
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simple and low-cost thiophene and benzene-conjugated triaryamines as hole-transporting materials for perovskite solar cells†
Jianchang Wu‡
 ab,
Chang Liu‡b,
Xiang Dengb,
Luozheng Zhangb,
Manman Hub,
Jun Tangab,
Wenchang Tan*a,
Yanqing Tian*b and
Baomin Xu*b
ab,
Chang Liu‡b,
Xiang Dengb,
Luozheng Zhangb,
Manman Hub,
Jun Tangab,
Wenchang Tan*a,
Yanqing Tian*b and
Baomin Xu*b
aSchool of Advanced Materials, Peking University Shenzhen Graduate School, Shenzhen, Guangdong 518055, P. R. China. E-mail: tanwch@pku.edu.cn
bDepartment of Materials Science and Engineering, Southern University of Science and Technology, No. 1088, Xueyuan Rd, Shenzhen, Guangdong 518055, P. R. China. E-mail: tianyq@sustc.edu.cn; xubm@sustc.edu.cn
First published on 25th September 2017
Abstract
Two novel electron-rich linear small-molecules, containing benzene and thiophene as the cores with arylamine side groups, named HTM1 and HTM2, respectively, were synthesized via short, easy and efficient synthetic routes. The influence of the π-linkers of the two materials on photophysical, electrochemical, and thermal properties, and hole mobility and photovoltaic performance was investigated. The compound with thiophene as π-linker (HTM2) shows better solubility and higher hole-transporting mobility than the compound with benzene as π-linker (HTM1). When these two materials were incorporated into perovskite solar cells as hole transporting materials (HTMs), short circuit photocurrent densities (Jscs) of 15.83 mA cm−2 and 21.1 mA cm−2, open circuit voltages (Vocs) of 0.79 V and 1.09 V, and fill factors (FFs) of 0.46 and 0.62, were obtained. These factors contributed to average overall power conversion efficiencies (PCEs) of 6.4% and 13.9% with the best PCEs of 7.5% and 14.7%, respectively. The performance of HTM2 is comparable to the PCE obtained using the current state-of-the-art HTM of 2,2′,7,7′-tetrakis(N,N′-di-p-methoxyphenylamine)-9,9′-spirobifluorene (spiro-OMeTAD) with the best PCE of 17.4% using a similar device preparation method and measurement conditions. These results showed that selecting a suitable π-linker is important for the performance of HTMs. And the simple HTM2 material is a promising HTM with the potential to replace the expensive spiro-OMeTAD due to its comparable performance with a much simpler synthesis route and much reduced cost (10 times less than that of spiro-OMeTAD). This study demonstrates that a compound with a suitable π-linker could be a low-cost and high performance HTM to replace spiro-OMeTAD.
Introduction
In recent years, the efficiency of organic–inorganic metal halide perovskite-based solar cells (PSCs) has improved rapidly, and is already comparable with current commercial technologies.1,2 Those perovskite materials, typically methylammonium lead iodides (CH3NH3PbI3)s, have excellent properties, including their absorption throughout the UV-vis-NIR spectrum, high extinction coefficients, large exciton diffusion lengths and ambipolar mobility. These characteristics make them excellent light harvesting materials.3–6 Their outstanding performance by showing an increase of the power conversion efficiency (PCE) from 3% to a certified 22% has been demonstrated during the past few years.1–3,7–9 A typical PSC device comprises a TiO2 anode, a perovskite absorbing layer, a hole transporting layer and a metallic counter electrode. In addition to perovskite absorbing materials, charge transporting layers also play an important role in charge extraction and interface modification, thus improving the performance of PSCs device.10,11 To date, 2,2′,7,7′-tetrakis(N,N′-di-p-methoxyphenylamine)-9,9′-spirobifluorene (spiro-OMeTAD) is the most extensively studied and applied HTM for perovskite devices.12–16 Many device characterization and optimization works have been centered on spiro-OMeTAD.9,15,17 However, in generally, the complex synthetic route and purification of spiro-OMeTAD render it expensive, which significantly limits potential large-scale application and commercialization of PSC devices in the future. The synthesis of the core of spiro-OMeTAD needs four steps, including a Suzuki–Miyaura cross-coupling reaction, a Grignard reaction, a cyclization reaction and a bromination reaction from commercially available raw materials, which limited its total yield (<30%) and cost (about 94 $ per g).18,19 Thus, significant research effort has been spent on designing and synthesizing alternative HTMs, which are more economical and are able to show device performance comparable to or even better than that of spiro-OMeTAD. Several conjugated polymers including poly(3-hexylthiophene-2,5-diyl) (P3HT)20 and poly[bis(4-phenyl)(2,4,6-trimethylphenyl)amine] (PTAA)8 have been employed as HTMs in perovskite solar cells. However their device results to date have been inferior to that of spiro-OMeTAD. What's more, the intrinsic batch to batch variation issues of polymers due to different polymerization degrees and polydispersity could hamper the performance reproducibility for possible commercialization of the device. On the other hand, a number of small molecule HTMs have also been reported, including spirobifluorenes derivatives,19,21–23 thiophene derivatives,24–26 triphenylamines,27,28 and triazatruxenes.29–31Most of them were namely decorated with diarylamines, triarylamines and/or carbazole derivatives. The best performance of those HTMs show similar or superior performance to spiro-OMeTAD in some aspects but no material has yet emerged clearly superior in all aspects of device performance, particularly in terms of cost, device efficiency and stability. Hence it remains a great challenge to develop new, easily synthesized, low cost and high performance HTMs for PSC devices.
An efficient way to simplify the synthesis of the HTMs is to change the spiro-core of the spiro-OMeTAD to a planar unit, such as diphenyl or terphenyl. In this way, only one or two synthetic steps are needed from the commercially available raw materials, this could reduce its cost significantly. In this article, 2,5-bis(4,4′-bis(methoxyphenyl)aminophen-4′′-yl)-benzene (HTM1, its structure is given in Fig. 1) was designed and synthesized using terphenyl to replace the spiro-core of the spiro-OMeTAD. It only needs two synthetic steps to get the final product. However, when the material changes from spiro-structure to a planar structure, a great decrease in solubility and slight decrease in mobility occur. Those factors affect the process of the HTM film, as a result the PCE of the devices based on HTM1 were much lower than that of spiro-OMeTAD. To solve the low mobility of HTM1, 2,5-bis(4,4′-bis(methoxyphenyl)aminophen-4′′-yl)-thiophene (HTM2) was designed and synthesized using thiophene unit to replace the center unit (benzene) of the HTM1. Thiophene moiety has been widely used in organic electronics because of its excellent properties due to its high charge mobility. Unfortunately, during the manuscript preparation, a recent paper reported the use of HTM2 as hole transporting layer in PSCs device32 with an extremely low PCE (4.31%). For this performance, it seems that HTM2 is far from being useful for high efficient devices and a candidate to replace spiro-OMeTAD. However, in our studies, we achieved much higher PCE of 14.7%. This result may be caused by the different components of perovskite layer. FAMAPbBrI3 was used in this article and MAPbI3 was used in the ref. 32. It is obvious that the device based on FAMAPbBrI3 could achieve higher performance. (Devices based on spiro-OMeTAD achieve an efficiency of 17.4% in this article; it was only 11.9% in the reference). Moreover, FAMAPbBrI3 has a deeper HOMO level compared with MAPbI3, which could match HTM2 better. Further, we compared the cost of the synthesis of HTM2 with spiro-OMeTAD. Because of the much shorter synthesis route of HTM2 than that of spiro-OMeTAD, the cost of HTM2 is about 1/10 fold of spiro-OMeTAD. Thus, our results demonstrated that the HTM2 has a great potential to replace the spiro-OMeTAD.
 | ||
| Fig. 1 Chemical structures of spiro-OMeTAD, HTM1 and HTM2. | ||
Results and discussions
The synthetic route (Scheme S1†) towards HTM1 and HTM2 only requires two steps and uses commercially available raw materials, which are much simpler than that of the synthesis of spiro-OMeTAD. Pd-catalyzed boronation of 4-bromo-N,N-bis(4-methoxyphenyl)aniline (compound 1, ESI Scheme S1†) with bis(pinacolato)diboron produced the key building block OMT BPIN (compound 2, Scheme S1†) in a yield of 80%. The two final HTMs were synthesized by Suzuki coupling reaction using OMT BPIN with dibromobenzene (compound 3, Scheme S1†) and dibromothiophene (compound 4, Scheme S1†), respectively. HTM2 has a good solubility in common solvents, such as chlorobenzene (200 mg mL−1); while the solubility of HTM1 is relatively low, about 60 mg mL−1 in chlorobenzene. The good solubility of HTM2 will benefit the smooth film formation. Differential scanning calorimetry (DSC) measurements show that HTM1 and HTM2 have a glass transition temperature (Tg) at 80 °C and 76 °C, respectively. Both are much lower than that of spiro-OMeTAD (around 125 °C).33 The lower Tg and higher solubility could increase the pore-filling of the HTM in mesoscopic architecture device, because the pore filling procedure begins by spin-coating a solution of the HTM onto the m-TiO2/perovskite film.33 As solvent evaporates and the solution concentration increases, HTM diffuses into the pores allowing for a higher degree of pore filling than that would be obtained from the initial concentration of the solution alone. When the concentration increased to a certain degree beyond the solubility of HTM, the diffusion stops, thus higher solubility contributes to better pore-filling of the device. And a lower Tg could help to increase the pore-filling through a melt infiltration process. The device with a high pore-filling may have a high coverage of perovskite with HTM, thus yields a higher charge injection efficiency and lower electron–hole recombination.33The absorption spectra (Fig. 2a and Table 1) indicate that HTM1 starts to absorb light (λonset) from λ at 405 nm, which is 8 nm blueshifted comparing to that of spiro-OMeTAD. While the λonset value of HTM2 is 451 nm, which is 38 nm redshifted from that of spiro-OMeTAD (46 nm redshifted from that of HTM1). This is attributed to the higher electron density of the backbone for HTM2 compared with HTM1.
 | ||
| Fig. 2 (a) Normalized UV-vis absorption spectra of HTM1, HTM2 and spiro-OMeTAD in THF solution; (b) cyclic voltammograms of HTM1, HTM2 and spiro-OMeTAD in dry dichloromethane–TBAPF6 (0.1 M); scan speed: 100 mV s−1, potentials vs. Fc/Fc+. | ||
| HTMs | Tg [°C] | λonset [nm] | Ega [V] | EHOMOb [eV] | ELUMOc [eV] |
|---|---|---|---|---|---|
| a Estimated from the absorption onset wavelength, Eg (eV) = 1240/λonset (nm).b Calculated from EHOMO = −(Eoxonset + 5.10) (eV).34c ELUMO calculated by ELUMO = EHOMO + Eg. | |||||
| HTM1 | 80 | 405 | 3.06 | −5.21 | −2.15 |
| HTM2 | 76 | 451 | 2.75 | −5.13 | −2.38 |
| Spiro-OMeTAD | 125 | 413 | 3.0 | −5.01 | −2.01 |
Cyclic voltammetric measurements were carried out to analyze and compare the energy levels of the two new HTMs and spiro-OMeTAD for the favourable hole injection process from the perovskite layer. Fig. 2b depicts the cyclic voltammograms (CV) in dichloromethane solution (1.0 × 10−3 mol L−1) with tetrabutylammonium hexafluorophosphate (TBAPF6, 0.1 mol L−1) at a scan rate of 100 mV s−1. As can be seen from this figure, the HOMO levels of HTM1 and HTM2 are −5.21 and −5.13 eV, respectively. Both are 0.10–0.20 eV deeper than that of spiro-OMeTAD (−5.01 eV). The HOMO level of HTM1 is 0.08 eV deeper than that of HTM2, which is attributed to the higher electron density of the backbone for HTM2. The deeper HOMO of the new HTMs could benefit the higher open-circuit voltage (Voc) of the device. In principle, Voc of a conventional solid-state dye sensitized solar cell (DSSC) is determined by the difference between the quasi-Fermi levels of the electrons in TiO2 and the holes in the HTM.35 And this is the same for PSCs devices. So from the electrochemical experiments data, we can expect that the device using those new HTMs would have a higher Voc than that of spiro-OMeTAD.
To investigate the charge-carrier mobility of HTM1, HTM2 and spiro-OMeTAD, hole-only devices were fabricated with device structure of fluorine-doped tin oxide (FTO) glasses/(poly(3,4-ethylenedioxythiophene):polystyrene, PEDOT:PSS)/doped HTM1 or doped HTM2 (or doped spiro-OMeTAD)/Au. Hole mobilities of HTM1 and HTM2 were measured via space charge limitation of current (SCLC) under dark conditions, as showed in Fig. S9.† The results calculated from the Mott–Gurney law shows that by replacing benzene unit with thiophene unit, the mobility of the material increased about three times (3.1 × 10−5 cm2 V−1 S−1 for HTM1 and 9.0 × 10−5 cm2 V−1 S−1 for HTM2). However, HTM1 and HTM2 both have a lower mobility than spiro-OMeTAD (3.7 × 10−4 cm2 V−1 S−1), which suggest that HTM1 and HTM2 based devices should show slightly lower fill factors.
The new semiconductors HTM1 and HTM2 were tested as HTMs in perovskite solar cells. The device structure can be seen from the scanning electron microscope (SEM) image of the device cross-section (Fig. 3a and b). At the anode, a compact and mesoporous layer of TiO2 was deposited on top of a fluorine-doped tin oxide (FTO) coated glass and used as the electron collector, while the cathode consists of a gold electrode thermally evaporated onto the HTM layer. The photoactive material FAMAPbBrI3 is sandwiched between the anode and the cathode. The current–voltage (J–V) characteristics of perovskite solar cells that employ HTM1, HTM2, and spiro-OMeTAD, respectively, as the HTMs are shown in Fig. 3d and summarized in Table 2. The PCEs of the best performing devices with HTM1, HTM2, and spiro-OMeTAD as HTMs are 7.5%, 14.7% and 17.4%, respectively. Correspondingly, the short-circuit current densities (Jsc) of the cells are 15.8, 21.1 and 23.5 mA cm−2, respectively. It is clear that Jsc value of HTM2 is as high as that spiro-OMeTAD, while Jsc of HTM1 is relatively low. The open-circuit voltage (Voc) of HTM2 (1.09 V) is also higher than that of spiro-OMeTAD. The only different parameters are the fill factors (FFs). For the device based on spiro-OMeTAD the FF could achieve 0.72, while FF is only 0.62 for HTM2, which contributes to the slightly low PCEs for HTM2. HTM1 has a deeper HOMO energy level, but the Voc has not been improved, only 0.79 V. And its FF is also relatively low, 0.46. Those may be caused by its poor mobility, limiting the charge transport in the film.
 | ||
| Fig. 3 Cross-sectional SEM pictures of PSCs devices based on HTM1 (a) and HTM2 (b); and the relative energy levels in the PSCs (c) and current–voltage curves (d) of PSCs based on different HTMs. | ||
| HTM | Jsc [mA cm−2] | Voc [V] | FF | PCE [%] |
|---|---|---|---|---|
| HTM1 | 15.83 ± 1.46 | 0.79 ± 0.06 | 0.46 ± 0.04 | 6.33 ± 1.21% |
| HTM2 | 21.13 ± 0.85 | 1.05 ± 0.04 | 0.61 ± 0.02 | 13.93 ± 0.77% |
| Spiro-MeOTAD | 22.22 ± 0.53 | 1.04 ± 0.02 | 0.69 ± 0.05 | 17.02 ± 0.38% |
To investigate the charge transfer at the interface between the perovskite and the hole-transport materials, steady-state and time-resolved photoluminescence (PL) spectra were recorded and these are shown in Fig. 4. From the steady-state PL spectra, the perovskite film without HTMs shows a strong PL signal due to the recombination between the photo-generated electrons and holes. After deposition of HTMs, a dramatic decrease of PL intensity is observed, especially for the HTM2 based devices. A more efficient quenching of the photo-generated charges can lead to faster interface charge separation in HTM2 based devices than that of HTM1 based devices, contributing to a better Jsc. However, the lower SCLC hole mobility limited its charge transporting compared to spiro-OMeTAD. Time-resolved PL measurements were carried out with excitation at 530 nm and monitoring the entire emission spectral range (Fig. 4b). Perovskite/spiro-OMeTAD samples showed a decay time of 3.5 ns, whereas perovskite/HTM1 and perovskite/HTM2 samples showed a longer decay lifetime of 4.3 ns and 4.2 ns, respectively. This indicates that HTM2 has slightly better hole extraction ability as compared to HTM1. The lower interface charge separation and poorer hole extraction ability of HTM1 could explain the low performance of the device based on HTM1 to some extent.
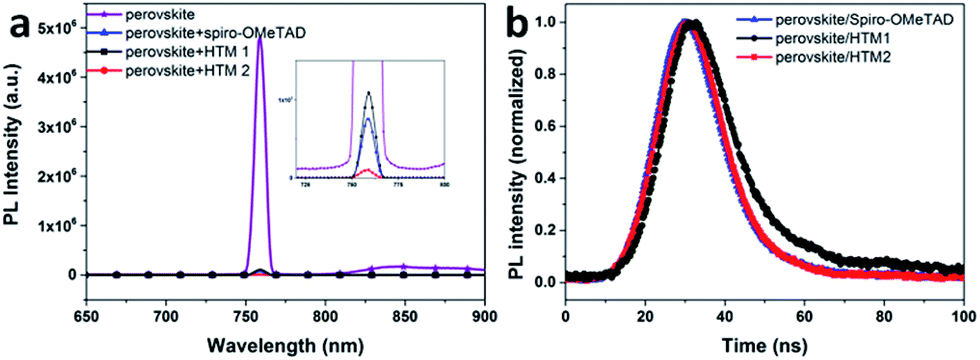 | ||
| Fig. 4 (a) Photoluminescence (PL) spectra of perovskite film, perovskite/spiro-OMeTAD film, perovskite/HTM1 film, perovskite/HTM2 film; (b) time-resolved PL spectra of perovskite/spiro-OMeTAD film, perovskite/HTM1 film, perovskite/HTM2 film. | ||
In order to compare the cost of the synthesized compound HTM1 and HTM2 with that of spiro-OMeTAD, estimations were made using a method published by Docampo et al.36 (see the Table S1†). As shown in Table 3, the cost of materials (including regents, solvents and other consumables) required for the synthesis of 1 g of HTM1 and HTM2 are 9.3 $ and 8.6 $, respectively, which are more than 10 times lower than that of spiro-OMeTAD. The main reason lies in the low cost starting materials and the simpler synthesis route. Hence it has a great potential to be commercialized for HTM2.
Conclusions
In conclusion, two simple triphenylamine-based HTMs were developed by varying the core structure. It is found that thiophene unit played an important role not only in increasing solubility, but also in enhancing hole mobility and charge extraction, thus improving the PSCs performance. The PSCs device based on HTM with thiophene (HTM2) achieved higher Voc, Jsc and FF values than that of HTM with benzene (HTM1). These results showed that selecting a proper π-linker is important for the performance of the HTMs. In addition, both the two HTMs could be synthesized simply with high yields by using two steps from commercially available starting materials, making those HTMs very promising for large-scale industrial production.Experimental section
Materials
All reagents and solvents, unless otherwise specified, were purchased from Aladdin, Alfa, Aldrich and J&K Scientific Ltd. and were used without further purification. 4-Bromo-N,N′-bis(4-methoxyphenyl)aniline was purchased from Sdyano fine chemical Co. Ltd. mesoporous TiO2 paste (Dyesol, 30NR-D), CsI, FAI, PbI2, MABr and PbBr2 were purchased from Xi'an Polymer Light Technology Corp., and 2,2′,7,7′-tetrakis(N,N-di-p-methoxyphenylamine)-9,9′-spirobifluorene (spiro-OMeTAD) was purchased from Feiming Chemical Limited.Measurement and characterization
Routine nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance III 400 MHz (400 and 100 MHz for 1H and 13C NMR, respectively). Chemical shifts were reported as δ values (ppm) with tetramethylsilane (TMS) as the internal standard. The splitting patterns are designated as follows: singlet (s), doublet (d), triplet (t), and multiplet (m). UV-vis absorption spectra were recorded on a PerkinElmer Lambda650 spectrophotometer from dilute solutions in THF. Cyclic voltammetry (CV) was obtained in a tetrabutylammonium hexafluorophosphate (TBAPF6, 0.1 mol L−1) supported CH2Cl2 solution at room temperature using a CHI600E electrochemical workstation (CH Instrument Ins.) operated at a scanning rate of 100 mV s−1. A Pt wire (ϕ = 1.0 mm) embedded in Teflon column was used as the working electrode, and a Pt sheet and Ag/AgCl electrodes were served as the counter and reference electrodes, respectively. Ferrocene/ferrocenium was used as the internal reference to calibrate the redox potentials. Thermo-gravimetric analysis (TGA) and differential scanning calorimetry (DSC) were carried out with a Mettler-Toledo TGA STARe/DSC1 thermal analyzer under purified nitrogen gas flow with a 10 °C min−1 heating rate. For time resolved fluorescence spectroscopy, a time-correlated single photon counting (TCSPC) system was used. The samples were excited with a picosecond diode laser (Edinburgh Instruments, EPL900) at 530 nm. Photocurrent density–voltage (J–V) curves were measured under AM 1.5G one sun illumination (100 mW cm−2) with a solar simulator (Enlitech SS-F7-3A) equipped with 300 W xenon lamp and a Keithley 2400 source meter. The light intensity was adjusted by an NREL-calibrated Si solar cell. The device active area was 0.08 cm−2. The J–V curves were recorded from −0.2 V to 1.2 V with 0.01 V steps, integrating the signal for 10 ms after a 10 ms delay. The average Voc, Jsc and FF were based on 10 devices.Device fabrication
Fluorine-doped tin oxide (FTO) glasses (Nippon Sheet Glass) were cleaned with detergent, deionized water, and acetone under sonication with ethanol in an ultrasonic bath for 30 minutes. After that, FTO glasses were treated in a UV cleaner for 30 minutes. The cleaned FTO glasses were coated with 0.15 M titanium diisopropoxide bis(acetylacetonate) in 1-butanol solution by the spin-coating method at 2000 rpm for 60 s, followed by heating at 125 °C for 5 minutes. The films were cooled down to room temperature and a 0.15 M titanium diisopropoxide bis(acetylacetonate) solution in 1-butanol was spin coated again to make a pin hole free dense TiO2 film. And then the substrates were calcined in a box furnace at 450 °C for 30 minutes. Mesoporous TiO2 (mp-TiO2) layer was deposited on the dense TiO2 layer by spin-coating the TiO2 ethanol solution containing 14.3 wt% TiO2 paste at 4000 rpm for 20 seconds, which was then calcined at 500 °C for 0.5 hour. CsI (0.05 m), FAI (1 M), PbI2 (1.1 M), MABr (0.2 M) and PbBr2 (0.2 M) was dissolved in anhydrous DMF:DMSO 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v), which was heated at 70 °C for 2 hours under magnetic stirring, and the mixture was then spin coated on the as-prepared TiO2 film at 4000 rpm for 40 seconds a nitrogen atmosphere glove box. During the spin coating step, 100 μL of chlorobenzene was poured on the spinning substrate 15 s prior to the end of the program. Afterward, the as-prepared films were heated at 100 °C for around 60 min until the color changed to dark red. The hole-transporting layer (HTL) was prepared by spin-coating an HTM solution at 4000 rpm for 20 s. The HTM solution were prepared by dissolving spiro-OMeTAD, HTM1 and HTM2 in chlorobenzene (60 mM) with 28.8 μL tBP solution and 17.5 μL Li-bis(trifluoromethanesulfonyl) imide (Li-TFSI)/acetonitrile (520 mg/1 mL). Devices were then left overnight in air. Finally, a 100 nm thick gold (Au) layer was thermally evaporated under vacuum of 4 × 10−5 Torr, at a rate of ∼0.05 nm s−1, to finish the device fabrication.
:1 (v/v), which was heated at 70 °C for 2 hours under magnetic stirring, and the mixture was then spin coated on the as-prepared TiO2 film at 4000 rpm for 40 seconds a nitrogen atmosphere glove box. During the spin coating step, 100 μL of chlorobenzene was poured on the spinning substrate 15 s prior to the end of the program. Afterward, the as-prepared films were heated at 100 °C for around 60 min until the color changed to dark red. The hole-transporting layer (HTL) was prepared by spin-coating an HTM solution at 4000 rpm for 20 s. The HTM solution were prepared by dissolving spiro-OMeTAD, HTM1 and HTM2 in chlorobenzene (60 mM) with 28.8 μL tBP solution and 17.5 μL Li-bis(trifluoromethanesulfonyl) imide (Li-TFSI)/acetonitrile (520 mg/1 mL). Devices were then left overnight in air. Finally, a 100 nm thick gold (Au) layer was thermally evaporated under vacuum of 4 × 10−5 Torr, at a rate of ∼0.05 nm s−1, to finish the device fabrication.
Synthesis
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Project funding from the Ministry of Science and Technology of China (Grants No. 2016YFA0202400 and 2016YFA0202404), and the Peacock Team Project funding from Shenzhen Science and Technology Innovation Committee (Grant No. KQTD2015033110182370).References
- J. Burschka, N. Pellet, S. J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Gratzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed.
- N. Research Cell Efficiency Records, http://www.nrel.gov/ncpv/, accessed: March 2016.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS PubMed.
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Gratzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344–347 CrossRef CAS PubMed.
- T. Salim, S. Sun, Y. Abe, A. Krishna, A. C. Grimsdale and Y. M. Lam, J. Mater. Chem. A, 2015, 3, 8943–8969 CAS.
- H. Zhou, Q. Chen, G. Li, S. Luo, T. B. Song, H. S. Duan, Z. Hong, J. You, Y. Liu and Y. Yang, Science, 2014, 345, 542–546 CrossRef CAS PubMed.
- W. S. Yang, J. H. Noh, N. J. Jeon, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Science, 2015, 348, 1234–1237 CrossRef CAS PubMed.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Gratzel, Energy Environ. Sci., 2016, 9, 1989–1997 CAS.
- L. Calio, S. Kazim, M. Gratzel and S. Ahmad, Angew. Chem., Int. Ed., 2016, 55, 14522–14545 CrossRef CAS PubMed.
- M. Salado, J. Idigoras, L. Calio, S. Kazim, M. K. Nazeeruddin, J. A. Anta and S. Ahmad, ACS Appl. Mater. Interfaces, 2016, 8, 34414–34421 CAS.
- U. B. Cappel, E. A. Gibson, A. Hagfeldt and G. Boschloo, J. Phys. Chem. C, 2009, 113, 6275–6281 CAS.
- I. K. Ding, N. Tétreault, J. Brillet, B. E. Hardin, E. H. Smith, S. J. Rosenthal, F. Sauvage, M. Grätzel and M. D. McGehee, Adv. Funct. Mater., 2009, 19, 2431–2436 CrossRef CAS.
- N. J. Jeon, H. G. Lee, Y. C. Kim, J. Seo, J. H. Noh, J. Lee and S. I. Seok, J. Am. Chem. Soc., 2014, 136, 7837–7840 CrossRef CAS PubMed.
- D. Liu and T. L. Kelly, Nat. Photonics, 2014, 8, 133–138 CrossRef CAS.
- W. H. Nguyen, C. D. Bailie, E. L. Unger and M. D. McGehee, J. Am. Chem. Soc., 2014, 136, 10996–11001 CrossRef CAS PubMed.
- F. Hao, C. C. Stoumpos, D. H. Cao, R. P. H. Chang and M. G. Kanatzidis, Nat. Photonics, 2014, 8, 489–494 CrossRef CAS.
- T. P. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker and J. Salbeck, Chem. Rev., 2007, 107, 1011–1065 CrossRef CAS PubMed.
- B. Xu, D. Bi, Y. Hua, P. Liu, M. Cheng, M. Grätzel, L. Kloo, A. Hagfeldt and L. Sun, Energy Environ. Sci., 2016, 9, 873–877 CAS.
- Y. Guo, C. Liu, K. Inoue, K. Harano, H. Tanaka and E. Nakamura, J. Mater. Chem. A, 2014, 2, 13827–13830 CAS.
- D. Q. Bi, B. Xu, P. Gao, L. C. Sun, M. Graetzel and A. Hagfeldt, Nano Energy, 2016, 23, 138–144 CrossRef CAS.
- M.-H. Li, C.-W. Hsu, P.-S. Shen, H.-M. Cheng, Y. Chi, P. Chen and T.-F. Guo, Chem. Commun., 2015, 51, 15518–15521 RSC.
- M. Saliba, S. Orlandi, T. Matsui, S. Aghazada, M. Cavazzini, J.-P. Correa-Baena, P. Gao, R. Scopelliti, E. Mosconi, K.-H. Dahmen, F. De Angelis, A. Abate, A. Hagfeldt, G. Pozzi, M. Graetzel and M. K. Nazeeruddin, Nat. Energy, 2016, 1, 15017 CrossRef CAS.
- H. Li, K. Fu, A. Hagfeldt, M. Grätzel, S. G. Mhaisalkar and A. C. Grimsdale, Angew. Chem., Int. Ed., 2014, 53, 4085–4088 CrossRef CAS PubMed.
- T. Krishnamoorthy, F. Kunwu, P. P. Boix, H. Li, T. M. Koh, W. L. Leong, S. Powar, A. Grimsdale, M. Grätzel, N. Mathews and S. G. Mhaisalkar, J. Mater. Chem. A, 2014, 2, 6305 CAS.
- A. Krishna, D. Sabba, H. Li, J. Yin, P. P. Boix, C. Soci, S. G. Mhaisalkar and A. C. Grimsdale, Chem. Sci., 2014, 5, 2702–2709 RSC.
- H. Choi, S. Park, S. Paek, P. Ekanayake, M. K. Nazeeruddin and J. Ko, J. Mater. Chem. A, 2014, 2, 19136–19140 CAS.
- H. Choi, S. Paek, N. Lim, Y. H. Lee, M. K. Nazeeruddin and J. Ko, Chem. – Eur. J., 2014, 20, 10894–10899 CrossRef CAS PubMed.
- F. J. Ramos, K. Rakstys, S. Kazim, M. Gratzel, M. K. Nazeeruddin and S. Ahmad, RSC Adv., 2015, 5, 53426–53432 RSC.
- K. Rakstys, A. Abate, M. I. Dar, P. Gao, V. Jankauskas, G. Jacopin, E. Kamarauskas, S. Kazim, S. Ahmad, M. Grätzel and M. K. Nazeeruddin, J. Am. Chem. Soc., 2015, 137, 16172–16178 CrossRef CAS PubMed.
- L. Calió, C. Momblona, L. Gil-Escrig, S. Kazim, M. Sessolo, Á. Sastre-Santos, H. J. Bolink and S. Ahmad, Sol. Energy Mater. Sol. Cells, 2017, 163, 237–241 CrossRef.
- X. Liu, F. Kong, F. Guo, T. Cheng, W. Chen, T. Yu, J. Chen, Z. A. Tan and S. Dai, Dyes Pigm., 2017, 139, 129–135 CrossRef CAS.
- T. Leijtens, I. K. Ding, T. Giovenzana, J. T. Bloking, M. D. McGehee and A. Sellinger, ACS Nano, 2012, 6, 1455–1462 CrossRef CAS PubMed.
- Y. Hua, B. Xu, P. Liu, H. Chen, H. Tian, M. Cheng, L. Kloo and L. Sun, Chem. Sci., 2016, 7, 2633–2638 RSC.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- M. L. Petrus, T. Bein, T. J. Dingemans and P. Docampo, J. Mater. Chem. A, 2015, 3, 12159–12162 CAS.
- L. Chen, B. H. Zhang, Y. X. Cheng, Z. Y. Xie, L. X. Wang, X. B. Jing and F. S. Wang, Adv. Funct. Mater., 2010, 20, 3143–3153 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra08052b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |