
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhysical and chemical dual-confinement of polysulfides within hierarchically meso-microporous nitrogen-doped carbon nanocages for advanced Li–S batteries†
Pan Wua,
Ming-Hui Suna,
Yong Yua,
Zhao Penga,
Shimeles T. Bulbulaa,
Yu Lia,
Li-Hua Chen *a and
Bao-Lian Su*abc
*a and
Bao-Lian Su*abc
aLaboratory of Living Materials, The State Key Laboratory of Advanced Technology for Materials Synthesis and Processing, Wuhan University of Technology, 122 Luoshi Road, 430070, Wuhan, China. E-mail: chenlihua@whut.edu.cn; baoliansu@whut.edu.cn
bLaboratory of Inorganic Materials Chemistry (CMI), University of Namur, 61 Rue de Bruxelles, B-5000 Namur, Belgium. E-mail: bao-lian.su@unamur.be
cDepartment of Chemistry and Clare Hall, University of Cambridge, Cambridge, CB2 1EW, UK
First published on 4th September 2017
Abstract
Lithium–sulfur (Li–S) batteries with high theoretical specific energy (1675 mA h g−1), environmental benignity and low cost are considered to be some of the most promising next-generation energy-storage systems compared with conventional lithium-ion batteries. However intrinsic large volume expansion and irreversible loss of activated materials seriously limit their commercial applications. To solve the problems, nitrogen-doped hierarchical carbon nanocages were synthesized and used as efficient sulfur hosts. The synthesized nitrogen doped hierarchically meso-microporous carbon (N-MMC) had large BET surface area and mesopore volumes, contributing to high sulfur loading and a reduced volume-change effect of sulfur during lithiation. Moreover, the strong physical adsorption in the micropores together with improved chemical adsorption caused by nitrogen doping lead to effective polysulfide trapping. The N-MMC/S exhibited high initial capacities of 1202.2 mA h g−1 at 0.2C and 1024 mA h g−1 at 0.5C. The capacity kept at as high as 780 mA h g−1 after 60 cycles at 0.2C and 623.8 mA h g−1 after 70 cycles at 0.5C.
Introduction
Lithium–sulfur (Li–S) batteries are considered as the most promising electrochemical energy storage technology due to their remarkably high theoretical specific capacity (1675 mA h g−1),1,2 abundant natural content, low cost and environmental benignity.3,4 Numerous research efforts have been devoted to the development of novel Li–S batteries.5–7 However, the undissolved final discharge product (Li2S2 and Li2S), highly soluble, in organic electrolyte, intermediate polysulfides (S4–62−) and serious volumetric expansion of sulfur on lithiation (up to 80%) cause low utilization of active materials, severe capacity fading and irreversible structural damage.8,9Recently, many efforts have been devoted to solving these problems, such as preparation of sulfur composites with conductive polymers,10,11 carbon nano tubes (CNT)5,12 or graphene.13,14 Among them, one effective and widely used approach is to compose sulfur with porous carbon matrices by a melting-diffusion strategy.15,16 The conductive carbon host can intimately contact with sulfur molecules, so the electronic conductivity of sulfur composites is dramatically enhanced. In addition, the diffusion of soluble intermediate polysulfides in the organic electrolyte can also be greatly suppressed due to the remarkable confinement of nanostructured porous carbon. Nazar and coworkers firstly used ordered mesoporous carbon (CMK-3) as matrix to host sulfur and restrain the loss of polysulfides.17 Other carbon materials, such as hollow carbon,18,19 mesoporous,20,21 microporous16,22 carbon and composites of the above materials,23,24 have also been used as sulfur supporting matrices.
In general, in order to obtain ideal electrochemical performance, the porous carbon host should meet the following demands: (1) high specific surface area and large pore volume to accommodate high rate sulfur loading; (2) high electrical conductivity to ensure an efficient conductive network in the sulfur cathode; (3) abundant micropores to suppress diffusion of the dissolved polysulfide; (4) enough space to alleviate volume expansion of sulfur electrode during charge and discharge reaction.25–28 But carbon materials that completely meet all the mentioned requirements have not been reported yet. There are always incompatibility between sulfur utilization rate and loading. It is likely that carbon host with single pore structure fails to bring both high sulfur loading and good electrochemical performance.29
Mesoporous carbon has high surface area and large volume, which can ensure high sulfur loading. However, large-size pores fail to prevent polysulfides diffusing out of the host pores since the organic electrolyte inevitably contacts the confined sulfur, which further caused inefficiently suppression of the diffusion of soluble polysulfides. Microporous carbon materials, strong physical adsorption to confine polysulfides, have also been used as sulfur supporting matrices. Guo et al. has already confirmed that sulfur exists as smaller chainlike S2–4 molecules in small-size micropores (<0.5 nm) because of the space limitation.30 Thus, the highly soluble polysulfides (S4–62−) would not be formed during discharging–charging process. The sulfur composite cathode with microporous carbon host exhibits an outstanding cycling stability. Nevertheless, small-size pores lead to slow kinetics and the low pore volume also restrict the sulfur loading in the electrode, thus caused relatively low energy capacity. So, just using mesoporous carbon or microporous carbon as sulfur host cannot live up to our expectation. But if combining these two kinds of carbon together to fabricate hierarchically meso-microporous carbon can inherit their individual intrinsic advantages. Additionally, recent studies have also shown that N-doping can enhance the carbon electrical conductivity, restrain the diffusion of soluble polysulfides and alleviate the “shuttle effect” of porous carbon.31,32
Herein, we synthesized nitrogen-doped hierarchically meso-microporous carbon nanocage and used it as sulfur host. The obtained sample possesses large specific surface area, high pore volume, appropriate nitrogen-doping and inner-connected nanometer pores. It provides enough space for high sulfur loading and volume expansion, facilitates electron and lithium ions transportation and confines the loss of soluble polysulfides, thus leading to high capacity and improvements in cycling stability.
Experimental section
Preparation of nitrogen-doped hierarchically meso-microporous carbon (N-MMC), mesoporous carbon (N-MesoC) and microporous carbon (N-MicroC)
All the chemicals were purchased from Aldrich Chemical Corporation and used without further purification. N-MMC was synthesized by co-precipitate. Firstly, 1 g of dopamine (PDA) and 20 mg of sodium dodecyl sulfate (SDS) were dispersed in 60 mL deionized water as the carbon precursors. About 10 min magnetic stirring at room temperature until PDA and SDS were totally dissolved, then 7.5 g of silica colloid (LUDOX AS-40%, 10 nm Sigma-Aldrich) and 2 g dilute sulphuric acid (concentration: 10%) were dropwise added to the solution. To guarantee the good dispersion of silica particles, another 30 min continuous drastic stirring and 1 h sonication was required after the addition of colloidal silica. In the subsequent process, semitransparent mixture solution was transferred into air drying oven at 60 °C for about 2 days until water evaporated completely to get light brown solid powder. Then place the solid powder at the center of furnace tube. The furnace temperature was increased from room temperature to 900 °C with a heating rate of 5 °C min−1 in the atmosphere of argon, then hold at 900 °C for 4 h. After pyrolysis, the furnace was cooled down to room temperature with argon flushing the sample. To attain the final product N-MMC, dilute hydrofluoric acid (20%) was applied to remove the SiO2. The resulting products after removing SiO2 were washed thoroughly with KOH followed by hot distilled water to remove hydrofluoric acid until the pH value of the washed solution ranged from 6.5 to 7.0. The detailed synthesis processes of N-MMC are made up of four key parts: (a) the dissolution of PDA; (b) dispersion of colloidal silica; (c) the temperature and a heating rate of carbonization; (d) SiO2 template etching. After dissolution of PDA, colloidal silica should be added dropwise into the PDA solution under drastic stirring.For comparison, N-MesoC was prepared by the same method, but without SDS. N-MicroC was prepared by abandoning silica colloid and other prescriptions and procedures followed preparation of N-MMC.
Preparation of N-MMC/S, N-MesoC/S, N-MicroC/S
The N-MMC/S, N-MesoC/S and N-MicroC/S were prepared by a melting-infusion process. N-MMC and sulfur were thoroughly mixed in agate mortar according to a mass ratio of mS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :mC = 3:1 to grind into a black mixture. The mixture was then sealed in a glass container and heated at 155 °C with 24 hours to ensure a complete migration of sulfur into N-MMC to obtain N-MMC/S. Then the composite was heated at 200 °C for 2 h under argon atmosphere to remove the excess sulfur and external sulfur of the porous carbon. The N-MesoC/S was also manufactured by same steps. The N-MicroC/S were prepared by compounding N-MicroC and sulfur according to a mass ratio of mS:mC = 1:5. Other procedures were the same as that of N-MMC/S.
:mC = 3:1 to grind into a black mixture. The mixture was then sealed in a glass container and heated at 155 °C with 24 hours to ensure a complete migration of sulfur into N-MMC to obtain N-MMC/S. Then the composite was heated at 200 °C for 2 h under argon atmosphere to remove the excess sulfur and external sulfur of the porous carbon. The N-MesoC/S was also manufactured by same steps. The N-MicroC/S were prepared by compounding N-MicroC and sulfur according to a mass ratio of mS:mC = 1:5. Other procedures were the same as that of N-MMC/S.
Characterizations
The surface morphology of as-prepared products were characterized with transmission electron microscopy (TEM; JEOL, JEM-2100F) operated at 200 kV and scanning electron microscopy (FESEM; JEOL, JSM-7600F) equipped with an energy dispersive X-ray spectrometer (EDX). XRD patterns were based on X-ray powder diffraction (XRD; Shimadzu XRD-6000, Cu Kα radiation) at a scan rate of 0.5° min−1. N2 adsorption/desorption isotherms, BET surface area, the pore size distribution and pore volume were acquired at −196 °C (77 K) with an ASAP-2020 surface area analyzer. The BET surface area was calculated from the adsorption branch using Brunauer–Emmet–Teller (BET) measurement and the pore size distribution was obtained based on desorption branch with Barrett–Joyner–Halenda (BJH) method and t-plot measurement. Sulfur contents of the carbon–sulfur composites were confirmed by thermo-gravimetric analysis (TGA, Q500), which was carried out from the room temperature to 800 °C at a heating rate of 5 °C min−1 under flowing argon atmosphere. X-ray photoelectron spectroscopy (XPS) measurements were performed on VG Multilab 2000.Electrochemical measurements
The electrochemical performances of the carbon–sulfur composites were evaluated by assembling into coin cells with Al foil metal as the current collector, Celgard 2400 as a separator and Li metal as anode. In order to make the cathode, the active material that we made, acetylene black and polyvinylidene fluoride (PVDF) binder based on a weight ratio of 75:15:10 were mixed in N-methylpyrrolidone (NMP) to form homogeneous slurry. The slurry was uniformly spread onto pure aluminium foil using a doctor blade cast and dried at 60 °C overnight. The electrodes then were dried at 60 °C for 12 h under vacuum to remove the NMP completely. Subsequently, the electrodes were cut into disks with a diameter of 12 mm. The CR2025 coin-type cells were fabricated using the working electrodes and lithium metal as the counter electrode. The electrolyte was 1.0 M lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) in 1,3-dioxolane and 1,2-dimethoxyethane (volume ratio, 1:1) with 1% LiNO3 as an additive. The coin cells were assembled in an argon atmosphere glove box. The galvanostatic charge/discharge tests were carried out on a LAND CT2001A battery tester in a potential range of 1.40 V and 2.80 V (versus Li/Li+). The cyclic voltammetry (CV) (scan rate: 0.20 mV s−1, cut-off voltage: 1.40–2.80 V) was studied using a CHI 660D electrochemical workstation. Electrochemical impedance spectra (EIS) were measured with an electrochemical workstation (Autolab PGSTAT 302N) in the frequency range from 200 KHz to 100 mHz.
Results and discussion
The morphology and structure of N-MMC are examined by SEM and TEM. Fig. S1a and b† displayed the SEM images of N-MMC at different resolution. Numerous ordered pores could be observed on the surface of N-MMC. TEM images at different resolution of N-MMC are displayed in Fig. 1a–d. Large amounts of uniform mesopores existed in the carbon matrix. Moreover, there were plenty of worm-like micropore tunnels with broken fringes (Fig. 1d), demonstrating the unsealed feature of the shells. The integration of the well-defined meso-micropore size distributions endowed the N-MMC a large specific surface area of 1187 m2 g−1 and a high total volume of 3.98 cm3 g−1 (Table 1). In addition, the network geometry of the inter-connected meso-micropores (red arrows) provided short transport pathway for both electrons and Li ions. The diameter of mesopores was about 8 nm, which was consistent with mesopore sized distribution obtained by N2 sorption method (Fig. 2b). TEM images of N-MesoC and N-MicroC were shown in Fig. S1a and b.† It was observed that only mesopores/micropores existed in N-MesoC/N-MicroC. | ||
| Fig. 1 TEM images of N-MMC. | ||
| Sample | SBETa (m2 g−1) | St-plotb (m2 g−1) | VBJHc (cm3 g−1) | Vt-plotd (cm3 g−1) | Vte (cm3 g−1) |
|---|---|---|---|---|---|
| a BET specific surface area.b t-Plot micropore surface area (<2 nm).c BJH mesopore volume (2–50 nm).d t-Plot micropore volume (<2 nm).e Total pore volume (P/P0 = 0.995). | |||||
| N-MMC | 1187 | 235 | 3.85 | 0.13 | 3.98 |
| N-MesoC | 759 | — | 2.97 | — | 2.97 |
| N-MicroC | 266 | 266 | — | 0.145 | 0.145 |
 | ||
| Fig. 2 (a) N2 adsorption–desorption isotherm plot of N-MMC, the inset is the corresponding micropore size distribution of N-MMC. (b) Mesopore size distribution of N-MMC. (c) XRD patterns of N-MMC, N-MMC/S and pure sulfur. (d) TG curves of N-MMC/S. | ||
N2 adsorption–desorption isotherm of N-MMC exhibited type I and IV isotherms with H1-type hysteresis loops at 0.6 < P/P0 < 0.9 (Fig. 2a).4 The micropores were about 0.45 nm (Fig. 2a inset) and mesopores were 7.4 nm (Fig. 2b), which were consistent with the TEM results (Fig. 1d). The N-MesoC and N-MicroC material were synthesized for comparison. The same precursor and carbonization temperature were applied to ensure that they had same degree of graphitization. Hysteresis loop nitrogen adsorption–desorption isotherms (Fig. S3a†) and the BJH pore size distribution (Fig. S3b†) of N-MesoC were almost the same as that of N-MMC. The t-plot pore size distribution of N-MicroC (Fig. S3c†) was also the similar to that of N-MMC. The more details about pore parameters of N-MMC, N-MesoC, N-MicroC were shown in Table 1.
The XRD patterns are shown in Fig. 2c. The pure sulfur showed strongly sharp diffraction peaks. However, after mixing with the N-MMC materials, no sulfur diffraction peaks could be observed in N-MMC/S, indicating that most of sulfur particles penetrated into nanopores of the carbon substrate.33,34 The actual content of sulfur in carbon materials was determined by thermogravimetric analysis (TGA). The sulfur loadings of N-MMC/S (Fig. 2d), N-MesoC/S (Fig. S4c†) and N-MicroC/S (Fig. S4d†) were as high as 72.7 wt%, 67.5 wt% and 22.9 wt%, respectively. It can be concluded that N-MicroC/S had a higher sulfur vaporizing temperature than N-MesoC/S due to the much more stable interaction between sulfur and carbon matrix in micropores than that in mesopores. It was noteworthy that sulfur content in N-MMC/S was almost five percent more than that in the N-MesoC/S, confirming that micropores surrounded around mesopores to form unsealed feature of the shells. Unlike ordered core–shell carbon material, sulfur must penetrate into the inner mesopores through the outer microporous shell, which inevitably led to sulfur loss and less practical content of sulfur in N-MMC/S than that in N-MesoC/S.
X-ray photoelectronic spectroscopy (XPS) is also used to investigate the influence of N-doping on electrochemical performance. The overall XPS spectrum of N-MMC, N-MMC/S, N-MesoC/S and N-MicroC/S are shown in Fig. 3a. Three distinct peaks of O 1s, N 1s and C 1s were obviously observed. The C 1s and N 1s peaks were attributed to the PDA carbonization. The existence of O 1s derived from the physicochemical adsorption of oxygen during the preparation process. After successfully permeating sulfur into porous structure of N-MMC, two other peaks appearing at 164.0 eV and 228.2 eV for N-MMC/S were attributed to S 2p and S 2s. The spectrum of C 1s (Fig. 3b) of N-MMC was divided into four individual peaks corresponding to C–C (286.4 eV),35 C–N (285.4 eV),31 C–O (286.5 eV),35 C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (288.7 eV).36 The high resolution N 1s peaks (Fig. 3c) could be divided into three components including pyridinic-N (398.2 eV),37 pyrrolic-N (399.7 eV)13 and graphitic-N (401.3 eV),37 respectively. These results further confirmed that N atoms were successfully doped in carbon framework and chemically combined with C atoms. Introducing nitrogen into the carbon could create more active sites than the non-doped carbons and the synergistic effects of dual activation of carbon atoms induced stronger chemical adsorption ability. In addition, pyridinic N and pyrrolic N were believed to be more effective in forming SxLi–N interactions via the N lone-pair electrons.38 This strong interaction may mitigate the dissolution of polysulfides into electrolyte and improved their redeposition process upon cycling. The resulting charge delocalization in N-MMC/S provided high conductivity and electrochemical performance.39 In order to identify the chemical state of sulfur molecules and distinguish the sulfur types in Li–S batteries, S 2p spectra of as-prepared materials was investigated. All three S 2p spectra of N-MMC/S (Fig. 3d) contained the characteristic sulfur splitting of the S 2p signal into two components S 2p3/2 and S 2p1/2 as a result of spin–orbital coupling effect at 164.0 eV and 165.0 eV, which were in good agreement with S8 molecules.5,40 Another peak at 168.7 eV indicated the existence of smaller S2–4 molecules in the microporous carbon and further proved the strengthened interaction between sulfur molecules and porous carbon hosts.24 As for N-MesoC/S (Fig. S5a†), no peak focused on 168.7 eV, but two peaks at 164.0 eV and 165.0 eV testified existence of S8. A strong peak at 168.7 eV demonstrated that smaller short-chain S2–4 molecules existed in N-MicroC/S (Fig. S5b†). N-MMC/S was also identified by energy-dispersion X-ray spectroscopy (EDX) (Fig. S6†). The linear EDX elemental maps obviously confirmed the existence of S, C, N, and O, which was consistent with data obtained by XPS in Fig. 3a. The EDX elemental distribution of S revealed that sulfur partials had been finely dispersed in carbon matrix of N-MMC.
O (288.7 eV).36 The high resolution N 1s peaks (Fig. 3c) could be divided into three components including pyridinic-N (398.2 eV),37 pyrrolic-N (399.7 eV)13 and graphitic-N (401.3 eV),37 respectively. These results further confirmed that N atoms were successfully doped in carbon framework and chemically combined with C atoms. Introducing nitrogen into the carbon could create more active sites than the non-doped carbons and the synergistic effects of dual activation of carbon atoms induced stronger chemical adsorption ability. In addition, pyridinic N and pyrrolic N were believed to be more effective in forming SxLi–N interactions via the N lone-pair electrons.38 This strong interaction may mitigate the dissolution of polysulfides into electrolyte and improved their redeposition process upon cycling. The resulting charge delocalization in N-MMC/S provided high conductivity and electrochemical performance.39 In order to identify the chemical state of sulfur molecules and distinguish the sulfur types in Li–S batteries, S 2p spectra of as-prepared materials was investigated. All three S 2p spectra of N-MMC/S (Fig. 3d) contained the characteristic sulfur splitting of the S 2p signal into two components S 2p3/2 and S 2p1/2 as a result of spin–orbital coupling effect at 164.0 eV and 165.0 eV, which were in good agreement with S8 molecules.5,40 Another peak at 168.7 eV indicated the existence of smaller S2–4 molecules in the microporous carbon and further proved the strengthened interaction between sulfur molecules and porous carbon hosts.24 As for N-MesoC/S (Fig. S5a†), no peak focused on 168.7 eV, but two peaks at 164.0 eV and 165.0 eV testified existence of S8. A strong peak at 168.7 eV demonstrated that smaller short-chain S2–4 molecules existed in N-MicroC/S (Fig. S5b†). N-MMC/S was also identified by energy-dispersion X-ray spectroscopy (EDX) (Fig. S6†). The linear EDX elemental maps obviously confirmed the existence of S, C, N, and O, which was consistent with data obtained by XPS in Fig. 3a. The EDX elemental distribution of S revealed that sulfur partials had been finely dispersed in carbon matrix of N-MMC.
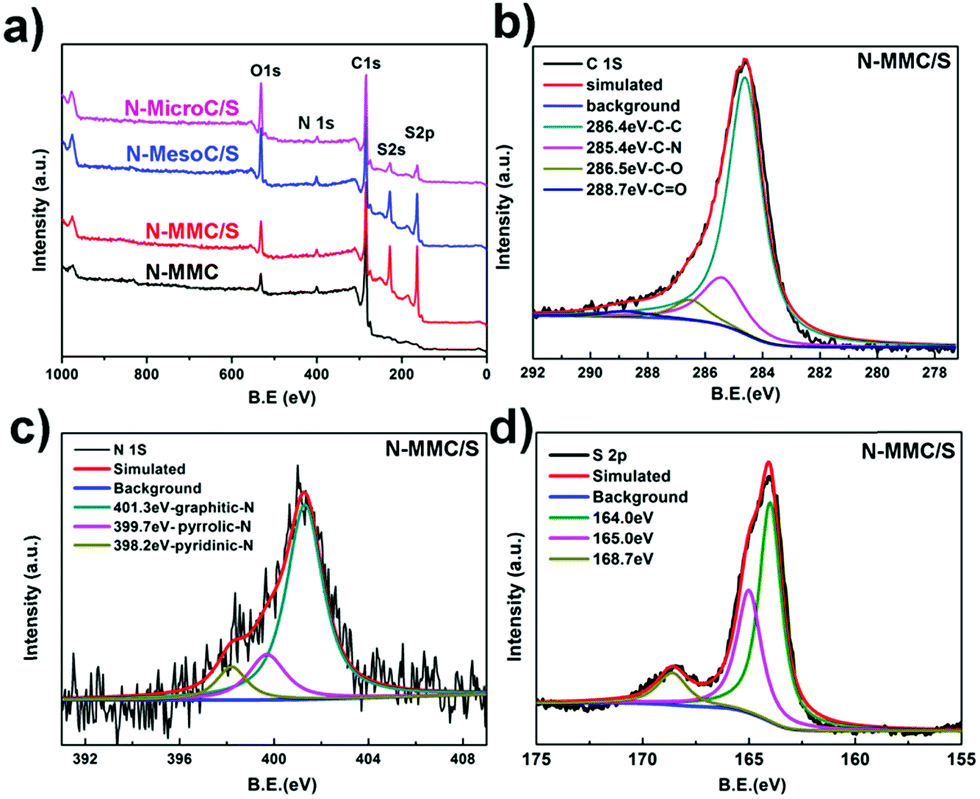 | ||
| Fig. 3 XPS survey of (a) as prepared carbon–sulfur and N-MMC, (b) C 1s spectra of N-MMC/S, (c) N 1s spectra of N-MMC/S, (d) S 2p spectra of N-MMC/S. | ||
The N-MMC/S, N-MesoC/S, N-MicroC/S and pristine S were evaluated as cathode materials for Li–S batteries by cyclic voltammograms (CV) and galvanostatic charging–discharging process in coin half-cell using lithium metal as an anode. Considering that the activation phenomenon always appears in the first cycle, we selected first three cycles CV curves that were obtained at a scanning rate of 0.20 mV S−1. In cathodic scanning, the N-MMC/S (Fig. 4a) presented two reduction peaks, which was assigned to the multistep reduction mechanism of element sulfur.41 The first peak in the range of 2.29–2.31 V related to the reduction of S8 to long-chain soluble lithium polysulfides (Li2Sx, 4≤ x ≤ 8).42 The second peak in the range of 2.01–2.10 V attributed to the further reduction of soluble polysulfide anions to short-chain polysulfides (Li2Sx, 2≤ x ≤ 4) and also an insoluble Li2S2/Li2S deposit.4 In the subsequent oxidation scan, an asymmetric broad peak around 2.60 V was owing to the conversion of Li2S2/Li2S into the sulfur. It was clear from the Fig. 4a that all the successive cycles of CV overlapped, proving the excellent durability of the hybrid sphere. As for N-MesoC/S (Fig. S7a†), two reduction peaks are around 2.20 V, 1.90 V and one oxidative peak is around 2.60 V.21 However, when it comes to N-MicroC/S (Fig. S7b†), it was interesting to notice that only one reduction peaks is around 1.50 V, which relates to the strong reduction of soluble intermediate sulfides to short-chain polysulfides and an insoluble Li2S2/Li2S deposit.43 Fig. 4b showed the charge/discharge curves of N-MMC/S, two typical plateaus around 2.30 V and 2.10 V correspond to a two-step reaction of sulfur with lithium during the discharge process, being in well agreement with the CVs. The charge/discharge curves of N-MesoC/S (Fig. S7c†) and N-MicroC/S (Fig. S7d†) showed typical plateaus, which are ultimately in line with their own galvanostatic charge–discharge curves.
 | ||
| Fig. 4 (a) CV curves of N-MMC/S electrode at a sweep rate of 0.2 mV s−1 in the potential range from 1.40 V to 2.80 V vs. Li/Li+. (b) The discharge/charge profiles of N-MMC/S electrode cycled at 0.2C. (c) The cycling performances at 0.2C. | ||
Fig. 4c showed different cycle behaviors of as-prepared materials and pure sulfur at 0.2C. It obviously proved the structural advantage of the N-MMC for sulfur capture. The N-MMC/S exhibited initial specific capacity of 1202 mA h g−1 and retained 780 mA h g−1 after 60 cycles with 64.9% capacity retention. The capacity of N-MesoC/S decreased to 510.3 mA h g−1 after 60 cycles, which was only 51% of that in the initial cycle, indicative of a high dissolution of polysulfides. The cycle stability of N-MicroC/S was almost as good as N-MMC/S, but it maintained lower initial specific capacity because of relatively low sulfur loading. Fig. 5a and b shows high current density of 0.5C and 1C. N-MMC/S electrode exhibited less pronounced capacity fading than the N-MesoC/S electrode. The improved cycle stability of the N-MMC/S electrode could be ascribed to the hierarchical porous carbon mixture. At high current, the process of redox moved faster accompanying with the high-speed loss of dissoluble polysulfides in mesopores. This part of active materials could make great contribution to capacity of N-MesoC/S, which gave rise to the better capacity than that of N-MMC/S at early 38 cycles. Then the capacity of N-MMC/S tended to be more stable for the reason that micropores acted as physical barrier to remarkably alleviate the fast loss of active materials. Both the N-MMC/S and N-MesoC/S electrodes outperformed the N-MicroC/S electrode in terms of capacity. This emphasized the effect of sulfur loading on increasing the capacity. All three samples show high coulombic efficiencies nearly to 100%.
 | ||
| Fig. 5 The cycling performances of N-MMC/S, N-MesoC/S, N-MicroC/S and pristine S electrodes at different current densities of (a) 0.5C and (b) 1C. | ||
Electrochemical impendence spectroscopy (EIS) was a common electrochemical method to reveal the inherent relationship between performance and materials. Fig. S8a† displays the Nyquist plots of the N-MMC/S before and after cycling. The impedance spectra of composites were consist of a semicircle in the high frequency region corresponding to the contact resistance and charge transfer resistance and an inclined line in the low frequency region due to the ion diffusion within the electrode. Before cycling, the N-MMC/S cathode exhibited resistance about 155 Ω. However a lower charge resistance with 35 Ω after cycle resistance, which could be attributed to permeation of electrolyte into porous carbon matrix and the reaction of polysulfide species with the lithium electrode, creating soluble polysulfides on the metal surface. Fig. S8b and c† shows EIS of the N-MesoC/S and N-MicroC/S before and after cycling. The same phenomenon was also observed in N-MMC/S electrode.
Conclusions
In summary, a nitrogen doping hierarchically meso-microporous carbon material was designed as a promising sulfur container for high-performance Li–S batteries. It inherits the advantages of both mesoporous and microporous carbon. The large mesopore volume in N-MMC/S assures high sulfur loading and the highly ordered conductive channels facilitate complete redox reaction of the active material. The micropores exhibited a strong adsorption of sulfur and thus efficiently confined the loss of polysulfides. These inner-connected hierarchical pores facilitate electron and lithium ions. Moreover, the nitrogen doping effectively promote chemical adsorption between sulfur atoms and oxygen functional groups on the carbon. All of these finally make a great contribution to an efficient way to realize excellent performance of Li–S batteries for practical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was carried out in the framework of a program for Changjiang Scholars and Innovative Research Team (IRT_15R52) of the Chinese Ministry of Education. B. L. SU acknowledges the Chinese Central Government for an “Expert of the State” position in the Program of the “Thousand Talents”, the Chinese Ministry of Education for a “Changjiang Chaire Professor” position and a Clare Hall Life Membership at the Clare Hall College and the financial support of the Department of Chemistry, University of Cambridge. L. H. CHEN acknowledges Hubei Provincial Department of Education for the “Chutian Scholar” program. This work was also financially supported by NFSC-21671155, SRF for ROCS SEM ([2015]311), Hubei Provincial Natural Science Foundation (2015CFB428).Notes and references
- Z. Lin and C. Liang, J. Mater. Chem. A, 2015, 3, 936–958 CAS.
- M. Liu, F. Ye, W. Li, H. Li and Y. Zhang, Nano Res., 2016, 9, 94–116 CrossRef CAS.
- S. Rehman, S. J. Guo and Y. L. Hou, Adv. Mater., 2016, 28, 3167–3172 CrossRef CAS PubMed.
- Z. Lyu, D. Xu, L. Yang, R. Che, R. Feng, J. Zhao, Y. Li, Q. Wu, X. Wang and Z. Hu, Nano Energy, 2015, 12, 657–665 CrossRef CAS.
- K. Mi, Y. Jiang, J. Feng, Y. Qian and S. Xiong, Adv. Funct. Mater., 2016, 26, 1571–1579 CrossRef CAS.
- L. Xiao, Y. Cao, J. Xiao, B. Schwenzer, M. H. Engelhard, L. V. Saraf, Z. Nie, G. J. Exarhos and J. Liu, Adv. Mater., 2012, 24, 1176–1181 CrossRef CAS PubMed.
- H. Q. Wang, S. Li, D. Li, Z. X. Chen, H. K. Liu and Z. P. Guo, Energy, 2014, 75, 597–602 CrossRef CAS.
- C. Wu, L. Yuan, Z. Li, Z. Yi, Y. Li, R. Zeng, W. Zhang and Y. Huang, RSC Adv., 2015, 5, 14196–14201 RSC.
- J.-G. Wang, K. Xie and B. Wei, Nano Energy, 2015, 15, 413–444 CrossRef CAS.
- F. Wu, J. Chen, R. Chen, S. Wu, L. Li, S. Chen and T. Zhao, J. Phys. Chem. C, 2011, 115, 6057–6063 CAS.
- Y. Fu and A. Manthiram, J. Phys. Chem. C, 2012, 116, 8910–8915 CAS.
- C. Wang, H. Chen, W. Dong, J. Ge, W. Lu, X. Wu, L. Guo and L. Chen, Chem. Commun., 2014, 50, 1202–1204 RSC.
- G. M. Zhou, E. Paek, G. S. Hwang and A. Manthiram, Nat. Commun., 2015, 6, 7760 CrossRef CAS PubMed.
- J.-L. Shi, H.-F. Wang, X. Zhu, C.-M. Chen, X. Huang, X.-D. Zhang, B.-Q. Li, C. Tang and Q. Zhang, Carbon, 2016, 103, 36–44 CrossRef CAS.
- D. Bresser, S. Passerini and B. Scrosati, Chem. Commun., 2013, 49, 10545–10562 RSC.
- Y. Xu, Y. Wen, Y. Zhu, K. Gaskell, K. A. Cychosz, B. Eichhorn, K. Xu and C. Wang, Adv. Funct. Mater., 2015, 25, 4312–4320 CrossRef CAS.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500–506 CrossRef CAS PubMed.
- N. Jayaprakash, J. Shen, S. S. Moganty, A. Corona and L. A. Archer, Angew. Chem., 2011, 50, 5904–5908 CrossRef CAS PubMed.
- Y. Wu, M. Gao, X. Li, Y. Liu and H. Pan, J. Alloys Compd., 2014, 608, 220–228 CrossRef CAS.
- J. Wang, Y. Wu, Z. Shi and C. Wu, Electrochim. Acta, 2014, 144, 307–314 CrossRef CAS.
- D. Wang, A. Fu, H. Li, Y. Wang, P. Guo, J. Liu and X. S. Zhao, J. Power Sources, 2015, 285, 469–477 CrossRef CAS.
- Z. Li, X. Li, Y. Liao, X. Li and W. Li, J. Power Sources, 2016, 334, 23–30 CrossRef CAS.
- S. Ungureanu, M. Birot, H. Deleuze, V. Schmitt, N. Mano and R. Backov, Carbon, 2015, 91, 311–320 CrossRef CAS.
- J. Shan, Y. Liu, Y. Su, P. Liu, X. Zhuang, D. Wu, F. Zhang and X. Feng, J. Mater. Chem. A, 2016, 4, 314–320 CAS.
- A. Manthiram, Y. Fu, S. H. Chung, C. Zu and Y. S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- Z. Li, Y. Huang, L. Yuan, Z. Hao and Y. Huang, Carbon, 2015, 92, 41–63 CrossRef CAS.
- R. Chen, T. Zhao and F. Wu, Chem. Commun., 2015, 51, 18–33 RSC.
- L. Ma, K. E. Hendrickson, S. Wei and L. A. Archer, Nano Today, 2015, 10, 315–338 CrossRef CAS.
- L. Qie, W. Chen, H. Xu, X. Xiong, Y. Jiang, F. Zou, X. Hu, Y. Xin, Z. Zhang and Y. Huang, Energy Environ. Sci., 2013, 6, 2497 Search PubMed.
- S. Xin, L. Gu, N. H. Zhao, Y. X. Yin, L. J. Zhou, Y. G. Guo and L. J. Wan, J. Am. Chem. Soc., 2012, 134, 18510–18513 CrossRef CAS PubMed.
- Z. Wang, L. Qie, L. Yuan, W. Zhang, X. Hu and Y. Huang, Carbon, 2013, 55, 328–334 CrossRef CAS.
- J. Qu, S. Lv, X. Peng, S. Tian, J. Wang and F. Gao, J. Alloys Compd., 2016, 671, 17–23 CrossRef CAS.
- Y. Qu, Z. Zhang, X. Zhang, G. Ren, Y. Lai, Y. Liu and J. Li, Carbon, 2015, 84, 399–408 CrossRef CAS.
- X. Gu, C. Lai, F. Liu, W. Yang, Y. Hou and S. Zhang, J. Mater. Chem. A, 2015, 3, 9502–9509 CAS.
- C. Reitz, B. Breitung, A. Schneider, D. Wang, M. von der Lehr, T. Leichtweiss, J. Janek, H. Hahn and T. Brezesinski, ACS Appl. Mater. Interfaces, 2016, 8, 10274–10282 CAS.
- M. Xiang, Y. Wang, J. Wu, Y. Guo, H. Wu, Y. Zhang and H. Liu, Electrochim. Acta, 2017, 227, 7–16 CrossRef CAS.
- H. Chen, Y. Wei, J. Wang, W. Qiao, L. Ling and D. Long, ACS Appl. Mater. Interfaces, 2015, 7, 21188–21197 CAS.
- F. Chen, J. Yang, T. Bai, B. Long and X. Zhou, Electrochim. Acta, 2016, 192, 99–109 CrossRef CAS.
- Z. Geng, Q. Xiao, D. Wang, G. Yi, Z. Xu, B. Li and C. Zhang, Electrochim. Acta, 2016, 202, 131–139 CrossRef CAS.
- Q. Li, Z. Zhang, Z. Guo, K. Zhang, Y. Lai and J. Li, J. Power Sources, 2015, 274, 338–344 CrossRef CAS.
- J. G. Werner, S. S. Johnson, V. Vijay and U. Wiesner, Chem. Mater., 2015, 27, 3349–3357 CrossRef CAS.
- Y. Zhang, Y. Zhao, A. Konarov, Z. Li and P. Chen, J. Alloys Compd., 2015, 619, 298–302 CrossRef CAS.
- M. Helen, M. A. Reddy, T. Diemant, U. Golla-Schindler, R. J. Behm, U. Kaiser and M. Fichtner, Sci. Rep., 2015, 5, 12146 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The HRTEM images of the samples, N2 adsorption and desorption isotherms and the pore size distributions, XRD patterns, XPS survey, EDX patterns and electronic performances. See DOI: 10.1039/c7ra07918d |
| This journal is © The Royal Society of Chemistry 2017 |