DOI:
10.1039/C7RA07886B
(Paper)
RSC Adv., 2017,
7, 44456-44462
The stability, characteristics and magnetic properties of iron carbide from an oolitic hematite†
Received
18th July 2017
, Accepted 4th September 2017
First published on 15th September 2017
Abstract
Iron carbide (Fe3C) is a magnetic material but it is not stable when it is prepared. Iron carbide was first prepared from high phosphorus oolitic hematite pellets using hydrogen reduction and was subsequently carburized with methane (CH4). The products were then cooled down to room temperature using three different cooling conditions: fast cooling, furnace cooling, and heat preservation for 2 h with subsequent fast cooling. The results showed that the optimal reaction conditions for the preparation of Fe3C from hydrogen reduced high phosphorus oolitic hematite are carburizing with CH4 for 15 min at 1023 K, then fast cooling with argon gas, thus a degree of carburization of 95.12% can be obtained. The heat preservation promotes the self-decomposition of Fe3C and the occurrence of multi-walled nano carbon fibers. The carbon nanotubes (CNTs) are in the form of rings, chains or nets, which can be attributed to the collisions between the CNTs. The nonuniform nano iron grains restrict the growth of CNTs deposited on the surface of metallic iron, and cause the bending of each CNT, which generates the chain or the net shape. Under fast cooling, the saturation magnetization declines from 82.59 to 61.97 emu g−1 as the time of carburization with CH4 increases from 10 to 30 min. The addition of the heat preservation and the control of the time of carburization with CH4 can give the desired saturation magnetization. The Fe3C prepared from high phosphorus oolitic hematite has relatively high magnetic properties.
Introduction
The high phosphorus oolitic hematite reserves (3–4 billion tons) in China cannot be utilized because of the difficulty of impurity removal.1 Recently, several technologies for the dephosphorization have been studied for the utilization of high phosphorus hematite, which were mainly: mineral processing,2,3 deep reduction4–8 and smelting technology.9 Generally, mineral processing has problems of low iron recovery and dephosphorization ratio. The deep reduction leads to the reduction of apatite to phosphorus, which will dissolve into the iron phase, therefore, the decline of phosphorus content in the iron phase needs to be further investigated. The immature technology of smelting for dephosphorization and the energy consumption mean that the technology is unable to be widely applied.
Iron carbide (Fe3C), a type of raw material used for steelmaking and also a functional material, can be widely used in many industrial fields.10–14 Nanosized Fe3C has been mainly considered as a side product during the synthesis of carbon (C) structures, where the metallic iron is used as a catalyst, for example, in chemical vapor deposition,15 ultrasonic spray pyrolysis,16 laser ablation in organic solvent17 and pyrolysis processes18 during the synthesis of carbon nanotubes (CNTs). Papers in the literature have presented countless procedures for the production of a plethora of nanoparticles and nanostructures (ranging from physical to chemical approaches, in water or solventless, by using a hard template or soft matter), thus, it is surprising that a synthetic pathway to produce Fe3C nanoparticles in a reproducible, simple, and fast manner is still missing. This should contribute to the stability of an interstitial compound of Fe3C, which can also be affected by the experimental conditions. Iron carbide in macro-scale was mainly prepared from high grade of iron ore such as Australian ore, Brazilian ore and other raw iron ores with little gangue, by reacting it with carburization gas, using a carburization process at low temperature (about 773–1123 K).19–21 The Fe3C can be prepared from the iron ores in the atmosphere of hydrogen/methane (H2/CH4) and the previous work by the Authors' research group has confirmed that 1023 K is the optimum temperature for the preparation of Fe3C from high phosphorus oolitic hematite by H2 reduction and subsequent carburization with CH4, and also verified that the phosphorus in apatite was not reduced during the preparation of Fe3C using thermodynamic calculations and related verification experiments.22–24 The results compensate well for the problem where phosphorus reduced from apatite can dissolve into the iron phase by the carbon-based reduction of high phosphorus hematite at relatively high temperature. Until now, the formation of Fe3C mainly consists of three steps: reduction of iron oxide, CH4 decomposition on the surface of the metallic iron, and the carburization of the metallic iron.20,21 Wang et al.24 has noted that the content of free carbon in the product prepared from high phosphorus oolitic hematite is relatively high because of the excessive time of CH4 decomposition and improper cooling treatment of samples after carburizing, which greatly decreased the quality and purity of the carburized specimens. Thus, if the key factor to make full use of the reaction time of CH4 decomposition and the cooling condition is well controlled, it may be promising in popularizing the use and the technology of Fe3C preparation in H2/CH4.
In order to study the stability and the properties of Fe3C from high phosphorus oolitic hematite at 1023 K, the high phosphorus oolitic hematite was first reduced in a H2 atmosphere for the stepwise reduction of hematite to metallic iron, and subsequently with CH4 for various reaction times (carburization of iron to Fe3C), before finally applying three different cooling conditions: (i) fast cooling with a large flow rate of argon (Ar); (ii) with furnace cooling under Ar protection; and, (iii) heat preservation for 2 h with subsequent fast cooling by Ar. X-ray diffraction (XRD) and Mössbauer spectroscopy were used for the qualitative and quantitative analysis of iron and Fe3C content in the products. Transmission electron microscopy (TEM) and vibrating sample magnetometry (VSM) were used for the characterization, and the magnetic property of the products from different cooling conditions were also investigated.
Experimental
Raw materials
A high phosphorus oolitic hematite from western Hubei province, China, was used as the raw material. Firstly, the iron ore was crushed into small pieces and then screened with a 100–120 particle size mesh. The fine ore powders were pelletized with some distilled water after an adequate mixing process in an agate mortar. The raw pellets were subsequently dried for 2 h at 373 K and then sintered for 15 min at 1173 K. The composition of the high phosphorus oolitic hematite is shown in Table 1.
Table 1 The composition of high phosphorus oolitic hematite (100–120 mesh) (mass%)
| Composition |
Fe |
SiO2 |
Al2O3 |
MgO |
CaO |
P |
| Content |
50.23 |
14.6 |
5.08 |
0.62 |
2.34 |
0.77 |
Experimental methods
Two-step process for the preparation of iron carbide. The preparation of Fe3C from high phosphorus oolitic hematite at 105 Pa was carried out using a two-step process, which was H2 reduction and subsequent carburization with CH4. Firstly, the basket with a pellet, which was hung on a thermobalance (METTLER TOLEDO, MS105), was placed in a quartz reaction tube. The specific apparatus has been described in previous papers.22–24 Then, the tube was placed into a vertical resistance furnace at the predetermined temperature of 1023 K, and the Ar gas was introduced to flush air in to the reaction tube for 5 min. Afterwards the gas was switched to H2 for the reduction of the pellet, with a flow rate of 150 cm3 min−1. When the pellet was reduced to a certain degree of 0.9 at 1023 K, the H2 gas was switched to CH4 gas with a flow rate of 300 cm3 min−1. When the carburization process finished, the sample was cooled down to room temperature with different cooling conditions. Then the sample obtained was ground in an agate mortar and vacuum packaged ready for the subsequent analysis. Additionally, these powder samples obtained from different cooling conditions and corresponding reaction times with CH4 were marked with specific numbers, as shown in Tables 2 and 3.
Table 2 The specimen numbers and the reaction times with CH4
| No. |
A |
B |
C |
D |
E |
F |
| Time (min) |
5 |
10 |
15 |
30 |
60 |
120 |
Table 3 The specimen numbers and the cooling condition
| No. condition |
A |
B |
C |
D |
E |
F |
| Fast cooling |
A-1 |
B-1 |
C-1 |
D-1 |
E-1 |
F-1 |
| Cooling in furnace |
A-2 |
B-2 |
C-2 |
D-2 |
— |
— |
| 1023 K holding and fast cooling |
A-3 |
B-3 |
C-3 |
D-3 |
E-3 |
F-3 |
Analytical apparatus. The powder samples obtained were analyzed using XRD (PANalytical X'Pert PRO MPD) with Cu Kα X-ray radiation. The Mössbauer spectra were recorded at room temperature with a constant acceleration and conventional standard transmission spectrometer (WissEl GmbH) with a 57Co(Pd) source. The drive velocity scale was calibrated with a 25 μm thick pure α-Fe foil. All isomer shifts were quoted relative to the center of the α-Fe calibration spectrum. The experimental 57Fe Mössbauer spectra were fitted using a least-squares regression fitting procedure. The TEM images were obtained using a 200 kV TEM (Tecnai G2 F20). Isothermal magnetization measurements at room temperature were performed in a commercial superconducting quantum interference device magnetometer (SQUID-VSM: Quantum Design).
Results and discussion
Methane reaction time and cooling conditions on Fe3C preparation – the weight change during the Fe3C preparation
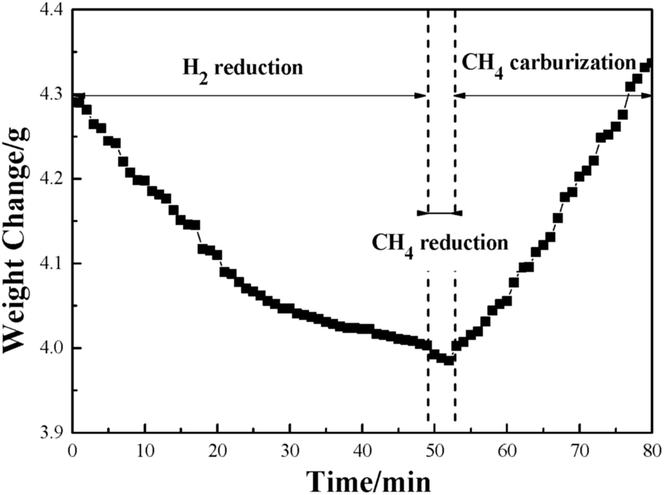
Fig. 1 illustrates the weight change of the pellet sample at 1023 K in the Fe3C preparation process. It shows that the weight of the pellet sample decreased with increase of time during the first 50 min of the H2 reduction. After that, the reduced metallic iron was reacted with CH4. In the first two minutes of reaction with CH4, the weight decreased at a relatively slow rate. Then the weight increased gradually with the CH4 reaction time from 52 to 80 min. It is interesting to find the phenomenon of the continuous decrease of the weight of pellet sample during the first 2 min of the reaction with CH4 gas. The result indicates that the reduction of iron(II) oxide (FeO) by the CH4 gas had occurred. In the previous work of our research group, a similar result was also achieved, and it is proposed that there is a mechanism of Fe3C formation which, with the additional process of the reduction of FeO to Fe by reaction with the CH4 gas, means that the Fe3C will not be generated.24 In order to evaluate the priority of process of FeO reacting with CH4, and the carburization of metallic Fe to Fe3C, the thermodynamics of reactions (1) to (3) has been calculated, and the results are shown in Table 4.25
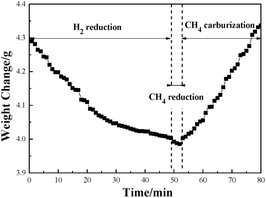 |
| | Fig. 1 The curve of the weight change versus reaction time at 1023 K. | |
Table 4 The possible reactions between FeO and CH4
| No. |
Reaction |
ΔG0 |
T at ΔG0 = 0 (K) |
| 1 |
3FeO + CH4 = 3Fe+2H2O + CO |
274![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 181.6–278.42T 181.6–278.42T |
984.8 |
| 2 |
4FeO + CH4 = 4Fe+2H2O + CO2 |
257494.6–257.86T |
998.6 |
| 3 |
3Fe + CH4 = Fe3C+2H2 |
120084–138.7T |
865.78 |
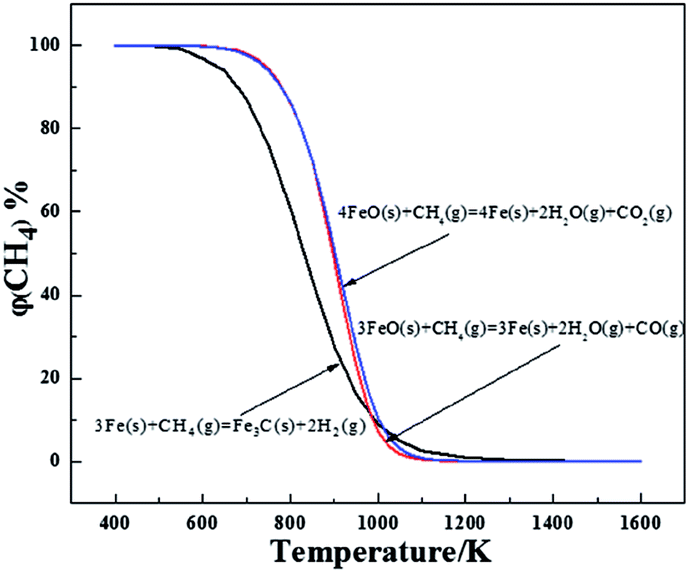
The three possible reactions that may occur are shown in Table 4. The temperatures at a Gibbs Free energy change (ΔG0) at 0 K show that the beginning temperature for reaction (3) is the lowest of 865.8 K and as the temperature is fixed at 1023 K, the Gibbs Free energy is −10212.4, −6296.2 and −21806.1 J mol−1, respectively, for reactions (1)–(3), which reveals that reaction (3) occurs most easily. To further evaluate the thermodynamics of each reaction, the volume percentage of CH4 for reactions (1) to (3) in equilibrium with FeO and Fe or Fe3C were plotted against temperature in Fig. 2. Fig. 2 shows that reaction (3) requires the minimum CH4 volume percentage at temperatures less than 975 K, which further illustrates that the formation of Fe3C by the carburization of metallic Fe with CH4 gas (reaction (3)) will occur after the reduction of the remaining FeO. Compared with reaction (3), reactions (1) and (2) require less volume percentage of CH4 in the temperature range from 975 to 1300 K. The results indicate that reactions (1) and (2) are more susceptible to occurring, which means that the carburization of metallic Fe to Fe3C is begun after the completion of the formation of metallic Fe from FeO. In addition, the required CH4 partial pressure for reaction (1) is lower than that of reaction (2), so the carbon monoxide (CO) gas is easier to generate than the CO2 gas during the deep reduction of FeO with CH4.
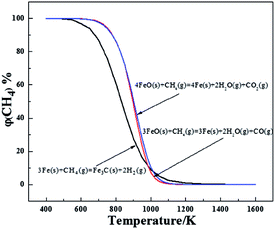 |
| | Fig. 2 The equilibrium relationship between the volume percentage of CH4 and the temperature of reactions (1) to (3). | |
XRD qualitative analysis
Fig. S1 (ESI†) shows the XRD patterns of sample groups A-1 to F-3 (referring to ASTM cards as shown in Table S1†). The relative intensity of each characteristic peak qualitatively reflects the Fe3C content and metallic Fe content after reacting with CH4 for different times and using different cooling conditions. Fig. S1(a) (ESI†) shows the XRD patterns of the specimens after reacting with CH4 for 5 min and using different cooling conditions. Two main phases: iron and quartz were in the three specimens. The analysis of the A- groups did not detect the phase of Fe3C, which means that Fe3C was not formed or not detected accurately in the measurements. In addition, the results also confirm that the deep reduction of reaction (2) exists, which is consistent with the results of previous research.24 After the unreacted FeO was totally converted into metallic Fe, reaction (3): 3Fe (s) + CH4 (g) = Fe3C (s) + 2H2 (g), occurred.
The XRD patterns for samples B-1 to D-3 are shown in Fig. S1(b) to S1(d).† Research by Narkiewicz et al.26 has shown that the 2θ = 37.63° belongs to the (1 2 1) reflection of Fe3C (PDF No. 00-035-0772), and the relative intensity of the characteristic peak reflects well the relative content of the Fe3C. The 2θ = 82.34° is assigned to the (2 1 1) reflection for the metallic Fe phase, and the intensity can qualitatively distinguish the relative content of Fe. From Fig. S1(b)–S1(d),† the XRD patterns show that with the increase of reaction time with CH4, the intensity of the characteristic peak for the Fe3C phase increases, whereas a relative lower intensity of Fe3C exists after the sample is furnace cooled or if the cooling condition is heat preservation for 2 h and subsequent fast cooling. In addition, based on the intensity of characteristic peak at 2θ = 82.34°, the relative content of metallic Fe of the samples from furnace cooling or heat preservation at 1023 K for 2 h plus fast cooling, is higher than that from the fast cooling. According to the intensity of the characteristic peaks for Fe3C in different samples, it was preliminarily deduced that the optimal cooling conditions for the Fe3C preparation from high phosphorus oolitic hematite should be the fast cooling. Meanwhile, the holding the temperature for 2 h plus the fast cooling could not improve the utilization of deposited carbon from the CH4 decomposition, but will lead to the self-decomposition of Fe3C instead.
XRD analysis of samples from E-1 to F-3 is shown in Fig. S1(e) (ESI†). The difference of relative intensity of the characteristic peak for Fe3C caused by the different cooling conditions in E-1 and E-3 or F-1 and F-3 was lower than that of B-1 to D-3, but the relative intensity of the Fe3C peak was still decreased if the holding time at 1023 K was extended for 2 h. Additionally, compared with the relative intensity of the characteristic peak for graphite in B-1 to D-3, the graphite peak was relatively wide and the intensity was high. It can be deduced that with the extension of the reaction time with CH4, the deposited carbon on the surface of the newly reduced metallic was increased, the self-decomposition of Fe3C was accelerated, and a relatively fine grained graphite with a low degree of crystallization was also easily generated.
Quantitative Mössbauer analysis
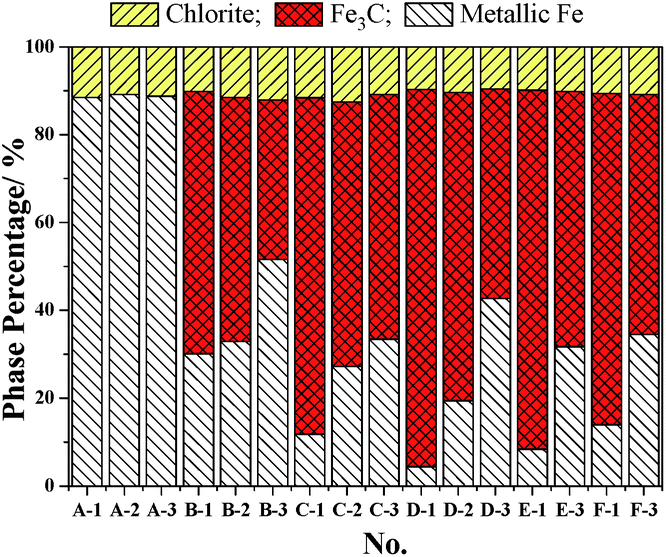
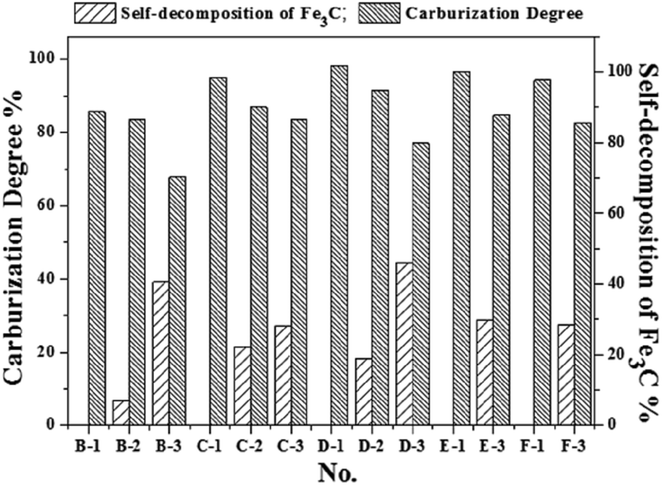
The Mössbauer analysis is an accurate detection method for the distinction of different Fe phases, especially with the relatively low content iron-bearing phases. Fig. 3 shows the results of the quantitative analysis of the content of Fe-containing phases under different cooling conditions using Mössbauer spectroscopy. The Fe-containing phases include metallic Fe, Fe3C and chlorite. The results in Fig. 3 reflect that the Fe3C cannot be formed in the initial 5 min of the reaction with CH4, which is consistent with the results from the XRD analysis shown in Fig. S1(a) (ESI†). Based on the results in Fig. 3, the Mössbauer analysis of the samples from A-1 to D-3, shows that when the cooling condition is fast cooling, the content of Fe3C increases with the increase of the reaction time with CH4. But the content of Fe3C in the E-1 and F-1 samples shows a small decline, which suggests that the extension of the CH4 reaction time from 30 min to 1 h and 2 h is not of benefit for the increase of the content of Fe3C. This phenomenon is because the formation rate of Fe3C is relatively slow when the CH4 reaction time is too long, and the Fe3C is a type of metastable interstitial compound. Chlorite also contains a small amount of iron, but the chlorite is chemically stable, so that the iron does not participate in the Fe3C formation. The effect of the cooling conditions and the reaction time with CH4 on the degree of carburization and the degree of decomposition of Fe3C can be deduced from Fig. 3 and 4. Fig. 4 shows that the degree of carburization during the first 10 min of the reaction with CH4 is up to 85.61%, which indicates that the carburization reaction is rapid after the reduction of FeO to metallic Fe completed. When the reaction time with CH4 was increased to 15 min with subsequent fast cooling, the degree of carburization was 95.12%. The degree of carburization was 98.32% after 30 min reaction with CH4 and then fast cooling. Compared with the fast cooling, the degree of self-decomposition of the samples obtained from the 10 min and 30 min reaction with CH4 and the subsequent heat preservation at 1023 K for 2 h plus the fast cooling was 39.20% and 44.47%, respectively. The degree of self-decomposition of the other samples from the other reaction times with CH4 and the subsequent heat preservation was approximately 28%.
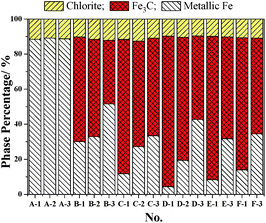 |
| | Fig. 3 The relationship between cooling conditions and the content of iron-bearing phases. | |
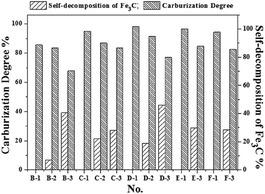 |
| | Fig. 4 The relationship between cooling conditions and the content of iron-bearing phases. | |
The results of the Mössbauer analysis confirmed that fast cooling is the optimal cooling condition for the preparation of Fe3C, and that the heat preservation at 1023 K for 2 h plus the fast cooling will promote the decomposition of Fe3C. At 1023 K, the product is a mixture of ferrite iron and cementite (Fe3C), and the heat preservation promotes the conversion of cementite to ferrite, resulting in the self-decomposition of Fe3C: Fe3C (s) = 3Fe (s) + C (s). Thus, the content of Fe3C decreases. Thus, the optimum conditions for the preparation of Fe3C from high phosphorus oolitic hematite are a temperature of 1023 K, reaction time of 15 min with CH4 and then fast cooling to room temperature. Under these conditions, the corresponding degree of carburization is 95.12%, and the atom ratio of carbon to iron is 0.51.
The characterization and magnetic properties of the iron carbide
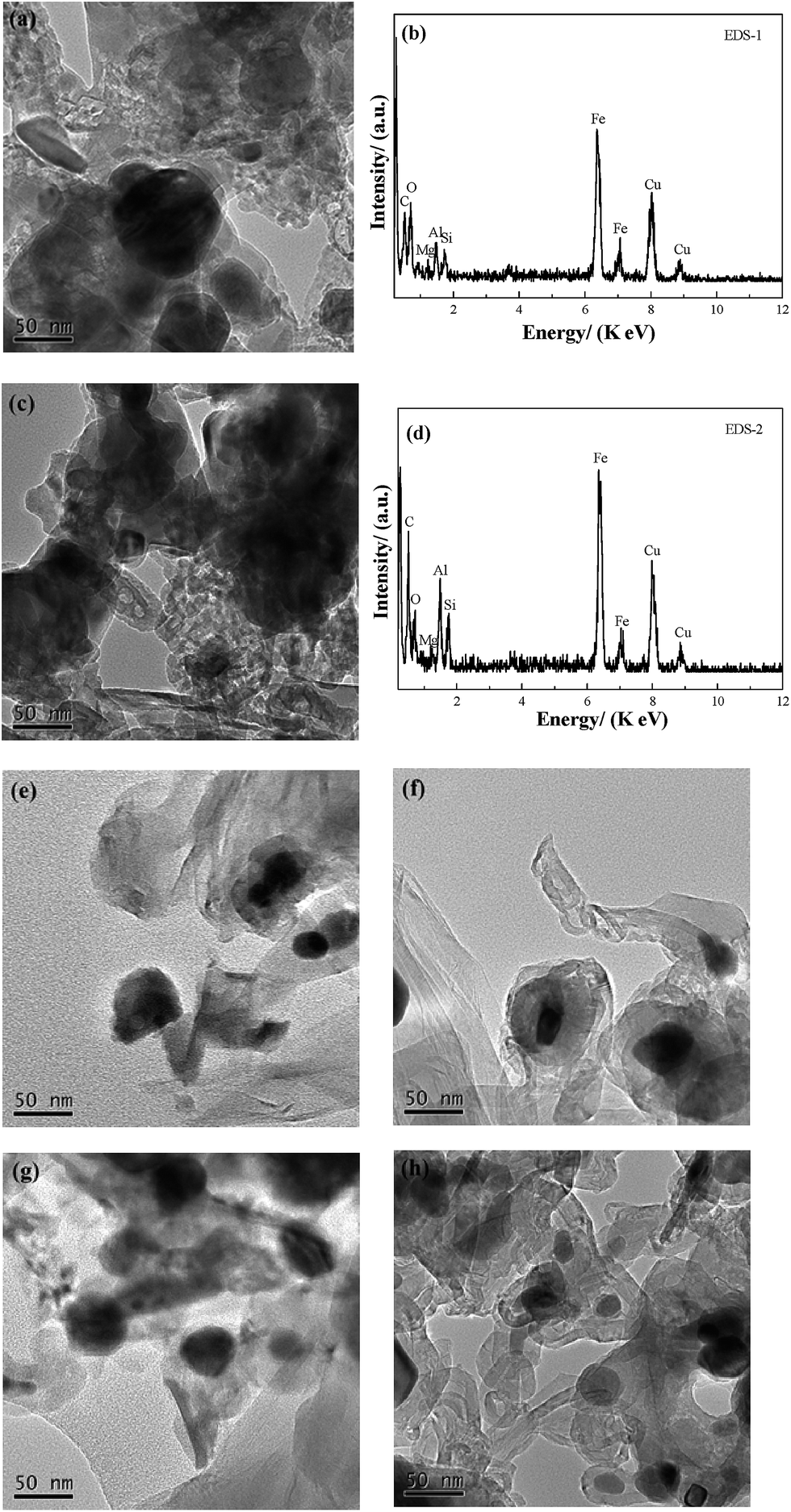
TEM analysis. Fig. 5(a) and (c) show the TEM images of specimens C-1 and C-3. Based on the dark area, which mainly consists of Fe3C, the relative content of Fe3C in C-1 is higher than that in C-3. The light area is mainly graphite. The number of multi-walled carbon nanotubes (MWCNTs) from CH4 decomposition in C-3 is obviously more than that in C-1, which is consistent with the results of the XRD analysis. However, the ring, and chain shaped MWCNTs in C-3 are different to that found by Helminiak et al.,27 because they used nanocrystal iron as the catalyst [aluminium oxide (Al2O3), silicon dioxide (SiO2) calcium oxide (CaO) and other oxides as the precursors] for the CH4 decomposition. In this study, the microstructure of the fine dissemination of gangue minerals and hematite in high phosphorus oolitic hematite was not damaged during the H2 reduction, and the Fe was mostly embedded in the ooide. Although the reduced metallic Fe was similar to the nano-iron grains, the iron was not evenly distributed. The non-uniform growth of nano iron grains contributed to the collisions of each CNT, and restricted the growth of CNTs deposited on the surface of metallic Fe, which caused the bending, and the rolling of the MWCNTs, and formed a chain or a ring shape during the aggregation of the nanotubes. From Fig. 5(b) and (d), the element in C-1 and C-3 samples is basically the same – they are both composed of Fe, C, silicon (Si), aluminium (Al) and oxygen (O), whereas the copper (Cu) element found comes from the standard Cu grid used in the TEM determination. Results from energy dispersive X-ray spectroscopy (EDS) analysis indicated that the Fe3C and graphite are the main phases in the C-1 and C-3 samples, and the Si, Al and O were the element sources of the gangue minerals in the high phosphorus oolitic hematite. The coexistence of the elements of Fe and C with Si, Al also revealed that the phase of Fe3C is still embedded closely with the gangue minerals, which explains why it is difficult to dephosphorize the Fe3C products using magnetic separation, which has been reported before by Wang et al.28 The morphology of the graphite of C-3 grows much better than that of C-1, and the nano carbon fibers are much clearer in C-3 than C-1. These results are in agreement with those from Helminiak et al.,27 Kameya et al.,29 Narkiewicz et al.,30 and Luo et al.,31 and they revealed that the carbon from the decomposition of Fe3C always presented as nano carbon fibers. Fig. 5(e) to 6(h) show the morphology of the samples that reacted with CH4 for 60 min and 120 min, respectively, under different cooling conditions (E-1 to F-3 samples). Comparing the TEM images of E-1, F-1 with C-1, the groups of E and F show a higher degree of graphitization, and the crystallinity of the samples is better. Based on the TEM images of E-3 and F-3, the product samples which were treated with heat preservation for 2 h plus the fast cooling, could form the MWCNTs and also some nanotubes coated around the Fe3C particles and the MWCNTs are onion-like. Because of self-decomposition of Fe3C, the newly generated carbon fibers were fine with a relatively low degree of crystallization and have many defects.
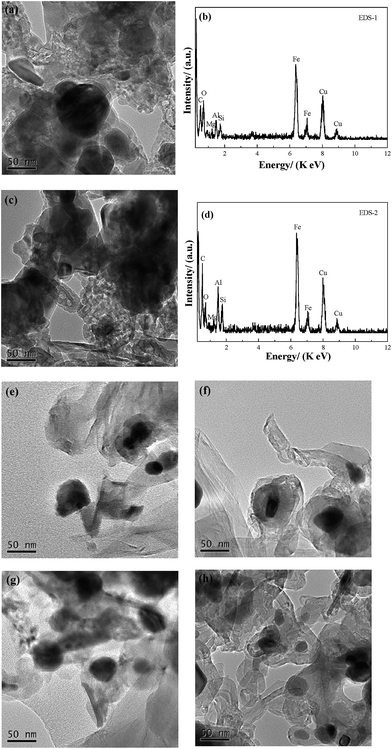 |
| | Fig. 5 Results of TEM and EDS analysis of the samples from C-1 to F-3; (a) and (c) are the TEM images of C-1 and C-3; (b) and (d) are the corresponding EDS analysis of (a) and (c); (e) to (h) are the samples of E-1, E-3, F-1 and F-3, respectively. | |
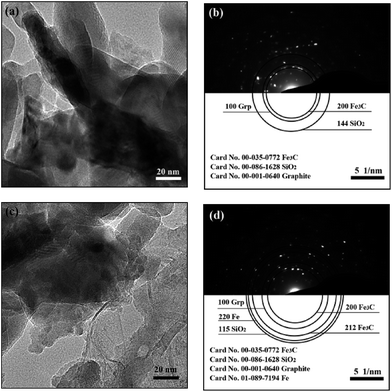 |
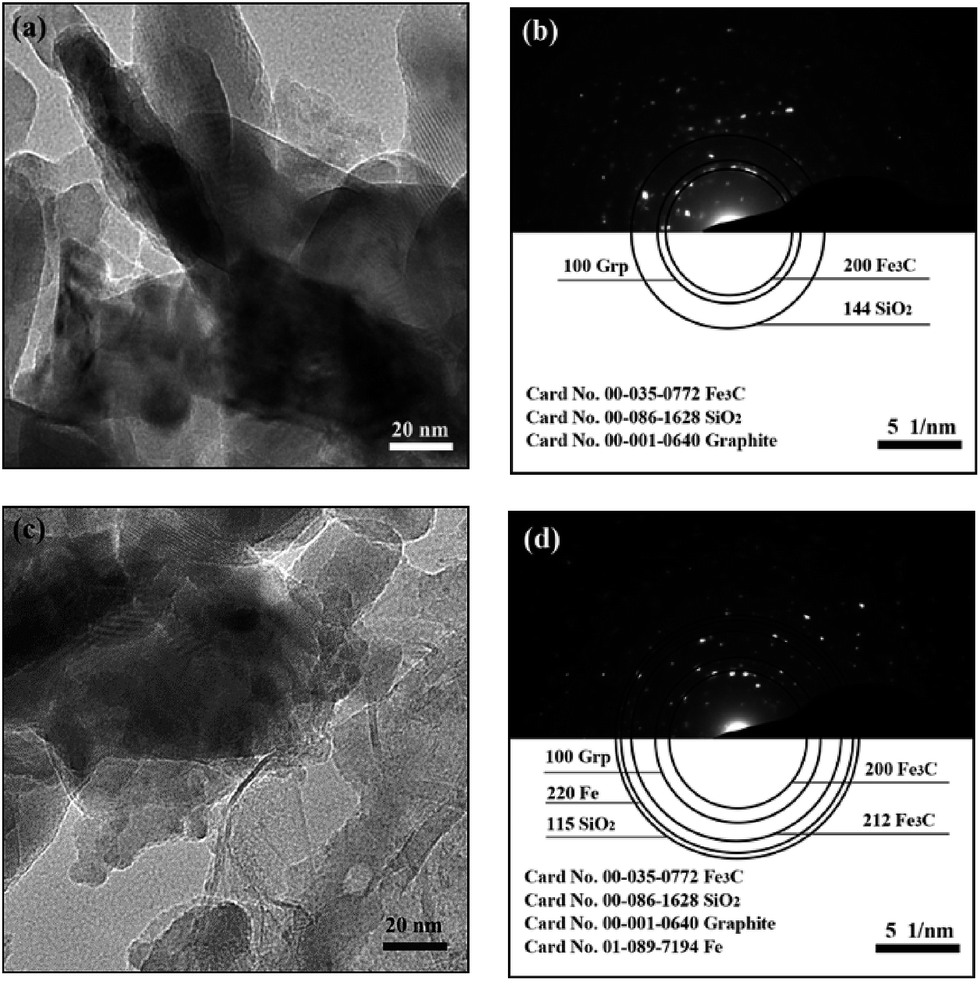
| | Fig. 6 The TEM images and the corresponding selected-area polycrystalline electron diffraction of C-1 (a, b) and C-3 (c, d) specimens. | |
Fig. 6(a) and (c) represent the TEM images of the product samples that were cooled using fast cooling and heat preservation for 2 h plus the fast cooling, respectively. The image for C-3 shows that the boundary in the samples which underwent fast cooling is more ambiguous, and the number of the carbon fibers is more than that in C-1. Fig. 6(b) and (d) are the corresponding selected-area polycrystalline electron diffraction (SAPED) images corresponding to Fig. 6(a) and (c). The Fe3C [(200) reflection, orthorhombic Fe3C, JCPDS No. 00-035-0772], SiO2 [(144) plane, hexagonal SiO2, JCPDS No. 00-086-1628] and graphite [(100) reflection, hexagonal C, JCPDS No. 00-001-0640] are the main phases in the C-1 sample. While the Fe3C [(200) and (212), orthorhombic], SiO2 [(115) plane, hexagonal], graphite [(100) reflection, hexagonal] and metallic iron [(200) plane, cubic, JCPDS No. 01-089-7194] exist in the C-3 sample. The index results of the presence of metallic Fe in C-3 further reveal that the self-decomposition of Fe 3C is caused by the heat preservation, which is in good agreement with the previously mentioned XRD and Mössbauer analysis of the corresponding samples. Meanwhile, the dark area in Fig. 6(a) is larger than that in Fig. 6(c), which can also reflect the self-decomposition of Fe3C by heat preservation.
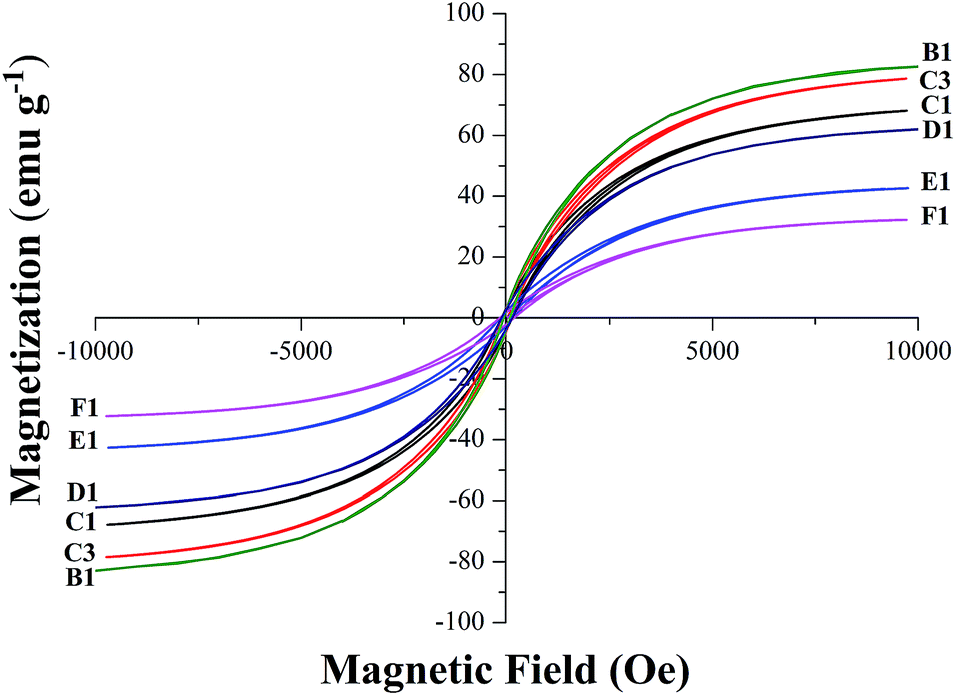
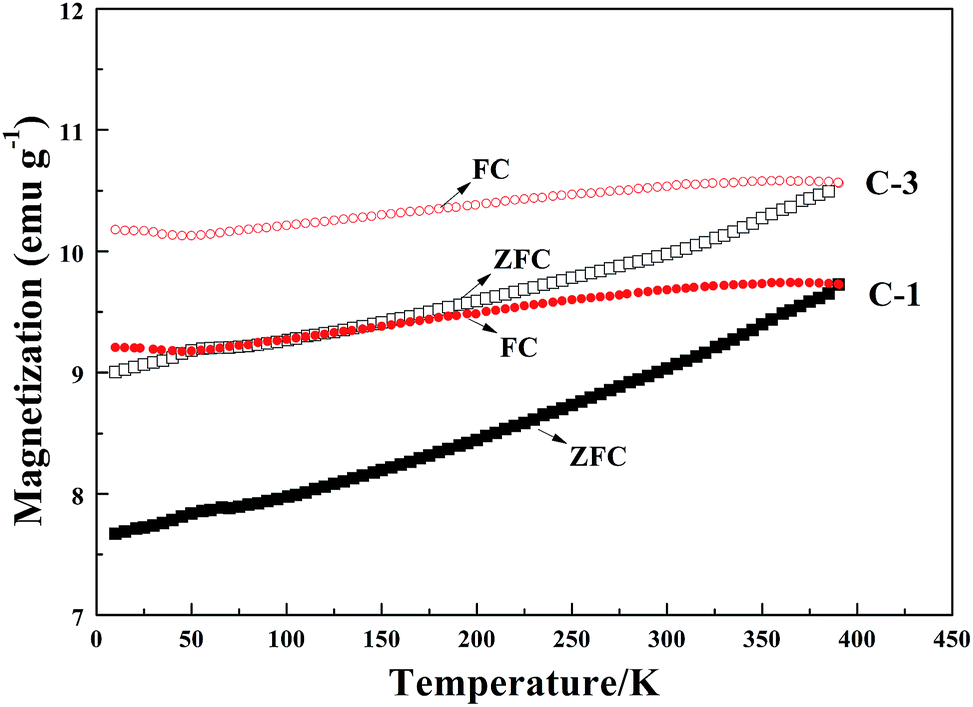
VSM magnetic analysis. The curves of the magnetization versus magnetic field and the magnetization versus temperature of the product samples under different cooling conditions are shown in Fig. 7 and 8, and the specific magnetic property is shown in Table 5. The results in Table 5 indicate that the saturation magnetization (Ms) decreases from 82.59 to 61.97 emu g−1 with the extension of the reaction time with CH4 from 10 min to 30 min (from B-1 to D-1). Compared with F-1, the Ms of F-3 increases from 68.08 to 78.64 emu g−1. The Ms of E-1 and F-1 is obviously less than that of B-1 and D-1, which may be attributed to the fact that much carbon was deposited from the CH4 decomposition wrapping the iron or the Fe3C particles (Fig. 5) when reacted with CH4 for too long. The coercive force (Hc) is approximately 20 Oe except for the result of 9.98 Oe obtained for B-1. The magnetic remanence (Mr) increases a little when the CH4 reaction lasts for more than 15 min, and the Mr is maintained at 0.2–0.3 emu g−1 if the reaction time with CH4 is longer than 30 min. If the reaction time with CH4 is too long, it contributes to the decrease of the magnetic property of the samples. The possible reason for this is that with the increase of reaction time with CH4, the carbon content of the sample will be increased, however, the Ms of metallic iron is higher than that of Fe3C. Fig. 8 represents the magnetization versus temperature of the different specimens (C-1 and C-3) and shows that the two branches: zero field cooling (ZFC) and field cooling (FC) of each curve, do not merge at any point below room temperature. The result proves that the specimens are ferromagnetic and not superparamagnetic, because superparamagnetism is a phenomenon in which hysteresis does not occur at temperatures in the range of room temperature and above.32 Magnetization curves (C-1) and (C-3) represent the Fe3C and the Fe3C with some metallic Fe, respectively. As the amount of the metallic Fe phase increases (summarized in Fig. 4), the Ms value of C-3 is larger than that of C-1, which is because of the self-decomposition of Fe3C by the different cooling conditions.
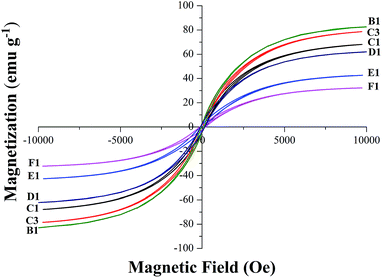 |
| | Fig. 7 The magnetic hysteresis loops recorded for the B1 to F1. | |
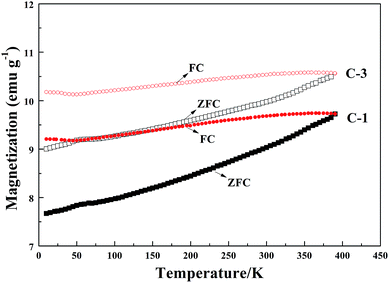 |
| | Fig. 8 The magnetization versus temperature of the specimens C-1 and C-3. | |
Table 5 The specific parameters of saturation magnetization (Ms), coercivity (Hc) and remanent magnetization (Mr)
| |
Ms (emu g−1) |
Hc (Oe) |
Mr (emu g−1) |
| B-1 |
82.59 |
9.98 |
0.39 |
| C-1 |
68.08 |
19.80 |
0.56 |
| C-3 |
78.64 |
20.08 |
0.72 |
| D-1 |
61.97 |
19.91 |
0.25 |
| E-1 |
42.43 |
19.89 |
0.29 |
| F-1 |
32.26 |
19.91 |
0.21 |
The magnetic property of the Fe3C prepared from high phosphorus oolitic hematite was also assessed in a previous study.24 The heat preservation for 2 h plus fast cooling caused the decline of the Fe3C content while the content of metallic iron increased, thus the Ms will increase, but the carbon from the self-decomposition of Fe3C will increase the degree of carbon coated on the surface of Fe3C, which will hinder the magnetic property of the sample. Table 5 shows that the increase of the content of metallic Fe is more beneficial than that of MWCNT coating on the surface of Fe3C. The magnetic property of Fe3C powder from the high phosphorus oolitic hematite is generally excellent.
Conclusions
(1) The optimal reaction conditions for the preparation of Fe3C from a high phosphorus oolitic hematite are a temperature of 1023 K, a CH4 reaction time of 15 min, and the cooling condition of fast cooling using Ar gas, and a degree of carburization of 95.12% can be obtained under these conditions.
(2) The heat preservation promotes the self-decomposition of Fe3C and the occurrence of nano carbon fibers. With the increase of the reaction time of CH4, the degree of graphitization increases, and the MWCNTs are in a ring, chain or net formation. The morphology is attributed to the fine dissemination of gangue minerals and the structure of hematite in the high phosphorus oolitic hematite was not damaged during the H2 reduction, which means that the Fe was mostly embedded in the ooide. The nonuniform growth of nano iron grains contributes to the collisions of each carbon nanotube, and restricts the growth of CNTs deposited on the surface of metallic Fe, resulting in the bending, and generates the chain or the net shape.
(3) With the increase of the reaction time with CH4, the saturation magnetization (Ms) of the Fe3C powder containing gangue minerals decreases. Under fast cooling, the saturation magnetization declines from 82.59 to 61.97 emu g−1 as the CH4 reaction time increases from 10 to 30 min. Whereas the addition of the heat preservation can promote the increase of the Ms. The results are because of the free carbon content increase with the increase of reaction time with CH4, and also the Ms of the metallic Fe is higher than that of Fe3C. The Fe3C prepared from high phosphorus oolitic hematite is a type of soft magnetic material with a relatively high magnetic property.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (No. 51374159) and the Open Foundation of State Key Laboratory of Advanced Metallurgy, University of Science and Technology Beijing (No. KF12-06).s
Notes and references
- D. Wei, Mod. Min., 2011, 5, 9 Search PubMed
 , in Chinese.
, in Chinese. - Y. B. Dong, M. Qiang, Z. Y. Duan, J. J. Xu and X. Wang, Metal. Mine, 2010, 404, 62 Search PubMed , in Chinese.
- Y. B. Long and Y. S. Zhang, Multipurp. Util. Miner. Resour., 2011, 1, 3 Search PubMed , in Chinese.
- H. Han, D. Duan, P. Yuan and S. Chen, Ironmaking Steelmaking, 2015, 42, 542 CrossRef CAS .
- G. F. Li, Y. X. Han, P. Gao and Y. S. Sun, Ironmaking Steelmaking, 2016, 43, 163 CrossRef CAS .
- Y. Sun, P. Gao, Y. Han and D. Ren, Ind. Eng. Chem. Res., 2013, 52, 2323 CrossRef CAS .
- Y. Li, T. Sun, J. Kou, Q. Guo and C. Xu, Miner. Process. Extr. Metall. Rev., 2014, 35, 66 CrossRef .
- Z. H. Li, Y. X. Han, Y. S. Sun and P. Gao, J. Northeast. Univ., 2015, 36, 1757 CAS , in Chinese.
- J. Wu, Z. J. Wen and M. J. Cen, Steel Res. Int., 2011, 82, 494 CrossRef CAS .
- A. N. Conejo and G. P. Martins, Ironmaking Steelmaking, 1999, 26, 111 CrossRef CAS .
- T. Akiyama, A. Miyazaki, H. Nakanishi, M. Hisa and A. Tsutsumi, Int. J. Hydrogen Energy, 2004, 29, 721 CrossRef CAS .
- A. N. Conejo and R. S. Estrada, Steel Res. Int., 2007, 78, 3 CrossRef CAS .
- T. Herranz, S. Rojas, F. J. Pérez-Alonso, M. Ojeda, P. Terreros and J. L. G. Fierro, J. Catal., 2006, 8199, 40199 Search PubMed .
- M. Bahgat, J. Mater. Sci. Technol., 2006, 22, 423 CAS .
- C. P. Deck and K. Vecchio, Carbon, 2006, 44, 267 CrossRef CAS .
- J. D. Atkinson, M. E. Fortunato, S. A. Dastgheib, M. Rostam-Abadi, M. J. Rood and K. S. Suslick, Carbon, 2011, 49, 587 CrossRef CAS .
- V. Amendola, P. Riello and M. Meneghetti, J. Phys. Chem. C, 2011, 115, 5140 CAS .
- A. K. Schaper, H. Q. Hou, A. Greiner and F. Phillipp, J. Catal., 2004, 222, 250 CrossRef CAS .
- G. Li, H. Ni, Q. Shen and F. Tsukihashi, ISIJ Int., 2006, 46, 981 CrossRef CAS .
- J. Zhang and O. Ostrovski, ISIJ Int., 2001, 41, 333 CrossRef CAS .
- H. W. Ni, F. S. Zhang, D. Q. Cang and J. P. Jiang, J. Iron Steel Res. Int., 2001, 13, 1 CAS , in Chinese.
- G. Q. Li, H. H. Wang, J. Yang and J. H. Ma, Adv. Mater. Res., 2014, 881–883, 98 Search PubMed .
- H. H. Wang, G. Q. Li, J. Yang, J. H. Ma and B. S. Khan, Metall. Mater. Trans. B, 2016, 47, 2571 CrossRef CAS .
- H. H. Wang, G. Q. Li, J. H. Ma and D. Zhao, RSC Adv., 2017, 7, 3921 RSC .
- D. Stull and H. Prophet, JANAF Thermochemical Tables, US Government Printing Office, Washington, DC, 2nd edn, 1971 Search PubMed .
- U. Narkiewicz, W. Arabczyk, W. Konicki and A. Pattek-Janczyk, J. Mater. Res., 2005, 20, 386 CrossRef CAS .
- A. Helminiak, E. Mijowska and W. Arabczyk, Mater. Sci., 2013, 31, 29 CAS .
- H. H. Wang, G. Q. Li, J. H. Ma, Y. X. Hu and D. Zhao, J. Wuhan Univ. Sci. Technol., 2017, 40, 11 Search PubMed , in Chinese.
- Y. Kameya and K. Hanamura, Carbon, 2012, 50, 3503 CrossRef CAS .
- U. Narkiewicz, W. Arabczyk, I. Pelech, N. Guskos, J. Typek, M. Maryniak, M. J. Woźniak, H. Matysiak and K. J. Kurzydłowski, Mater. Sci., 2006, 24, 1067 CAS .
- N. Luo, X. Li, X. Wang, H. Yan, C. Zhang and H. Wang, Carbon, 2010, 48, 3858 CrossRef CAS .
- D. L. Huber, Small, 2005, 1, 482 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra07886b |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
J. H. Mab and
D. Zhaoa
*ab,
J. H. Mab and
D. Zhaoa