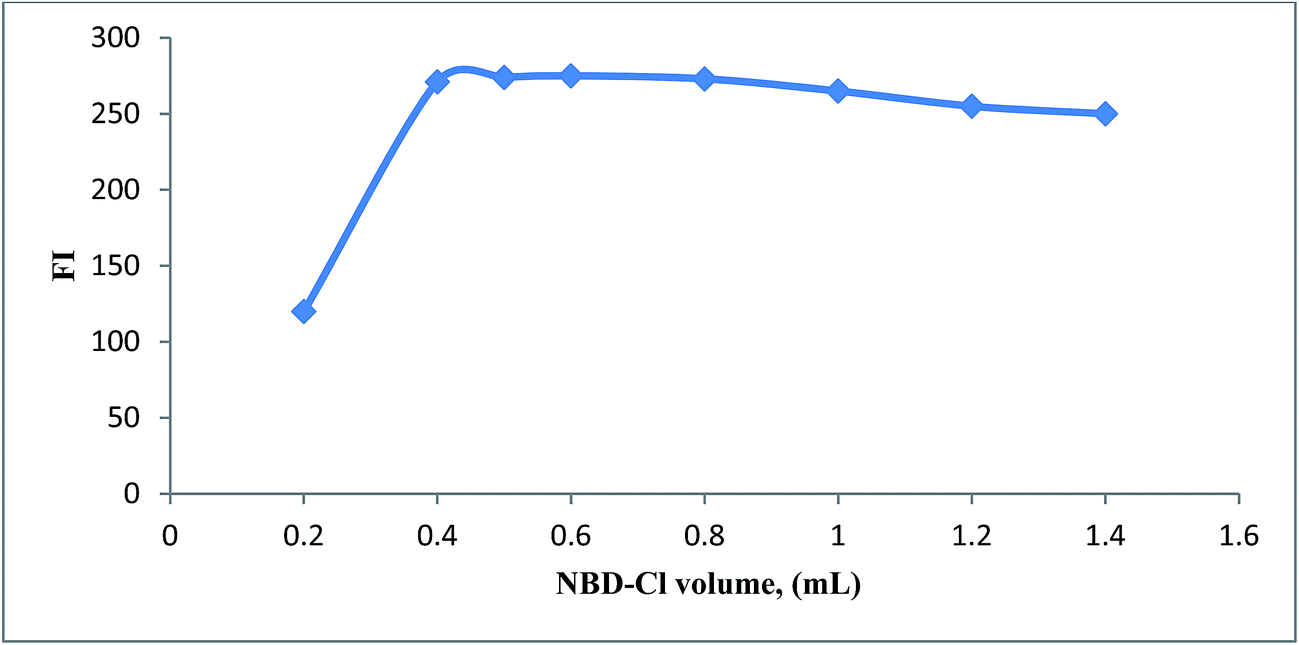
3.1.3. Effect of volume of NBD-Cl. The fluorescence intensity of reaction product of SAL was studied using different volumes of NBD-Cl (2.5 × 10−3 M) in the range of (0.2–1.4 mL). Maximum fluorescence intensity obtained upon using 0.5 ± 0.1 mL of NBD-Cl reagent (Fig. 5), after which the fluorescence intensity started to decline probably due to inner filter effect.25
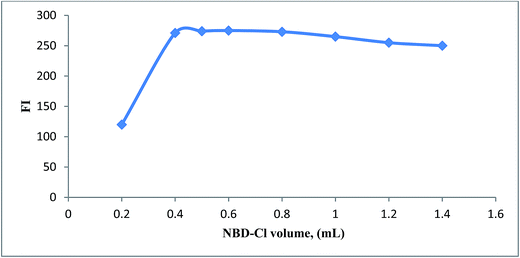 |
| | Fig. 5 Effect of NBD-Cl volume (2.5 × 10−3 M) on the reaction with SAL (1.0 μg mL−1). | |
3.1.4. Effect of temperature and time. The effect of temperature on fluorescence intensity of reaction product of SAL with NBD-Cl was studied, at different temperatures over different time intervals. It was observed that, the reaction product started to form at 50 °C and the fluorescence intensity reached its maximum at 80 °C ± 5 °C after 20 min.
3.1.5. Effect of diluting solvent. In order to select the most appropriate solvent for the reaction, many solvents were studied such as: water, methanol, ethanol, acetonitrile, acetone and DMF. Methanol was found to be the best solvent for dilution as it gave the highest fluorescence readings with good reproducibility and low blank readings; water gave poor readings, while other organic solvents gave high blank readings compromising the sensitivity of the method or gave poor results relative to methanol.
3.1.6. Effect of HCl concentration. Under these reaction conditions, relatively high fluorescence background was observed. This was attributed to the bi product formed during the reaction namely 4-hydroxy-7-nitrobenzo-2-oxa-1,3-diazole (NBD-OH), which come from the hydrolysis of NBD-Cl.25 The fluorescence of NBD-OH was found to be quenched by decreasing the pH of the reaction medium to less than one, this was done by the addition of 1 mL of 2 M HCl prior to measure. The volume of 1 mL was sufficient to quench the fluorescence of the NBD-OH without affecting the fluorescence of the reaction product.
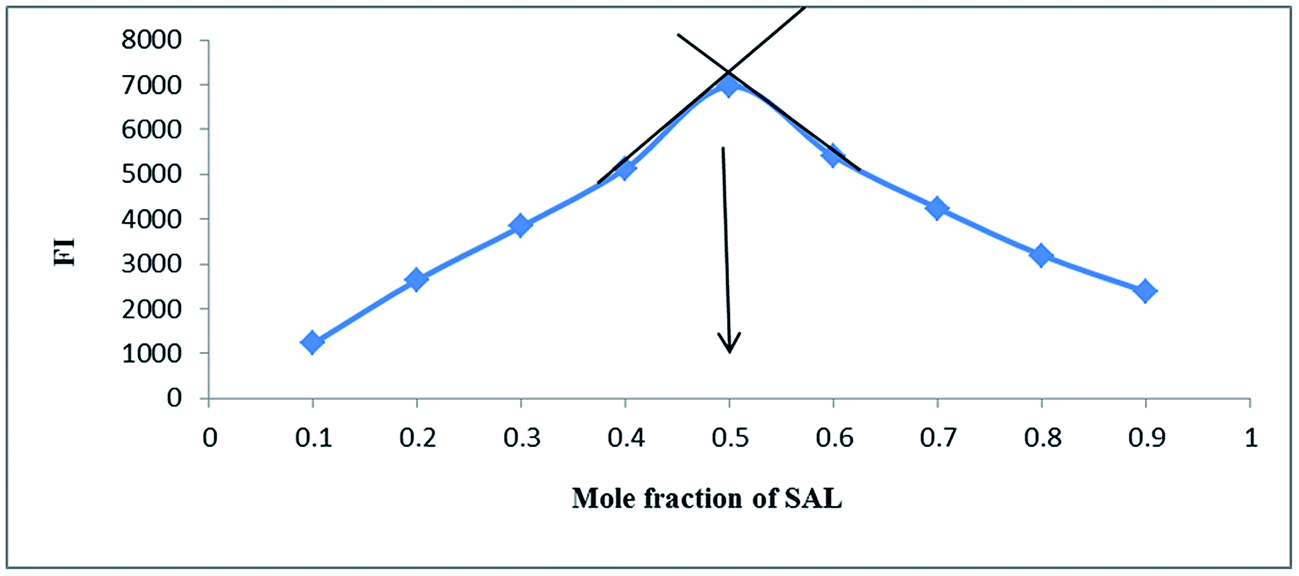
3.1.7. Stoichiometry and mechanism of the reaction. The stoichiometry of the derivatization reaction was studied through Job's method of continuous variation,26 using equimolar solutions (1.0 × 10−3 M) of both SAL and NBD-Cl. Then portions of mixture of master solutions of the drug and reagent were made up comprising different complementary proportions (0.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.9, 0.2:0.8, 0.3:0.7⋯0.7:0.3, 0.8:0.2, 0.9:0.1). It was found that the reaction proceeds in 1:1 ratio and this result agrees with the suggested reaction mechanism,17–22 Fig. 6.
:0.9, 0.2:0.8, 0.3:0.7⋯0.7:0.3, 0.8:0.2, 0.9:0.1). It was found that the reaction proceeds in 1:1 ratio and this result agrees with the suggested reaction mechanism,17–22 Fig. 6.
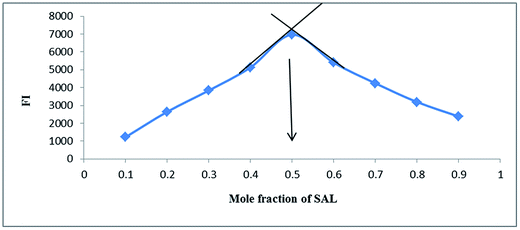 |
| | Fig. 6 Method of continuous variation for determination of stoichiometry of the reaction between SAL & NBD-Cl (5 × 10−3) M. | |
3.2. Validation of the proposed procedure
The proposed spectrofluorimetric method was validated according to the ICH guide line27 regarding accuracy, precision, limit of quantification, limit of detection and robustness.
3.2.1. Linearity and concentration range. In this work, the general analytical procedures were performed on a series of standard drug solutions having concentrations ranging from 100 to 1500 ng mL−1 for SAL. The whole set of experiments were carried out within this range to ensure the validation of the proposed procedure. Linear calibration was obtained by plotting the fluorescence intensity (FI) of the reaction product between SAL and NBD-Cl, subtracted from blank reading and the concentration of SAL. Statistical treatment of the data was carried out using linear regression analysis and the analytical parameters were calculated. The correlation coefficient was 0.9998 over the concentration range (100–1500) ng mL−1 indicating good linearity of the proposed method, Table 1.
Table 1 Analytical parameters of proposed spectrofluorimetric method between SAL and NBD-Cla
| Parameter |
Value |
| LOD: limit of detection, LOQ: limit of quantitation. |
| λex (nm) |
473 |
| λem (nm) |
543 |
| Concentration range (ng mL−1) |
(100–1500) |
| Correlation coefficient (r) |
0.9998 |
| Determination coefficient (r2) |
0.9996 |
| Slope |
0.2649 |
| Intercept |
11.116 |
| SD the intercept (Sa) |
1.567 |
| SD of slope (Sb) |
0.0021 |
| LOD (ng mL−1) |
19 |
| LOQ (ng mL−1) |
59 |
3.2.2. Accuracy and precision. Accuracy was checked at five concentration levels within the specified range. Three replicate measurements were recorded at each concentration level. The results were presented as percentage recovery ± standard deviation. The obtained results showed good agreement between the measured and true value indicating high accuracy of the proposed method, Table 2.
Table 2 Evaluation of the accuracy of the proposed spectrofluorimetric methodb
| Sample no. |
Taken (ng mL−1) |
Found (ng mL−1) |
% recoverya |
| Mean of three determinations. SD: standard deviation, RSD: relative standard deviation. |
| 1 |
100 |
100.19 |
100.19 |
| 2 |
300 |
302.19 |
100.73 |
| 3 |
500 |
496.89 |
99.37 |
| 4 |
700 |
700.43 |
100.06 |
| 5 |
1000 |
1007.65 |
100.76 |
| Mean |
|
|
100.22 |
| SD |
|
|
0.56 |
| RSD |
|
|
0.55 |
Precision was examined at three concentration levels; three replicate measurements were recorded at each concentration level; both inter-day and intra-day precision were evaluated and their results were summarized in Table 3. The calculated relative standard deviations were below 2% indicating excellent precision of the proposed procedure at both level of repeatability and intermediate precision.
Table 3 Evaluation of intraday and interday precision of the proposed spectrofluorimetric methodb
| Precision level |
Conc. (ng mL−1) |
% recoverya ± SD |
RSD (%) |
| Mean of three determinations. SD: standard deviation, RSD: relative standard deviation. |
| Intraday precision |
500 |
99.49 ± 0.42 |
0.42 |
| 700 |
100.63 ± 0.50 |
0.50 |
| 1000 |
99.83 ± 0.21 |
0.21 |
| Interday precision |
500 |
99.59 ± 1.53 |
1.53 |
| 700 |
100.51 ± 1.32 |
1.32 |
| 1000 |
99.87 ± 1.53 |
1.53 |
3.2.3. Limits of detection (LOD) and quantitation (LOQ). The limits of detection (LOD) and quantification (LOQ) were calculated based on standard deviation of response and the slope of calibration curve using the equations; LOD = 3.3 σ/S and LOQ = 10 σ/S, where S is the slope of the calibration curve and σ is the standard deviation of intercept. The results were presented in Table 1. The limit of quantitation was 19 ng mL−1 indicating high sensitivity of the proposed method compared with the reported HPLC method for SAL in dosage form [9], The reversed phase HPLC method for determination of SAL in metered dose inhalers represent detection level of 2 μg mL−1 using expensive HPLC solvents. Therefore, our proposed spectrofluorimetric method is considered highly sensitive and economic compared to the reported one.
3.3. Application to pharmaceutical dosage forms
The proposed method was successfully applied for determination of SAL in its pharmaceutical dosage form. The selectivity of the method was investigated by observing any interference encountered from the excipients of the pharmaceutical preparation. It was shown that these compounds did not interfere with the proposed method and the results of the proposed method were statistically compared with these of the reported method8 using student's t-test and F-test. There was no significant difference between the proposed and the reported method as the calculated values did not exceed the theoretical values at 95% confidence level, Table 5. This indicates good level of precision and accuracy of the proposed spectrofluorimetric method.
Table 5 Application of proposed spectrofluorimetric method to dosage form
| Dosage form |
% recoverya ± SD |
t-Valueb |
F-Valueb |
| Proposed |
Reported [8] |
| Mean of five determinations. The tabulated values of t- and F-values at 95% confidence limit are 2.78 and 6.39, respectively. |
| Metrovent® inhaler (25 μg SAL/actuation) |
100.6 ± 1.3 |
99.4 ± 0.94 |
1.7 |
1.8 |
3.4. Application to spiked human plasma
The high sensitivity attained by the proposed method allowed the possible determination of salmeterol in spiked human plasma. Inter-day, intra-day precision and accuracy in spiked plasma samples were determined by analyzing the biological samples at three concentration levels (100, 200 and 300 (ng mL−1)) on three separated days. The precision was defined as the relative standard deviation of spiked sample concentrations determined at three replicates, whereas accuracy was assessed as the percentage to the nominal concentration (%).
Precision and accuracy data for inter- and intra-day human plasma samples are presented in Table 6. The results for all samples (inter- and intra-day) were found to be within the acceptance criteria for the method validation. The concentration of the studied drug was computed from its corresponding regression equation. The mean percentage recovery for SAL ranged from 98.9% to 100.5% at the three spiked concentrations applied. The relative standard deviation of the obtained recoveries did not exceed 2% which fall within the acceptable limits of analytical method variability arising from different matrix effects.
Table 6 Intra-day, inter-day precision and accuracy study of proposed method for determination the studied drug in spiked human plasma sample
| Concentration (ng mL−1) |
Intra-day precision |
Inter-day precision |
| % recoverya ± SD |
RSD |
% recoverya ± SD |
RSD |
| Average of three determinations. |
| 100 |
99.49 ± 0.61 |
0.61 |
99.44 ± 0.26 |
0.26 |
| 200 |
98.90 ± 0.22 |
0.22 |
100.5 ± 0.41 |
0.41 |
| 300 |
99.83 ± 0.35 |
0.35 |
99.8 ± 0.38 |
0.62 |
Although the proposed method has been successfully applied for determination of SAL in spiked plasma, it is recommended to be applied only in real plasma for those patients who are being treated with SAL and not receiving other drugs containing primary or secondary amine moieties due to the unselective reaction of NBD-Cl towards these drugs.
3.5. Stability indicating study
Our proposed spectrofluorimetric procedure is one of few methods that could determine SAL in presence of its degradation products. The reported methods,28,29 used high cost techniques such as HPLC & UPLC, while this method is cost effective technique. It was reported that the main degradation pathway of phenolic compounds that have the same nucleus as studied drug is through oxidation under different conditions, especially under alkaline conditions30,31 and these results agree with that of the proposed method.
Application of analytical proposed method for stability indicating application using the previously mentioned degradation conditions indicated that 46%, 36% and 37% of SAL remained intact after acid, base induced and oxidative degradation, respectively, while no degradation was observed in case of exposing stock solution to direct artificial day light for 12 hours.
4. Conclusion
The present study describes the first spectrofluorimetric attempt to determine salmeterol xinafoate in its pharmaceutical dosage form as well as spiked human plasma without being affected by surrounding matrices depending on derivatization reaction with NBD-Cl. The proposed method is simple, rapid and accurate and it has been validated according to ICH guidelines. Furthermore, the developed method is extended to determine the intact drug in the presence of its degradation products. Therefore, the suggested method is suitable for analysis of the investigated drug in quality control and clinical laboratories.
Conflicts of interest
There are no conflicts to declare.
References
- S. Sweetman, Martindale: the complete drug reference, Royal Pharmaceutical Society of Great Britain, Pharmaceutical Press, London, 36th edn, 2009 Search PubMed.
- K. P. Chowdary and G. Devala Rao, Indian Drugs, 1997, 34, 606–607 Search PubMed.
- S. Thea Sufernent, Indian J. Pharm. Sci., 1998, 60, 294–296 Search PubMed.
- K. P. Chowdary and G. Devala Rao, Indian J. Pharm. Sci., 1999, 61, 246–247 CAS.
- M. N. Reddy, K. V. Rao Kanna, S. S. Rao and M. E. Rao, Indian J. Pharm. Sci., 2000, 62, 193–195 Search PubMed.
- M. N. Reddy, K. K. Rao, S. S. Rao and M. E. Rao, Indian Drugs, 2000, 37, 105–107 CAS.
- M. S. Kondawar, J. J. Waghmare, M. K. Malusare and N. D. Shah, Int. J. PharmTech Res., 2011, 3, 1801–1806 CAS.
- A. Samir, H. Salem and M. Abdelkawy, Bull. Fac. Pharm. (Cairo Univ.), 2012, 50, 121–126 CAS.
- V. G. Nayak, S. G. Belapure, C. D. Gaitonde and A. A. Sule, J. Pharm. Biomed. Anal., 1996, 14, 511–513 CrossRef CAS PubMed.
- P. S. Jain, A. P. Gorle, S. S. Patil, R. S. Chavan, P. R. Bari and S. J. Surana, Int. J. Pharmaceut. Chem. Anal., 2015, 2, 28–33 Search PubMed.
- L. Kasaye, A. Hymete and A. M. Mohamed, Saudi Pharm. J., 2010, 18, 153–159 CrossRef CAS PubMed.
- M. A. Omar, D. M. Nagy, M. A. Hammad and A. A. Aly, AAPS PharmSciTech, 2013, 14, 828–837 CrossRef CAS PubMed.
- S. M. Derayea, M. A. Omar, I. M. Mostafa and M. A. Hammad, RSC Adv., 2015, 5, 78920–78926 RSC.
- A. M. Mohamed, M. A. Omar, M. A. Hammad and A. A. Mohamed, Spectrochim. Acta, Part A, 2015, 149, 934–940 CrossRef CAS PubMed.
- M. A. Omar, H. M. Ahmed, M. A. Hammad and S. M. Derayea, Spectrochim. Acta, Part A, 2015, 135, 472–478 CrossRef CAS PubMed.
- M. A. Omar, M. A. Hammad, D. M. Nagy and A. A. Aly, Spectrochim. Acta, Part A, 2015, 136, 1760–1766 CrossRef CAS PubMed.
- A. A. Al-Majed, Anal. Chim. Acta, 2000, 408, 169–175 CrossRef CAS.
- A. A. Al-Majed and J. Al-Zehouri, Il Farmaco, 2001, 56, 291–296 CrossRef CAS PubMed.
- I. A. Darwish, S. M. Amer, H. H. Abdine and L. I. Al-Rayes, J. Fluoresc., 2009, 19, 463 CrossRef CAS PubMed.
- T. Sevgi, Chin. J. Chem., 2010, 28, 2209–2215 CrossRef CAS.
- T. Sevgi and K. Elif, J. Chromatogr. Sci., 2011, 49, 417–421 Search PubMed.
- M. Walash, N. El-Enany and H. Askar, Luminescence, 2015, 30, 1119–1124 CrossRef CAS PubMed.
- M. Pesez and J. Bartos, Colorimetric and Fluorimetric Analysis of Organic Compounds and Drugs, Marcel Dekker Inc, New York, 1974 Search PubMed.
- M. A. Omar, M. A. Hammad, B. I. Salman and S. M. Derayea, Spectrochim. Acta, Part A, 2016, 157, 55–60 CrossRef CAS PubMed.
- H. Miyano, T. Toyooka and K. Imai, Anal. Chim. Acta, 1985, 170, 81–87 CrossRef CAS.
- J. S. Renny, L. L. Tomasevich, E. H. Tallmadge and D. B. Collum, Angew. Chem., 2013, 52, 11998–12013 CrossRef CAS PubMed.
- ICH Harmonized
Tripartite Guideline, Validation of Analytical Procedures: Text and Methodology, Q2(R1), Current Step 4 Version, Parent Guidelines on Methodology Dated November 6 1996, Incorporated in November 2005, http://www.ich.org/LOB/ media/MEDIA417.pdf, accessed 15 February 2008.
- P. Pratibha, S. Kapendra, K. Chandrabose, N. Narayana Moorthy and T. Piuesh, Lat. Am. J. Pharm., 2011, 30, 985–990 Search PubMed.
- B. Akmese, S. Sanli, N. Sanli and A. Asan, J. Anal. Chem., 2014, 69, 563–573 CrossRef CAS.
- G. A. Jacobson and G. M. Peterson, J. Pharm. Biomed. Anal., 1994, 12, 825–832 CrossRef CAS PubMed.
- H. Al-Malaq, A. Al-Majed and F. Belal, Anal. Lett., 2000, 33, 1961–1974 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Mohamed A. Hammad*a and
Mohamed Awadb
*a,
Mohamed A. Hammad*a and
Mohamed Awadb